
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-performance protonic ceramic fuel cell cathode using protophilic mixed ion and electron conducting material†
Dingyue
Hu
,
Junyoung
Kim
,
Hongjun
Niu
,
Luke M.
Daniels
 ,
Troy D.
Manning
,
Ruiyong
Chen
,
Bowen
Liu
,
Richard
Feetham
,
John B.
Claridge
and
Matthew J.
Rosseinsky
*
,
Troy D.
Manning
,
Ruiyong
Chen
,
Bowen
Liu
,
Richard
Feetham
,
John B.
Claridge
and
Matthew J.
Rosseinsky
*
Department of Chemistry, Materials Innovation Factory, University of Liverpool, 51 Oxford Street, Liverpool, L7 3NY, UK. E-mail: m.j.rosseinsky@liverpool.ac.uk
First published on 23rd December 2021
Abstract
Protonic ceramic fuel cells (PCFCs) are attractive energy conversion devices for intermediate-temperature operation (400–600 °C), however widespread application of PCFCs relies on the development of new high-performance electrode materials. Here we report the electrochemical and protonic properties of a self-assembled nanocomposite, Ba0.5Sr0.5(Co0.7Fe0.3)0.6875W0.3125O3−δ (BSCFW) consisting of a disordered single perovskite and an ordered double perovskite phase, as a PCFC cathode material. BSCFW shows thermodynamic and kinetic protonic behaviour conducive to PCFC application with favourable proton defect formation enthalpy (ΔH = −35 ± 7 kJ mol−1) comparable to existing proton conducting electrolyte materials. BSCFW presents an excellent polarization resistance (Rp) of 0.172(2) Ω cm2 at 600 °C and a high power density of 582(1) mW cm−2 through single-cell measurement, which is a comparable performance to current state-of-the-art cathode materials. BSCFW exhibits good chemical and thermal stability against BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb) electrolyte with a low Rp degradation rate of 1.0(1) × 10−6 Ω cm2 min−1. These performance and stability figures represent an advance beyond those of Ba0.5Sr0.5Co0.7Fe0.3O3−δ (BSCF), which is unstable under the same conditions and is incompatible with the electrolyte material. Our comprehensive characterization of the protonic properties of BSCFW, whose performance and stability are ensured via the interplay of the single and double perovskite phases, provides fundamental understanding that will inform the future design of high-performance PCFC cathodes.
Introduction
Amongst the various type of fuel cells, ceramic fuel cells have been extensively studied as eco-friendly alternative energy devices as a result of high energy conversion efficiency and continuous production of electricity without pollutant emissions.1 The high operating temperature (>800 °C) of conventional O2− conducting ceramic fuel cells (solid oxide fuel cells, SOFCs), however, leads to serious issues such as chemical inter-diffusion and high thermal stress.2–5 Protonic ceramic fuel cells (PCFCs) using proton-conducting oxides (PCOs) are promising candidates for intermediate to low temperature operation (400–600 °C) because of the relatively high ionic conductivity and low activation energy for H+ transport compared with O2− transport in conventional O2− conducting oxides under the same operating conditions.6The first report of high-temperature proton conductivity in oxide materials by Iwahara and co-workers in the 1980s garnered significant research interest for developing PCOs based on barium-zirconates and -cerates as electrolyte materials.7,8 Aliovalent-doped BaCe1−xZrxO3−δ (0 < x < 1) have demonstrated high proton conductivity overcoming the major issues such as poor sinterability of BaZrO3−δ and low proton conductivity of BaCeO3−δ under practical operating conditions.9–11 A barium zirconate-cerate co-doped with Y and Yb, BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb), is reported with high ionic conductivity (∼0.01 S cm−1 in a humidified O2 atmosphere), and excellent tolerance to sulfur poisoning and coking12 while BZCYYb with A-site barium deficiency showed chemical instability under both CO2 and H2S atmosphere.13 However, despite such performance from the electrolyte, the limiting factor that prevents the widespread application of PCFCs is the limited availability of cathode materials with high activity and/or stability.
Generally, mixed ionic (O2−) and electronic conductors (MIECs) have been used as the cathode material for conventional SOFC systems because of their excellent electronic conductivity and catalytic activity for the oxygen reduction reaction. However, for MIECs with poor proton conductivity, electrochemical activity is localized to the interface between cathode and electrolyte, as protons diffuse from the PCO electrolyte to the cathode material in order to generate electricity. Triple conducting oxides (TCOs), that allow simultaneous transport of H+, O2− and e−,14,15 with high chemical stability are required to effectively extend the electrochemically active sites to the entire surface of the cathode,16 and two different approaches have been used to increase the electrochemical performance of TCOs. The first approach is to dope various aliovalent elements (Co, Fe, Sc, Y) into pure proton-conducting oxides (i.e. BaZrO3−δ or BaCeO3−δ) to enhance their electrochemical conductivity of O2− and e−.17–19 Trivalent cations are doped into the B-site of perovskite PCOs to increase oxygen-ion kinetics, and the O'Hayre group reported that transition-metal cations (Co and Fe) doped into BaZrxY1−xO3−δ significantly improve its catalytic properties as a PCFC cathode by activating electronic conduction while maintaining existing ionic conductivity.20 The second approach is to target existing MIEC (O2−/e−) materials with protophilic properties.21 This approach has the advantage that the material is already well-studied and its reaction mechanisms of O2− and e− are understood. Several researchers have investigated the proton properties of typical MIEC perovskite-related materials, such as BaCoO3, which exhibit excellent electrochemical properties.22–24
Among those BaCoO3-based materials, Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) exhibits excellent electrochemical performance as a SOFC cathode.25 However, BSCF has been limited for use as a PCFC cathode because of its chemical instability under PCFC operating conditions (e.g., after hydration treatment)26,27 and degradation of the electrochemical performance below 650 °C in humidified air.28 We previously reported the self-assembled dynamic perovskite composite material, Ba0.5Sr0.5(Co0.7Fe0.3)0.6875W0.3125O3−δ (BSCFW), as an SOFC cathode.29 By incorporating W6+ into BSCF, BSCFW self-assembles into a composite where the interplay between a majority phase (∼71%) of stable oxygen-stoichiometric and electronically conducting double perovskite (DP), and a relatively minor phase (∼29%) of highly electro-catalytically active MIEC single perovskite (SP), ensures a combination of low polarization resistance (Rp) with chemical and microstructural stability via coherent, quasi–epitaxial interfaces between the phases. The weight ratio of DP and SP phase in BSCFW is ∼7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. This enhanced stability and high performance provided by this self-assembly identifies BSCFW as a candidate for PCFC cathode materials.
:3. This enhanced stability and high performance provided by this self-assembly identifies BSCFW as a candidate for PCFC cathode materials.
Here we report the high electrochemical performance of BSCFW as a PCFC cathode material with comprehensive characterization of protonic properties. The thermodynamic protonation enthalpy of BSCFW is evaluated by thermogravimetric analysis (TGA). The electrochemical performance of BSCFW as a PCFC cathode is examined through single-cell tests. Our findings highlight the excellent performance of the perovskite composite BSCFW and detailed characterization of the material provides insight and understanding towards the development of more efficient high-performance PCFC electrode materials.
