
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotential roles of hyaluronic acid in in vivo CAR T cell reprogramming for cancer immunotherapy
Chavee
Laomeephol
 a,
Sudartip
Areecheewakul
b,
Supannikar
Tawinwung
bc,
Koramit
Suppipat
cd,
Preedakorn
Chunhacha
e,
Nuno M.
Neves
fg and
Jittima Amie
Luckanagul
*ah
a,
Sudartip
Areecheewakul
b,
Supannikar
Tawinwung
bc,
Koramit
Suppipat
cd,
Preedakorn
Chunhacha
e,
Nuno M.
Neves
fg and
Jittima Amie
Luckanagul
*ah
aDepartment of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok 10330, Thailand. E-mail: Jittima.L@pharm.chula.ac.th; Fax: +662-218-8401; Tel: +662-218-8400
bDepartment of Pharmacology and Physiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok 10330, Thailand
cChulalongkorn University Cancer Immunology Excellence Center, Faculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand
dChulalongkorn University Stem Cell and Cell Therapy Research Center, Department of Pharmacology, Faculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand
eDepartment of Biochemistry and Microbiology, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok 10330, Thailand
f3B's Research Group, I3Bs – Research Institute on Biomaterials, Biodegradables and Biomimetics, University of Minho, Headquarters of the European Institute of Excellence on Tissue Engineering and Regenerative Medicine, AvePark – Parque de Ciência e Tecnologia, Zona Industrial da Gandra, 4805-017 Barco, Guimarães, Portugal
gICVS/3B's – PT Government Associate Laboratory, Braga, Guimarães, Portugal
hCenter of Excellence in Plant-produced Pharmaceuticals, Chulalongkorn University, Bangkok 10330, Thailand
First published on 9th November 2022
Abstract
Chimeric antigen receptor (CAR) T cell therapy has recently shown unprecedented clinical efficacy for cancer treatment, particularly of hematological malignancies. However, the complex manufacturing processes that involve ex vivo genetic modification of autologous T cells limits its therapeutic application. CAR T cells generated in vivo provide a valid alternative immunotherapy, “off-the-shelf”, for cancer treatment. This approach requires carriers for the delivery of CAR-encoding constructs, which are plasmid DNA or messenger RNA, to T cells for CAR expression to help eradicate the tumor. As such, there are a growing number of studies reporting gene delivery systems for in vivo CAR T cell therapy based on viral vectors and polymeric nanoparticles. Hyaluronic acid (HA) is a natural biopolymer that can serve for gene delivery, because of its inherent properties of cell recognition and internalization, as well as its biodegradability, biocompatibility, and presence of functional groups for the chemical conjugation of targeting ligands. In this review, the potential of HA in the delivery of CAR constructs is discussed on the basis of previous experience of HA-based nanoparticles for gene therapy. Furthermore, current studies on CAR carriers for in vivo-generated CAR T cells are included, giving an idea of a rational design of HA-based systems for the more efficient delivery of CAR to circulating T cells.
1. Introduction
Chimeric antigen receptor (CAR) T cells offer a paradigm shift in clinical cancer immunotherapy using genetically modified T cells with synthetic receptors that recognize target antigens to mediate tumor rejection.1,2 CAR T cell products have been commercially available since 2017 for the treatment of blood cancers, including tisagenlecleucel (Kymriah®, Novartis), axicabtagene ciloleucel (Yescarta®, Gilead), brexucabtagene autoleucel (Tecartus®, Gilead) and mostly recent lisocabtagene maraleucel (Breyanzi®, Juno Therapeutics).3 All products share the same characteristics in which the patient's T cells are collected, genetically programmed and expanded in a lab, before being reinfused into the patient, so-called adoptive cell transfer (ACT).4,5 Individualized approaches with complex and labor-intensive manufacturing processes and the requirement for accredited GMP laboratories to control and ensure the quality of the products lead to a costly treatment course that limits accessibility for some patients.4,6 In addition, cytokine release syndrome could occur up to 25% of patients after receiving ACT, leading to multiple organ failures, neurologic disorders, or severe immune reactions.7,8 To address these limitations, a novel concept of in situ or in vivo programming of CAR T cells has been introduced in which CAR constructs are administered to patients, allowing de novo genetic modification, expansion, and then attack of tumor cells.4,6 Due to this ‘off-the-shelf’ approach, individualization can be overcome, and the products can be manufactured in a large batch and delivered to a large number of patients. The comparison scheme between adoptive CAR T cell transfer and in vivo CAR T cell programming is illustrated in Fig. 1.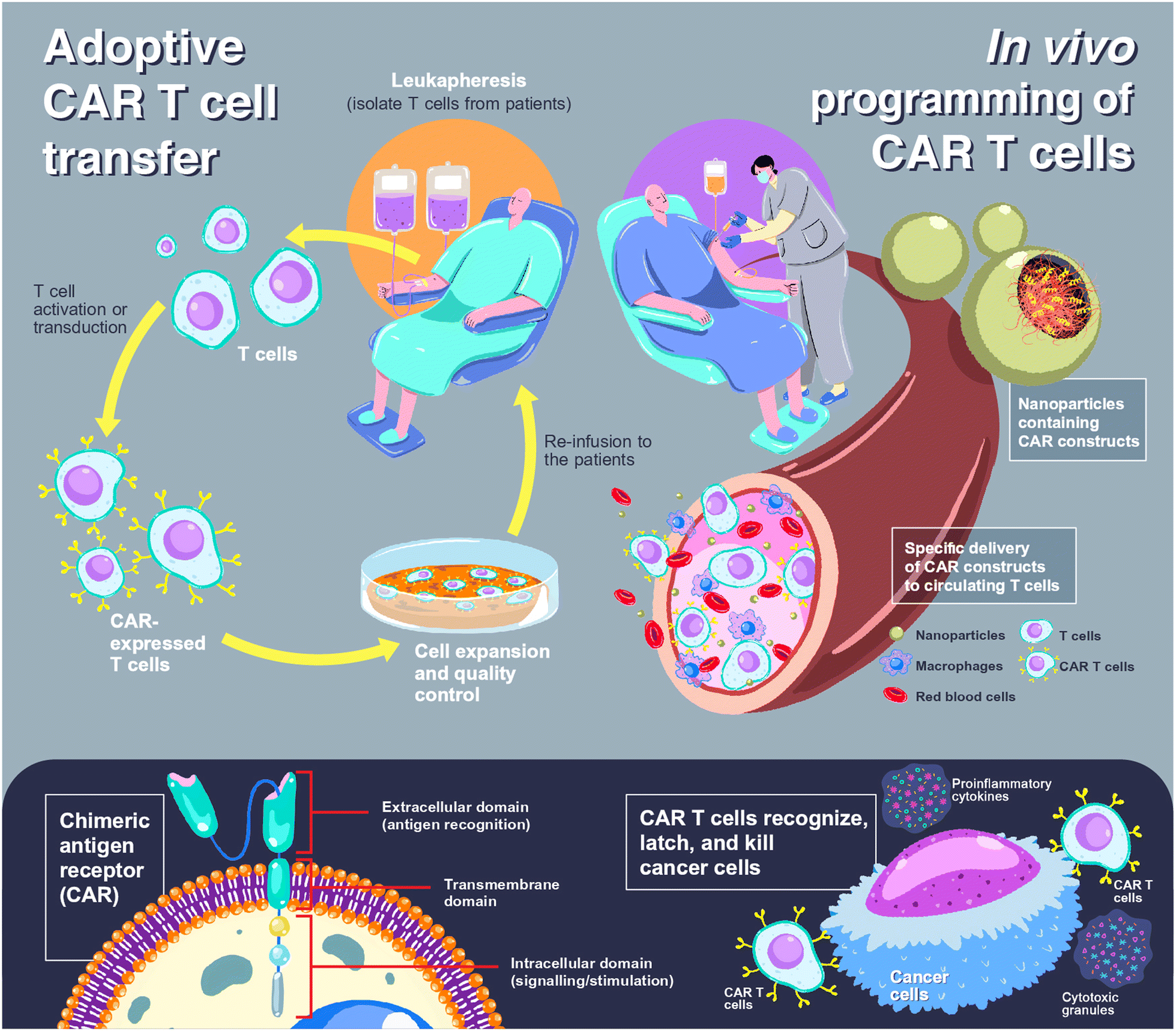 | ||
| Fig. 1 Comparison between adoptive cell transfer and in vivo cell programming in CAR T cell therapy. For adoptive CAR T cell transfer (upper left), T cells are isolated from patients, so-called leukapheresis, using an apheresis machine. The obtained T cells are then transfected to express CAR (lower left), before expanding, controlling the cell quality in a certified laboratory, and re-administration to the patients. In the case of in vivo programming of CAR T cells (upper right), the CAR constructs are loaded into nanoparticles, which can be either viral or non-viral vectors, and the nanoparticles are administered to the patients. The nanoparticles are designed to specifically bind to circulating T cells and release CAR constructs to the cytosol or nucleus of T cells. Transduced T cells express CAR to target and eliminate cancer cells (lower right). | ||
Expression of the CAR transgene in primary T cells can be achieved by the cellular delivery of CAR constructs, which can be plasmid DNA (pDNA), messenger RNA (mRNA) or CRISPR/Cas (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein). Entrapment of loaded genes into a complex using polymers is required to protect them from endogenous enzymes and facilitate cellular internalization. CAR-loaded nanoparticles are typically administered intravenously, which means that the nanoparticles should enable specific delivery to circulating T cells. A rational carrier design can prolong the retention time in the bloodstream by preventing the early elimination of nanoparticles.9,10 After cell uptake of the nanoparticles through receptor-mediated endocytosis, delivery systems could aid in cell trafficking within the cells by releasing payloads from endosomes into the cytosol before CAR translation and expression (Fig. 2).
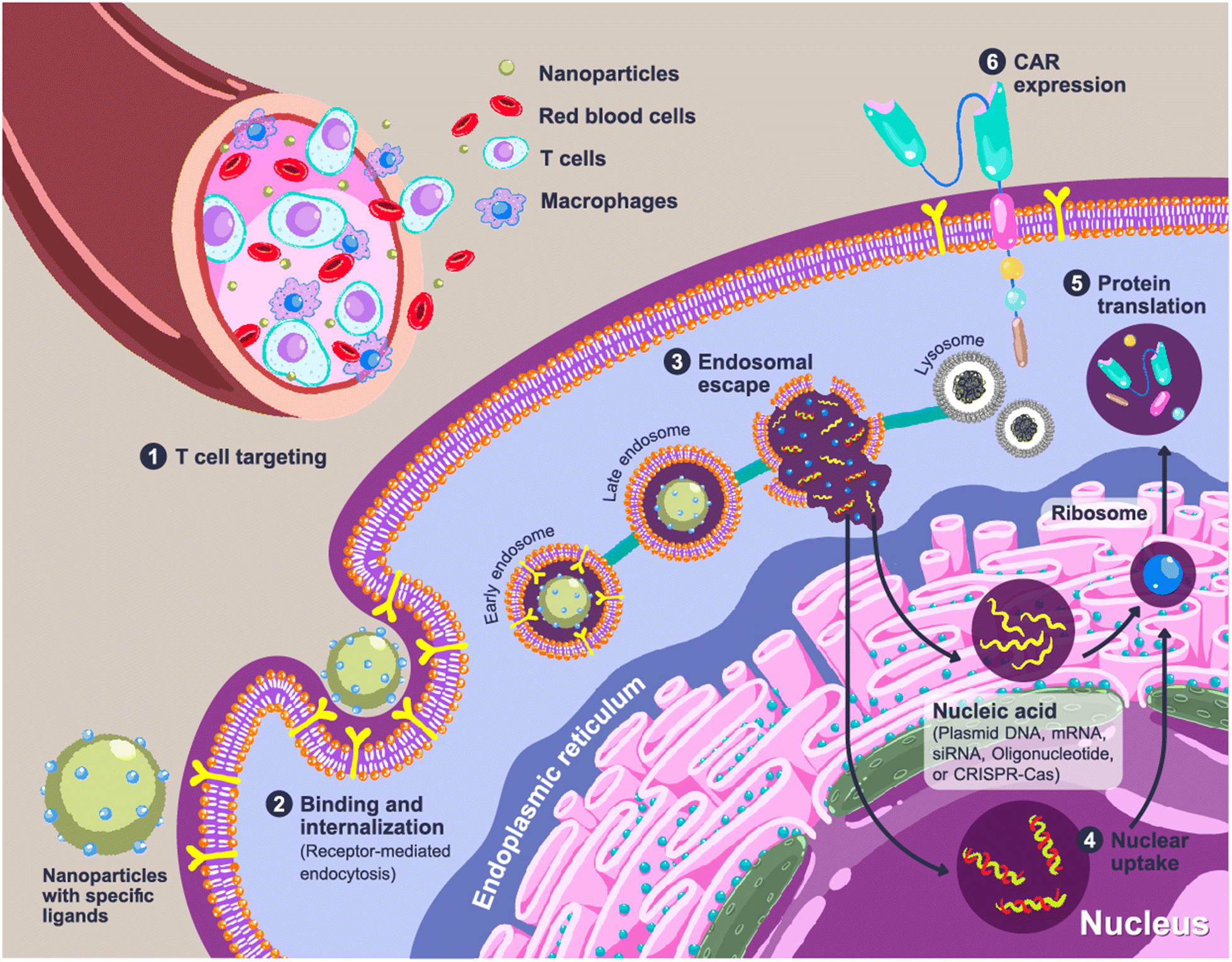 | ||
| Fig. 2 Cellular uptake and CAR expression of CAR after nanoparticle administration. Step 1: The nanoparticles are injected intravenously and are specifically recognized by circulating T cells. Step 2: Cellular binding and internalization of nanoparticles occurs via receptor-mediated endocytosis, and the nanoparticles are entrapped in endosomes. Step 3: The nanoparticles mediate the disruption of the endosomes to release the loaded gene cargos into the cytosol. The remaining nanoparticles are packed in lysosomes as a cellular waste. Step 4: The loaded genes are taken up by the target organelles. Step 5: The mRNA is translated to proteins by the ribosomes. Step 6: The expression of the CAR receptor on the surface of T cells. | ||
Typically, in vivo applications of nanoparticles should be aware of the biodistribution and accumulation in the major organs, such as the lungs, liver, spleen, and kidneys, which are dictated by the physical properties of the nanoparticles, including their size, surface charge, and shape.11 For T cell reprogramming, nanoparticle delivery could be limited by the innate characteristics of T cells, including the small cell size, the high nucleus-cytoplasm ratio, and the non-phagocytic nature.12 Internalization through receptor-mediated endocytosis could be limited due to the low expression of surface receptors in a naïve state.13 More importantly, circulating T cells, which are the main target of CAR programming, are present in very small numbers in the blood,14 resulting in a low population of reprogrammed cells.