Experimental
Synthesis of electrolyte and electrode powders
The solid–state reaction method was used to synthesize Ba0.5Sr0.5(Co0.7Fe0.3)0.6875W0.3125O3−δ (BSCFW). Stoichiometric amounts of BaCO3 (99.995%), SrCO3 (99.994%), Co3O4 (99.7%), Fe2O3 (99.99%), and WO3 (99.9%) were weighed and ball milled in 15 mL propan-2-ol for 14 h with ZrO2 balls (10 mm diameter) using a Fritsch Pulverisette 7 Planetary Mill (350 rpm). The milled powder was dried and calcined at 700 °C for 6 h and 900 °C for 8 h with heating and cooling rates of 5 °C min−1 in air. The calcined BSCFW powder was pressed into 10 mm diameter pellet and sintered at 1200 °C for 12 h in a zirconia crucible. The relative density of sintered BSCFW pellet is 94%. BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb) powder was synthesized by the solid-state reaction. Stoichiometric amounts of BaCO3 (99%, Sigma Aldrich), CeO2 (99.9%, Sigma Aldrich), ZrO2 (99%, Alfa Aesar), Y2O3 (99.99%, Alfa Aesar), and Yb2O3 (99.9%, Alfa Aesar) were mixed by the ball milling process, followed by sintering at 1200 °C for 12 h in air.Preparation of samples for SEM and XRD analysis
BSCFW-BZCYYb mixtures are sintered at 950 °C in air for 4 h, to investigate the chemical reactivity between the BSCFW and BZCYYb. The chemical reactivity of BSCFW and BZCYYb powders was checked by an X-ray diffractometer (Panalytical X'Pert Pro Bragg–Brentano geometry laboratory X-ray diffractometer with Co Kα1 radiation, λ = 1.78898 Å). Synchrotron X-ray diffraction data of powder samples were recorded on Beamline I11 with a wide-angle position sensitive detector (λ = 0.826596(10) Å) at Diamond Light Source with samples loaded in borosilicate capillaries. The microstructures and cross-section images of the NiO-BZCYYb|BZCYYb|BSCFW single cell were observed using scanning electron microscopy (Hitachi S4800).Fabrication of single-cell
The NiO-BZCYYb anode-supported cell was fabricated for fuel cell durability and performance. NiO-BZCYYb supported cells were fabricated by the drop-coating method to form the electrolyte layer. The NiO-BZCYYb hydrogen electrode was prepared by a mixture of NiO and BZCYYb (weight ratio of 6.5:3.5) after being ball-milled (5 mm balls, 100 rpm) for 24 h in ethanol. The BZCYYb suspension was applied to the NiO-BZCYYb support by drop-coating, followed by drying in air and co-sintering at 1400 °C for 4 h. The diameter of the single cell after sintering is 1.025 cm (shown in Fig. S1†). The calcined BSCFW powder was blended with a binder (Heraeus V006) at a weight ratio of 1:0.7 for air electrode slurries. Air electrode slurries were screen-printed onto the surface of the BZCYYb electrolyte and were finally sintered at 950 °C in air for 1 h.
Thermogravimetric analysis
The concentration of protonic defects was determined by thermogravimetric analysis (TGA). To remove all water from the structure, the powders (60–70 mg) were heated to 800 °C in dry air and held until thermal equilibrium was reached. The weight changes of samples under dry (Fig. S4a†) and wet air (PH2O = 0.023 atm, Fig. S4b†) were collected every 100 °C from 800 °C to 400 °C with a 2 h stabilization time at each temperature. TGA data were logged using a Q500 device from TA. Measurements were carried out on samples in platinum crucibles under dry air and wet air saturated by bubbling a constant air with a flow rate of 60 mL min−1 through water at 25 °C. The difference of sample weight in dry and hydrated conditions at various temperatures is interpreted as the water uptake in BSCFW.Electrochemical performances
The preparation procedure of the symmetrical cells began with pressing 0.6 g BZCYYb into 10 mm diameter pellets that were sintered at 1400 °C for 14 h in air with a heating rate and cooling rate of 2 °C min−1 and 3 °C min−1, respectively. The cathode slurry was screen printed onto both sides of the BZCYYb electrolyte pellet with an Aremco Screen Printer (model 3230, from Aremco) and fired in air at 950 °C for 1 h. After firing, silver electrodes were adhered to both faces of the symmetrical cell and heated at 600 °C for 1 h with a heating rate 3 °C min−1. The symmetrical cell was held at 600 °C under the 3 vol% H2O-containing air with a flow rate of 20 mL min−1, collecting the impedance spectra every 30 min for a total time of 60 h. For single-cell tests, Ag wires were attached at the cathode and anode side using Ag paste as a current collector. The NiO-BZCYYb anode-supported single-cell was sealed fully onto one end of the alumina tube using a ceramic adhesive (Aremco, Ceramabond 552). Humidified H2 (3 vol% H2O-containing H2) was applied to the anode side as a fuel through a water bubbler with a flow rate of 200 mL min−1, while dry air was supplied as an oxidant with a flow rate of 100 mL min−1 to the cathode during the single cell test. The cell operational stability test was carried out at 600 °C using BZCY721 (BaZr0.7Ce0.2Y0.1O3−δ, Fuel Cell Materials Ltd) or BZCYYb with a constant 0.6 V potential applied for 8 h. Impedance spectra and I–V curves were acquired with Solartron 1260 frequency response analyzer in combination with a Solartron 1287 potentiostat. The resistance is determined by using the fitting tool in the software ZView v.3.2b (Scribner Associates).Results and discussion
The powder X-ray diffraction (XRD) patterns of phase pure cathode Ba0.5Sr0.5(Co0.7Fe0.3)0.6875W0.3125O3−δ (BSCFW) and electrolyte BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb) materials and their composite powders are presented in Fig. 1a. Compatibility between BSCFW and BZCYYb is investigated by heating the mixtures at 950 °C for 4 h. Fig. 1a shows there is no reaction between BSCFW and BZCYYb, indicating that BSCFW is chemically compatible with BZCYYb under these processing conditions. As shown in the fits against synchrotron XRD data (Fig. 1b, c and S2†), the lattice parameters of BSFCW do not significantly change between dry (aSP = 3.98593(5) Å, aDP = 8.01198(4) Å) and humidified air (3 vol%) at 300 °C (aSP = 3.98357(10) Å, aDP = 8.01115(7) Å). BSCFW, stabilized due to the self-assembly effect by incorporating W6+ doping, shows excellent chemical stability after exposure to 3 vol% H2O in air for 1 h at 300 °C, while BSCF powder treated under the same conditions decomposed, forming Co3O4 as a secondary phase (Fig. S3†). Moreover, the thermal expansion coefficient (TEC) difference between BSCFW (15 × 10−6 K−1)29 and BZCYYb (100–500 °C: 14.2 × 10−6 K−1, 600–700 °C: 5.5 × 10−6 K−1, indicating there is probably structure transformations or phase change in BZCYYb at ∼500–600 °C)30 is smaller (5.6% at 500 °C) than that between BSCF (approximately 20 × 10−6 K−1)31 and BZCYYb (40.8% at 500 °C), suggesting BSCFW exhibits superior mechanical compatibility with the electrolyte material compared to BSCF, which will minimize or avoid cell degradation, delamination or fracture.2,25,32,33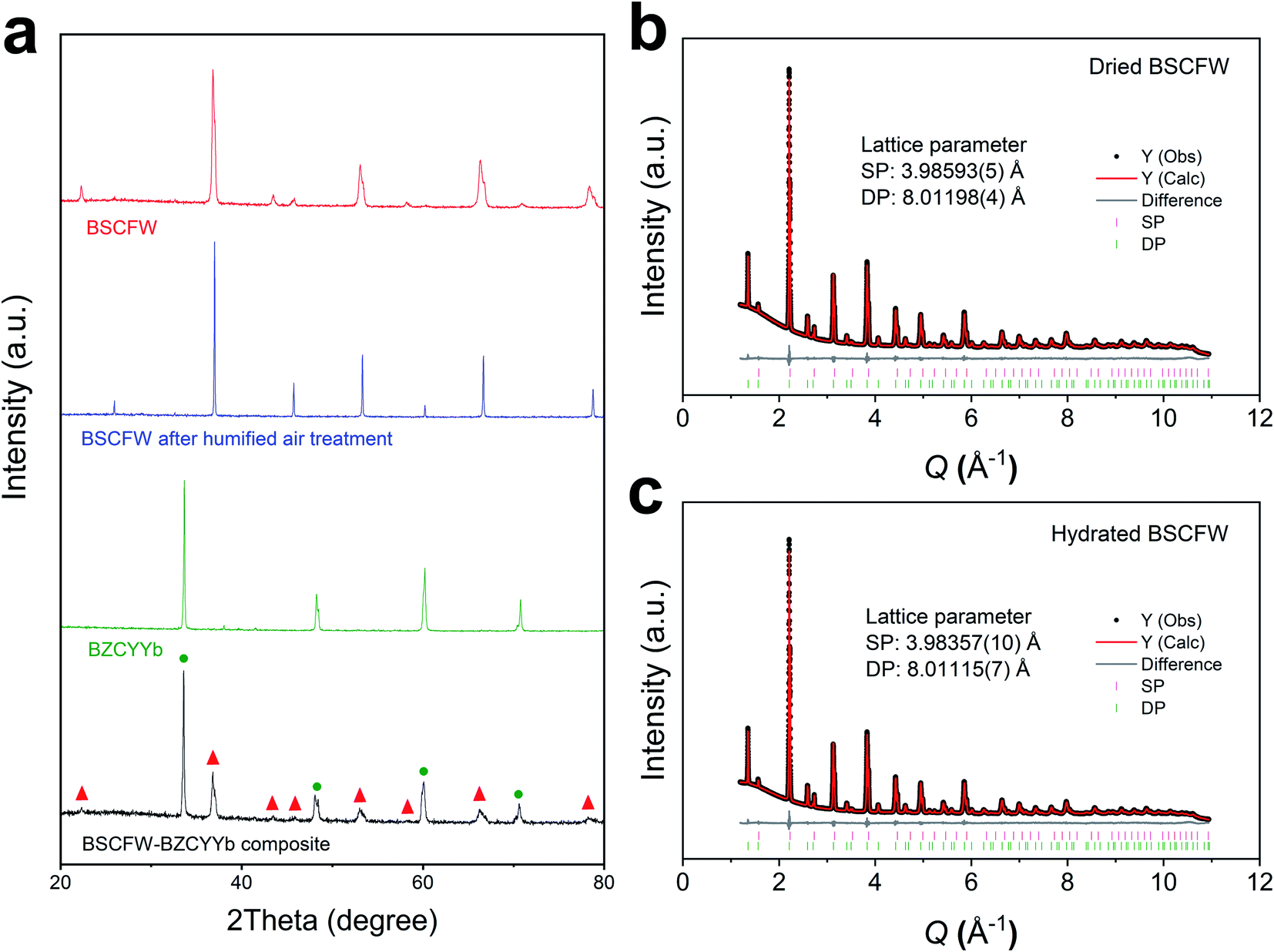 | ||
| Fig. 1 (a) XRD diffraction pattern of BSCFW sintered at 1200 °C for 12 h (red), BSCFW after humidified air exposure (flowing 3 vol% H2O containing air) for 1 h at 300 °C (blue), BZCYYb electrolyte sintered at 1400 °C for 4 h (green), and a mixture of BSCFW and BZCYYb sintered at 950 °C 4 h (black). Peaks corresponding to BSCFW and BZCYYb are labeled with red triangles and green circles, respectively. Observed (black points), calculated (red line), and difference (grey line) profiles from Pawley fits of synchrotron data (beamline I11, Diamond Light Source, λ = 0.826596(10) Å) for (b) dried and (c) hydrated BSCFW (heated to 300 °C for 1 h under flowing 3 vol% H2O air) at room temperature. Tick marks are shown for the SP (pink) and DP (green) phases in both. B-site cations occupancy and average cation radii for BSCFW are given in Table S1.† | ||
To determine the thermodynamic protonic properties of BSCFW, the extent of H2O uptake into BSCFW was measured by thermogravimetric analysis (TGA) in the temperature range of 400–800 °C. The mass of the material was recorded as a function of temperature under dry (PO2 = 0.19 atm, balanced N2) and under humidified synthetic air (PH2O = 0.023 atm). When exposed to humidified air (3 vol% H2O) under an isothermal condition (sample held for 2 h in 100 °C steps in the range 400–800 °C), the BSCFW sample absorbed water due to the hydration reaction as described in Kröger–Vink notation below (eqn (1), Fig. S4†).
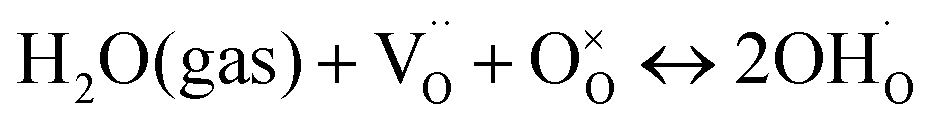 | (1) |
Thus, the equilibrium constant (KOH) for the hydration reaction can be expressed as,
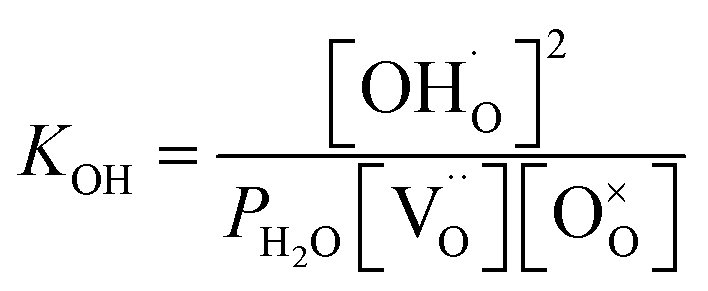 | (2) |
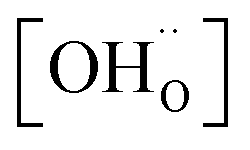 ,
, 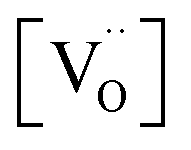 , and
, and 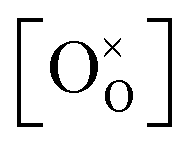 are, respectively, the proton, oxygen vacancy, and oxygen concentrations of the sample in the hydrated state. Following the previous study of BSCFW, the oxidation state of B-site cations Co, Fe and W are +2/+3, +3 and +6 respectively.29 Then, the BSCFW composition Ba0.5Sr0.5Co0.48125Fe0.20637W0.3125O3−δ is approximated as Ba0.5Sr0.5B0.79Fe0.21O3−δ (while keeping the total chemical stoichiometry of B-site unchanged, B = Co2+/3++W6+). Thus, the relevant reactions in BSCFW for vacancy creation and the water uptake reaction are given as
are, respectively, the proton, oxygen vacancy, and oxygen concentrations of the sample in the hydrated state. Following the previous study of BSCFW, the oxidation state of B-site cations Co, Fe and W are +2/+3, +3 and +6 respectively.29 Then, the BSCFW composition Ba0.5Sr0.5Co0.48125Fe0.20637W0.3125O3−δ is approximated as Ba0.5Sr0.5B0.79Fe0.21O3−δ (while keeping the total chemical stoichiometry of B-site unchanged, B = Co2+/3++W6+). Thus, the relevant reactions in BSCFW for vacancy creation and the water uptake reaction are given as | (3) |
From TGA data, only 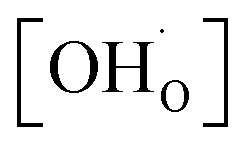 could be obtained while both
could be obtained while both 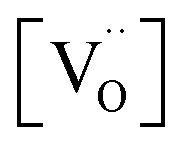 and
and 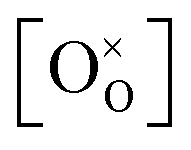 are unknown. Under oxidising conditions, the acceptor (Fe3+)-doped perovskites can be oxidized in the process of consuming oxygen vacancies and generating electron holes.
are unknown. Under oxidising conditions, the acceptor (Fe3+)-doped perovskites can be oxidized in the process of consuming oxygen vacancies and generating electron holes.