In this paper, nanoparticle-based delivery systems for gene delivery targeting T cells are reviewed, focusing on polymeric nanoparticles. Hyaluronic acid (HA) is one of the polymers that has potential as a component of delivery systems for targeting T cells. An addition of HA to the formulations of gene complexes can reduce the cytotoxicity, improve transduction activity in vitro and in vivo, guide the nanoparticles to the target sites, and prolong the stability of the gene products. Additionally, HA is a versatile biomaterial that allows a rational design of nano-formulations with desired physicochemical and biological properties.15 Although to our knowledge, there are no HA-based carriers designed specifically for T cell delivery, especially for in vivo CAR T cell immunotherapy, the roles and functions of HA as a gene carrier, as well as the strategies to apply HA to enhance T cell targeting properties, will be discussed. Studies relating to nucleic acid-loaded HA-based nanoparticles, as well as the delivery systems serving in vivo CAR T cells, will be mentioned to give an idea of the development of an efficient carrier for in vivo programming of T cells.
2. Immunotherapy based on T cells for cancer treatment
The concept of cellular immunotherapy is to use host immune systems in tumor eradication. Typically, immune cells are responsible for the elimination of foreign bodies, including pathogenic microorganisms, as well as cells with abnormal proliferation, termed cancer immunosurveillance or cancer immunoediting. The fundamental concept is that the expression of cancer cells’ antigens is different from normal cells, serving as rejection antigens in provoking CD8+ effector cells and natural killer (NK) cells to eliminate transformed cells together with innate immune cells, such as macrophages.16,17 However, the genetic adaptation of cancer cells and the development of variants could reduce tumor immunogenicity,18 leading to a high rate of tumor evasion and a poor prognosis. Tumor cells could suppress the expression of non-self-antigens or overly express a particular ligand to avoid immune recognition. The activities of cytotoxic T lymphocytes (CTL) can be debilitated because of altered tumor microenvironments, namely the release of cytokines that suppress CTL activities or increase the number of regulatory T cells.19 These are the reasons that limit the attractiveness of immune cell-based therapy, even with its high specificity for tumor cells compared with traditional therapies, such as chemotherapeutic agents or radiation. Therefore, immune cell reprogramming is used to enhance their activities in the recognition and cytotoxicity of cancer cells, and T cells have been focused on in their key role in cancer eradication.To date, T-cell-based therapies are commonly based on an ACT approach, as mentioned above. ACT of tumor-infiltrating lymphocytes (TILs), which refer to T cells located in tumor tissues, has been successful in the treatment of melanoma and other epithelial cancers. TILs can also recognize neoantigens, indicating their efficacy against cancer mutations.20 However, the number of isolated TILs varies according to an individual variability in tumor characteristics.18 Therefore, other approaches have focused on circulating T cells, which provide more sufficient numbers of antitumor cells.
Isolated circulating T cells can be genetically reprogrammed ex vivo for the expression of T cell receptors (TCR) or chimeric antigen receptors (CAR). TCR and CAR are designed to recognize tumor-associated or tumor-specific antigens for tumor-targeted therapy. The difference in tumor association between TCR and CAR T cells is that activation of TCR T cells is needed for an association with the major histocompatibility complex (MHC) molecules. Intracellular antigen can be detected by TCR-engineered T cells, and immune processing through MHC pathways could be advantageous in tumor cytotoxicity. However, excessive immune activation could induce cytokine-related toxicities, known as cytokine storms.21 Additionally, off-target toxicities could occur in cells that express antigens identical to those of target tumors, provoking an autoimmune response that could lead to a fatal destruction of normal cells.21,22
Instead of expressing the full length α and β chains as TCRs, the antigen binding domain of CAR presents only a single chain variable fragment (scFv). Therefore, the binding of CAR-engineered T cells is independent of the MHC molecules. This could be beneficial for the treatment of resistant tumors in an escape phase, where immune evasion develops due to the loss of presentation of the MHC-associated antigen.23 As shown in Fig. 1, the composition of CAR can be simply divided into 3 elements: (1) extracellular domains, which are responsible for antigen recognition and binding, linked to (2) transmembrane domains and (3) intracellular domains, which impart intracellular signaling and T cell activation.
The evolution of CAR is based on the design of intracellular signaling domains to achieve high efficiency and sustainability in tumor cytotoxicity by enhancing cytokine secretion and to facilitate a persistent expansion of T cells. The first-generation CAR was designed to express a single signaling molecule, such as CD3ζ. In the subsequent generation, co-stimulatory signals, namely CD28 or 4-1BB, have been added.18,23 CAR T cell therapy has been reported to be successful in hematologic cancers18 and some have been commercialized, namely Kymriah® for acute lymphoblastic leukemia, Yescarta® for large B cell lymphoma, Tecartus® for mantle cell lymphoma, and Breyanzi® for relapsed or refractory large B cell lymphoma.3 However, efficient CAR T cell therapy for solid tumors has not yet been reported because irregular antigen expression and an adaptation of the tumor microenvironment can hinder target recognition.24 The newly designed CAR currently under investigation can recognize more than one antigen to cope with the antigen heterogeneity of solid tumors or activate T cell cytotoxicity under specific conditions to be specifically active in tumor tissues.18,24
3. Gene delivery systems
Direct administration of naked genetic materials is limited by their anionic characteristics, which prevent an association with a negatively charged cell surface, resulting in low and uncertain transgene expression. Furthermore, their instability to physiological enzymes in serum, namely nucleases, as well as rapid clearance through renal excretion, drastically reduces the retention time in the systemic circulation.25,26 In some cases, pDNA, mRNA, or short-strand RNA can be recognized as exogenous danger signals by host cells and trigger innate or humoral immune responses, as well as early clearance through the reticuloendothelial system.27,28 Therefore, two types of gene delivery system, namely viral-based and non-viral-based delivery systems, have been introduced as they potentially serve greater efficiency in mammalian cells. The virus-based delivery system is known for its high transfection efficiency due to the inherent properties of viruses in promoting cell uptake and penetrating the host nucleus. However, the immunogenic properties, random recombination, and potential in a recurrence of pathogenic variants are of great concerned. In addition, the development of neutralizing antibodies against viral vectors by the hosts could limit the clinical applications.29 The highly recognized and commercialized DNA viral vectors are Azd1222 and AD5-nCOV, which are vaccine platforms against SARS-CoV-2 viruses developed by Oxford/AstraZeneca, UK and Cansino Biologics, China, respectively. Non-replicating adenovirus-based cargos were used to deliver DNA for the translation of viral spike proteins, and acceptable vaccine effectiveness was reported for both vaccines with mild-to-moderate side effects.30To overcome the limitations of virus-based delivery systems, non-viral approaches, namely cationic lipids and polycations, offered a safer alternative for gene transfer. Most of the non-viral cargos comprise positively charged moieties, such as protonated amines, which are capable of interaction with the negative phosphate groups of nucleic acids, resulting in a condensation of nanosized complexes. The positive surface charge of the complexes also enhances cellular uptake through electrostatic binding to the negatively charged cell membrane. Furthermore, the vectors can be chemically modified with targeting ligands to improve their specific recognition. However, the relatively low transfection efficiency, nonspecific binding to serum proteins, and cytotoxicity of cationic molecules are the major issues in the development of an effective non-viral-based delivery system.29 BNT162b1 and mRNA-1273, the SARS-CoV-2 vaccines developed by Pfizer/BioNTech and Moderna, respectively, are mRNA-based vaccines encapsulated in lipid nanoparticles, and can serve as examples of non-viral gene delivery systems. Lipid nanoparticles not only improve the cellular internalization of mRNA, but also act as a shelter for mRNA, allowing the practical use of very labile mRNA and extending its shelf life.30
Shielding the cationic complexes of DNA or RNA and lipids (lipoplexes) or polycations (polyplexes) with uncharged or counter-charged polymers is an approach for a reduction of surface charge density, resulting in a lower non-specific binding, longer retention time in the bloodstream, and reduced toxicities. The most common polymer for complex shielding is polyethylene glycol (PEG), so-called PEGylation, and the obtained nanoparticles are referred to as stealth nanoparticles. Even though PEG does not possess a specific ligand of biological receptors, the introduction of targeting groups by a chemical conjugation of PEG chains could enable the targetability of nanoparticles.31 In addition to PEG, other negatively charged polymers, namely hyaluronic acid (HA), chondroitin sulfate, polyesters, and polyglutamic acid, are utilized as shielding polymers by either simple electrostatic interaction or chemical conjugation with cationic lipids or polymers. Among these polymers, HA-based DNA or RNA complexes have been widely explored for their in vitro and in vivo efficiency and safety,32 confirming a promising non-viral polymeric gene delivery system for clinical uses.
4. Designing HA-based nanoparticles for gene delivery
HA is a naturally derived polysaccharide with a disaccharide monomeric unit that contains N-acetyl-D-glucosamine and D-glucuronic acid residues. Biological HA serves as a component of the extracellular matrix in mammals, and is highly present in synovial fluid, vitreous humor, and skin. Its physiological functions include supporting cell proliferation, differentiation, migration, and tissue repair.33 The lists of studies that involve HA in gene delivery are presented in Tables 1 and 2, categorized by the forms of HA (original versus chemically modified formats) used for nanoparticle formation.| Nanoparticle formation | Nanoparticle characteristics | Evaluation of transfection efficiency | Proposed applications | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Payloads | Cationic lipids/polymers | HA (MW) | Size (nm) | Charge (mV) | In vitro | In vivo | ||
| a 4T1 (mouse breast cancer cells), AsPC-1 (human pancreas adenocarcinoma cells), B16 (murine melanoma cells), BMSCs (bone-marrow derived stem cells), COS-7 (African green monkey kidney fibroblast-like cells), HCE (human corneal epithelial cells), HCT-116 (human colorectal carcinoma cells), HDF (human dermal fibroblasts), HEK-293T (human embryonic kidney cells), HeLa (human cervical cancer cells), HT-29 (human colorectal adenocarcinoma cells), Huh-7 (human hepatocyte-derived carcinoma cells), HUVEC (human umbilical vein endothelial cells), IOBA-NHC (human conjunctiva cells), MCF-7 (human breast cancer cells), MDA-MB-231 (human breast cancer cells), MDA-MB-435 (human breast cancer cells), NIH/3T3 (murine embryonic fibroblasts), NSCs (neural stem cells), PANC-1 (human pancreatic cancer cells), SW-480 (human colon adenocarcinoma cells), T98G (human glioblastoma cells), TK-1 (human myeloid leukemia cells), THP-1 (human monocytic cells), U251 (human glioblastoma cells), U87MG (human glioblastoma cells). Abbreviations: APC (adenomatous polyposis coli), CRISPR/Cas9 (clustered, regularly interspaced short palindromic repeats)/CRISPR-associated protein (9), DGL (dendrigraft poly-L-lysine), GFP (green fluorescent protein), GM-CSF (granulocyte macrophage-colony stimulating factor), IL-1 (interleukin-1), KO (knock out), Luc (luciferase), miRNA (microRNA), mRNA (messenger RNA), PBA (phenylboronic acid), PDA (polydopamine), pDNA (plasmid DNA), PEI (polyethyleneimine), PLA (polylactic acid), PLK1 (polo-like kinase 1), Ptpn2 (protein tyrosine phosphatase non-receptor type 2), siRNA (small interfering RNA). | ||||||||
| pDNA (SOX9) | Linear PEI (25 kDa) | 5.8 kDa | 70–150 | +5.8 to +20.3 | Human BMSCs | N/A | Cartilage tissue engineering | 35 |
| pDNA (GFP, Luc) | Linear PEI (25 kDa) | N/A | 300–800 | −46.3 to −37.3 | B16 | Tumor-bearing mice | Cancer treatment | 43 |
| pDNA (GFP, Luc, GM-CSF) | Linear PEI (25 kDa) | N/A | 70–200 | −48 to −40 | B16 | Tumor-bearing mice | Cancer treatment | 45 |
| pDNA (GFP, GM-CSF) | Linear PEI (40 kDa) | N/A | 200 | N/A | B16 | Tumor-bearing mice | Cancer treatment | 96 |
| pDNA (GFP) | Branched PEI (25 kDa) | 17 kDa | 218–389 | −18.9 to +27.0 | MDA-MB-231, MDa-MB-435, MCF-7, COS-1 | N/A | Breast cancer treatment | 38 |
| pDNA (IL-1 receptor antagonist) | Chitosan (5 kDa) | 35 kDa | 140–500 | +15 to +35 | Primary rat synoviocytes | N/A | Treatment of osteoarthritis | 70 |
| pDNA (Luc) | Chitosan | N/A | 242.7–372.3 | +39.4 to +57.8 | Mouse NSCs | Rats | Gene delivery to the spinal cord | 62 |
| pDNA (Luc) | Chitosan (46.4 kDa) | 400–1300 kDa | 333.5–381.3 | −39.6 to −44.6 | COS-7, Huh-7 | N/A | Solid-phase reverse transfection | 120 |
| pDNA (GFP) | Chitosan (50 kDa) | <10 kDa | 115.6–364.8 | −34.5 to +24.0 | Rabbit chondrocytes | N/A | Treatment of osteoarthritis | 128 |
| pDNA (GFP, β-galactosidase) | Chitosan (10–12 kDa & 110 kDa) | 170 kDa | 152–199 | +19 to +27 | HCE, NIH/3T3 | Rabbits | Ocular gene delivery | 129 |
| pDNA (GFP, β-galactosidase) | Chitosan (10–12 kDa & 170 kDa) | <10 kDa & 110 kDa | 103–235 | −30 to +28 | HCE, IOBA-NHC | N/A | Ocular gene delivery | 41 |
| pDNA (GFP) | Chitosan (5 kDa) | 17, 35 & 64 kDa | 146–175 | +15 to +32 | HEK-293T | N/A | N/A | 130 |
| pDNA (GFP, TGF-β1) | Chitosan (50 kDa) | 160 kDa | 155.4 | +22.14 | Rabbit chondrocytes | N/A | Cartilage tissue engineering | 74 |
| pDNA (GFP), siRNA (anti GFP, anti Snail1) | Chitosan-grafted-PEG | 170 kDa | 150.8–250.6 | −20.1 to +31.9 | HEK-293T | N/A | Cancer treatment | 131 |
| siRNA (anti STAT3) | PEI-PLA-lipoic acid | 9 kDa | 195 | −23 | 4T1, MDA-MB-231 | Tumor-bearing mice | Cancer treatment | 132 |
| Paclitaxel | ||||||||
| siRNA (anti KRAS) | Chitosan (656 kDa), poly hexamethylene biguanide (3.2 kDa) | 183 kDa | 120–275 | −34.5 to +52.0 | HCT-116, HDF | N/A | Cancer treatment | 34 |
| siRNA (anti cyclophilin B) | Chitosan (35 & 656 kDa) | 180 kDa | 250 | −40 to −35 | AsPC-1, HCT-116, HDF, HT-29, HUVEC, PANC-1, THP-1 | N/A | N/A | 82 |
| siRNA (anti PLK1) | PBA modified DGL | N/A | 100–150 | −16.9 | N/A | Tumor-bearing mice | Cancer treatment | 133 |
| siRNA (anti PLK1) | Lipid nanoparticles | 5 kDa | 100.7 | −8.2 | T98G, U251, U87MG | Glioma mouse model | Treatment of glioma | 77 |
| siRNA (anti Luc) | Liposomes, protamine | N/A | 114.3–117.9 | +21.2 to +27.5 | B16F10 | Tumor-bearing mice | Cancer treatment | 134 |
| siRNA (anti cyclin D1) | Liposomes, protamine | N/A | 144–161 | −17.9 to −19.8 | TK-1, mouse splenocytes | Mice | Intestinal inflammation, colitis | 71 |
| mRNA (Luc), siRNA (anti Luc) | Chitosan (685 kDa) | 180 kDa | 195–350 | −44.0 to −36.0 | HCT-116 | N/A | N/A | 48 |
| miRNA (antimir-138) | Chitosan (100 kDa) | 10 kDa | 153.0–290.7 | +21.5 to +45.5 | Rat BMSCs | N/A | Osteogenic differentiation | 72 |
| CRISPR/Cas9 (Ptpn2 KO) | PEI (1.8 kDa) | 35 kDa | 115.4 | −32.9 | B16F10 | Tumor-bearing mice | Melanoma | 37 |
| CRISPR/Cas9 (Nanog KO) | Dexamethasone-loaded PDA and PEI (10, 25 kDa) | <10 kDa | 200–550 | +15 to +20 | HeLa | N/A | Cancer treatment | 135 |
| CRISPR/Cas9 (KRAS KO) | β-Cyclodextrin conjugated PEI | N/A | 200 | −16.1 | HEK-293T, SW-480 | Tumor-bearing mice | Treatment of colorectal cancer | 76 |
| CRISPR/Cas9 (APC and KRAS KO) | Phenylboronic dendrimer | N/A | 220 | −9.1 | SW-480 | Tumor-bearing mice | Treatment of colorectal cancer | 136 |
| Nanoparticle formation | Nanoparticle characteristics | Evaluation of transfection efficiency | Proposed applications | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Payloads | Cationic lipids/polymers | Modified HA | Size (nm) | Charge (mV) | In vitro | In vivo | ||
| a 4T1 (murine breast cancer cells), A549 (human non-small-cell lung cancer cells), AML2 (mouse hepatocyte cells), B16 (murine melanoma cells), CHO (Chinese hamster ovary cells), CV-1 (green monkey kidney cells), GES-1 (human gastric epithelial cells), H1299 (human non-small cell lung carcinoma cells), H69 (human small-cell lung cancer cells), HCT-116 (human colorectal carcinoma cells), HEK-293 (human embryonic kidney cells), Hep3B (human hepatoma cells), HeLa (human cervical cancer cells), HepG2 (human liver cancer cells), HFF (human foreskin fibroblasts), HT-29 (human colorectal adenocarcinoma cells), J774A.1 (murine monocytes), MCF-7 (human breast cancer cells), MCF-10A (human breast epithelial cells), MDA-MB-231 (human breast cancer cells), MDA-MB-468 (human breast cancer cells), MSCs (mesenchymal stem cells), NIH/3T3 (murine embryonic fibroblasts), OVCAR-8 (human ovarian cancer cells), RAW 264.7 (mouse macrophage cells), SGC-7901 (human gastric cancer cells). Abbreviations: AS1411 (anti-nucleolin aptamer), BMP-2 (bone morphogenetic protein 2), COX-2 (cyclooxygenase-2), GFP (green fluorescent protein), KO (knock out), miRNA (microRNA), MDR1 (multidrug resistance 1 gene), OEI (oligoethyleneimine), p53 (tumor protein 53), PAMAM (poly(amidoamine)), PBAE (poly (β-amino ester)), PDA (2-(2-pyridyldithio)ethylamine), pDMAEMA (poly[2-(dimethylamino)ethyl methacrylate]), PPM1D (protein phosphatase, Mg2+/Mn2+ dependent 1D), RFP (red fluorescent protein), TAT (trans-activator of transcription), TMSP (tumor microenvironment-sensitive peptide), VEGF (vascular endothelial growth factor). | ||||||||
| pDNA (Luc) | N/A | PEI or PEG conjugated HA | 200–400 | −15 to −30 | A549, HeLa | N/A | N/A | 53 |
| pDNA (IL-4 and IL-10) | N/A | PEI conjugated HA | 185.9 | −11.6 | J774A.1 | Mice | Macrophage repolarization | 73 |
| pDNA (GFP, endostatin) | N/A | PEI conjugated HA with cleavable disulfide bonds | 82.26–154.80 | +32.3 to +70.3 | Human MSCs | N/A | Chondrogenic differentiation | 59 |
| pDNA (Luc, GFP, p53) | N/A | OEI and β-cyclodextrin conjugated HA | 100–150 | +25 to +40 | MDA-MB-231, MCF-7 | N/A | Cancer treatment | 40 |
| pDNA (Luc) | N/A | Spermine conjugated HA | 200–300 | −6.1 | CHO | Mice | N/A | 65 |
| siRNA (anti MDR1) | N/A | PEI or PEG conjugated HA | 173.3 | −22.5 | OVCAR-8 | Tumor-bearing mice | Treatment of ovarian cancer | 56 |
| siRNA (anti survivin) | N/A | PEI or PEG conjugated HA | 90–100 | −15 | A549, H69 | Tumor-bearing mice | N/A | 52 |
| siRNA (anti Luc & anti VEGF) | N/A | PEI conjugated HA | 42.7 | N/A | B16F1 | Tumor-bearing mice | Cancer treatment | 64 |
| siRNA (anti Luc) | N/A | PEI conjugated HA | 21 | −21.5 to −15.3 | B16F1, HEK-293 | N/A | N/A | 122 |
| siRNA (anti VEGF) | N/A | PEI conjugated HA with cleavable disulfide bonds | 110 | +1.8 to +12.1 | B16F1 | Tumor-bearing mice | Cancer treatment | 60 |
| siRNA (anti PLK1) | N/A | Polyamine (PEI, spermine) conjugated HA | 85–190 | −5.5 to +16.5 | A549, H69, MDA-MB-468, Hep3B, B16F10 | Tumor-bearing mice | Cancer treatment | 54 |
| siRNA (anti COX-2) | N/A | Alkyl chain and spermine conjugated HA | 200–500 | N/A | SGC-7901, GES-1 | N/A | Treatment of gastric cancer | 137 |
| siRNA | N/A | Alkyl chain and spermine conjugated HA | 125.6–555.1 | +18.4 to +21.3 | N/A | N/A | N/A | 138 |
| siRNA (anti RFP) | N/A | pDMAEMA and PDA conjugated HA | 314.4 | −6.02 | B16F10, CV-1, NIH/3T3 | Tumor-bearing mice | Cancer treatment | 10 |
| siRNA (anti TNF-α) | PBAE | Dopamine-grafted HA | 150 | <0 | RAW 264.7, NIH/3T3 | Inflammation-stimulated mice | Treatment of acute lung injury | 46 |
| pDNA (GFP, Luc) | Branched PEI (25 kDa) | HA with cleavable disulfide bonds | 200 | −23.4 to +5.0 | HepG2, B16F10, NIH/3T3 | Tumor-bearing mice | Cancer treatment | 39 |
| siRNA (anti GFP) | N/A | HA with cleavable disulfide bonds | 198 | N/A | HCT-116, NIH/3T3 | N/A | N/A | 50 |
| siRNA (anti KRAS) | PAMAM | HA with cleavable diselenide bonds | 181 | −24.6 | A549 | Tumor-bearing mice | Cancer treatment | 61 |
| Antisense oligonulcleotide (anti GFP) | Protamine | HA with cleavable disulfide bonds | 166–200 | N/A | HEK-293 | N/A | N/A | 51 |
| siRNA (anti RFP | 2b RNA-binding protein | Cholesterol conjugated HA | 190–410 | −64.3 to −45.1 | B16F10, HFF | N/A | Cancer treatment | 42 |
| siRNA (anti survivin) | Azide-modified liposomes with cleavable disulfide bonds | Alkyne functionalized HA | 130 | −26 | A549 | Tumor-bearing mice | Cancer treatment | 36 |
| pDNA (BMP-2), miRNA (mir148b) | Calcium phosphate | Catechol (dopamine) functionalized HA | 71 | −12 | Human MSCs | N/A | Osteogenic differentiation | 139 |
| siRNA (anti Luc) | Calcium phosphate | Catechol functionalized HA | 63–278 | −20 to −70 | HT29 | Tumor-bearing mice | Cancer treatment | 44 |
| siRNA (PD-L1, STAT3) | Chitosan (modified with trimethyl and thiol groups) | TAT conjugated HA | 110 | +20 | B16F10, 4T1 | Tumor-bearing mice | Cancer treatment | 140 |
| pDNA (IL-12) | Fluorinated PEI | TMSP conjugated HA | 220–250 | −19.7 to −9.06 | B16F10 | Tumor-bearing mice | Cancer immunotherapy | 141 |
| CRISPR/Cas9 (CD47 KO) | ||||||||
| CRISPR/Cas9 (PPM1D KO) | Histone, KALA peptide | AS1411 conjugated HA | 134–177 | −18.2 to +23.5 | H1299, MCF-7, MCF-10A | N/A | Cancer treatment | 79 |
| CRISPR/Cas9 (β-catenin KO) | CaCO3, protamine | TAT and AS1411 conjugated HA | 230–320 | −20 to −10 | H1299, HeLa, HEK-293T | N/A | Cancer treatment | 78 |
| CRISPR/Cas9 (GFP KO) | N/A | PEI and/or mannose conjugated HA | 115–118 | −21.2 to −17.9 | AML2 | Mice | Liver targeted delivery | 142 |
Because of the negatively charged characteristic of HA, it is difficult to form polyplexes with DNA or RNA unless the polymer is formed with other cationic molecules prior to the addition of HA or there is modification of the HA structure to enable the potential for complexation. As shown in Table 1, cationic polyplexes or lipoplexes are formed before shielding with HA, which results in a lower surface charge of the complexes.34–38 Minimizing the positively charged complexes dramatically decreases cytotoxicity39–41 and immunogenicity,42 and prevents coagulation with serum proteins,43–45 avoiding complement activation and a rapid clearance from the systemic circulation. In addition, anionic shielding improves penetration through the layer of negatively charged glycoproteins, such as mucin, to the target tissues.46 The most common polycation for polyplex formation is polyethyleneimine (PEI), a polymer containing amine groups that can be protonated over a wide range of pH, allowing electrostatic interaction with the phosphate groups of DNA or RNA9 and facilitating endosomal escape from the proton sponge effect.47
However, due to the inherent cytotoxicity and non-biodegradability of PEI, chitosan, a naturally derived cationic polysaccharide with biodegradability and biocompatibility profiles, offers a safer alternative to use with HA in the formation of gene complexes.9 The major limitations of chitosan are the variability in molecular weight and the degree of deacetylation, which affect the affinity for the entrapped gene. Tirelli, N. and colleagues have used chitosan for the entrapment of RNA and have observed the targeted tumor treatment when the polyplexes are shielded with HA. Their findings revealed that chitosan with a low degree of deacetylation, related to a lower positive charge, possessed a lower affinity for RNA, resulting in a loose complexation and a high exposure to physiological nucleases. In contrast, a high molecular weight chitosan could better stabilize RNA, but a strong electrostatic interaction led to poor decomplexation, lowering RNA release and transfection efficiency. The size of the entrapped gene can also affect the affinity for chitosan, with long-stranded mRNA binding to polyplexes tighter than short-stranded siRNA.34,48 Balancing the molecular weight and degree of deacetylation of chitosan with DNA or RNA of different sizes to achieve high transfection efficiency and stability could be a major issue in designing HA- and chitosan-based delivery systems.