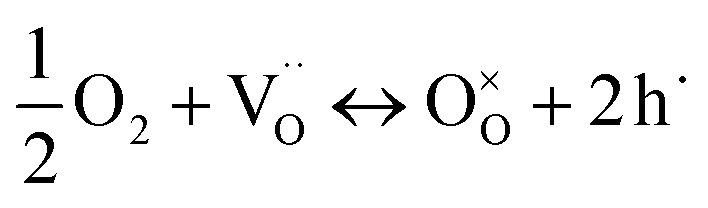 | (4) |
Considering the overall electro-neutrality, then based on eqn (1), (3) and (4) above, eqn (5) could be obtained:
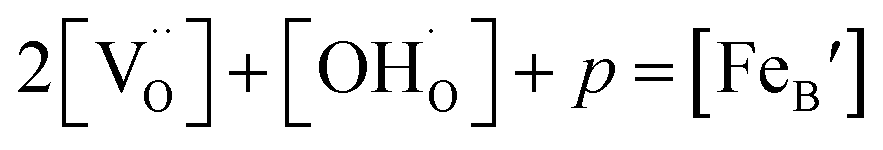 | (5) |
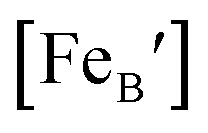 is the concentration of Fe on B-site
is the concentration of Fe on B-site 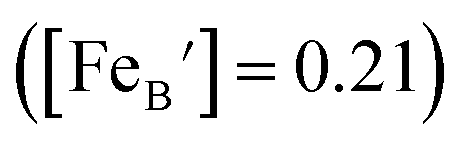 and p is the unknown hole concentration. Note there is also a constraint for the number of oxygen sites to sum to 3.
and p is the unknown hole concentration. Note there is also a constraint for the number of oxygen sites to sum to 3.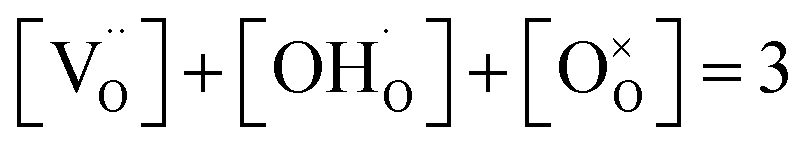 | (6) |
Using the above eqn (5) and (6), and as some studies reported, the hole concentration p is negligible.34,35 Therefore the unknown 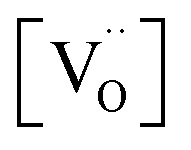 and
and 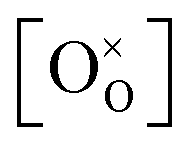 can be expressed as
can be expressed as 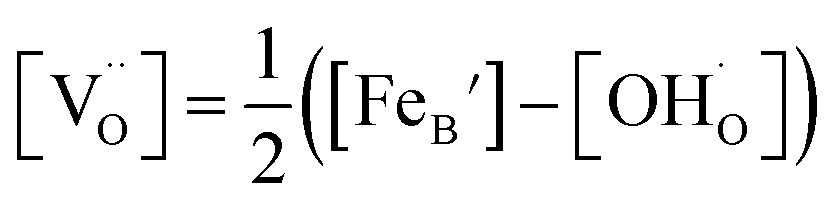 and
and 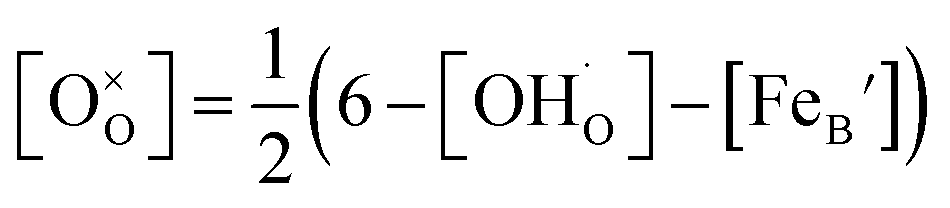 , respectively.
, respectively.
Then the eqn (2) now could be expressed as
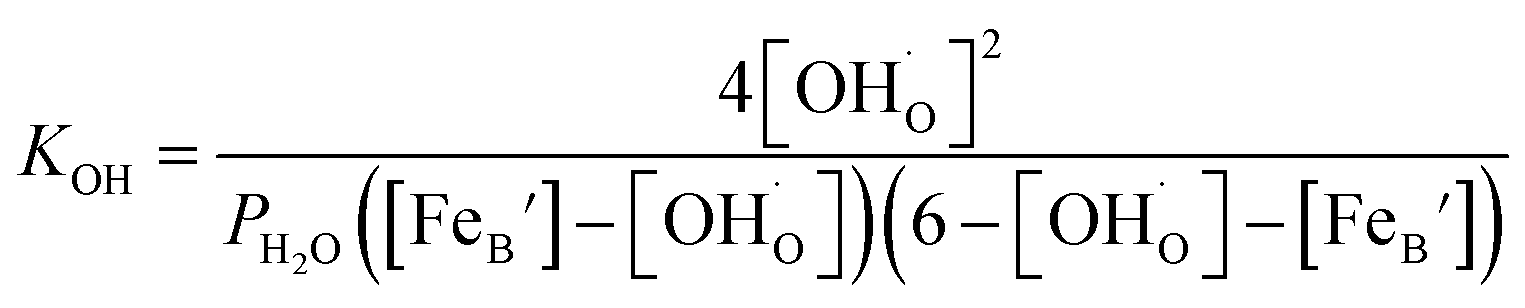 | (7) |
Therefore, the equilibrium constant (KOH) could be solved from known parameters. The proton uptake reaction is a thermally activated process, so the enthalpy of proton defect formation (ΔH) for BSCFW can be determined from the slope of the Arrhenius plot of equilibrium constants (KOH) at different temperatures (Fig. 2a) using the following Van't Hoff equation,35
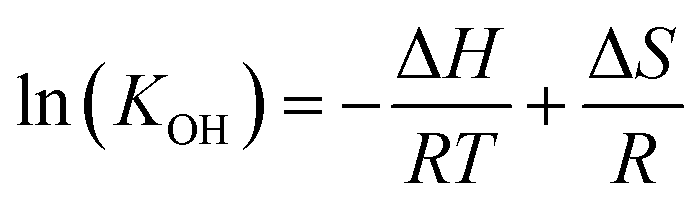 | (8) |
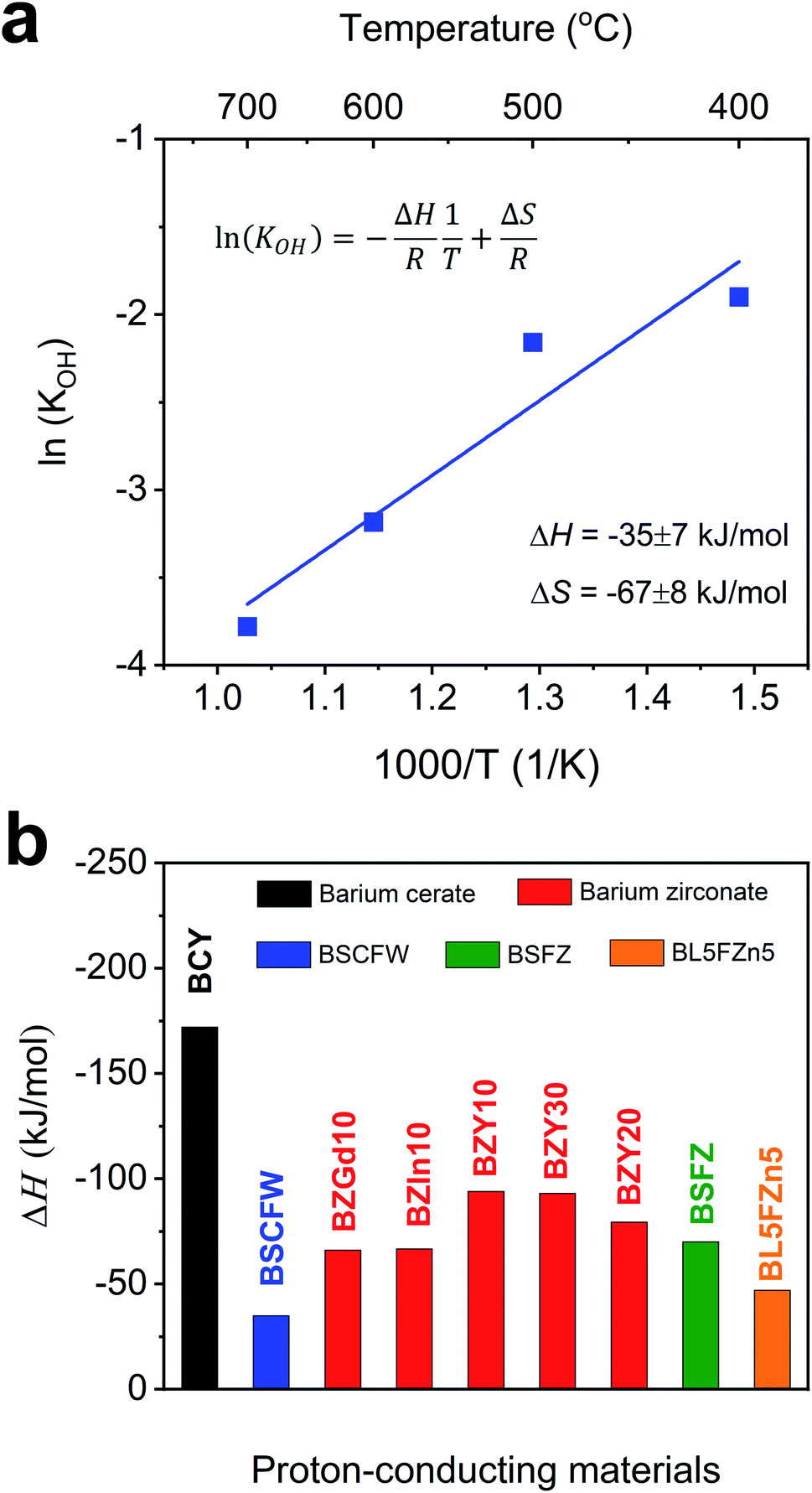 | ||
| Fig. 2 (a) Arrhenius plot showing the equilibrium constant of hydration reaction as a function of temperature for BSCFW. The slope yields the hydration enthalpy (ΔH) of −35 ± 7 kJ mol−1. (b) Comparison of ΔH values with representative state-of-the-art proton-conducting oxides for electrolyte: BaCe0.81Y0.19O3−δ (BCY),44 BaZr0.9Gd0.1O3−δ (BZGd10),35 BaZr0.9In0.1O3−δ (BZIn10),35 BaZr0.7Y0.3O3−δ (BZY30),41 BaZr0.8Y0.2O3−δ (BZY20),40 and BaZr0.9Y0.1O3−δ (BZY10)39 and proton-conducting oxides used as electrodes: Ba0.5Sr0.5Fe0.8Zn0.2O3−δ (BSFZ)16 and Ba0.95La0.05Fe0.95Zn0.05O3−δ (BL5FZn5).15 | ||
Fig. 3a shows impedance spectroscopy data measured from a BSCFW|BZCYYb|BSCFW symmetrical cell at t = 0 (which is when the atmosphere was switched from dry air to humidified 3% vol-H2O air) and at t = 30 min, highlighting a rapid response in cathodic performance of BSCFW to humidified air. The area specific resistance (ASR) as measured in symmetrical cells decreases dramatically to 0.572(6) Ω cm2 at t = 30 min before reaching 0.092 Ω cm2 after a break-in equilibration period of 205 min, as observed in Fig. 3b which shows the evolution of ASR with time for a BSCFW|BZCYYb|BSCFW symmetrical cell under 3 vol% H2O-containing air at 600 °C. After the break-in period, the ASR of the cell gradually increases from 0.092(1) Ω cm2 to 0.097(2) Ω cm2 over 60 h (Fig. 3c compares the corresponding impedance spectra at t = 205 and 3925 min) yielding an ASR degradation rate of 1.0(1) × 10−6 Ω cm2 min−1. For comparison, an equivalent experiment was attempted using BSCF but due to the large TEC mismatch between BSCF and BZCYYb, as described above, measurements were not possible because of delamination. Fig. S5† shows the symmetrical cells of BSFCW and BSCF after preparation, clearly demonstrating delamination of the BSCF coating.
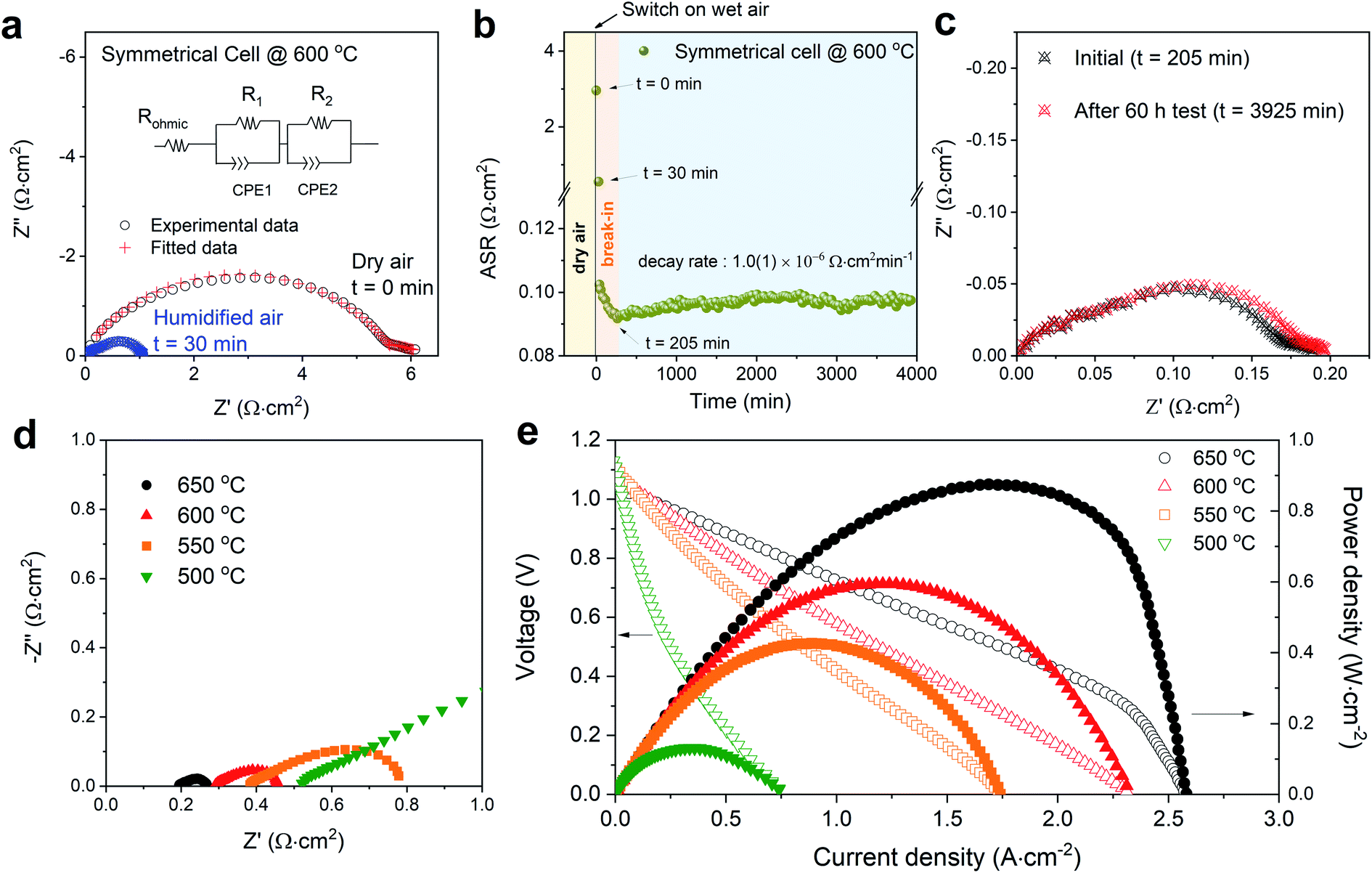 | ||
| Fig. 3 (a) Impedance spectra (real vs. imaginary impedance) of a BSCFW|BZCYYb|BSCFW symmetrical cell under dry (black data points) and humidified 3 vol% H2O-containing air (blue datapoints) conditions at 600 °C. The ohmic resistance (Rohmic) including the contribution from the instrument and electrolyte is subtracted (the original impedance data is shown in Fig. S8†). The ASR of the cathode material is determined by the total resistance which is the sum of R1 and R2 (shown in the equivalent circuit diagram) (b) ASR of the symmetrical cell in 3 vol% water-containing air showing a 205 min break-in period after which the ASR stabilised and increases with a decay rate of 1.0(1) × 10−6 Ω cm2 min−1. (c) Impedance spectra plot of the symmetrical cell in Fig. 3bt = 205 min (after break-in period) and after the 60 h test (t = 3925 min). (d) Impedance spectra of the single cell (BSCFW|BZCYYb|NiO-BZCYYb) measured under open-circuit condition, A reduced scale is displayed to show detail at higher temperature (full-scale plot is shown in Fig. S9†). (e) I–V curve (open symbols) and corresponding power density curves (filled symbols) of the BSCFW-BZCYYb single cell in the 500–650 °C temperature range. | ||
Next, a BSCFW|BZCYYb|NiO-BZCYYb single cell was assembled and tested. Fig. 3d and e present the electrochemical performance of a single cell with a configuration of BSCFW|BZCYYb|NiO-BZCYYb. Generally, the measured impedance spectra of a single cell are used to describe total resistances associated with components such as electrode and electrolyte. The intercept with the real axis at high frequencies represents the ohmic resistance which mainly originates from the electrolyte, whereas the difference between the high- and low-frequency intercepts with the real axis indicates the sum of the electrode polarization resistance particularly from the cathode. As expected from the favourable protonic kinetic and thermodynamic properties presented by BSCFW, a small Rp of 0.172(2) Ω cm2 and a maximum power density of 0.582(1) W cm−2 were obtained at 600 °C (Fig. 3d and e) from the single cell measurement. Fig. S6a† shows the cross-section scanning electron microscopy image of the cell after the single cell test. There is no evidence of delamination or change of microstructure of the BSCFW cathode (Fig. S6b and c†).
The operational stability of the cell using BSCFW as cathode material was also examined at 600 °C for 8 h with a constant voltage of 0.6 V applied to the cell. For the cell stability test, 3 vol% H2O-containing H2 with a flow rate of 200 mL min−1 was supplied to the NiO-BZCY721 anode while air with a flow rate of 100 mL min−1 was supplied to the BSCFW cathode. Two different electrolytes (BZCY721 and BZCYYb) were used to preliminarily examine their influence on the stability of cell. Note that the cell durability depends on not only the cathode–electrolyte compositional combinations, but also anode, working gas, temperature and the applied voltage.45–47 More detailed evaluation with longer tests is required. As shown in Fig. S7,† the power density of the cell using BZCY721 electrolyte decreased slightly (at the rate of 2.2(1) × 10−5 W cm−2 min−1), which is lower that reported for LSCF-BZCY cell (La0.6Sr0.4Co0.2Fe0.8O3−δ cathode – BZCY electrolyte, approximately 3.3 × 10−5 W cm−2 min−1, Table S2†). In addition, the power density decay rate of the cell using BSCFW cathode and BZCYYb electrolyte is 3.0(2) × 10−4 W cm−2 min−1 (Fig. S7†), which is larger than that using BZCY721 electrolyte since the high Ce:Zr ratio (7:1) of BZCYYb increases the instability of the cell.10,48