Nanoparticle formation using counterion complexation is a simple, one-pot approach, but the weak and transient electrostatic interaction can be easily disturbed. An early dissociation of the entrapped gene in the presence of negatively charged serum proteins leads to an exposure of naked genes to circulating endonucleases, resulting in lower gene availability at target sites.10,36,49 Therefore, nucleotide-loaded polymer-based nanoparticles, such as the covalent conjugation of nucleotides to polymers50,51 or polycation-modified HA, as shown in Table 2, result in a predictable gene encapsulation efficiency as well as a reliable transfection efficiency. Furthermore, the size of the gene-loaded modified HA nanoparticles was in a range of 100 to 200 nm, which is suitable for cell internalization and in vivo biodistribution to target cells,52 compared with larger electrostatically formed HA nanoparticles. Polyamines, namely PEI, spermine and pDMAEMA (poly[2-(dimethylamino)ethyl methacrylate]), were popularly grafted onto HA to provide protonated amines for electrostatic binding to DNA or RNA. Furthermore, some studies revealed that the negative carboxyl groups of HA can loosen the electrostatic complexation between nucleic acids and cationic molecules.49 Decreasing the negative charge density by introducing non-ionic moieties, such as PEG, was applied, and the findings showed an enhanced transfection efficiency and a longer retention time in the circulatory system.52–56
Stimulus-responsive moieties were introduced to enhance the site-specific release of the entrapped genes. Cleavable disulfide or diselenide linkages with redox responsiveness were widely used, because of the different reductive environment between the extracellular space and the cytosolic compartment. The intracellular glutathione is in the range of 1–10 mM, which is much higher than the amount in the extracellular matrix (in the micromolar range).57,58 Redox-sensitive bonds can be directly linked to DNA or RNA,50,51 bonded between HA and polycations,59–61 or cross-linked nanoparticles,10,39 leading to bond cleavage after internalization and subsequent payload release.
5. Roles of HA in gene delivery systems
HA could play a vital role in nanoparticle-based formulae for gene delivery due to its anionic characteristics and inherit physicochemical properties. As illustrated in Fig. 3, the size of HA-based nanoparticles is tunable depending on the polymer chemistry and the fabrication process, as discussed previously. The advantages of HA in improving the safety of delivery systems and the stability of products and directing to target sites are discussed below.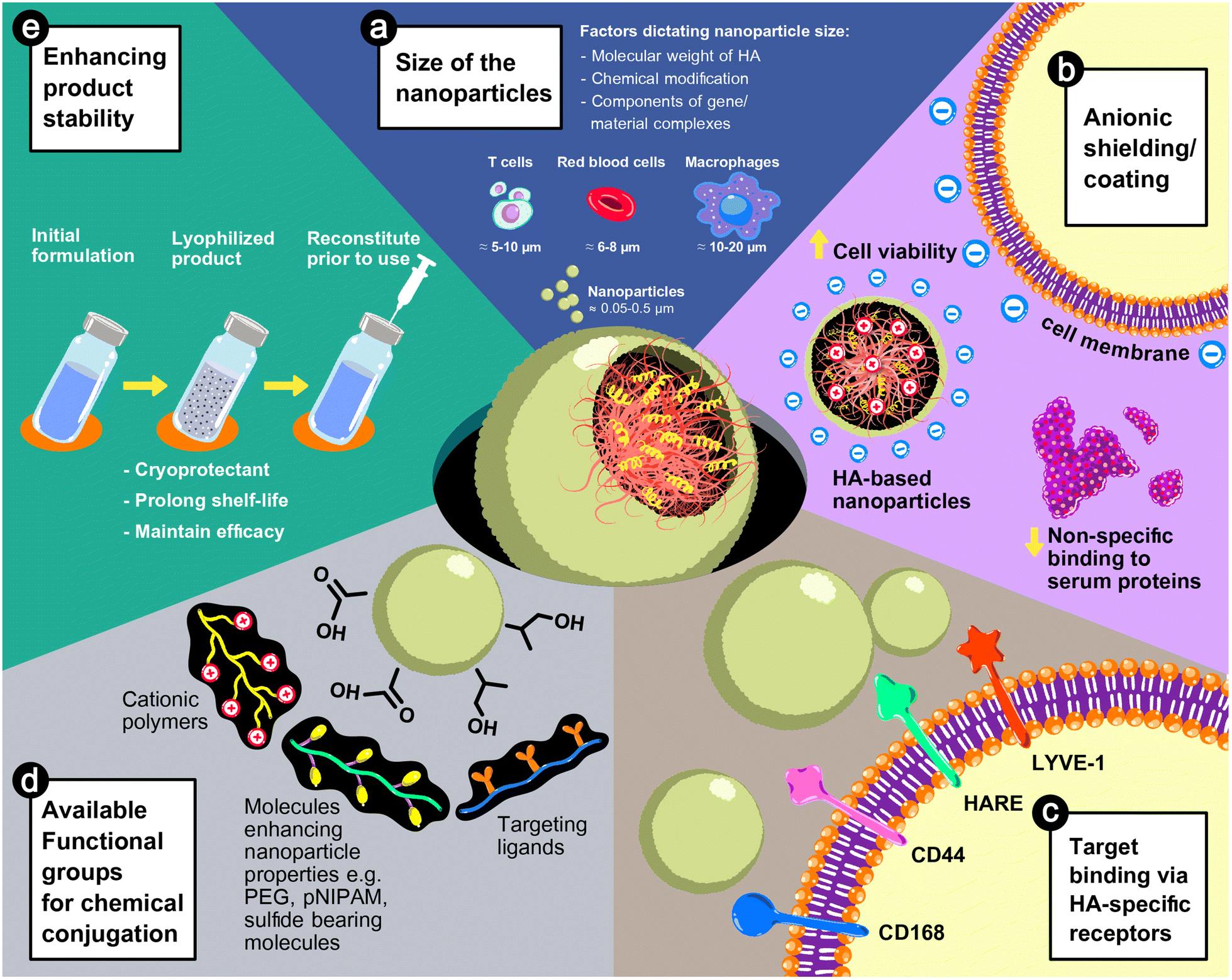 | ||
| Fig. 3 The advantages of HA in nanoparticle-based gene delivery systems: (a) the size of the nanoparticles is adjustable from 50 to 500 nm based on the physicochemical properties of the HA used in the formulations or the fabrication process. (b) The anionic characteristic of HA can reduce the cytotoxicity and non-specific interactions with serum proteins. (c) HA possesses innate properties in the recognition of cell surface receptors, e.g., CD168 or RHAMM (receptor for hyaluronan-mediated motility), CD44, HARE (hyaluronic acid receptor for endocytosis), and LYVE-1 (lymphatic vessel endothelial hyaluronic acid receptor 1). (d) The carboxyl (–COOH) and hydroxyl (–OH) groups can be chemically conjugated with functional moieties, such as cationic polymers, targeting ligands, or molecules enhancing the properties of nanoparticles (e.g., stimulus-responsiveness), and (e) HA-based nanoparticle formulations can be lyophilized, by which the shelf-life of the products is extended without damaging the efficacy of the systems. | ||
5.1. Cell viability and transfection efficiency
Higher transfection efficiency has been reported in cultured cells when the gene complexes were shielded with HA.39,53,64,65 Anionic shielding of the polyplexes can prevent particle aggregation or coagulation with serum proteins.39,43,64 Therefore, the size of the particle is uniform and suitable for uptake by cells. Indeed, the negative charge of HA can neutralize the positive charge of cationic polymers and loosen the complexes. So, the release of loaded genes from endosomes increases after internalization into the cytosol.65 Furthermore, the uptake of HA-coated nanoparticles is mediated by cell surface receptors, such as CD44, of which the transfection efficiency depends on the amount of expression and the activation state of the receptors.38,66 High expression of CD44 relates to cell pathophysiology, such as tumor progression, infection, or inflammation, and HA has been used as a ligand for targeted treatment.67 Therefore, HA-based nanoparticles have been used for broad applications, including cancer treatment,68,69 anti-inflammatory,70,71 tissue regeneration,59,72–74 and vaccine formulation.75 Target cells with high expression of CD44 are efficiently transduced from receptor-mediated internalization of HA nanoparticles.
An addition of HA to the gene complexes with cationic polymers or lipids improved the in vivo transduction due to the anionic characteristics and receptor recognition of HA. Nanoparticles with a negative surface charge show no coagulation with proteins present in the blood or extracellular spaces. Thus, the size and shape of the nanoparticles are maintained as prepared, resulting in predictable distribution kinetics.43,52 The negative charge surface can also reduce early elimination by the complement system or capture by innate immune cells. Therefore, the nanoparticles can circulate in the bloodstream for a longer period, improving the transgene expression of the target cells.36,76 Furthermore, HA can guide the nanoparticles to cells with a high expression of CD44 and mediate endocytosis through the receptors.73,77 A conjugation of extra targeting ligands to HA can reduce the delivery to non-target CD44-overexpressed cells, leading to high accumulation, and transduction activity of the target cells.37,71,78,79
5.2. Targetability of HA-based nanoparticles
A major concern is that the upregulation of CD44 is not only limited to immune cells during the inflammation state, but elevated expression of CD44 is also evidenced in various types of tumor cell, including acute lymphoblastic leukemia, acute myeloid leukemia, and multiple myeloma,91,92 which are the main targets of CAR T cell treatment. HA, as a CD44 ligand, has been used as a component in the targeting of cancer delivery systems since HA can detect tumor cells with a high expression of CD44 receptors in an activated state, before the cells internalize them to deliver chemotherapeutic agents with high specificity.68,69 Therefore, the development of HA-based delivery systems to target circulating T cells based only on the inherent CD44 binding properties of HA could not be used and could result in undesirable transduction of tumor cells. Indeed, a modification of HA for a higher specificity for T cells could be required, in which the chemical structure of HA is feasible for chemical conjugation of the targeting ligands.