Fig. 4 presents a comparison of Rp for BSCFW against representative PCFC cathodes materials. According to Steele et al., the total Rp of cell components (electrolyte, anode, and cathode) should be below 0.50 Ω cm2 to ensure high power densities, with targets of 1 kW dm−3 and 1 kW kg−1.49 Assuming that 30% of the total cell Rp is attributed to the electrolyte (i.e., a typical value of 0.15 Ω cm2),50 an adequate performance is obtained from a PCFC if the electrode's Rp is less than 0.35 Ω cm2. Notably, the Rp measured from the BSCFW single cell gives a value of 0.172(2) Ω cm2, which is less than half the recommended value and meets the requirement for efficient operation of a PCFC. Also, BSCFW shows a power density of 0.426(1) W cm−2 even at 550 °C, which is higher than the required power density of 0.25 W cm−2 for efficient operation51 (Table 1 and Fig. 4). Such efficient performance in the intermediate-to-low temperatures regime (∼600 °C) enables cost-effective manufacturing by widening the choice of materials (e.g., metallic interconnects, non-ceramic seals) and eliminating major performance degradation.32
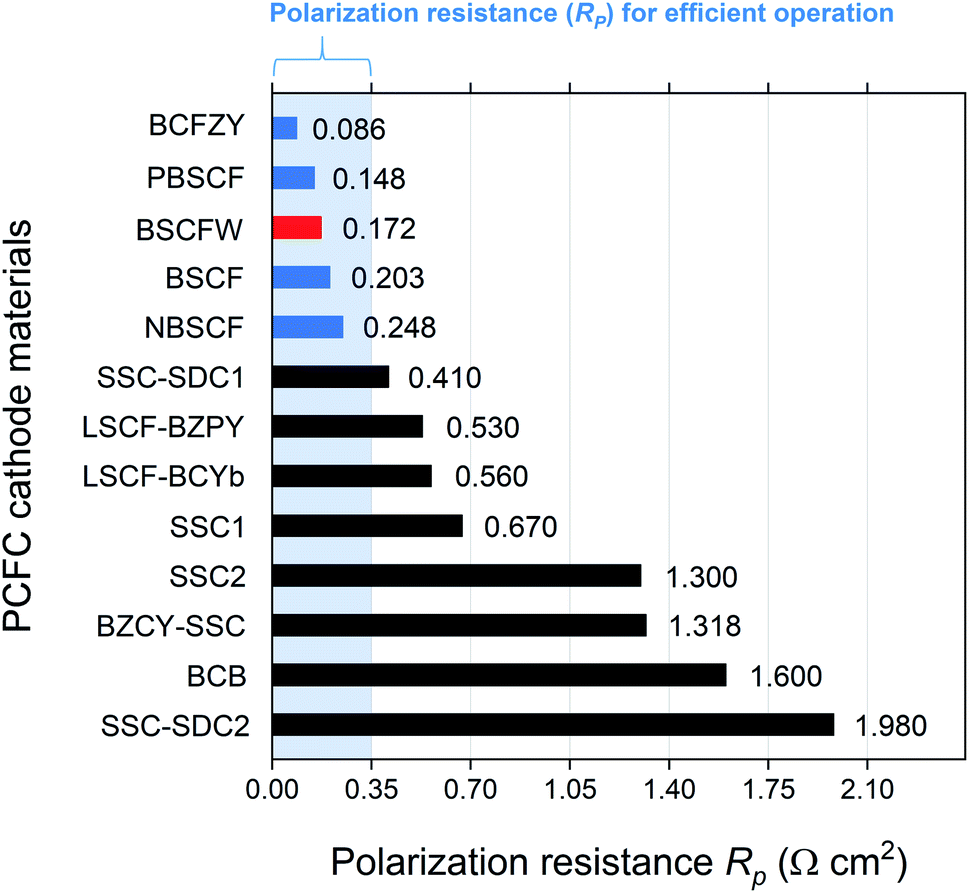 | ||
| Fig. 4 Comparison of Rp of BSCFW (red) with other representative PCFC cathodes at 600 °C. The values coloured in blue are the cathodes that have a value of lower than 0.35 Ω cm2 which allows efficient operation at a given temperature. *(BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY),20 PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF),52 NdBa0.5Sr0.5Co1.5Fe0.5O5+δ (NBSCF),53 Sm0.5Sr0.5CoO3−δ–Ce0.8Sm0.2O2−δ on BaZr0.7Sr0.1Y0.2O3−δ electrolyte (SSC–SDC1),54 La0.6Sr0.4Fe0.8Co0.2O3−δ–BaZr0.7Pr0.1Y0.2O3−δ (LSCF–BZPY),55 La0.6Sr0.4Fe0.8Co0.2O3−δ–BaCe0.9Yb0.1O3−δ (LSCF–BCYb),22 Sm0.5Sr0.5CoO3−δ on BaCe0.8Sm0.2O3−δ electrolyte (SSC1),56 Sm0.5Sr0.5CoO3−δ on BaZr0.8Y0.2O3−δ electrolyte (SSC2),57 BaZr0.1Ce0.7Y0.2O3−δ–Sm0.5Sr0.5CoO3−δ (BZCY–SSC),58 BaCe0.5Bi0.5O3−δ (BCB),59 Sm0.5Sr0.5CoO3−δ–Ce0.8Sm0.2O2−δ on BaZr0.1Ce0.7Y0.2O3−δ electrolyte (SSC–SDC2)60). | ||
| Cathode | Electrolyte (thickness) | Anode | Ohmic resistance (Ω cm2) | R p (Ω cm2) | Maximum power density (W cm−2) | Ref. |
|---|---|---|---|---|---|---|
| a (BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY), BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb), PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF), BaZr0.4Ce0.4Y0.1Yb0.1O3−δ (BZCYYb4411), BaZr0.1Ce0.7Y0.2O3−δ (BZCY), NdBa0.5Sr0.5Co1.5Fe0.5O5+δ (NBSCF), Sm0.5Sr0.5CoO3−δ (SSC), Ce0.8Sm0.2O2−δ (SDC), BaZr0.7Sr0.1Y0.2O3−δ (BZSY), La0.6Sr0.4Fe0.8Co0.2O3−δ (LSCF), BaZr0.7Pr0.1Y0.2O3−δ (BZPY), BaZr0.8Y0.2O3−δ (BZY), BaCe0.9Yb0.1O3−δ (BCYb), BaCe0.8Sm0.2O3−δ (BCS), BaCe0.5Bi0.5O3−δ (BCB)). | ||||||
| BCFZY | BZCYYb (20–30 μm) | Ni-BZCYYb | 0.295 | 0.086 | 0.65 | 20 |
| PBSCF | BZCYYb4411 (15 μm) | Ni-BZCYYb4411 | 0.178 | 0.148 | 0.873 | 52 |
| BSCFW | BZCYYb (15 μm) | Ni-BZCYYb | 0.272(3) | 0.172(2) | 0.582(1) | This work |
| BSCF | BZCY (25–40 μm) | Ni-BZCY | 1.226 | 0.203 | 0.544 | 23 |
| NBSCF | BZCYYb (15 μm) | Ni-BZCYYb | 0.128 | 0.248 | 0.7 | 53 |
| SSC-SDC | BZSY (12 μm) | Ni-BZSY | 0.460 | 0.410 | 0.29 | 54 |
| LSCF-BZPY | BZPY (12 μm) | Ni-BZY | 0.530 | 0.530 | 0.163 | 55 |
| LSCF-BCYb | BZY (20 μm) | Ni-BZY | 1.850 | 0.560 | 0.112 | 22 |
| SSC | BCS (70 μm) | Ni-BCS | 0.100 | 0.670 | 0.42 | 56 |
| SSC | BZY (20 μm) | Ni-BZCY | 1.400 | 1.300 | 0.07 | 57 |
| BZCY-SSC | BZCY (20 μm) | Ni-BZCY | 0.357 | 1.318 | 0.24 | 58 |
| BCB | BZCY (25 μm) | Ni-BZCY | 0.670 | 1.600 | 0.125 | 59 |
| SSC-SDC | BZY (25 μm) | Ni-BZCY | 3.240 | 1.980 | 0.055 | 60 |
Conclusions
BSCFW, a self-assembled dynamic perovskite composite cathode, shows excellent chemical stability in contact with humidified air and the solid electrolyte (BZCYYb) at high temperatures, and electrochemical stability under PCFC operating conditions with a symmetrical cell showing a Rp degradation rate of 1.0(1) × 10−6 Ω cm2 min−1 over more than 60 h at 600 °C. A single-cell test showed operational stability over 8 h, with a power density decay rate of 2.2(1) × 10−5 W cm−2 min−1. These initial stability tests give decay rates less than those found for LSCF electrodes in equivalent single cell configurations and require more detailed evaluation with longer tests. This performance is a significant improvement over BSCF, which was not stable under the same conditions and its TEC mismatch with BZCYYb prevented symmetrical cell testing. Thermodynamically, BSCFW has a favourable enthalpy value (ΔH) of proton defect formation (−35 ± 7 kJ mol−1) comparable to leading proton-conducting oxides. Such favourable protonic properties highlight the potential of BSCFW as a PCFC cathode material with high activity for operation at low temperatures (e.g. 550 °C). Our identification of the self-assembled nanocomposite BSCFW as a high-performance PCFC cathode material, and comprehensive characterisation of thermodynamic behaviour provides fundamental scientific insight for the rational design of more efficient electrode materials for high-performance PCFCs.Data availability
Underlying data is available at http://datacat.liverpool.ac.uk/id/eprint/1409.Author contributions
JK and DH designed and conducted the material synthesis, characterisation and electrochemical measurements and prepared the first draft of the manuscript. HN assisted with construction and operation of single-cell measurements. RF and BL assisted with TGA measurements. LD, TM, RC participated in the design of the experiment and provided assistance with data analysis and revision of the manuscript. All authors contributed to the discussion of the results and approved the final version of the manuscript. MJR directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the EPSRC Programme Grant EP/N004884/1. DH and BL thank the China Scholarship Council (CSC) for studentship funding (201706280042 and 2018064500).References
- S. Sengodan, S. Choi, A. Jun, T. H. Shin, Y.-W. Ju, H. Y. Jeong, J. Shin, J. T. Irvine and G. Kim, Nat. Mater., 2015, 14, 205–209 CrossRef CAS PubMed.
- Y. Zhang, B. Chen, D. Guan, M. Xu, R. Ran, M. Ni, W. Zhou, R. O'Hayre and Z. Shao, Nature, 2021, 591, 246–251 CrossRef CAS PubMed.
- J. S. Ahn, S. Omar, H. Yoon, J. C. Nino and E. D. Wachsman, J. Power Sources, 2010, 195, 2131–2135 CrossRef CAS.
- F. Baumann, J. Maier and J. Fleig, Solid State Ionics, 2008, 179, 1198–1204 CrossRef CAS.
- E. Bucher, A. Egger, G. Caraman and W. Sitte, J. Electrochem. Soc., 2008, 155, B1218 CrossRef CAS.