5.3. HA as stability enhancers and cryoprotectants
Rapid clearance after a bolus injection of naked plasmid DNA or oligonucleotides was reported in vivo, due to the presence of circulatory nucleases as a biological barrier to prevent exogenous genetic materials,93,94 resulting in transfection failure unless the effective viral or non-viral vectors were applied. For non-viral vectors, the in vitro studies confirmed that complexation with polycations or cationic lipid nanoparticles significantly enhances stability against nucleases,36,44,71,95 which can be implied for in vivo applications.10 The addition of HA showed a greater protection effect against enzyme degradation, resulting in a higher transfection efficiency over polycations and nucleotide complexes.44,50 The stability of HA-coated nanoparticles, determined by physical characteristics, e.g., size, charge, or morphology, or transfection efficiency, was preserved at 4 °C for at least one week in a solution state,34,53 and for a longer period after lyophilization.45,74,96 From the work of Ito, T. et al.,45,96 HA can act as a cryoprotectant, allowing the nanoparticles to be lyophilized. The reconstituted nanoparticles performed a transfection efficiency similar to that of the freshly prepared nanoparticles. Hence, the HA-shielded nanoparticles can possess a long shelf-life in a lyophilized form, enabling a practical use in the clinical setting.6. Current studies on the delivery systems for in vivo CAR T cell therapy
To our knowledge, there are a few studies related to a design of a CAR-containing delivery system to target circulating T cells since the concept of CAR T cells generated in situ was recently proposed, in the past few years. Viral vectors were introduced for in vivo delivery of the CAR construct, since stable transgene expression and long-term expansion of transduced cells can be achieved.97,98 Additionally, viral vectors, namely retrovirus, lentivirus (LV) or adeno-associated virus (AAV)-based vectors, have been subjected to more than a thousand clinical trials, confirming their efficacy and safety in clinical uses.99 From the study of Nawaz, W. and colleagues, viral vectors from AAV without targeting ligands were used to package CD4 CAR genes and then administered intravenously to murine models with acute T-cell leukemia. High transduction of circulating T cells with CD4 CAR was observed along with an apparent depletion of CD4+ leukemia T cells, which was comparable to conventional ACT therapy using ex vivo reprogrammed CD4 CAR T cells.100 However, the broad tropism of the viral vectors could contribute to a non-selective cell transduction, resulting in an off-target transgene expression, a highly controversial issue when used in vivo. To date, several strategies have been applied to solve the nonspecificity issues, including a mutation of envelope proteins to ablate cell surface receptor binding or a direct conjugation of targeting ligands to the vectors. Huckaby, J. T. and colleagues developed a pseudotyping of LV with the E2 glycoprotein of Sindbis virus, of which the glycoprotein was genetically modified to eliminate its tropism, to deliver the CD19 CAR plasmid to treat CD19+ B cell lymphoma. The bispecific antibody of E2 glycoprotein and αCD3 was mixed stoichiometrically with mutant LV, and in vitro tests using activated human peripheral blood mononuclear cells (PBMC) showed a specific delivery of the CAR construct to CD3+ cells, leading to a significant reduction in B cell lymphoma tumor cell lines in coculturing systems. The efficacy was confirmed in immunodeficient mice xenografted with luciferase BV-173 tumor B cells for the luminescence tracking of tumors. Total flux, which indicates the tumor distribution, was drastically reduced in the treatment group, confirming a depletion of CD19+ B cells, as well as a high expression of CAR+/CD3+ cells, and extended overall survival.101Buchholz, C. J.'s groups also used receptor-targeted viral vectors for the in vivo delivery of CD19 CAR plasmid to circulating T cells in a treatment of B cell lymphoma. Due to the broad tropism of the viral vectors, the surface of the vectors was genetically modified by pseudotyping with structural glycoproteins of measles or Nipah virus to eliminate the innate nonspecific binding abilities or decrease the affinity to cell surface receptors of the LV or AAV vectors. Additionally, targeting ligands, CD3,102 CD4,103 and CD8,104–107 were displayed to achieve a specific recognition of circulating T cells. Interestingly, even the genome integration of viral vectors has been of great concern as mutagens, engineered viral vectors with targeting ligands, showed lower gene insertion into the host genome than conventional vectors.107 Cytotoxicity to CD19+ B cells was observed in mice that received LV vectors containing CAR genes, which shared the same effectiveness as conventional ACT treatment. Cytokine release syndrome, which is a major concern in adoptive CAR T cell therapy,7 occurred with a delayed onset in a group treated with in vivo CAR delivery, as the surge of cytokines and lymphocyte-related symptoms of the lungs, brain, spleen, and bone marrow, were slower than with ex vivo CAR T cell infusion.106 Interestingly, the vectors decorated with different antibodies possessed different functions that could lead to a rational design of the vectors. For example, LV-conjugated CD3 antibody can transduce circulating T cells without the preactivation of T cells using monoclonal antibodies or exogeneous cytokines.102 CD4-targeted vectors were found to outperform their CD8 counterparts in a faster kinetic of CAR expression and enhanced efficacy in a high tumor burden, since it was proposed to exhaust CD8+ T cells to limit their transduction. However, a combination of CD4 and CD8-lentiviral vectors resulted in the complete elimination of CD19+ B cells, while a treatment using only CD4-viral vectors could lead to tumor relapse, since a high ratio of regulatory T cells generated by the CD4 vectors can inhibit the functions of effector T cells. The addition of CD8 vectors with slower CAR expression could be beneficial in the complete eradication of tumor cells.103
Non-viral polymeric nanoparticles offered an alternative approach to avoid an off-target transduction of the viral vectors, and the nanoparticle surface can be conjugated with antibodies specific to T-cell ligands. Based on the studies by Stephan, M. T. and colleagues, nanoparticles made from poly(β-amino ester), or PBAE, and poly(glycolic acid), or PGA, were developed for the nuclear delivery of CAR pDNA108 or the cytosolic delivery of mRNA.109 Cationic PBAE was used to compact CAR-encoding genes into a nano-sized complex as well as facilitate endosomal escape to release the payload to the nucleus or cytoplasm. The complex was coated with anionic PGA to ablate non-selective binding and cytotoxicity of the cationic moieties, and the PGA was functionalized with monoclonal antibodies to target CD3+ T cells. CAR was selectively expressed solely in CD3+ T cells and tumor depletion was comparable to ex vivo T cell infusion. The advantage of CAR-containing nanoparticles is the predictable pharmacokinetics as in small molecule drugs, with the expression of CAR increasing after administration before reducing to the base line within a week. Repeated administration of non-viral vectors resulted in stable CAR expression, while a bolus injection of ACT cells could cause T cell exhaustion, suggesting reduced effectiveness.109
To date, most registered CAR T cell clinical trials have focused on blood tumors, with few studies focusing on the treatment of solid tumors.24 Since solid tumors are typically protected by a local immunosuppression environment and showed a heterogeneity of surface receptors, adoptive CAR T cells are less effective in complete elimination of the target tumors.110 From the study of Parayath, N. N. et al., mRNA-loaded PBAE–PGA nanoparticles were designed to deliver anti-ROR1 CAR constructs to treat prostate cancer using an in situ T cell programming approach.109 Mice receiving the treatment showed a low tumor clearance, which was comparable to a control group that were treated with ex vivo CAR T cells. A reduction in the expression of the ROR1 antigen of tumor cells was observed, suggesting the development of low or negative variants of ROR1 after exposure to T cells targeting ROR1. Therefore, improved strategies or combination approaches are needed to effectively tackle the hostile conditions of solid tumors to broaden the utilization of CAR T cell therapy for a wide variety of tumors.
Most of the CAR construct used for in vivo T cell programming is pDNA, which requires nuclear transport from the cytosolic compartment after escape from the endosomes. Typically, viral vectors can deliver the cargo to the nucleus with ease because of their inherent features for nuclear integration, but nuclear fusion can be a major limitation of polymeric nanoparticle systems. CAR-encoding pDNA-loaded PBAE–PGA nanoparticles require the aid of microtubule-associated sequence (MTAS) and nuclear localization signal (NLS) peptides to improve their nuclear uptake via microtubule-mediated nuclear transport mechanisms.108 Furthermore, random integration into host chromosomes after nuclear internalization could be a main limitation for the launch of products containing pDNA, which require a long-term study of genotoxicity, mutagenicity or developmental toxicity.111 Hence, mRNA, which requires a delivery and a protein translation in the cytosolic compartment, can offer a safer alternative for CAR-encoding constructs. The study of Stephan, M. T.'s groups showed that a small backbone of mRNA, compared with pDNA, resulted in a greater and faster transgene expression after transduction, leading to a controllable pharmacokinetic of CAR-expressed T cells.109
The concept of CAR T cell therapy can also be used in the treatment of diseases caused by overly activated pathological cells, such as fibrosis, in which the quiescence state of cells is disabled in maintaining a regular extracellular matrix structure, resulting in an uncontrollable production of structural proteins.112 Engineered CAR T cells against fibroblast activation protein (FAP) have been reprogrammed ex vivo for the treatment of cardiac fibrosis, and, after reinfusion into mouse hypertensive models, the cardiac functions of the injured mice were restored and remodeling of the extracellular matrix of the heart muscle was normal.113 Later, lipid nanoparticles containing CAR mRNA against FAP were engineered with a targetability to T cells using anti-CD5 antibody conjugation for in vivo reprogramming. In vitro and in vivo results using cardiac-injured mice showed that the developed nanoparticles possessed a high and selective transduction, and the reprogrammed T cells were able to ablate FAP+ cells in comparison with those of a retroviral vector. Two weeks after the administration of the nanoparticles to the mice, heart functions and overall extracellular matrix burdens had improved and were almost comparable to normal anatomy and physiological conditions.114
7. Challenges and future perspectives
The conditions of the patients dictate the feasibility of in situ CAR T cell therapy, of which the effectiveness of the treatment depends on sufficient and functional T cells. Patients with a low T cell count or lymphocytopenia could not receive the treatment.109 Additionally, activated T cells are needed for CAR programming, which is not limited to the ex vivo transduction of T cells. Conjugation of anti-CD3 to vectors can prime circulating T cells, but with much lower activity than the pre-administration of soluble anti-CD3 antibody.102 Pretreatment can be an injection of anti-CD3 or CD28 monoclonal antibodies or exogenous IL-2, IL-7 or IL-15.102,105,115 Furthermore, the expression of CD44 receptors in T cells after activation increases,85 which could be beneficial for the uptake of HA-decorated nanoparticles.The major concern of the utilization of HA-based nanoparticles in in vivo engineered CAR T cells is the retention time in the bloodstream after IV administration to allow a sufficient transfection period for the circulated T cells. Since a rapid clearance from the circulatory system was reported in the first few hours, the nanoparticles subsequently accumulated in tumor tissues and major organs, such as the lungs, spleen, liver, and kidneys.52 Furthermore, macrophages and leukocytes are known as CD44-overexpressing cells55,73,80 in which HA nanoparticles presented in the blood can be phagocytosed and rapidly eliminated through reticuloendothelial systems. An approach to increase the retention time is the PEGylation of nanoparticles to acquire stealth properties for higher systemic distribution and lower clearance.52,55 PEG can be simply mixed with HA for shielding nanoparticles55 or conjugated to the HA backbone together with polyamine conjugation.52,53,56 Therefore, an extended systemic distribution can increase the exposure period to T cells, resulting in a greater chance of delivering entrapped genetic materials to engineer the targeted T lymphocytes.
With direct administration to the bloodstream, the nanoparticles undergo a rapid elimination, resulting in an insufficient retention time and a low transfection efficiency. Gene-containing nanoparticles can be immobilized to the solid substrate, and the gene-containing prosthesis can be implanted. Hence, circulating cells can be latched onto substrates and the cell can internalize the embedded gene cargos. This approach is called ‘solid phase reverse transfection’, which has been widely applied in in vitro assay platforms116,117 and tissue-engineered scaffolds.118 From a proof-of-concept study by Agarwalla, P. and colleagues using modified alginate scaffolds loaded with retroviral vectors containing CD19 CAR-encoding genes, in vitro results demonstrated an equivalent amount of CAR transduced cells compared with the conventional approach as well as a non-inferior capability in tumor cell eradication. In vivo generation of CAR T cells using subcutaneous implants in tumor xenograft mice showed sufficient tumor-lytic effects on CD19+ tumor cells, and mice exhibited an equal survival rate to those treated with conventional ACT therapy.119
The immobilization of the gene-delivery platform could not only reduce the clearance from the circulatory system compared with injectable nanoparticles, but the transfection efficiency was also found to be higher than that of the conventional transfection method due to higher efficiency in the nuclear uptake of loaded genes.120 HA-coated pDNA polyplexes were grafted to the surface of metallic implants121 or embedded in resorbable scaffolds,74 and it was found that a higher transfection efficiency can be achieved. HA can reduce the cytotoxicity of polyplexes and prevent aggregation when exposed to serum proteins. Indeed, coating the pDNA complexes on a substrate facilitated the cells to be exposed to pDNA at a higher concentration and for a longer period than an exposure to the pDNA in the solution since low cell transduction has been reported to be improved by a prolonged exposure time of cells to gene complexes.122 Furthermore, embedding transfection nanoparticles in biodegradable matrices could allow a sustained release of the genetic materials, avoiding a repeated administration, for extended transgene expression.74
The clinical translation of CAR loaded nanoparticles could take a long time, as they are needed to fulfill the considerations of both nanomaterials and products containing genetic materials, which are classified as advanced therapy medicinal products, or ATMPs. In addition to confirming proposed indications of the products, including preclinical proof-of-concept efficacy, a dose–response relationship, and a level of transgene expression, as well as a controlled quality of product manufacturing,123 toxicities, including infusion reactions, immune-mediated responses, and genotoxicity, which could be transient or persistent, are of great concern. In an aspect of nanomaterial considerations, in vitro and in vivo models are required to evaluate immune compatibility after administration to confirm the feasibility of the products, such as hemocompatibility tests, complement activation, oxidative stress to T cells, and the suppression of immune cells or bone marrow,109,124 which can refer to protocols developed by the Nanotechnology Characterization Laboratory (NCL) (reviewed in ref. 125 and 126). Furthermore, genome alteration due to the nuclear internalization of exogenous genetic materials mediated by viral or non-viral vectors should be addressed, and preclinical tests are required to confirm genotoxicity, mutagenicity, and sustained genetic modification, which could affect reproductive systems or the development of descendants.123,127 Hence, researchers working on the development of delivery systems for CAR constructs should consider not only proving the efficacy but also ensuring the biocompatibility of the systems, which could pave the way to accelerate the clinical translation of the developed systems.
8. Conclusion
In vivo CAR T cell engineering showed equivalent efficacy to ACT in a preclinical evaluation for the treatment of hematologic malignancies. Lentiviral vectors decorated with T cell targeting ligands were developed to abrogate their broad tropism and avoid off-target delivery of the gene cargo. However, a risk of insertional mutagenesis, a relapse of pathogenic variants, and persistent genomic alteration could raise awareness and delay the clinical translation of viral vectors. Non-viral, polymeric nanoparticles offer a safer alternative. Cationic polymers, such as PEI, PBAE, chitosan, or lipids, are used to condense DNA or RNA and facilitate the release of payloads from the endosomes to the cytosolic compartment. Typically, the complexes are electrostatically coated or chemically conjugated with anionic polymers to reduce a non-specific binding to the cell surface or serum proteins, as well as enhance the biocompatibility of the systems. To date, only CAR-loaded nanoparticles using PBAE and coated with antibody-conjugated PGA have been introduced for in vivo T cell engineering. On the basis of the same approach, HA can also be used for the fabrication of nanoparticles targeting T cells, due to its negative charge, which enables counterion conjugation to the cationic core polymers. The available carboxyl groups of HA allow for polymer functionalization and a chemical bonding to the targeting antibodies of T cells. More importantly, the inherent properties of HA as a ligand of cell surface receptors, such as CD44, could improve the targetability to T cells, due to elevated HA-binding CD44 receptors of T cells in correspondence to immunological activation. Hence, HA-based nanoparticles demonstrate promising platforms for the delivery of CAR constructs specifically to circulating T cells for the in vivo programming of CAR T cell therapy.Conflicts of interest
J.A.L. is the co-founder of Nabsolute Co., Ltd., Bangkok, Thailand.Acknowledgements
This research is funded by Chulalongkorn University (grant number: ReinUni_65_03_33_20). Additionally, this research was partially supported by the Asahi Glass Foundation (grant number: RES_65_530_33_026). This research project is supported by the Second Century Fund (C2F), Chulalongkorn University.References
- C. H. June, Chapter 30 - Adoptive Cellular Therapy With Synthetic T Cells as an “Instant Vaccine” for Cancer and Immunity, in The Vaccine Book, ed. B. R. Bloom and P.-H. Lambert, Academic Press, 2nd edn, 2016, pp. 581–596 Search PubMed.
- X. Wang and M. I. Nishimura, Chapter 18 - Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy, in Gene Therapy of Cancer, ed. E. C. Lattime and S. L. Gerson, Academic Press, San Diego, 3rd edn, 2014, pp. 259–271 Search PubMed.
- A. Mullard, FDA approves fourth CAR-T cell therapy, Nat. Rev. Drug Discovery, 2021, 20(3), 166–166 Search PubMed.
- N. N. Parayath and M. T. Stephan, In Situ Programming of CAR T Cells, Annu. Rev. Biomed. Eng., 2021, 23(1), 385–405 CrossRef PubMed.
- S. Guedan, H. Calderon, A. D. Posey and M. V. Maus, Engineering and Design of Chimeric Antigen Receptors, Mol. Ther.–Methods Clin. Dev., 2019, 12, 145–156 CrossRef PubMed.
- R. Mhaidly and E. Verhoeyen, The Future: In Vivo CAR T Cell Gene Therapy, Mol. Ther., 2019, 27(4), 707–709 CrossRef PubMed.
- C. L. Bonifant, H. J. Jackson, R. J. Brentjens and K. J. Curran, Toxicity and management in CAR T-cell therapy, Mol. Ther.–Oncolytics, 2016, 3, 16011 CrossRef CAS PubMed.
- T. Xin, L. Cheng, C. Zhou, Y. Zhao, Z. Hu and X. Wu, In vivo Induced CAR-T Cell for the Potential Breakthrough to Overcome the Barriers of Current CAR-T Cell Therapy, Front. Oncol., 2022, 12, 1–11 Search PubMed.
- J. K. Vasir and V. Labhasetwar, Polymeric nanoparticles for gene delivery, Expert Opin. Drug Delivery, 2006, 3(3), 325–344 CrossRef CAS PubMed.
- H. Y. Yoon, H. R. Kim, G. Saravanakumar, R. Heo, S. Y. Chae, W. Um, K. Kim, I. C. Kwon, J. Y. Lee, D. S. Lee, J. C. Park and J. H. Park, Bioreducible hyaluronic acid conjugates as siRNA carrier for tumor targeting, J. Controlled Release, 2013, 172(3), 653–661 CrossRef CAS PubMed.
- R. Rotem, D. Prosperi and M. Colombo, Chapter 10 - Targeted delivery of nanoparticles, in Frontiers of Nanoscience, ed. W. J. Parak and N. Feliu, Elsevier, 2020, pp. 253–264 Search PubMed.
- P. M. Cevaal, A. Ali, E. Czuba-Wojnilowicz, J. Symons, S. R. Lewin, C. Cortez-Jugo and F. Caruso, In Vivo T Cell-Targeting Nanoparticle Drug Delivery Systems: Considerations for Rational Design, ACS Nano, 2021, 15(3), 3736–3753 CrossRef CAS PubMed.
- S. S. M. Lee-Sayer, Y. Dong, A. A. Arif, M. Olsson, K. L. Brown and P. Johnson, The where, when, how, and why of hyaluronan binding by immune cells, Front. Immunol., 2015, 6, 150–150 Search PubMed.
- B. V. Kumar, T. J. Connors and D. L. Farber, Human T Cell Development, Localization, and Function throughout Life, Immunity, 2018, 48(2), 202–213 CrossRef CAS PubMed.
- M. M. A. Abdel-Mottaleb, H. Abd-Allah, R. I. El-Gogary and M. Nasr, Chapter 1 - Versatile hyaluronic acid nanoparticles for improved drug delivery, in Drug Delivery Aspects, ed. R. Shegokar, Elsevier, 2020, pp. 1–18 Search PubMed.
- D. Schreiber Robert, J. Old Lloyd and J. Smyth Mark, Cancer Immunoediting: Integrating Immunity's Roles in Cancer Suppression and Promotion, Science, 2011, 331(6024), 1565–1570 CrossRef PubMed.
- D. Mittal, M. M. Gubin, R. D. Schreiber and M. J. Smyth, New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape, Curr. Opin. Immunol., 2014, 27, 16–25 CrossRef PubMed.
- C. Hayes, Cellular immunotherapies for cancer, Ir. J. Med. Sci., 2021, 190(1), 41–57 CrossRef PubMed.
- R. Saleh and E. Elkord, Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression, Semin. Cancer Biol., 2020, 65, 13–27 CrossRef PubMed.
- S. A. Rosenberg and N. P. Restifo, Adoptive cell transfer as personalized immunotherapy for human cancer, Science, 2015, 348(6230), 62–68 CrossRef PubMed.
- Y. Liu, X. Yan, F. Zhang, X. Zhang, F. Tang, Z. Han and Y. Li, TCR-T Immunotherapy: The Challenges and Solutions, Front. Oncol., 2022, 11, 1–14 CAS.
- Q. Zhao, Y. Jiang, S. Xiang, P. J. Kaboli, J. Shen, Y. Zhao, X. Wu, F. Du, M. Li, C. H. Cho, J. Li, Q. Wen, T. Liu, T. Yi and Z. Xiao, Engineered TCR-T Cell Immunotherapy in Anticancer Precision Medicine: Pros and Cons, Front. Immunol., 2021, 12, 1–12 Search PubMed.
- H. June Carl, S. O'Connor Roddy, U. Kawalekar Omkar, S. Ghassemi and C. Milone Michael, CAR T cell immunotherapy for human cancer, Science, 2018, 359(6382), 1361–1365 CrossRef PubMed.
- A. Holzinger, M. Barden and H. Abken, The growing world of CAR T cell trials: a systematic review, Cancer Immunology, Immunotherapy, 2016, 65(12), 1433–1450 Search PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Design and development of polymers for gene delivery, Nat. Rev. Drug Discovery, 2005, 4(7), 581–593 CrossRef PubMed.
- S. Kawakami, Y. Higuchi and M. Hashida, Nonviral approaches for targeted delivery of plasmid DNA and oligonucleotide, J. Pharm. Sci., 2008, 97(2), 726–745 CrossRef PubMed.
- M. A. Liu and J. B. Ulmer, Human Clinical Trials of Plasmid DNA Vaccines, Advances in Genetics, Academic Press, 2005, pp. 25–40 Search PubMed.
- A. L. Jackson and P. S. Linsley, Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application, Nat. Rev. Drug Discovery, 2010, 9(1), 57–67 CrossRef CAS PubMed.
- D. Ibraheem, A. Elaissari and H. Fessi, Gene therapy and DNA delivery systems, Int. J. Pharm., 2014, 459(1), 70–83 CrossRef CAS PubMed.
- Y. H. Chung, V. Beiss, S. N. Fiering and N. F. Steinmetz, COVID-19 Vaccine Frontrunners and Their Nanotechnology Design, ACS Nano, 2020, 14(10), 12522–12537 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, PEGylation as a strategy for improving nanoparticle-based drug and gene delivery, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- G. Huang and H. Huang, Hyaluronic acid-based biopharmaceutical delivery and tumor-targeted drug delivery system, J. Controlled Release, 2018, 278, 122–126 CrossRef CAS PubMed.
- G. Kogan, L. Šoltés, R. Stern and P. Gemeiner, Hyaluronic acid: a natural biopolymer with a broad range of biomedical and industrial applications, Biotechnol. Lett., 2007, 29(1), 17–25 CrossRef CAS PubMed.
- A. Tirella, K. Kloc-Muniak, L. Good, J. Ridden, M. Ashford, S. Puri and N. Tirelli, CD44 targeted delivery of siRNA by using HA-decorated nanotechnologies for KRAS silencing in cancer treatment, Int. J. Pharm., 2019, 561, 114–123 CrossRef CAS PubMed.
- J. S. Park, S. W. Yi, H. J. Kim and K.-H. Park, Receptor-mediated gene delivery into human mesenchymal stem cells using hyaluronic acid-shielded polyethylenimine/pDNA nanogels, Carbohydr. Polym., 2016, 136, 791–802 CrossRef CAS.
- Q. Sun, Z. Kang, L. Xue, Y. Shang, Z. Su, H. Sun, Q. Ping, R. Mo and C. Zhang, A Collaborative Assembly Strategy for Tumor-Targeted siRNA Delivery, J. Am. Chem. Soc., 2015, 137(18), 6000–6010 CrossRef CAS PubMed.
- C. Yang, Y. Fu, C. Huang, D. Hu, K. Zhou, Y. Hao, B. Chu, Y. Yang and Z. Qian, Chlorin e6 and CRISPR-Cas9 dual-loading system with deep penetration for a synergistic tumoral photodynamic-immunotherapy, Biomaterials, 2020, 255, 120194 CrossRef PubMed.
- X. Sun, P. Ma, X. Cao, L. Ning, Y. Tian and C. Ren, Positive hyaluronan/PEI/DNA complexes as a target-specific intracellular delivery to malignant breast cancer, Drug Delivery, 2009, 16(7), 357–362 CrossRef PubMed.
- Y. He, G. Cheng, L. Xie, Y. Nie, B. He and Z. Gu, Polyethyleneimine/DNA polyplexes with reduction-sensitive hyaluronic acid derivatives shielding for targeted gene delivery, Biomaterials, 2013, 34(4), 1235–1245 CrossRef PubMed.
- H. Yin, F. Zhao, D. Zhang and J. Li, Hyaluronic acid conjugated β-cyclodextrin-oligoethylenimine star polymer for CD44-targeted gene delivery, Int. J. Pharm., 2015, 483(1), 169–179 CrossRef PubMed.
- M. de la Fuente, B. a. Seijo and M. J. Alonso, Novel Hyaluronic Acid-Chitosan Nanoparticles for Ocular Gene Therapy, Invest. Ophthalmol. Visual Sci., 2008, 49(5), 2016–2024 CrossRef PubMed.
- K.-M. Choi, M. Jang, J. H. Kim and H. J. Ahn, Tumor-specific delivery of siRNA using supramolecular assembly of hyaluronic acid nanoparticles and 2b RNA-binding protein/siRNA complexes, Biomaterials, 2014, 35(25), 7121–7132 CrossRef CAS PubMed.
- T. Ito, N. Iida-Tanaka and Y. Koyama, Efficient in vivo gene transfection by stable DNA/PEI complexes coated by hyaluronic acid, J. Drug Targeting, 2008, 16(4), 276–281 CrossRef CAS PubMed.
- M. S. Lee, J. E. Lee, E. Byun, N. W. Kim, K. Lee, H. Lee, S. J. Sim, D. S. Lee and J. H. Jeong, Target-specific delivery of siRNA by stabilized calcium phosphate nanoparticles using dopa–hyaluronic acid conjugate, J. Controlled Release, 2014, 192, 122–130 CrossRef CAS PubMed.
- T. Ito, C. Yoshihara, K. Hamada and Y. Koyama, DNA/polyethyleneimine/hyaluronic acid small complex particles and tumor suppression in mice, Biomaterials, 2010, 31(10), 2912–2918 CrossRef CAS PubMed.
- J. Zhu, M. Guo, Y. Cui, Y. Meng, J. Ding, W. Zeng and W. Zhou, Surface Coating of Pulmonary siRNA Delivery Vectors Enabling Mucus Penetration, Cell Targeting, and Intracellular Radical Scavenging for Enhanced Acute Lung Injury Therapy, ACS Appl. Mater. Interfaces, 2022, 14(4), 5090–5100 CrossRef CAS PubMed.
- R. V. Benjaminsen, M. A. Mattebjerg, J. R. Henriksen, S. M. Moghimi and T. L. Andresen, The Possible “Proton Sponge” Effect of Polyethylenimine (PEI) Does Not Include Change in Lysosomal pH, Mol. Ther., 2013, 21(1), 149–157 CrossRef CAS PubMed.
- E. Lallana, J. M. Rios de la Rosa, A. Tirella, M. Pelliccia, A. Gennari, I. J. Stratford, S. Puri, M. Ashford and N. Tirelli, Chitosan/Hyaluronic Acid Nanoparticles: Rational Design Revisited for RNA Delivery, Mol. Pharm., 2017, 14(7), 2422–2436 CrossRef CAS PubMed.
- H. Yan, O. P. Oommen, D. Yu, J. Hilborn, H. Qian and O. P. Varghese, Chondroitin Sulfate-Coated DNA-Nanoplexes Enhance Transfection Efficiency by Controlling Plasmid Release from Endosomes: A New Insight into Modulating Nonviral Gene Transfection, Adv. Funct. Mater., 2015, 25(25), 3907–3915 CrossRef CAS.
- H. Lee, H. Mok, S. Lee, Y.-K. Oh and T. G. Park, Target-specific intracellular delivery of siRNA using degradable hyaluronic acid nanogels, J. Controlled Release, 2007, 119(2), 245–252 CrossRef CAS PubMed.
- H. Mok, J. W. Park and T. G. Park, Antisense Oligodeoxynucleotide-Conjugated Hyaluronic Acid/Protamine Nanocomplexes for Intracellular Gene Inhibition, Bioconjugate Chem., 2007, 18(5), 1483–1489 CrossRef CAS PubMed.
- S. Ganesh, A. K. Iyer, F. Gattacceca, D. V. Morrissey and M. M. Amiji, In vivo biodistribution of siRNA and cisplatin administered using CD44-targeted hyaluronic acid nanoparticles, J. Controlled Release, 2013, 172(3), 699–706 CrossRef CAS PubMed.
- H. M. Aldawsari, H. K. Dhaliwal, B. M. Aljaeid, N. A. Alhakamy, Z. M. Banjar and M. M. Amiji, Optimization of the Conditions for Plasmid DNA Delivery and Transfection with Self-Assembled Hyaluronic Acid-Based Nanoparticles, Mol. Pharm., 2019, 16(1), 128–140 CrossRef CAS.
- S. Ganesh, A. K. Iyer, D. V. Morrissey and M. M. Amiji, Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors, Biomaterials, 2013, 34(13), 3489–3502 CrossRef CAS PubMed.