- K. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- H. Iwahara, T. Esaka, H. Uchida and N. Maeda, Solid State Ionics, 1981, 3, 359–363 CrossRef.
- H. Iwahara, Solid State Ionics, 1996, 86, 9–15 CrossRef.
- C. Zhang and H. Zhao, Solid State Ionics, 2012, 206, 17–21 CrossRef CAS.
- P. Sawant, S. Varma, B. Wani and S. Bharadwaj, Int. J. Hydrogen Energy, 2012, 37, 3848–3856 CrossRef CAS.
- K. Xie, R. Yan, X. Chen, D. Dong, S. Wang, X. Liu and G. Meng, J. Alloys Compd., 2009, 472, 551–555 CrossRef CAS.
- L. Yang, S. Wang, K. Blinn, M. Liu, Z. Liu, Z. Cheng and M. Liu, Science, 2009, 326, 126–129 CrossRef CAS PubMed.
- A. VahidMohammadi and Z. Cheng, J. Electrochem. Soc., 2015, 162, F803–F811 CrossRef CAS.
- R. Zohourian, R. Merkle and J. Maier, Solid State Ionics, 2017, 299, 64–69 CrossRef CAS.
- R. Zohourian, R. Merkle, G. Raimondi and J. Maier, Adv. Funct. Mater., 2018, 28, 1801241 CrossRef.
- D. Poetzsch, R. Merkle and J. Maier, Faraday Discuss., 2015, 182, 129–143 RSC.
- H. Kawamori, I. Oikawa and H. Takamura, Chem. Mater., 2021, 33, 5935–5942 CrossRef CAS.
- Y. Meng, J. Gao, Z. Zhao, J. Amoroso, J. Tong and K. S. Brinkman, J. Mater. Sci., 2019, 54, 9291–9312 CrossRef CAS.
- N. L. R. M. Rashid, A. A. Samat, A. A. Jais, M. R. Somalu, A. Muchtar, N. A. Baharuddin and W. N. R. W. Isahak, Ceram. Int., 2019, 45, 6605–6615 CrossRef CAS.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O'Hayre, Science, 2015, 349, 1321–1326 CrossRef CAS PubMed.
- D. Poetzsch, R. Merkle and J. Maier, Phys. Chem. Chem. Phys., 2014, 16, 16446–16453 RSC.
- J. Kim, S. Sengodan, S. Kim, O. Kwon, Y. Bu and G. Kim, Renewable Sustainable Energy Rev., 2019, 109, 606–618 CrossRef CAS.
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355–4369 RSC.
- T. Norby, Solid State Ionics, 1999, 125, 1–11 CrossRef CAS.
- Z. Shao and S. Haile, Nature, 2004, 431, 170–173 CrossRef CAS PubMed.
- N. Bausá, C. Solís, R. Strandbakke and J. M. Serra, Solid State Ionics, 2017, 306, 62–68 CrossRef.
- A. Grimaud, F. Mauvy, J. Bassat, S. Fourcade, L. Rocheron, M. Marrony and J. Grenier, J. Electrochem. Soc., 2012, 159, B683 CrossRef CAS.
- S. Sun and Z. Cheng, J. Electrochem. Soc., 2016, 164, F81 CrossRef.
- J. F. Shin, W. Xu, M. Zanella, K. Dawson, S. N. Savvin, J. B. Claridge and M. J. Rosseinsky, Nat. Energy, 2017, 2, 16214 CrossRef CAS.
- X. Zhou, L. Liu, J. Zhen, S. Zhu, B. Li, K. Sun and P. Wang, J. Power Sources, 2011, 196, 5000–5006 CrossRef CAS.
- W. Zhou, R. Ran, Z. Shao, W. Jin and N. Xu, J. Power Sources, 2008, 182, 24–31 CrossRef CAS.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, Energy Environ. Sci., 2016, 9, 1602–1644 RSC.
- Y. Yamazaki, P. Babilo and S. M. Haile, Chem. Mater., 2008, 20, 6352–6357 CrossRef CAS.
- K. Kreuer, W. Münch, A. Fuchs, U. Klock and J. Maier, Solid State Ionics, 2001, 145, 295–306 CrossRef CAS.
- R. Haugsrud and T. Norby, Nat. Mater., 2006, 5, 193–196 CrossRef CAS.
- T. Norby, M. Widerøe, R. Glöckner and Y. Larring, Dalton Trans., 2004, 3012–3018 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355 Search PubMed.
- C. Kjølseth, L.-Y. Wang, R. Haugsrud and T. Norby, Solid State Ionics, 2010, 181, 1740–1745 CrossRef.
- S. Eisele, F. M. Draber and S. Grieshammer, Phys. Chem. Chem. Phys., 2021, 23, 4882–4891 RSC.
- H. Zhu, S. Ricote, C. Duan, R. P. O'Hayre, D. S. Tsvetkov and R. J. Kee, J. Electrochem. Soc., 2018, 165, F581 CrossRef CAS.
- K.-D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- S. Ricote, N. Bonanos and G. Caboche, Solid State Ionics, 2009, 180, 990–997 CrossRef CAS.
- K. Kreuer, T. Dippel, Y. M. Baikov and J. Maier, Solid State Ionics, 1996, 86, 613–620 CrossRef.
- K. Zhu, Y. Yang, D. Huan, X. Hu, N. Shi, Y. Xie, X. Li, C. Xia, R. Peng and Y. Lu, ChemSusChem, 2021, 14, 3876–3886 CrossRef CAS PubMed.
- D. Huan, L. Zhang, X. Li, Y. Xie, N. Shi, S. Xue, C. Xia, R. Peng and Y. Lu, ChemSusChem, 2020, 13, 4994–5003 CrossRef CAS PubMed.
- M. Hakim, C.-Y. Yoo, J. H. Joo and J. H. Yu, J. Power Sources, 2015, 278, 320–324 CrossRef CAS.
- E. Fabbri, A. D'Epifanio, E. Di Bartolomeo, S. Licoccia and E. Traversa, Solid State Ionics, 2008, 179, 558–564 CrossRef CAS.
- B. Steel and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef PubMed.
- D. J. Brett, A. Atkinson, N. P. Brandon and S. J. Skinner, Chem. Soc. Rev., 2008, 37, 1568–1578 RSC.
- A. Weber and E. Ivers-Tiffée, J. Power Sources, 2004, 127, 273–283 CrossRef CAS.
- S. Choi, C. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H. Ji and S. Haile, Nat. Energy, 2018, 3, 202–210 CrossRef CAS.
- J. Kim, S. Sengodan, G. Kwon, D. Ding, J. Shin, M. Liu and G. Kim, ChemSusChem, 2014, 7, 2811–2815 CrossRef CAS PubMed.
- W. Sun, M. Liu and W. Liu, Adv. Energy Mater., 2013, 3, 1041–1050 CrossRef CAS.
- E. Fabbri, L. Bi, J. L. Rupp, D. Pergolesi and E. Traversa, RSC Adv., 2011, 1, 1183–1186 RSC.
- T. Wu, R. Peng and C. Xia, Solid State Ionics, 2008, 179, 1505–1508 CrossRef CAS.
- W. Sun, L. Yan, Z. Shi, Z. Zhu and W. Liu, J. Power Sources, 2010, 195, 4727–4730 CrossRef CAS.
- S. Lee, I. Park, H. Lee and D. Shin, Int. J. Hydrogen Energy, 2014, 39, 14342–14348 CrossRef CAS.
- Z. Tao, L. Bi, L. Yan, W. Sun, Z. Zhu, R. Peng and W. Liu, Electrochem. Commun., 2009, 11, 688–690 CrossRef CAS.
- J. Xiao, W. Sun, Z. Zhu, Z. Tao and W. Liu, Mater. Lett., 2012, 73, 198–201 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta07113k |
| This journal is © The Royal Society of Chemistry 2022 |