- H. S. S. Qhattal, T. Hye, A. Alali and X. Liu, Hyaluronan Polymer Length, Grafting Density, and Surface Poly(ethylene glycol) Coating Influence in Vivo Circulation and Tumor Targeting of Hyaluronan-Grafted Liposomes, ACS Nano, 2014, 8(6), 5423–5440 CrossRef CAS PubMed.
- X. Yang, A. K. Iyer, A. Singh, L. Milane, E. Choy, F. J. Hornicek, M. M. Amiji and Z. Duan, Cluster of Differentiation 44 Targeted Hyaluronic Acid Based Nanoparticles for MDR1 siRNA Delivery to Overcome Drug Resistance in Ovarian Cancer, Pharm. Res., 2015, 32(6), 2097–2109 CrossRef PubMed.
- H. J. Forman, H. Zhang and A. Rinna, Glutathione: overview of its protective roles, measurement, and biosynthesis, Mol. Aspects Med., 2009, 30(1–2), 1–12 CrossRef PubMed.
- G. Saito, J. A. Swanson and K.-D. Lee, Drug delivery strategy utilizing conjugation via reversible disulfide linkages: role and site of cellular reducing activities, Adv. Drug Delivery Rev., 2003, 55(2), 199–215 CrossRef PubMed.
- P.-H. Yeh, J.-S. Sun, H.-C. Wu, L.-H. Hwang and T.-W. Wang, Stimuli-responsive HA-PEI nanoparticles encapsulating endostatin plasmid for stem cell gene therapy, RSC Adv., 2013, 3(31), 12922–12932 RSC.
- K. Park, M.-Y. Lee, K. S. Kim and S. K. Hahn, Target specific tumor treatment by VEGF siRNA complexed with reducible polyethyleneimine-hyaluronic acid conjugate, Biomaterials, 2010, 31(19), 5258–5265 CrossRef.
- D. Chen, P. Zhang, M. Li, C. Li, X. Lu, Y. Sun and K. Sun, Hyaluronic acid-modified redox-sensitive hybrid nanocomplex loading with siRNA for non-small-cell lung carcinoma therapy, Drug Delivery, 2022, 29(1), 574–587 CrossRef CAS PubMed.
- S. J. Gwak, J. K. Jung, S. S. An, H. J. Kim, J. S. Oh, W. A. Pennant, H. Y. Lee, M. H. Kong, K. N. Kim, D. H. Yoon and Y. Ha, Chitosan/TPP-hyaluronic acid nanoparticles: a new vehicle for gene delivery to the spinal cord, J. Biomater. Sci., Polym. Ed., 2012, 23(11), 1437–1450 CrossRef CAS PubMed.
- L. Gan, J. Wang, Y. Zhao, D. Chen, C. Zhu, J. Liu and Y. Gan, Hyaluronan-modified core–shell liponanoparticles targeting CD44-positive retinal pigment epithelium cells via intravitreal injection, Biomaterials, 2013, 34(24), 5978–5987 CrossRef CAS PubMed.
- G. Jiang, K. Park, J. Kim, K. S. Kim and S. K. Hahn, Target Specific Intracellular Delivery of siRNA/PEI–HA Complex by Receptor Mediated Endocytosis, Mol. Pharm., 2009, 6(3), 727–737 CrossRef CAS PubMed.
- T. Ito, N. Iida-Tanaka, T. Niidome, T. Kawano, K. Kubo, K. Yoshikawa, T. Sato, Z. Yang and Y. Koyama, Hyaluronic acid and its derivative as a multi-functional gene expression enhancer: Protection from non-specific interactions, adhesion to targeted cells, and transcriptional activation, J. Controlled Release, 2006, 112(3), 382–388 CrossRef CAS.
- A. Spadea, J. M. Rios de la Rosa, A. Tirella, M. B. Ashford, K. J. Williams, I. J. Stratford, N. Tirelli and M. Mehibel, Evaluating the Efficiency of Hyaluronic Acid for Tumor Targeting via CD44, Mol. Pharm., 2019, 16(6), 2481–2493 CrossRef CAS PubMed.
- A. R. Jordan, R. R. Racine, M. J. P. Hennig and V. B. Lokeshwar, The Role of CD44 in Disease Pathophysiology and Targeted Treatment, Front. Immunol., 2015, 6, 182–182 Search PubMed.
- J. H. Kim, M. J. Moon, D. Y. Kim, S. H. Heo and Y. Y. Jeong, Hyaluronic Acid-Based Nanomaterials for Cancer Therapy, Polymer, 2018, 10(10), 1–15 CrossRef.
- S. Y. Lee, M. S. Kang, W. Y. Jeong, D. W. Han and K. S. Kim, Hyaluronic Acid-Based Theranostic Nanomedicines for Targeted Cancer Therapy, Cancers, 2020, 12(4), 1–17 Search PubMed.
- R.-H. Deng, B. Qiu and P.-H. Zhou, Chitosan/hyaluronic acid/plasmid-DNA nanoparticles encoding interleukin-1 receptor antagonist attenuate inflammation in synoviocytes induced by interleukin-1 beta, J. Mater. Sci.: Mater. Med., 2018, 29(10), 155 CrossRef PubMed.
- D. Peer, E. J. Park, Y. Morishita, C. V. Carman and M. Shimaoka, Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target, Science, 2008, 319(5863), 627–630 CrossRef PubMed.
- G. Wu, C. Feng, G. Hui, Z. Wang, J. Tan, L. Luo, P. Xue, Q. Wang and X. Chen, Improving the osteogenesis of rat mesenchymal stem cells by chitosan-based-microRNA nanoparticles, Carbohydr. Polym., 2016, 138, 49–58 CrossRef PubMed.
- T.-H. Tran, R. Rastogi, J. Shelke and M. M. Amiji, Modulation of Macrophage Functional Polarity towards Anti-Inflammatory Phenotype with Plasmid DNA Delivery in CD44 Targeting Hyaluronic Acid Nanoparticles, Sci. Rep., 2015, 5(1), 16632 CrossRef CAS PubMed.
- H. Lu, L. Lv, Y. Dai, G. Wu, H. Zhao and F. Zhang, Porous Chitosan Scaffolds with Embedded Hyaluronic Acid/Chitosan/Plasmid-DNA Nanoparticles Encoding TGF-β1 Induce DNA Controlled Release, Transfected Chondrocytes, and Promoted Cell Proliferation, PLoS One, 2013, 8(7), e69950 CrossRef CAS PubMed.
- X. Liang, X. Li, J. Duan, Y. Chen, X. Wang, L. Pang, D. Kong, B. Song, C. Li and J. Yang, Nanoparticles with CD44 Targeting and ROS Triggering Properties as Effective in Vivo Antigen Delivery System, Mol. Pharm., 2018, 15(2), 508–518 CrossRef CAS PubMed.
- T. Wan, Y. Chen, Q. Pan, X. Xu, Y. Kang, X. Gao, F. Huang, C. Wu and Y. Ping, Genome editing of mutant KRAS through supramolecular polymer-mediated delivery of Cas9 ribonucleoprotein for colorectal cancer therapy, J. Controlled Release, 2020, 322, 236–247 CrossRef CAS PubMed.
- Z. R. Cohen, S. Ramishetti, N. Peshes-Yaloz, M. Goldsmith, A. Wohl, Z. Zibly and D. Peer, Localized RNAi Therapeutics of Chemoresistant Grade IV Glioma Using Hyaluronan-Grafted Lipid-Based Nanoparticles, ACS Nano, 2015, 9(2), 1581–1591 CrossRef PubMed.
- X.-Y. He, X.-H. Ren, Y. Peng, J.-P. Zhang, S.-L. Ai, B.-Y. Liu, C. Xu and S.-X. Cheng, Aptamer/Peptide-Functionalized Genome-Editing System for Effective Immune Restoration through Reversal of PD-L1-Mediated Cancer Immunosuppression, Adv. Mater., 2020, 32(17), 2000208 CrossRef PubMed.
- X.-H. Ren, X.-Y. He, B.-Y. Liu, C. Xu and S.-X. Cheng, Self-Assembled Plasmid Delivery System for PPM1D Knockout to Reverse Tumor Malignancy, ACS Appl. Bio Mater., 2020, 3(11), 7831–7839 CrossRef PubMed.
- D. Jiang, J. Liang and P. W. Noble, Hyaluronan as an immune regulator in human diseases, Physiol. Rev., 2011, 91(1), 221–264 CrossRef PubMed.
- S. Misra, V. C. Hascall, R. R. Markwald and S. Ghatak, Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer, Front. Immunol., 2015, 6, 201 Search PubMed.
- J. M. Rios de la Rosa, P. Pingrajai, M. Pelliccia, A. Spadea, E. Lallana, A. Gennari, I. J. Stratford, W. Rocchia, A. Tirella and N. Tirelli, Binding and Internalization in Receptor-Targeted Carriers: The Complex Role of CD44 in the Uptake of Hyaluronic Acid-Based Nanoparticles (siRNA Delivery), Adv. Healthcare Mater., 2019, 8(24), 1901182 CrossRef CAS.
- G. Bachar, K. Cohen, R. Hod, R. Feinmesser, A. Mizrachi, T. Shpitzer, O. Katz and D. Peer, Hyaluronan-grafted particle clusters loaded with Mitomycin C as selective nanovectors for primary head and neck cancers, Biomaterials, 2011, 32(21), 4840–4848 CrossRef CAS PubMed.
- N. Maeshima, G. F. T. Poon, M. Dosanjh, J. Felberg, S. S. M. Lee, J. L. Cross, D. Birkenhead and P. Johnson, Hyaluronan binding identifies the most proliferative activated and memory T cells, Eur. J. Immunol., 2011, 41(4), 1108–1119 CrossRef CAS PubMed.
- H. C. DeGrendele, M. Kosfiszer, P. Estess and M. H. Siegelman, CD44 activation and associated primary adhesion is inducible via T cell receptor stimulation, J. Immunol., 1997, 159(6), 2549 CAS.
- J. Lesley, N. Howes, A. Perschl and R. Hyman, Hyaluronan binding function of CD44 is transiently activated on T cells during an in vivo immune response, J. Exp. Med., 1994, 180(1), 383–387 CrossRef CAS PubMed.
- S. P. Evanko, S. Potter-Perigo, P. L. Bollyky, G. T. Nepom and T. N. Wight, Hyaluronan and versican in the control of human T-lymphocyte adhesion and migration, Matrix Biol., 2012, 31(2), 90–100 CrossRef CAS PubMed.
- R. L. Camp, A. Scheynius, C. Johansson and E. Puré, CD44 is necessary for optimal contact allergic responses but is not required for normal leukocyte extravasation, J. Exp. Med., 1993, 178(2), 497–507 CrossRef CAS PubMed.
- H. C. DeGrendele, P. Estess and M. H. Siegelman, Requirement for CD44 in Activated T Cell Extravasation into an Inflammatory Site, Science, 1997, 278(5338), 672–675 CrossRef CAS PubMed.
- A. Ariel, O. Lider, A. Brill, L. Cahalon, N. Savion, D. Varon and R. Hershkoviz, Induction of interactions between CD44 and hyaluronic acid by a short exposure of human T cells to diverse pro-inflammatory mediators, Immunology, 2000, 100(3), 345–351 CrossRef PubMed.
- L. J. Bendall, K. F. Bradstock and D. J. Gottlieb, Expression of CD44 variant exons in acute myeloid leukemia is more common and more complex than that observed in normal blood, bone marrow or CD34+ cells, Leukemia, 2000, 14(7), 1239–1246 CrossRef PubMed.
- P. Liebisch, S. Eppinger, C. Schöpflin, G. Stehle, G. Munzert, H. Döhner and M. Schmid, CD44v6, a target for novel antibody treatment approaches, is frequently expressed in multiple myeloma and associated with deletion of chromosome arm 13q, Haematologica, 2005, 90(4), 489–493 Search PubMed.
- K. Kawabata, Y. Takakura and M. Hashida, The Fate of Plasmid DNA After Intravenous Injection in Mice: Involvement of Scavenger Receptors in Its Hepatic Uptake, Pharm. Res., 1995, 12(6), 825–830 CrossRef PubMed.
- S. Akhtar and S. Agrawal, In vivo studies with antisense oligonucleotides, Trends Pharmacol. Sci., 1997, 18(1), 12–18 CrossRef PubMed.
- K. Hagiwara, M. Nakata, Y. Koyama and T. Sato, The effects of coating pDNA/chitosan complexes with chondroitin sulfate on physicochemical characteristics and cell transfection, Biomaterials, 2012, 33(29), 7251–7260 CrossRef CAS PubMed.
- T. Ito, Y. Koyama and M. Otsuka, Preparation of Calcium Phosphate Nanocapsule Including Deoxyribonucleic Acid-Polyethyleneimine-Hyaluronic Acid Ternary Complex for Durable Gene Delivery, J. Pharm. Sci., 2014, 103(1), 179–184 CrossRef CAS PubMed.
- S. Bobisse, P. Zanovello and A. Rosato, T-cell receptor gene transfer by lentiviral vectors in adoptive cell therapy, Expert Opin. Biol. Ther., 2007, 7(6), 893–906 CrossRef CAS PubMed.
- B. L. Levine, J. Miskin, K. Wonnacott and C. Keir, Global Manufacturing of CAR T Cell Therapy, Mol. Ther.–Methods Clin. Dev., 2017, 4, 92–101 CrossRef CAS PubMed.
- J. T. Bulcha, Y. Wang, H. Ma, P. W. L. Tai and G. Gao, Viral vector platforms within the gene therapy landscape, Signal Transduction Targeted Ther., 2021, 6(1), 53 CrossRef CAS PubMed.
- W. Nawaz, B. Huang, S. Xu, Y. Li, L. Zhu, H. Yiqiao, Z. Wu and X. Wu, AAV-mediated in vivo CAR gene therapy for targeting human T-cell leukemia, Blood Cancer J., 2021, 11(6), 119 CrossRef PubMed.
- J. T. Huckaby, E. Landoni, T. M. Jacobs, B. Savoldo, G. Dotti and S. K. Lai, Bispecific binder redirected lentiviral vector enables in vivo engineering of CAR-T cells, J. Immunother. Cancer, 2021, 9(9), e002737 CrossRef PubMed.
- A. M. Frank, A. H. Braun, L. Scheib, S. Agarwal, I. C. Schneider, F. Fusil, S. Perian, U. Sahin, F. B. Thalheimer, E. Verhoeyen and C. J. Buchholz, Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors, Blood Adv., 2020, 4(22), 5702–5715 CAS.
- S. Agarwal, J. D. S. Hanauer, A. M. Frank, V. Riechert, F. B. Thalheimer and C. J. Buchholz, In Vivo Generation of CAR T Cells Selectively in Human CD4(+) Lymphocytes, Mol. Ther., 2020, 28(8), 1783–1794 CrossRef CAS PubMed.
- S. Agarwal, T. Weidner, F. B. Thalheimer and C. J. Buchholz, In vivo generated human CAR T cells eradicate tumor cells, OncoImmunology, 2019, 8(12), e1671761 CrossRef PubMed.
- A. Jamali, L. Kapitza, T. Schaser, I. C. D. Johnston, C. J. Buchholz and J. Hartmann, Highly Efficient and Selective CAR-Gene Transfer Using CD4- and CD8-Targeted Lentiviral Vectors, Mol. Ther.–Methods Clin. Dev., 2019, 13, 371–379 CrossRef PubMed.
- A. Pfeiffer, F. B. Thalheimer, S. Hartmann, A. M. Frank, R. R. Bender, S. Danisch, C. Costa, W. S. Wels, U. Modlich, R. Stripecke, E. Verhoeyen and C. J. Buchholz, In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome, EMBO Mol. Med., 2018, 10(11), e9158 Search PubMed.
- A. Michels, A. M. Frank, D. M. Günther, M. Mataei, K. Börner, D. Grimm, J. Hartmann and C. J. Buchholz, Lentiviral and adeno-associated vectors efficiently transduce mouse T lymphocytes when targeted to murine CD8, Mol. Ther.–Methods Clin. Dev., 2021, 23, 334–347 CrossRef PubMed.
- T. T. Smith, S. B. Stephan, H. F. Moffett, L. E. McKnight, W. Ji, D. Reiman, E. Bonagofski, M. E. Wohlfahrt, S. P. S. Pillai and M. T. Stephan, In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers, Nat. Nanotechnol., 2017, 12(8), 813–820 CrossRef PubMed.
- N. N. Parayath, S. B. Stephan, A. L. Koehne, P. S. Nelson and M. T. Stephan, In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo, Nat. Commun., 2020, 11(1), 6080 CrossRef PubMed.
- F. Marofi, R. Motavalli, V. A. Safonov, L. Thangavelu, A. V. Yumashev, M. Alexander, N. Shomali, M. S. Chartrand, Y. Pathak, M. Jarahian, S. Izadi, A. Hassanzadeh, N. Shirafkan, S. Tahmasebi and F. M. Khiavi, CAR T cells in solid tumors: challenges and opportunities, Stem Cell Res. Ther., 2021, 12(1), 81 CrossRef.
- S. R. Husain, J. Han, P. Au, K. Shannon and R. K. Puri, Gene therapy for cancer: regulatory considerations for approval, Cancer Gene Ther., 2015, 22(12), 554–563 CrossRef PubMed.
- R. J. Vagnozzi, A. K. Z. Johansen and J. D. Molkentin, CARdiac Immunotherapy: T Cells Engineered to Treat the Fibrotic Heart, Mol. Ther., 2019, 27(11), 1869–1871 CrossRef PubMed.
- H. Aghajanian, T. Kimura, J. G. Rurik, A. S. Hancock, M. S. Leibowitz, L. Li, J. Scholler, J. Monslow, A. Lo, W. Han, T. Wang, K. Bedi, M. P. Morley, R. A. Linares Saldana, N. A. Bolar, K. McDaid, C.-A. Assenmacher, C. L. Smith, D. Wirth, C. H. June, K. B. Margulies, R. Jain, E. Puré, S. M. Albelda and J. A. Epstein, Targeting cardiac fibrosis with engineered T cells, Nature, 2019, 573(7774), 430–433 CrossRef PubMed.
- J. G. Rurik, I. Tombácz, A. Yadegari, P. O. Méndez Fernández, S. V. Shewale, L. Li, T. Kimura, O. Y. Soliman, T. E. Papp, Y. K. Tam, B. L. Mui, S. M. Albelda, E. Puré, C. H. June, H. Aghajanian, D. Weissman, H. Parhiz and J. A. Epstein, CAR T cells produced in vivo to treat cardiac injury, Science, 2022, 375(6576), 91–96 CrossRef PubMed.
- T. Weidner, S. Agarwal, S. Perian, F. Fusil, G. Braun, J. Hartmann, E. Verhoeyen and C. J. Buchholz, Genetic in vivo engineering of human T lymphocytes in mouse models, Nat. Protoc., 2021, 16(7), 3210–3240 CrossRef PubMed.
- S. Fujita, E. Ota, C. Sasaki, K. Takano, M. Miyake and J. Miyake, Highly efficient reverse transfection with siRNA in multiple wells of microtiter plates, J. Biosci. Bioeng., 2007, 104(4), 329–333 CrossRef PubMed.
- H. Erfle, B. Neumann, P. Rogers, J. Bulkescher, J. Ellenberg and R. Pepperkok, Work Flow for Multiplexing siRNA Assays by Solid-Phase Reverse Transfection in Multiwell Plates, J. Biomol. Screening, 2008, 13(7), 575–580 CrossRef PubMed.
- A. Okazaki, J.-I. Jo and Y. Tabata, A Reverse Transfection Technology to Genetically Engineer Adult Stem Cells, Tissue Eng., 2007, 13(2), 245–251 CrossRef PubMed.
- P. Agarwalla, E. A. Ogunnaike, S. Ahn, K. A. Froehlich, A. Jansson, F. S. Ligler, G. Dotti and Y. Brudno, Bioinstructive implantable scaffolds for rapid in vivo manufacture and release of CAR-T cells, Nat. Biotechnol., 2022, 40(8), 1250–1258 CrossRef PubMed.
- T. Arai, Y. Aiki and T. Sato, Accelerated transgene expression of pDNA/polysaccharide complexes by solid-phase reverse transfection and analysis of the cell transfection mechanism, Polym. J., 2022, 54(4), 603–613 CrossRef.
- T. G. Kim, Y. Lee and T. G. Park, Controlled gene-eluting metal stent fabricated by bio-inspired surface modification with hyaluronic acid and deposition of DNA/PEI polyplexes, Int. J. Pharm., 2010, 384(1), 181–188 CrossRef PubMed.
- G. Jiang, K. Park, J. Kim, K. S. Kim, E. J. Oh, H. Kang, S. E. Han, Y. K. Oh, T. G. Park and S. Kwang Hahn, Hyaluronic acid-polyethyleneimine conjugate for target specific intracellular delivery of siRNA, Biopolymers, 2008, 89(7), 635–642 CrossRef PubMed.
- M. White, R. Whittaker, C. Gándara and E. A. Stoll, A Guide to Approaching Regulatory Considerations for Lentiviral-Mediated Gene Therapies, Hum. Gene Ther: Methods, 2017, 28(4), 163–176 CrossRef PubMed.
- J. Szebeni, D. Simberg, Á. González-Fernández, Y. Barenholz and M. A. Dobrovolskaia, Roadmap and strategy for overcoming infusion reactions to nanomedicines, Nat. Nanotechnol., 2018, 13(12), 1100–1108 CrossRef.
- M. A. Dobrovolskaia and S. E. McNeil, Understanding the correlation between in vitro and in vivo immunotoxicity tests for nanomedicines, J. Controlled Release, 2013, 172(2), 456–466 CrossRef PubMed.
- R. M. Crist, J. H. Grossman, A. K. Patri, S. T. Stern, M. A. Dobrovolskaia, P. P. Adiseshaiah, J. D. Clogston and S. E. McNeil, Common pitfalls in nanotechnology: lessons learned from NCI's Nanotechnology Characterization Laboratory, Integr. Biol., 2013, 5(1), 66–73 CrossRef.
- R. D. Jayant, D. Sosa, A. Kaushik, V. Atluri, A. Vashist, A. Tomitaka and M. Nair, Current status of non-viral gene therapy for CNS disorders, Expert Opin. Drug Delivery, 2016, 13(10), 1433–1445 CrossRef.
- H.-D. Lu, H.-Q. Zhao, K. Wang and L.-L. Lv, Novel hyaluronic acid–chitosan nanoparticles as non-viral gene delivery vectors targeting osteoarthritis, Int. J. Pharm., 2011, 420(2), 358–365 CrossRef.
- M. de la Fuente, B. Seijo and M. J. Alonso, Bioadhesive hyaluronan–chitosan nanoparticles can transport genes across the ocular mucosa and transfect ocular tissue, Gene Ther., 2008, 15(9), 668–676 CrossRef.
- N. Duceppe and M. Tabrizian, Factors influencing the transfection efficiency of ultra low molecular weight chitosan/hyaluronic acid nanoparticles, Biomaterials, 2009, 30(13), 2625–2631 CrossRef.
- M. Raviña, E. Cubillo, D. Olmeda, R. Novoa-Carballal, E. Fernandez-Megia, R. Riguera, A. Sánchez, A. Cano and M. J. Alonso, Hyaluronic Acid/Chitosan-g-Poly(ethylene glycol) Nanoparticles for Gene Therapy: An Application for pDNA and siRNA Delivery, Pharm. Res., 2010, 27(12), 2544–2555 CrossRef PubMed.
- K. Luo, Y. Gao, S. Yin, Y. Yao, H. Yu, G. Wang and J. Li, Co-delivery of paclitaxel and STAT3 siRNA by a multifunctional nanocomplex for targeted treatment of metastatic breast cancer, Acta Biomater., 2021, 134, 649–663 CrossRef.
- L. Ye, H. Liu, X. Fei, D. Ma, X. He, Q. Tang, X. Zhao, H. Zou, X. Chen, X. Kong and P. Liu, Enhanced endosomal escape of dendrigraft poly-L-lysine polymers for the efficient gene therapy of breast cancer, Nano Res., 2022, 15(2), 1135–1144 CrossRef.
- S. Chono, S.-D. Li, C. C. Conwell and L. Huang, An efficient and low immunostimulatory nanoparticle formulation for systemic siRNA delivery to the tumor, J. Controlled Release, 2008, 131(1), 64–69 CrossRef PubMed.
- K. Ma, W. Li, G. Zhu, S. Sun, H. Chi, Y. Yin, H. Diao, X.-J. Xing, Z. Guo, L. Wang, W. Xu, C. Cui and J. Xu, Functionalized PDA/DEX-PEI@HA nanoparticles combined with sleeping-beauty transposons for multistage targeted delivery of CRISPR/Cas9 gene, Biomed. Pharmacother., 2021, 142, 112061 CrossRef PubMed.
- T. Wan, Q. Pan, C. Liu, J. Guo, B. Li, X. Yan, Y. Cheng and Y. Ping, A Duplex CRISPR-Cas9 Ribonucleoprotein Nanomedicine for Colorectal Cancer Gene Therapy, Nano Lett., 2021, 21(22), 9761–9771 CrossRef.
- Y. Shen, B. Wang, Y. Lu, A. Ouahab, Q. Li and J. Tu, A novel tumor-targeted delivery system with hydrophobized hyaluronic acid-spermine conjugates (HHSCs) for efficient receptor-mediated siRNA delivery, Int. J. Pharm., 2011, 414(1–2), 233–243 CrossRef PubMed.
- Y. Shen, Q. Li, J. Tu and J. Zhu, Synthesis and characterization of low molecular weight hyaluronic acid-based cationic micelles for efficient siRNA delivery, Carbohydr. Polym., 2009, 77(1), 95–104 CrossRef.
- J. E. Lee, Y. Yin, S. Y. Lim, E. S. Kim, J. Jung, D. Kim, J. W. Park, M. S. Lee and J. H. Jeong, Enhanced Transfection of Human Mesenchymal Stem Cells Using a Hyaluronic Acid/Calcium Phosphate Hybrid Gene Delivery System, Polymer, 2019, 1–12 Search PubMed.
- S. Bastaki, S. Aravindhan, N. Ahmadpour Saheb, M. Afsari Kashani, A. Evgenievich Dorofeev, F. Karoon Kiani, H. Jahandideh, F. Beigi Dargani, M. Aksoun, A. Nikkhoo, A. Masjedi, A. Mahmoodpoor, M. Ahmadi, S. Dolati, S. Namvar Aghdash and F. Jadidi-Niaragh, Codelivery of STAT3 and PD-L1 siRNA by hyaluronate-TAT trimethyl/thiolated chitosan nanoparticles suppresses cancer progression in tumor-bearing mice, Life Sci., 2021, 266, 118847 CrossRef.
- M. Lin, Z. Yang, Y. Yang, Y. Peng, J. Li, Y. Du, Q. Sun, D. Gao, Q. Yuan, Y. Zhou, X. Chen and X. Qi, CRISPR-based in situ engineering tumor cells to reprogram macrophages for effective cancer immunotherapy, Nano Today, 2022, 42, 101359 CrossRef CAS.
- C. Francis, L. Wroblewska, P. Pegman and M. Amiji, Systemic biodistribution and hepatocyte-specific gene editing with CRISPR/Cas9 using hyaluronic acid-based nanoparticles, Nanomedicine, 2022, 40, 102488 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |