
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent developments on the transformation of CO2 utilising ligand cooperation and related strategies
Ramaraj
Ayyappan†
 ,
Issam
Abdalghani†
,
Rosenildo C.
Da Costa†
and
Gareth R.
Owen
*
,
Issam
Abdalghani†
,
Rosenildo C.
Da Costa†
and
Gareth R.
Owen
*
School of Applied Science, University of South Wales, Treforest, CF37 4AT, UK. E-mail: gareth.owen@southwales.ac.uk
First published on 1st July 2022
Abstract
A portfolio of value-added chemicals, fuels and building block compounds can be envisioned from CO2 on an industrial scale. The high kinetic and thermodynamic stabilities of CO2, however, present a significant barrier to its utilisation as a C1 source. In this context, metal–ligand cooperation methodologies have emerged as one of the most dominant strategies for the transformation of the CO2 molecule over the last decade or so. This review focuses on the advancements in CO2 transformation using these cooperative methodologies. Different and well-studied ligand cooperation methodologies, such as dearomatisation–aromatisation type cooperation, bimetallic cooperation (M⋯M′; M′ = main group or transition metal) and other related strategies are also discussed. Furthermore, the cooperative bond activations are subdivided based on the number of atoms connecting the reactive centre in the ligand framework (spacer/linker length) and the transition metal. Several similarities across these seemingly distinct cooperative methodologies are emphasised. Finally, this review brings out the challenges ahead in developing catalytic systems from these CO2 transformations.
Introduction
Importance of utilising CO2 as a chemical feedstock
The utilisation of fossil fuels to meet global energy demands and feedstock chemicals leads to the continuous rise in carbon dioxide (CO2) emissions. The CO2 level in the atmosphere is currently 417.21 parts per million, as measured at the Mauna Loa Baseline Observatory on 10th May 2022.1 The steep rise in CO2 levels over many decades is attributable to the industrial processing of non-renewable fossil fuels (coal, oil, natural gas), which are still utilised to produce electricity, commodity chemicals and materials. Activities such as deforestation, expansion of vehicle use and other demands due to population increase have exacerbated this further.2 Thus, the alarming levels of anthropogenic CO2 have led to significant climate change and global warming. A substantial paradigm shift in approach towards green, sustainable and renewable technologies is required.3–5Currently, CO2 is used on a large scale for the synthesis of urea, salicylic acid and polycarbonates by the chemical industry. In addition, CO2 is utilised in oil recovery, the beverage industry, low temperature storage applications and for fire extinguishers. It is introduced in syngas feeds in quantities of up to 30% for the synthesis of CH3OH. Together, carbon capture utilisation and storage (CCUS) technologies only reduce a fraction (ca. 299 million tonnes per year) of annual CO2 emissions, which amount to 33 Gtonnes (2019).6,7 Electricity and heat production usages are the most significant contributors to CO2 emissions, amounting to just over 14 Gtonnes (40% of the total emissions) during 2019.8,9 A comprehensive switch to renewable energy sources such as geothermal, wind, solar and biomass-based resources for hydrogen production would make a substantial impact, helping to mitigate CO2 levels and providing part of the solution.
A further aspect that needs to be addressed is resourcing raw materials needed for the chemical and manufacturing industries. CO2 is considered as a sustainable alternative C1 feedstock to fossil fuel derived CO because it is abundant, inexpensive, and non-toxic. A portfolio of commodity chemicals can be envisaged from CO2, including base chemicals, polymers and fuels.10–12 The promise of using CO2 as a C1 synthon, however, faces significant practical challenges in terms of chemical activation due to its high thermodynamic and kinetic stabilities. The CO2 molecule is centrosymmetric, non-polar and a weak electrophile. The bond energies of its C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds are 127 kcal mol−1 and its CO bond lengths are 1.16 Å.13,14
O bonds are 127 kcal mol−1 and its CO bond lengths are 1.16 Å.13,14
Many transformations of CO2 involve its reduction with hydrogen leading to various oxidation levels of the carbon centre.15,16 As an example, the formation of formic acid is thermodynamically unfavourable in the gas phase due to large negative entropic contributions (ΔS298 K −216 J mol−1 K−1, ΔG298 K 7.8 kcal mol−1). This becomes favourable in aqueous media (ΔG298 K −2.3 kcal mol−1) and in the presence of a base to form the corresponding formate salts. On the other hand, reduction to methanol is thermodynamically favourable (ΔH298 K −11.9 kcal mol−1, ΔG298K −2.3 kcal mol−1), yet selective reduction and the development of efficient catalytic pathways to this molecule remain a challenge.17–21 Methanol is one of the most important building blocks for industry and can be used for hydrogen storage and fuel cell applications similar to formic acid.22
In this context, homogeneous catalysis plays a vital role in tackling the challenges associated with rising CO2 levels.23 Transition metal catalysts provide various bond activation tools to functionalise CO2. They can activate CO2 in a number of different ways and many different coordination modes of CO2 are observed.24,25 The development of stable, low cost, active and selective catalysts operating under mild conditions is imperative for industry from the long-term perspective in terms of sustainability. The chemical industries need to be inspired by nature where plants use atmospheric CO2 from the air to synthesise molecules. Typically, nature employs multifunctional strategies for this, where the transformation at the transition metal centre is supported by additional sites on a given enzyme to facilitate the transformation. This cooperation between multiple sites appears to be an essential factor. Here, we focus on recent developments where metal ligand cooperation (MLC) and related strategies have been employed to open up pathways leading to the transformation of CO2.
Metal ligand cooperation related to CO2
A key strategy and theme for research on CO2 activation is focused on MLC methodologies. For the reasons outlined above, CO2 is a challenging molecule to activate, and thus multicentre activation strategies are required. MLC is a term utilised to describe bond activations and other processes in which the metal and ligand work together in synergy to achieve them. Ligand participation is key for the bond formation and bond breaking steps at the metal centre. Such processes can occur either in a concerted or stepwise manner. For example, in the context of CO2 activation, the presence of either a basic site on the ligand backbone, which can attack the electrophilic carbon centre, or an acidic site, which coordinates to one of the Lewis basic oxygen centres, would decrease the activation barrier for CO2 activation. Essentially, any interaction between the ligand and CO2 in the inner/outer sphere or ligand centred photo or redox transformations in the metal complex could be classified as MLC.26 One ground-breaking example in this category belongs to Noyori's bifunctional asymmetric hydrogenation catalysts where H2 is activated across a metal–amido bond (i.e. M–NR2 + H–H → H–M–NHR2).27In general, metal-centred activation of small molecules is predominant across catalysis. Bond breaking and bond making steps are the cornerstones of many processes, such as hydrogenations, hydroformylations, C–C couplings, olefin metathesis and polymerisations.28 Oxidative addition is one of the most important fundamental steps in many catalytic processes (Fig. 1A). It requires a metal centre with the ability to increase its oxidation state by two units. In contrast, MLC methodology emerged as an alternative avenue for bond activation where cooperation of a ligand was a key feature.29,30 These methodologies provide unprecedented pathways for the cleavage of strong carbon–oxygen bonds and transformations on CO2.31 There have been many promising developments across a range of MLC methodologies involving CO2 in recent times. Several interesting cooperative pairs have been identified across the periodic table. This can be attributed to the rise in the number of ligands from simple innocent Lewis bases to versatile multifunctional scaffolds, modulating the structural rigidity and electronic and steric properties of the metal complex. Insights into the different binding modes of CO2 in MLC would further enrich our knowledge of CO2 functionalisation.
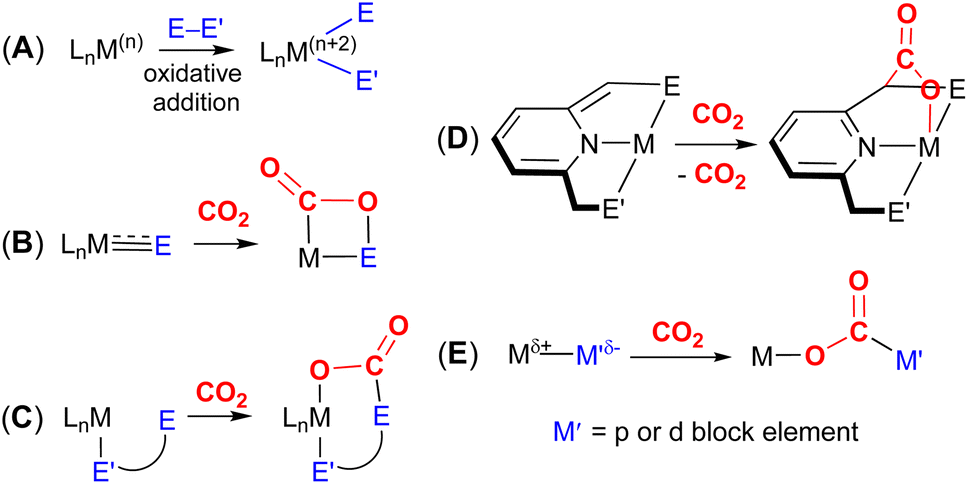 | ||
| Fig. 1 Bond activation via oxidative addition (A) and an overview of the main MLC strategies for the activation of CO2, including [2 + 2] cycloaddition across ME multiple bonds (one-atom bridges) (B); MLC activation of CO2 across transition metal and ligand centres (two- and three-atom bridges) (C); dearomatisation–aromatisation (D) and bimetallic activations (E); (M indicates a metal, L indicates a ligand and E and E′ stand for different donor elements containing various substituents). | ||
The most common cooperative methodologies are shown in Fig. 1. Firstly, [2 + 2] cycloaddition of CO2 across unsaturated ME units (i.e. one atom; where E = CR2, SiR2, NR or O) is an alternative method (Fig. 1B). These form a four-membered metallacyclic species of the type [![[upper bond 1 start]](https://www.rsc.org/images/entities/char_e010.gif) MC(O)OE
MC(O)OE![[upper bond 1 end]](https://www.rsc.org/images/entities/char_e011.gif) ]. A significant bond polarity between M and E facilitates the activation of CO2. Secondly, a remote or lateral reactive site on a ligand can work in cooperation with the metal for the activation of CO2 (Fig. 1C). When the reactive centre is distanced two or three atoms away from the metal centre, this leads to the formation of stable (five or six) chelate rings upon reaction with CO2, which serve to stabilise the resulting products. Thirdly, Milstein-type dearomatisation–aromatisation transformations (Fig. 1D) have been widely used in the last decade or so for the activation of CO2. This was applied in catalytic transformations where cooperation typically occurred between a dearomatised ligand containing a deprotonated methylene “side-arm” and the metal centre. This is a subclass of the two-atom or three-atom bridged systems but is considered in a separate section here since it also involves the dearomatisation–aromatisation transformation alongside CO2 activation. Finally, the bimetallic cooperation between two contrasting metal centres (Fig. 1E). For example, early–late transition metal combinations or cooperation between a metal and main group metalloid species (e.g. Al, Ga, In). Here, we explore the various developments in these key MLC types; each are outlined in the following sections.
]. A significant bond polarity between M and E facilitates the activation of CO2. Secondly, a remote or lateral reactive site on a ligand can work in cooperation with the metal for the activation of CO2 (Fig. 1C). When the reactive centre is distanced two or three atoms away from the metal centre, this leads to the formation of stable (five or six) chelate rings upon reaction with CO2, which serve to stabilise the resulting products. Thirdly, Milstein-type dearomatisation–aromatisation transformations (Fig. 1D) have been widely used in the last decade or so for the activation of CO2. This was applied in catalytic transformations where cooperation typically occurred between a dearomatised ligand containing a deprotonated methylene “side-arm” and the metal centre. This is a subclass of the two-atom or three-atom bridged systems but is considered in a separate section here since it also involves the dearomatisation–aromatisation transformation alongside CO2 activation. Finally, the bimetallic cooperation between two contrasting metal centres (Fig. 1E). For example, early–late transition metal combinations or cooperation between a metal and main group metalloid species (e.g. Al, Ga, In). Here, we explore the various developments in these key MLC types; each are outlined in the following sections.
Recent developments in metal ligand cooperation methodologies involving CO2
Activation of CO2 across a one-atom bridge (Fig. 1B)
E (or M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) E); where E = CR2, SiR2, O, NR containing functional groups] can undergo [2 + 2] cycloaddition reactions with CO2. This is a class of complexes where extensive π-bonding between metal and E component renders them nucleophilic, depending on the nature of E.36 These compounds are typically highly reactive and often quite unstable. The reactivity is further influenced by bond polarity due to the difference in electronegativity between the metal and E. Thus, the bond is polarised either as Mδ+Eδ− or Mδ−Eδ+ and this will dictate the regiochemistry of CO2 activation. It should be noted that the MLC of ME systems can be comparable to bimetallic M–M or M–E′ (where E′ = main group element) cooperation. This is covered in the last section.
E); where E = CR2, SiR2, O, NR containing functional groups] can undergo [2 + 2] cycloaddition reactions with CO2. This is a class of complexes where extensive π-bonding between metal and E component renders them nucleophilic, depending on the nature of E.36 These compounds are typically highly reactive and often quite unstable. The reactivity is further influenced by bond polarity due to the difference in electronegativity between the metal and E. Thus, the bond is polarised either as Mδ+Eδ− or Mδ−Eδ+ and this will dictate the regiochemistry of CO2 activation. It should be noted that the MLC of ME systems can be comparable to bimetallic M–M or M–E′ (where E′ = main group element) cooperation. This is covered in the last section.
Four major classes of metal–ligand multiple bonds are discussed in this section. The ligands can be carbenes, silylenes, nitrenes and oxo functionalities. Firstly, Fischer and Schrock carbenes are the two major classes of carbenes extensively investigated in this family.37–39 Fischer carbenes are electrophilic, L-type, two-electron donors with weak back-bonding from metal to ligand. On the other hand, Schrock carbenes are nucleophilic. Secondly, silylenes (R2Si:) are heavier analogues of their lighter congener carbenes; however, due to the low electronegativity of silicon (Si: 1.8 vs. C: 2.5 on the Pauling scale), and its ability to stabilise incipient positive charge, silylenes are inherently electrophilic. In addition, they have the tendency to form hypervalent bonds, making them distinct when compared to carbenes. Finally, the bonding in terminal oxo (MO) and imido (MNR) complexes involves extensive π-bonding interactions with the metal due to lone pairs on the oxygen and nitrogen centres. The hybridisation of E changes from sp2 for carbenes and silylenes to sp for imido. In terms of oxo and imido compounds, those examples that react with CO2 tend to be high valent early transition metals where the reactivity observed involves nucleophilic attack at the electrophilic carbon centre of CO2.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CR2 and CO2.
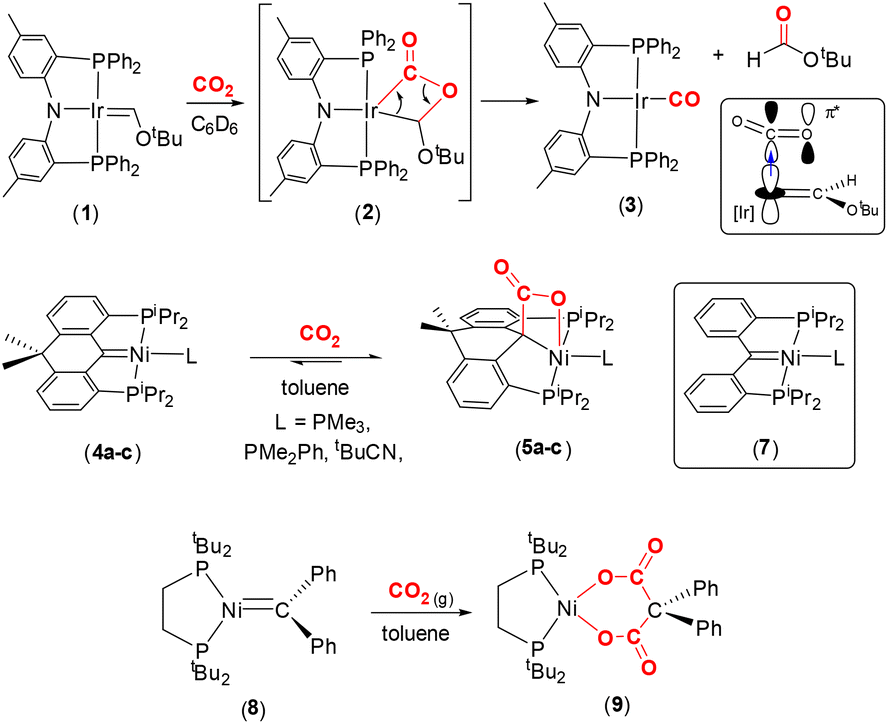
Representative carbene complexes exhibiting [2 + 2] cycloaddition reactions with CO2 are highlighted in Scheme 1. Grubbs and co-workers characterised the first nucleophilic Fischer carbene iridium(I) complex supported by Ozerov's amidophosphine PNP pincer ligand.40–42 The square planar complex, [(PNP)IrC(H)OtBu] (1), which contains a short IrC bond [1.882(4) Å], reacts with CO2 in C6D6 to form the stable carbonyl complex (3) along with tbutylformate as a by-product. The reactivity was explained by nucleophilic attack of the dz2 HOMO electrons at the iridium centre on the π* orbital of the CO2 moiety. The population of the π* orbital of CO2 results in the reduction of the bond order of the C–O bonds. Furthermore, the proximity of the electron rich oxygen centre to the IrC bond also serves to reduce this bond order by interrupting the π backdonation, thereby facilitating the formation of the intermediate metallalactone, [IrC(O)OC] complex 2 (Scheme 1, top). The formation of this species was supported by low temperature NMR studies and density functional theory (DFT) calculations. In the DFT optimised structure of 2, the former iridium–carbon double bond is elongated to 2.003 Å and the IrC(O)–O distance elongated from 1.16 Å to 1.348 Å.
CR2 and CO2.
Representative carbene complexes exhibiting [2 + 2] cycloaddition reactions with CO2 are highlighted in Scheme 1. Grubbs and co-workers characterised the first nucleophilic Fischer carbene iridium(I) complex supported by Ozerov's amidophosphine PNP pincer ligand.40–42 The square planar complex, [(PNP)IrC(H)OtBu] (1), which contains a short IrC bond [1.882(4) Å], reacts with CO2 in C6D6 to form the stable carbonyl complex (3) along with tbutylformate as a by-product. The reactivity was explained by nucleophilic attack of the dz2 HOMO electrons at the iridium centre on the π* orbital of the CO2 moiety. The population of the π* orbital of CO2 results in the reduction of the bond order of the C–O bonds. Furthermore, the proximity of the electron rich oxygen centre to the IrC bond also serves to reduce this bond order by interrupting the π backdonation, thereby facilitating the formation of the intermediate metallalactone, [IrC(O)OC] complex 2 (Scheme 1, top). The formation of this species was supported by low temperature NMR studies and density functional theory (DFT) calculations. In the DFT optimised structure of 2, the former iridium–carbon double bond is elongated to 2.003 Å and the IrC(O)–O distance elongated from 1.16 Å to 1.348 Å.
 | ||
| Scheme 1 Selected examples involving [2 + 2] cycloaddition between a carbene moiety and the CO2 molecule. The inset in the top reaction provides a simplistic representation of the interaction between complex 1 and CO2 in this transformation (see text for details). | ||
Later, Piers and co-workers showed that square planar nickel pincer complexes, [(PCP)Ni(L)] (4a–c), where the PCP ligand was based on a 10,10-dimethyl-9,10-dihydroanthracene scaffold and L = PMe3, PMe2Ph or tBuCN, underwent reversible [2 + 2] cycloaddition with CO2 in toluene (or C6D6) to yield square pyramidal nickellalactone complexes 5a–c (Scheme 1, middle).43 In the reaction involving the PMe2Ph ligand, new C–C and Ni–O bonds were formed with distances of 1.571(2) Å and 2.146(1) Å, respectively. The corresponding carbon–oxygen distances were found to be 1.276(2) Å and 1.226(2) Å. Interestingly, the related higher oxidation state complex, [(PCP)NiBr][SbF6] (6), did not react with CO2. This is due to the carbene centre becoming more electrophilic as a result of the Ni(II) oxidation state, as compared to Ni(0) in 4a–c. As expected, the nickel–carbon distance in the pincer ligand is shorter in the Ni(II) complex when compared to the corresponding distance in Ni(0) complex, cf. 1.874(4) Å in 6 and 1.906(1) Å in 4c (tBuCN). The 13C{1H} NMR chemical shift also reflects the change in electronic parameters at the metal centre from Schrock-type, δ 114.6 ppm (THF-d8) in complex 4c, to more Fischer-type, δ 250.8 ppm (CD2Cl2) in 6 in terms of carbene-type character.
A closely related complex, [(PCP)Ni(NCtBu)], 7 (shown in the box in Scheme 1, middle), where the PCP ligand in this case is [C{o-C6H4P(iPr)2}2], gave intractable mixtures upon reaction with CO2 unlike the 10,10-dimethyl-9,10-dihydroanthracene based complexes 4a–c.44 The key difference between these two ligand scaffolds was attributed to the change in torsion angles involving the aryl rings and rigidity of the ligand backbones. These examples demonstrate the effect of tailoring the ligand platform and the charge at the metal centre on the propensity for CO2 activation.
Hillhouse and co-workers previously isolated a nucleophilic, trigonal planar, zerovalent nickel complex containing a “CPh2” carbene fragment, complex 8, as shown in Scheme 1, bottom.45 It was found that the nickel–carbon distance was 1.836(2) Å. NMR spectroscopy revealed a signal in the 13C{1H} NMR spectrum at δ 222 ppm (d, 2JPC = 51 Hz). When a solution of 8 was placed under a CO2 atmosphere in toluene, new cyclic complex 9 was formed as a bright yellow solid in 90% yield. This complex contained a dicarboxylate {[OC(O)C(Ph)2C(O)O]2−} unit coordinated to the nickel centre. The formation of 9 was explained by the initial [2 + 2] cycloaddition to form the nickellalactone, i.e. NiC(Ph)2C(O)O, followed by a second insertion of CO2 into the Ni–C(Ph)2 bond. It was rationalised that the second insertion was driven by relaxation of the strain associated with the initial four-membered ring. In 9, the two Ni–O distances were found to be 1.8696(13) Å and 1.8773(13) Å, the CO bond lengths were 1.221(2) Å and 1.225(2) Å. The corresponding O–C(O) distances were 1.292(2) Å and 1.287(2) Å and the C–C(Ph)2 distances were 1.554(3) Å and 1.559(3) Å. It appeared that there was significant double bond character in the O–C(O) units.
SiR2 and CO2.
More recently a number of research groups have investigated cycloadditions involving LnMSiR2 species and CO2 (Scheme 2). Emslie and co-workers reported the reaction of manganese–hydride complexes containing terminal silylene units (10a, 10b) with CO2 to give trans-[MnH(CO)(dmpe)2] (11) and the corresponding polysiloxane [R2SiO]n products (Scheme 2, top).46 The proposed mechanism for this transformation was via a [2 + 2] cycloaddition between the MnSiR2 unit and one of the double bonds of the CO2 molecule. The formation of a strong Si–O bond was proposed to be the driving force for the formation of the polysiloxane product.
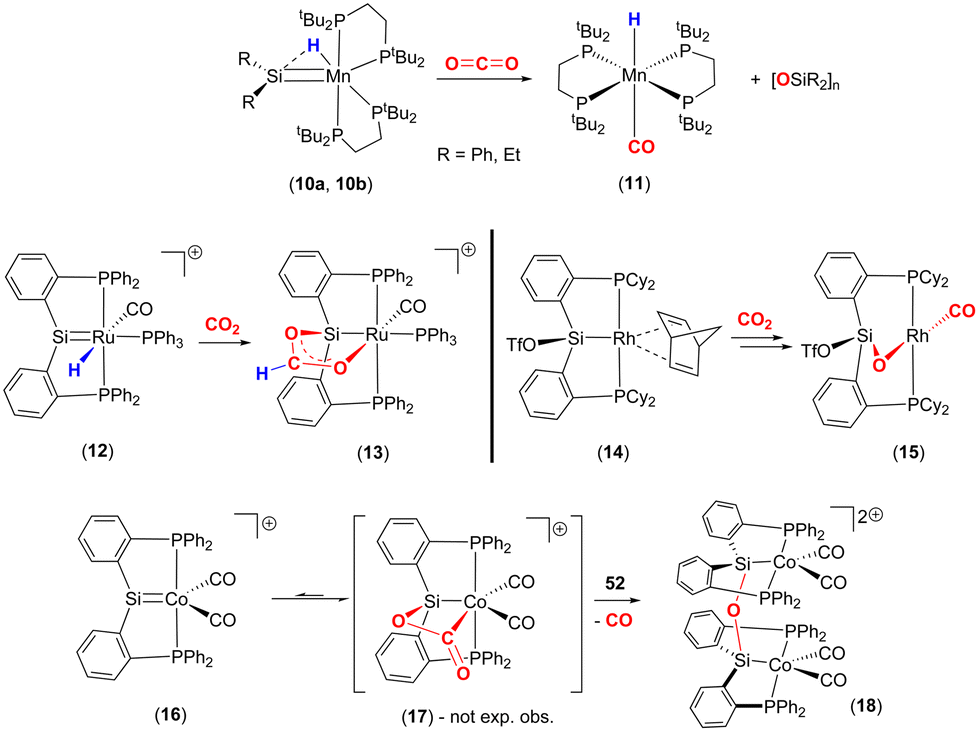 | ||
| Scheme 2 Selected examples of reactions involving silylene complexes with CO2. | ||
Over a number of years, the Whited group has worked extensively on metal silylene complexes, where the silylene unit is embedded in a pincer scaffold. In one such example, they highlighted a MLC process in [RuH(SiP2Ph)(CO)(PPh3)]+ (12), where PhP2Si is Si(o-C6H4PPh2)2 reacted with CO2 to form complex 13 (Scheme 2, middle-left).47 The new complex was found to contain a new metallacycle featuring a RuOC(H)OSi ring. The transformation was attributed to Ru–Si cooperative bond activation. The first step involved the insertion of a CO2 molecule into the Ru–H bond to form the corresponding formato [RuOC(O)H] complex. The carbonyl oxygen of this new species was then stabilised by interaction with the Lewis acidic silicon centre, thus forming the RuOC(H)OSi cycle. It was shown that the transformation could be monitored by 29Si NMR, which revealed a clean transition from δ = 278 ppm, in 12, to δ = 118 ppm in the product, consistent with a change in hybridisation at silicon. This mode of cooperative activation (i.e. activation involving a TM and a Lewis acid) shares a resemblance with FLP type activation where activation occurs through the cooperation of the M–H functionality and a p-block Lewis acid.
Developing on this, the same group reported a contrasting mechanism for the reductive cleavage of CO2 mediated by a related rhodium silyl [Rh(CyP2SiOTf)(NBD)] (14, Scheme 2, middle-right).48 They showed that 14 readily reacted with CO2 in CH2Cl2 to form the square planar complex 15, which featured a new CO ligand and a bridging oxygen atom between the rhodium and silicon centres. Here, the CO2 molecule has undergone reductive cleavage to form a Si–O bond and a rhodium(I) carbonyl ligand. The 2-electron reduction of CO2 to Rh–CO and Si–O is facilitated by MLC and the polarity difference between the rhodium and silicon centres. Among others, two main mechanisms were considered by DFT calculations (Scheme 3). The first one involved the formation of a NBD free complex with a silylene (RhSi) species formed via migration of triflate to the metal centre and subsequent reaction with CO2via [2 + 2] cycloaddition. This pathway, however, required a significantly large activation barrier (37.0 kcal mol−1, 298 K). Whereas the second pathway proceeded via an “anomalous” insertion into the Rh–Si bond to form the siloxyacyl species, RhC(O)OSi. This was found to have a more reasonable activation barrier of 23.5 kcal mol−1. The RhC(O)OSi species then underwent de-insertion of CO to form the Rh–CO unit with the remaining oxygen bridging the silicon and rhodium centres. Incidentally, the “normal” insertion pathway, leading to RhOC(O)Si, was also ruled out since it was found to be a high energy species.
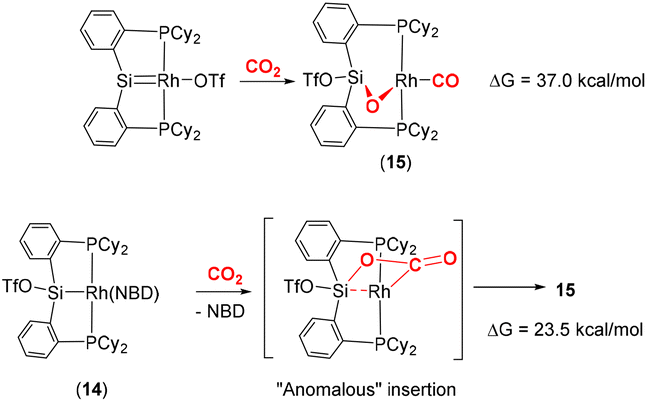 | ||
| Scheme 3 DFT calculated pathways for CO2 reductive cleavage of CO2 into CO and siloxide by complex 14. Top: silylene pathway, bottom: anomalous insertion pathway. | ||
Later, Whited and co-workers also reported the reactivity of CO2 on a 3d8 cationic cobalt(I) silylene complex [Co(SiP2Ph)(CO)2]+ (16, Scheme 2, bottom).49,50 The reaction leads to dinuclear complex 18, which features Co(II) centres and the silicon centres are bridged (μ2) by an oxo species. Again, this arises due to the 2-electron reduction of CO2 to CO (which is eliminated) and where each cobalt centre undergoes a one electron oxidation. The reaction was investigated by DFT calculations, which suggested that it proceeded via an initial [2 + 2] cycloaddition of CO2 across the cobalt–silicon double bond. Interestingly, when the substituents at phosphorus were changed to iso-propyl groups, the same transformation involving CO2 was not observed. This suggests that the influence of the ligand's electronic and steric properties are also important parameters.
The reactivity highlighted in the two examples above is reminiscent of the bimetallic cooperative activation of CO2 leading to the formation of metal oxo M–O and M–CO bonds (vide infra). In these examples, MLC is facilitated by the formation of strong Si–O bonds, which provide a thermodynamic driving force for the transformations. It becomes evident that in different situations the deoxygenation of CO2 is strongly driven by the formation of E–O bonds (for example, where E = Si, B and Al) in addition to the formation of stable transition metal–CO species.
Reaction of metal–oxo complexes with CO2
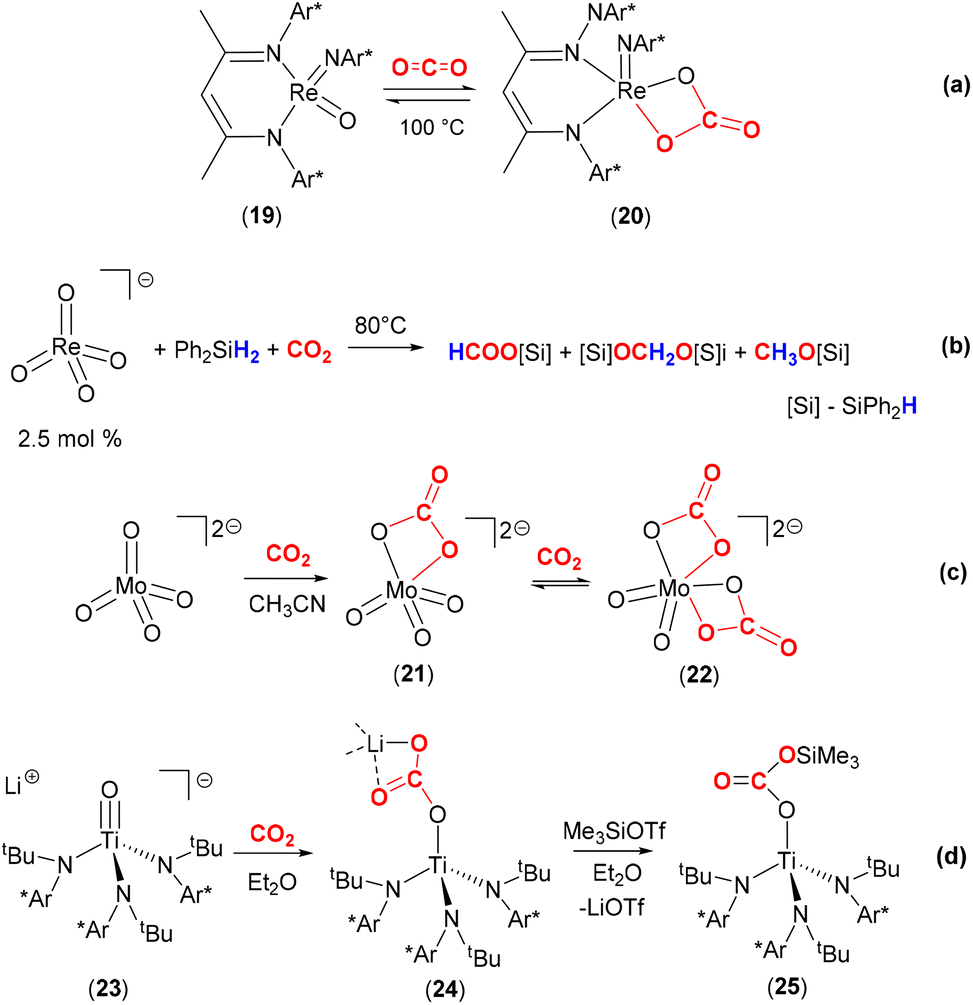
The reactivity of carbon dioxide with transition metal oxo complexes has been explored; selected examples are shown in Scheme 4. Recently, a nucleophilic rhenium(V) oxo imido complex, [Re(NDipp)(O)(BDI)] (19, where Dipp = 2,6-diisopropylphenyl and BDI = N,N′-bis-(Dipp)-3,5-dimethyl-β-diketiminate) was shown to reversibly bind CO2 to form the carbonate complex 20 [Scheme 4(a)].51 The carbonate ligand in this complex coordinates with a κ2-O,O coordination mode, forming a four-membered ring motif. Calculations revealed the weak entrapment of the CO2 molecule with a binding strength of 2.2 kcal mol−1. A crystal structure revealed that Re–O distances involving the carbonate ligand were 2.018(4) Å and 2.021(4) Å.
 | ||
| Scheme 4 Reaction of high valent early transition oxo complexes with CO2. Ar* = 2,6-diisopropylphenyl (Dipp). | ||
It is of interest to explore the further transformation of CO2 once it has been activated at a metal centre. One of the many examples is shown in Scheme 4(b).52 Love and co-workers demonstrated that the air stable, high valent perrhenate complex [N(hexyl)4][(ReO4)] could be utilised as a catalyst for the hydrosilylation of CO2. Utilising this complex with a catalytic loading of 2.5 mol%, with Ph2SiH2 and 1 atm pressure of CO2 gave the products silylformate, silylacetal and methoxysilane. The product distribution and selectivity were strongly dependent on the nature of the specific silane, solvent, temperature, and reaction duration. CH3OH was obtained from CH3OSiPh2H after quenching with H2O in almost quantitative yield. The selectivity for the formation of CH3OSiPh2H was improved under optimised reaction conditions (DMF, Ph2SiH2, 80 °C). The different pathways for the catalytic hydrosilylation of CO2 using [ReO4]− were investigated by experimental and DFT methods. Experimental results clearly ruled out direct [2 + 2] cycloaddition between CO2 and ReO to form the corresponding carbonate species, [ReO3(CO3)]−. Alternative mechanistic pathways proposed by DFT calculations suggested the activation of PhSiH3 by ReO in the first step. This hydrosilane can be activated via two different routes; either (i) 1,2-addition of the Si–H bond across the ReO bond to form a rhenium hydride siloxide species, Re(H)(OSiH2Ph), with an activation barrier of ΔG = 27 kcal mol−1. This is followed by insertion of the CO2 molecule into the resulting Re–H bond to generate the formato species, Re{OC(O)H}(OSiH2Ph); or (b) through a ReO⋯SiH3Ph interaction, forming a hypervalent “ReO–SiH3Ph” species followed by insertion of CO2 directly into one of the Si–H bonds to form the silylformate adduct [ReOSiPhH2{OC(O)H}]. This latter pathway has a lower activation barrier (ΔG = 19.4 kcal mol−1). Subsequent Si–H bond activation in the same way would explain the observed silyl acetal and methoxysilane products.
Interestingly, the [N(hexyl)4][(ReO4)] complex was also found to serve as a catalyst for the N-methylation of amines (including iPr2NH, pyrrolidine, morpholine, piperidine) using CO2 as a C1 source and silanes as a reducing agent. Similarly, a simple environmentally benign inorganic oxometallate, K2WO4 (10 mol% catalytic loading), was also shown to act as a catalyst for the N-methylation or N-formylation of amines using PhSiH3 as a reducing agent under the following reaction conditions: CH3CN, 70 °C, 12 h.53
Furthermore, the soluble monomeric molybdate dianion [PPN]2[MoO4] {PPN = (Ph3P)2N+} reacted with CO2 to form well defined mono- and bicarbonate complexes [MoO3(κ2-O,O-CO3)]2− (21) and [MoO2(κ2-O,O-CO3)2]2− (22, Scheme 4c).54 In monocarbonate complex 21, the C–O bonds were found to be 1.3357(14) and 1.3048(13) whilst the Mo–O bond was 2.2191(9) Å. The CO bond [1.2258(13) Å] was elongated with respect to free CO2 as expected (cf. 1.162 Å in CO2). Stoichiometric reaction of the monocarbonate, [PPN]2[MoO3(κ2-O,O-CO3)], with Et3Si–H resulted in the clean conversion to formate [PPN][OC(O)H] and [PPN][MoO3(OSiEt3)] in good yields. Isolation and characterisation of discrete homogeneous cycloaddition intermediates resulting from the reaction between CO2 and MoO (such as 21 and 22) provides an insight into the mechanism of activation of their heterogeneous counterparts.
The Cummins group also demonstrated the nucleophilicity of the oxo ligand, and the effect of a lithium cation, on the reactivity of a titanium complex with CO2 [Scheme 4(d)].55 The anionic titanium oxo complex, [Li(Et2O)2][Ti(O){N(tBu)(3,5-Me2C6H3)}3] (23), rapidly reacted with one equivalent of CO2 in Et2O to generate complex 24. As shown in the scheme, this new complex features a titanium–carbonate moiety where the carbonate is coordinated in a rare κ1-O coordination mode to the titanium centre. The negative charge resides on the other two oxygen atoms and is stabilised by Li–O interactions with an unsymmetrical κ2-O,O mode (Li–O distances 1.911(5) Å and 2.257(5) Å). The lithium cations also form a hexamer type structure where they connect the carbonate moieties of other complexes within a hexagonal motif. The binding of CO2 was found to be reversible in solution under dynamic vacuum and was additionally assisted by sequestering the lithium cations with 12-crown-4. The Ti–O bond length in 24 was found to be 1.849(2) Å, which was a significant elongation when compared to the titanium–oxygen distance in starting material 23 (1.712(2) Å). This infers relatively weak π-bonding between titanium and oxygen centres following CO2 activation. On the other hand, the weak binding of CO2 was also supported by the bond lengths found in 24. The TiO–C distance was 1.319(3) Å, the CO distance was 1.232(3) Å and the C–OLi distance 1.292(3) Å. Complex 24 could be further reacted as a means of transforming activated CO2. The addition of Me3SiOTf to 24 resulted in the formation of silyl carbonate complex [Ti{OC(O)OSiMe3}{N(tBu)-(3,5-Me2C6H3)}3] (25) and elimination of LiOTf. Heating this new complex above 80 °C results in the loss of CO2 and the formation of siloxide complex [Ti(OSiMe3){N(tBu)(3,5-Me2C6H3)}3]. Similarly, complex 24 reacts with pivaloyl chloride, tBuC(O)Cl, to form [Ti{OC(O)tBu}{N(tBu)(3,5-Me2C6H3)}3] with the elimination of LiCl and CO2. An analogous nitride system is outlined in the following section.
Reaction of metal–nitrido and –imido complexes with CO2
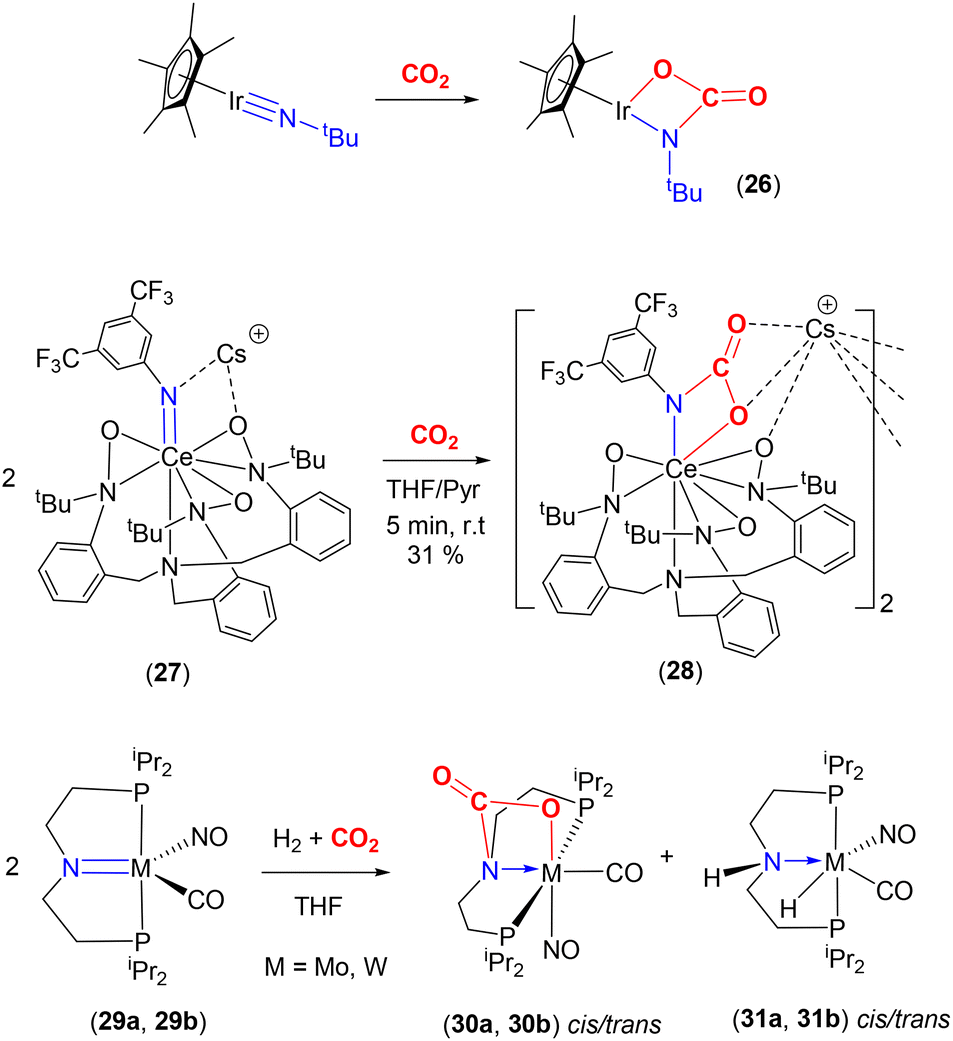
Many of the developments observed for metal–oxo complexes with CO2 are also found for the corresponding nitrido and imido complexes. A range of selected examples are shown in Scheme 5. Transformations similar to those found for oxo complex 23 above were also observed for the nitride complex, [Na][Nb(N){N(tBu)(3,5-Me2C6H3)}3]. This complex reacted with CO2 to form the corresponding carbamate complex, [Na][Nb{N → CO2}{N(tBu)(3,5-Me2C6H3)}3]. This further reacted with Ac2O to give the isocyanate–acetate complex [Nb(OAc)(OCN){N(tBu)(3,5-Me2C6H3)}3] with the elimination of sodium acetate. Finally, reduction with SmI2, followed by reaction with Na/Hg (two electron reduction) resulted in the extrusion of CO and regeneration of the starting niobium nitride. This series of transformations provided a closed cycle for the transformation of CO2 into CO mediated by a nitride complex.56
 | ||
| Scheme 5 The [2 + 2] reaction of selected amido or imido complexes with CO2. | ||
The Bergman group was one of the pioneers in the development of the area surrounding metal–ligand multiple bond activation, first demonstrating [2 + 2] activation of CO2 across an iridium–imido triple bond (Scheme 5, top). They synthesised the first “one legged piano stool” complex, containing an iridium(III) centre, a η5-C5Me5 ligand and a nucleophilic imido ligand containing bulky substituents at nitrogen.57,58 Complexes of the type [Ir(Cp*)(NR)] (where R = tBu, SiMe2tBu, 2,6-Me2-C6H3, 2,6-iPr2-C6H3) were investigated. The presence of strong π-donor interactions between the metal and imido ligand were confirmed by short Ir–N distances [e.g. 1.712(7) Å for Ir(Cp*)(NtBu)] and linear arrangements [a Ir–N–C angle of 177.2(5)° for the same complex]. As shown in the scheme, the reaction of [Ir(Cp*)(NtBu)] with CO2 led to [2 + 2] cycloaddition across the iridium–nitrogen bond and the formation of the carbamate complex, [(Cp*)Ir{OC(O)NtBu}] (26), isolated as a red crystalline solid. The Ir–N bond length, in this carbamate complex was found to be 1.943(6) Å. This was longer than the corresponding distance for the starting material (vide supra). Furthermore, the reduction of the Ir–N–C bond angle to 141.6(5)° confirmed the change in hybridisation of imido nitrogen from sp to sp2.
More recent examples exhibiting similar reactivity were reported by a number of research groups. For example, in 2018 Schneider and co-workers reported the reaction of a square planar pincer complex, [Ir(NtBu)(PNP)] {PNP = N(CHCHPtBu2)2}, with CO2 at 283 K in THF, resulting in [2 + 2] cycloaddition to form the corresponding carbamate complex, [Ir(OC(O)NtBu)(PNP)].59
Similarly, a set of nickel(II) imido complexes of the type [Ni(NR)(dtbpe)], analogous to carbene complex 8 (Scheme 1, bottom), were found to react with CO2 (R = 2,6-iPr2C6H3 and 1-adamantyl, and dtbpe = tBu2PCH2CH2PtBu2). For complexes where R = 2,6-iPr2C6H3, square planar nickel metallacycles of the type, [Ni{OC(O)N(2,6-iPr2C6H3)}(dtbpe)] were formed via a [2 + 2] addition, as a pale-yellow solid. This complex was structurally characterised. The key distances were Ni–O 1.9334(15) Å, Ni–N 1.8912 Å, OC 1.229(2) Å and C–O 1.340(2) Å. It was found, however, that the corresponding electron rich and sterically hindered adamantyl complex directly formed a bicarbamate product of the form [Ni{OC(O)N(1-Ad)C(O)O}-(dtbpe)]. The substituents on the imido nitrogen influence the reactivity of NiN–R complexes. Again, this is analogous to related carbene complex 8, which forms dicarboxylate complex 9 (vide supra). The isolated adamantyl bicarbamate complex shows two carbonyl stretching bands at 1667 cm−1 and 1624 cm−1 in the IR spectrum. Thus, it can be stated that the reactivity of carbene and imido species on a Ni(dtbpe) fragment is analogous.60
[2 + 2] cycloaddition reactivity was also reported for palladium(II) complexes. Munz and co-workers reported that terminal sulfonimido complex [Pd(NTs)(CAAC)] {where Ts = p-toluenesulfonyl and CAAC = cyclic(alkyl)(amino)carbene} also reacted with CO2. The authors indicated that the zwitterionic character of the palladium–nitrogen bond (Pdδ+ and Nδ−) made the nitrogen centre strongly nucleophilic thus facilitating its reactivity with CO2. The resulting product was [Pd{OC(O)NTs}(CAAC)] containing a four-membered cyclic palladacarbamate metallacycle.61
Another recent example involving an anionic rare earth lanthanide cerium(IV) imido complex was reported by Schelter and co-workers (Scheme 5, middle).62 They demonstrated that cerium complex 27, shown in the scheme, reacted with CO2 to form complex 28, which existed as a dinuclear structure featuring carbamate functional groups. This, again, formed via a [2 + 2] cycloaddition.
An interesting example was reported by Berke and co-workers that showed competing reactions involving H2 and CO2 with group 6 transition metal pincer type complexes (Scheme 5, bottom).63 The pincer ligand used in this investigation was [N(CH2CH2PiPr2)2]−, (P′NP′), where the central functional group was an amido species. The starting complexes, [M(CO)(NO)(P′NP′)] {M = Mo (29a), W (29b)}, reacted with CO2 (at 2 atm pressure, 298 K) to form the [2 + 2] cycloaddition product (across the metal–nitrogen bond) to form octahedral carbamate complexes (30a, 30b, respectively). In the X-ray structure of the tungsten complex (trans isomer), the W–N distance was found to be 2.2603(19) Å. This is elongated by 0.2 Å with respect to the precursor complex. The W–O distance was recorded as 2.2155(18) Å, the C(O)–O distance 1.283(3) Å, and the CO distance 1.208(3) Å. During this study, the cycloaddition product was found to be an “off-cycle step”, interfering with the productive catalytic hydrogenation of CO2 in the presence of base (which occurs via complexes 31a or 31b, respectively). This example demonstrates the competition between the 1,2-addition of H–H and the [2 + 2] cycloaddition of CO2 across the M–amido (M = Mo, W) reactive site. Due to the strong, irreversible binding of CO2, complexes 30a and 30b were found not to react further with H2, thus blocking the catalytic activity. On the other hand, it should be noted that the same ligand platform supporting the late transition metal complex, [(P′N(H)P′)IrH3] (the analogue to 31 with a proton at the nitrogen centre), reported by Hazari and co-workers was an excellent catalyst for the production of formate.64 Here, [2 + 2] cycloaddition across the iridium–amido bond was not observed in the catalytic cycle involving the iridium–amido bond in [(P′NP′)IrH2]. The key differences between these two systems can be traced back to their tendency to form strong M–O bonds and the presence of π-acceptor co-ligands, NO and CO, on the M(0) metal centres, making the [2 + 2] pathway irreversible.
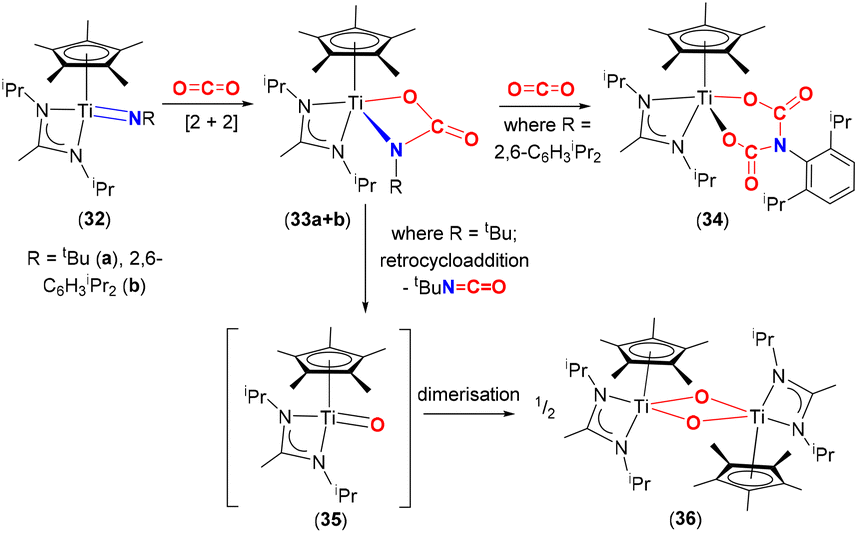
Mountford and co-workers studied the reactivity of three-legged piano stool complexes [Ti(NR)(η5-C5Me5){MeC(NiPr2)2}], containing imido ligands, NR; R = tbutyl (32a) and 2,6-diisopropylphenyl (Dipp, 32b) with CO2 (Scheme 6).65,66 The reactivity of these complexes was dependent on the substituents on the imido nitrogen. Reactions of 32a and 32b with CO2 proceed through a [2 + 2] cycloaddition involving the titanium–imido bond to form the corresponding carbamate complexes, [Ti{O(CO)NR}(η5-C5Me5)(MeC(NiPr2)2)] (33a and 33b, respectively). The carbamate complexes reacted in two different ways depending on the R substituents. In the case of 33b, the carbamate reacted further with an additional equivalent of CO2 to form bicarbamate species [Ti{OC(O)N(C6H3iPr2)C(O)O}}(η5-C5Me5){MeC(NiPr2)2}] (34), where a second molecule of CO2 inserted into the Ir–N bond. Whereas for 33a, the carbamate underwent a retrocycloaddition reaction eliminating tBuNCO as a by-product and forming transient titanium oxo species [Ti(O)(η5-C5Me5){MeC(NiPr)2}] (35), which dimerised quickly forming a Ti2O2 (μ2-O) bridged structure (36). It was postulated that the difference in reactivity between 33a and 33b was due to the electron donating (tBu) and withdrawing (2,6-diisopropylphenyl) properties of the substituents, which influenced π-bonding in TiNR species as well as stabilisation of the resulting bicarbamate species, in the case of 34. The aryl group on nitrogen is able to reduce the electron density on the resultant “OC(O)N(C6H3iPr2)(CO)O” moiety thereby forming a more stable complex, resistant to retrocycloaddition. For the case of tBu complex 33a, the tendency of early transition metals to form strong M–O bonds yet again acts as a thermodynamic sink for the formation of 36.67
 | ||
| Scheme 6 Reactivity of titanium imido complex with CO2. | ||
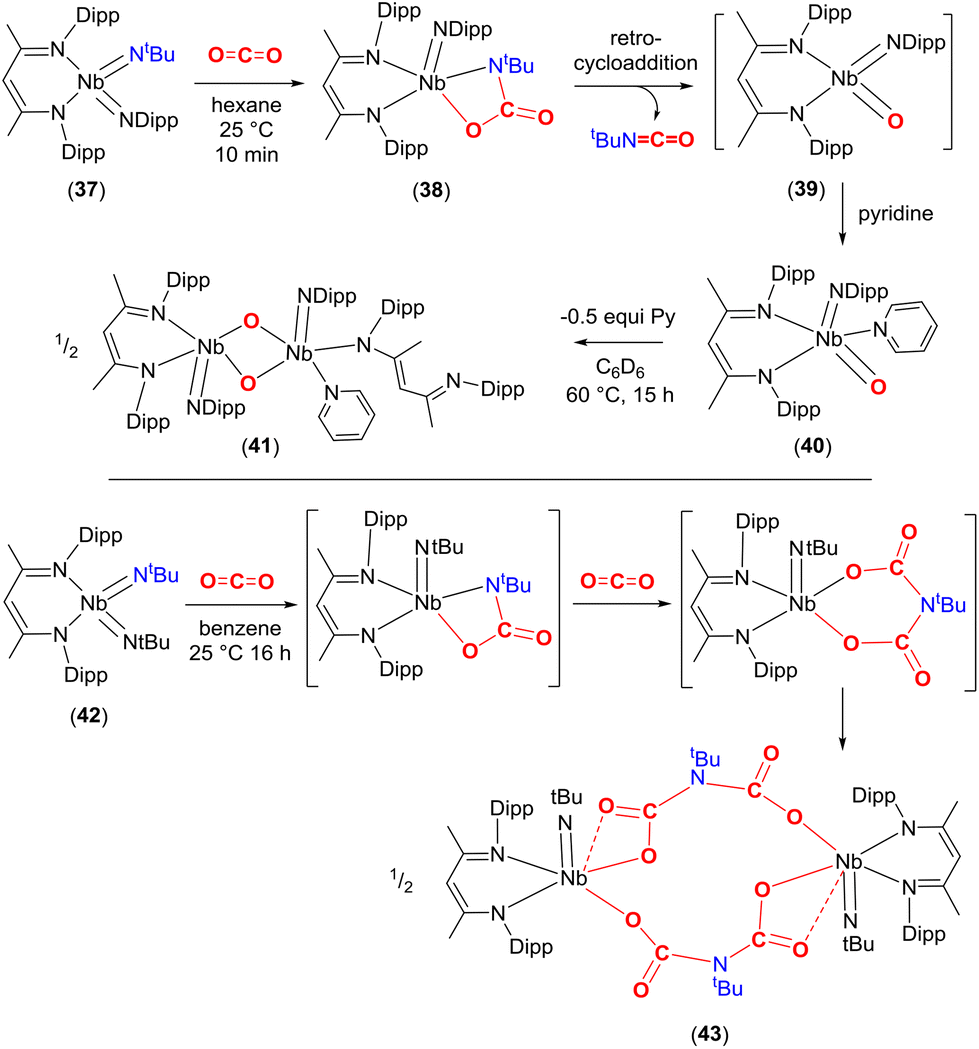
A similar reactivity pattern was found in a set of bis-imido complexes containing niobium(V) metal centres of the type [Nb(NtBu)2(BDI)] and [Nb(NtBu)(Ndipp)(BDI)] (37, Scheme 7). They undergo [2 + 2] cycloaddition with CO2 at 298 K. The effect of the substituent on the second spectator imido ligand NR (R = tBu or Dipp) played a crucial role in terms of reactivity. For example, in the case of 37, the carbamate complex [Nb(OC(O)NtBu)(Ndipp)(BDI)] (38) was isolated from hexane (Scheme 7, top). The newly formed bond lengths in this complex were found to be Nb–O 2.070(3), tBuN–CO 1.391(5), C(O)–O 1.345(5) and OC 1.205(5) Å. In the same way as that outlined for complex 33a above, complex 38 also underwent a [2 + 2] retrocycloaddition to eliminate tBuNCO from the carbamate and generates transient oxo species, [Nb(O)(Ndipp)(BDI)] (39), which could be trapped with pyridine to form [Nb(O)(Ndipp)(BDI)(pyridine)] (40). Finally, complex 40 was found to transform further by dimerising to form the bis-μ-oxo complex [Nb(Nar)(κ1-BDI)(pyridine)(μ-O)Nb(Nar)(BDI)] (41). In contrast, bis-imido complex [Nb(NtBu)2(BDI)] (42) formed iminodicarboxylate bridged dimer [Nb(NtBu){μ-O2CN(tBu)CO2}(BDI)]2 (43) through the proposed successive carbamate [Nb{OC(O)N(tBu)}(NtBu)(BDI)] and bicarbamate [Nb{OC(O)N(tBu)C(O)O)}(NtBu)(BDI)] intermediates after reaction with CO2 (Scheme 7, bottom). The extensive reactivity was ascribed to the high energy d-valence electrons of group 5 and the π-loading methodology using MNR multiple bonds.68
 | ||
| Scheme 7 The [2 + 2] cycloaddition reaction of niobium imido complexes with CO2 and their further transformations. | ||
In summary, the [2 + 2] bond activation of CO2 represents a suitable and promising reaction step for the activation of CO2. The key transformations for each of the species, CR2, SiR2, NR and O, are summarised in Scheme 8. Each shares a similar initial mode of CO2 addition via reactivity of one of the CO bonds in CO2, though the regiochemistry is dependent on the polarisation of the ME bond. This mode of reactivity is consistently observed across a range of transition metal systems. Subsequent steps, when observed, then follow different pathways. In many cases, the CO2 binding is reversible and weak within the complexes. This may preclude further functionalisation of this moiety since it is not sufficiently activated. On the other hand, in some examples, the stability of the resulting product is too high, thus precluding their use within a catalytic process. Some fine tuning of the ME species is required specifically with regard to the polarity of the bond and the nature of π-bonding within the ME bond. The two most common deactivation pathways have been found: (a) disproportionation of CO2 and (b) the formation of stable M–CO and/or M–O bonds. Thus, to date, transformations of CO2 utilising ME systems are limited to stoichiometric ones.
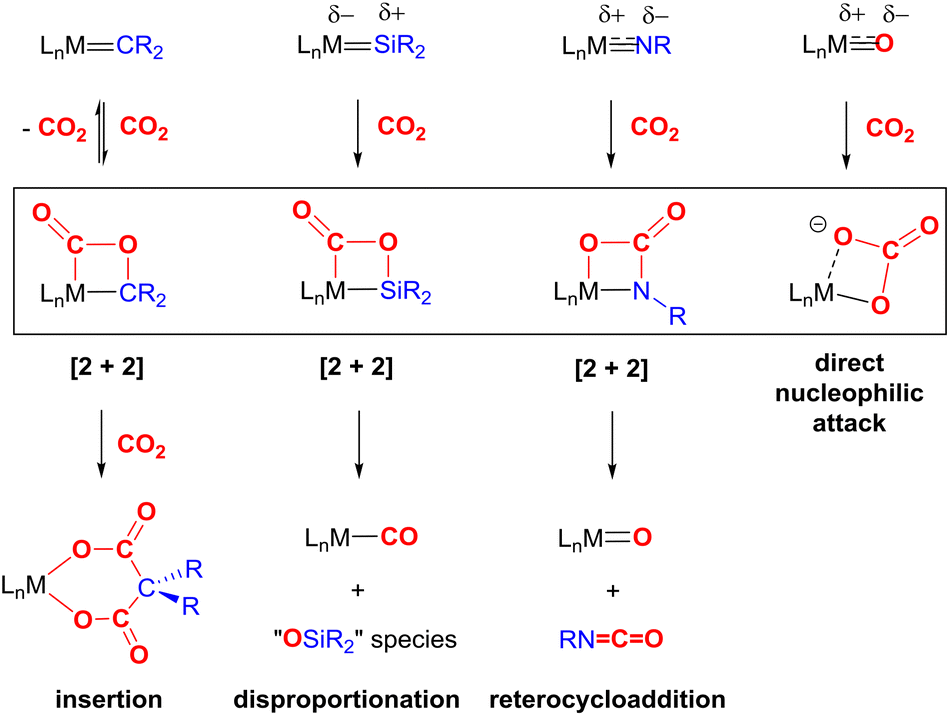 | ||
| Scheme 8 Summary of the reactivity of ME (E = CR2, SiR2, NR, O) multiple bonds with CO2. L = auxiliary ligands, R = alkyl/aryl substituents. | ||
Activation of CO2 in systems containing a two-atom bridge (Fig. 1C)
This section focuses on systems that present a two-atom spacer between activated CO2 and the transition metal centre in the final product. As shown in Scheme 9, we outline three key modes of CO2 activation involving systems containing two-atom (AB or EH) bridges. For the purposes of defining this reactivity we consider two scenarios, either two centres A and B (where A is connected to the metal centre) or two centres containing an electronegative atom and hydrogen atom (i.e.E–H). In the case of A and B centres, there are two types of activation mode. Type 1 is typically found when there is a degree of unsaturation between the A and B centres. In this case, an oxidation state increase by two at the metal centre is observed. In type 2, the B functional group is a nucleophilic heteroatom. In both of these cases, the activation can be classed as a 1,3-addition of CO2. This is analogous to the 1,3-addition observed in the dearomatisation/aromatisation systems that are outlined in a separate section below (vide infra). This mode of activation “traps” the CO2 molecule within a 5-membered metallacycle where the outer centre B attacks the electrophilic carbon centre and a new M–O bond is formed.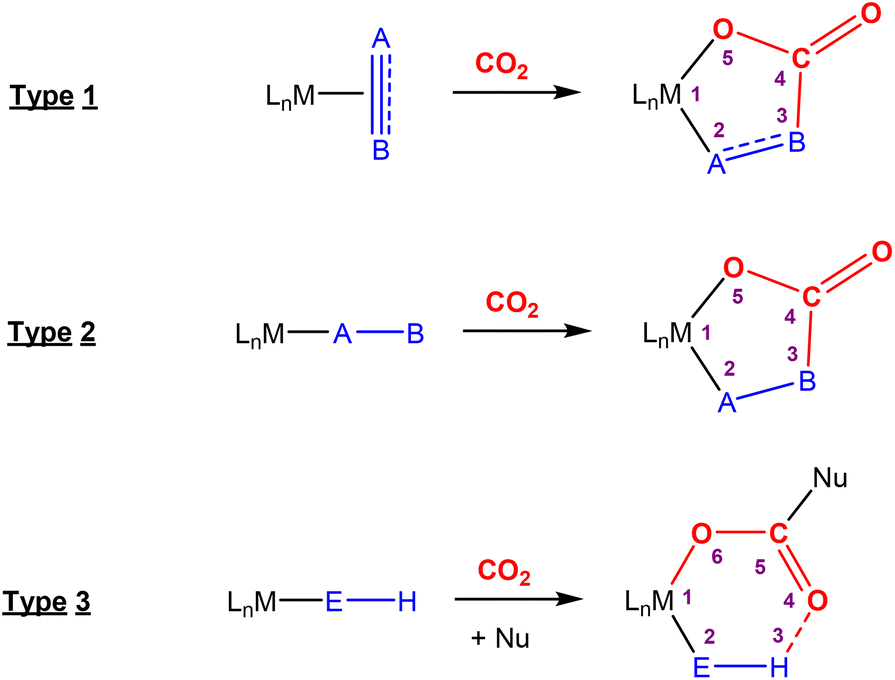 | ||
| Scheme 9 Three different types of CO2 fixation involving a two-atom bridge. For type 1, the oxidation state of the metal centre increases by two. | ||
An alternative and quite different mode of two-atom bridge activation is also possible. This third type of transformation occurs when the two atoms are an E–H unit. In this case, there is an attack of the electrophilic carbon with an external nucleophile. Upon activation, a covalent bond between the hydrogen of the E–H unit and the oxygen of the former CO2 molecule is not possible. However, a six membered arrangement is feasible via hydrogen bonding interactions between the hydrogen and oxygen atoms. This mode of transformation, in particular, has found application in the hydrogenation of CO2 to formates. A number of relevant examples for each type of activation are outlined below.
Type 1 activation
There are several examples where the AB ligand unit is an unsaturated alkene or alkyne moiety (Scheme 10). The first examples typically employed zerovalent nickel-based precursors, which led to the formation of five-membered nickellalactones; a representative original example reported by Hoberg, Schaefer and co-workers is shown in Scheme 10.69 Such systems represent one of the first examples of stoichiometric CO2 activation and transformation.70–73 Activation is expected to be facilitated by the cooperation of the pre-coordinated alkene (or alkyne), which bridges the metal and CO2 moiety, furnishing the stable five membered metallacycles. This is shown in the scheme where [Ni(cdt)] is reacted with ethene and CO2 in the presence of bidentate ligands, bipy or dcpe, where the products 44 and 45 are formed. Nickel's original tridentate ligand is lost from the coordination sphere and is replaced by the bidentate ligand and a new κ2-C,O-CH2CH2C(O)O ligand is formed via the coupling of one ethene and one CO2 molecule. During the transformation, the oxidation state of the metal increases by two and the new organic fragment acts as an X2-type ligand (i.e. an oxidative coupling reaction). In a number of related examples involving nickel metal centres, it was found that further transformation of the metallalactones was possible, leading to the formation of organic functionalities such as carboxylic acids and organic formates.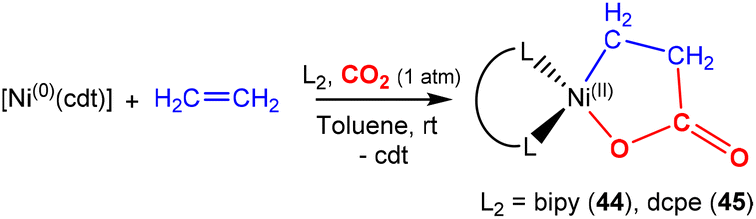 | ||
| Scheme 10 First example of an isolated nickellalactone from oxidative coupling involving ethene and CO2 [cdt = 1,5,9-cyclododecatriene; bipy = bipyridine and dcpe = bis(dicyclohexylphosphino)ethane]. | ||
The corresponding iron-based metallalactones were also prepared (Schemes 11 and 12). The five-membered rings formed in these systems are less stable than the nickel(II) systems, which means that they are able to undergo multiple insertions of either CO2 or alkenes. This is exemplified in the reactions reported by Hoberg and co-workers (Scheme 11).72 They reported the transformation of [Fe(CH2CH2)2(PEt3)2] with ethene and CO2 gases in the presence of various co-ligands. The initial oxidative coupling transformation led to ferralactone complexes 46a and 46b. As shown in the scheme, the co-ligands influenced the subsequent reactivity and selectivity of the insertion of a second equivalent of CO2. When the co-ligand was the bidentate ligand dcpe, the complex [Fe{OC(O)CH2CH2C(O)O}(dcpe)(PEt3)x] (47a) was formed. It should be noted that in the original article, the authors indicated that it was likely that some PEt3 ligand remained within the coordination sphere of the metal centre within these transformations. The formation of 47a is the result of a formal insertion of a second CO2 molecule into the Fe–C bond of 46a. Complex 47a contains a seven-membered metallacycle. On the other hand, when the co-ligands were monodentate Pme3 ligands, a different product selectivity was observed. In this case [Fe{OC(O)CH(CH3)C(O)O}(Pme3)2(PEt3)x] (47b) was formed. Complex 47b contains the smaller six-membered metallacycle. The proposed mechanism for this transformation involves a prior “ring contraction” process where the five-membered ferralactone is transformed into the four-membered equivalent (i.e.46bint, in the scheme) via a set of β-hydride elimination/migratory insertion, isomerisation steps. The insertion of a second CO2 molecule into the new Fe–C bond occurs. Neither 47a or 47b were isolated, however, their identities were confirmed by their subsequent reaction with acidic methanol solutions, which liberated the methyl esters of succinic acid and methylmalonic acid, respectively.
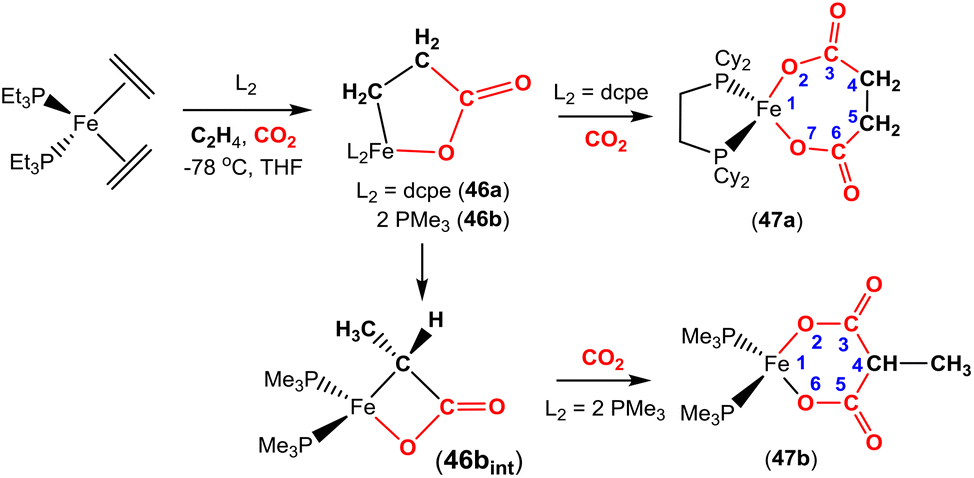 | ||
| Scheme 11 Ligand controlled insertion of CO2 into ferralactones. | ||
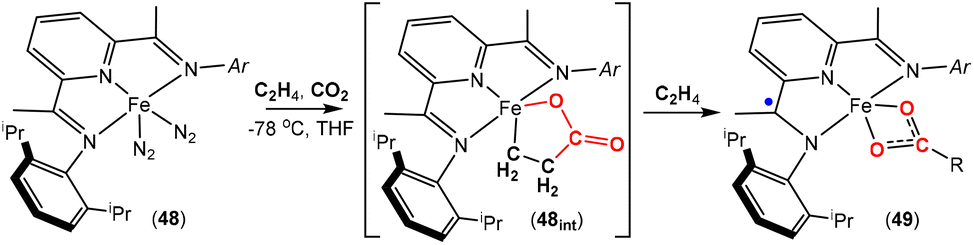 | ||
| Scheme 12 Unsaturated iron carboxylate products via proposed intermediate species 48int. The R group is variable depending on the conditions (see text for details). | ||
More recently, an interesting iron system was reported by Chirik and co-workers (Scheme 12).74 In this case, instead of double insertion of CO2 to form a ferralactone, there was evidence of multiple insertions of ethene moieties. Their investigation focused on the reactivity of pyridine(diimine) iron complex 48 with ethene and CO2 under various conditions. These reactions led to the formation of a range of iron carboxylate products of the type of complex 49, where the R group (as shown in the scheme) was found to have even chain lengths up to 20 carbon atoms long. This arises from multiple insertions of the ethene. By examining the product distributions, carrying out stoichiometric experiments with hydrogen and deuterium labelling studies, it was rationalised that the five-membered metallalactone species, 48int, was the common intermediate in all transformations and multiple insertions into this species was possible, leading to further macrocyclic metallalactone intermediates. It was proposed that competitive β-hydride elimination steps with ethene insertion were important in determining the size of the chain. Furthermore, both saturated and unsaturated chains (as well as dicarboxylate species bridging two iron complexes) were observed, providing evidence for metal–hydride species. It was also found that one of the imine functional groups on the ligand underwent a single electron reduction.
Bernskoetter and co-workers recently reported the synthesis of Fe(0) complex 50 containing a coordinated alkene, which promoted CO2 fixation leading to the formation of the stable ferralactone 51 (Scheme 13).75 The authors also prepared a Fe(0) complex containing coordinated CO2 instead of an alkene. The use of KC8 as a reducing agent in the presence of CO2 (1.5 atm) led to the stoichiometric formation of acrylate salts.
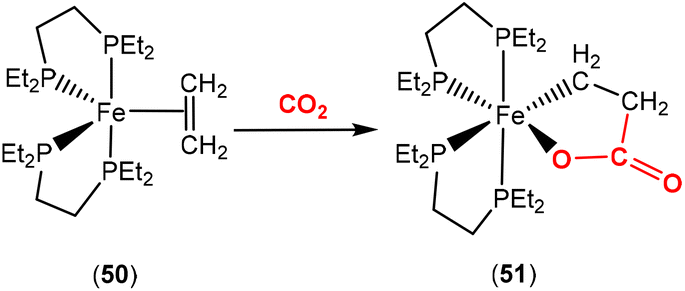 | ||
| Scheme 13 Ferralactone formed from direct CO2–ethylene coupling. | ||
Earlier this year, Field and co-workers reported the isolation and structural characterisation of unsaturated ferralactone complexes derived from phenylacetylene and CO2 (Scheme 14).76 Their investigations revealed some interesting selectivity. For example, when trans-[Fe(H)(CCPh)(dmpe)2] [52; dmpe = bis(dimethylphosphino)ethane] was reacted with CO2, the initial product was the corresponding formate complex (53), which resulted from the formal insertion of the CO2 unit into the Fe–H bond. This fast insertion step, which was observed immediately after addition, was found to be reversible, however, and it was found that the thermodynamic product from this reaction mixture was the ferralactone complex, cis-[Fe{C(H)C(Ph)C(O)O}(dmpe)2] (54). Partial conversion to complex 54 was achieved after extended periods. In this case, the former hydride was located on the α-carbon in the product. An analogous complex is found in the case of a tbutyl group in place of the phenyl substituent. Interestingly, regio-isomer complex 56 was obtained by using the η2-bound alkyne precursor 55 instead of complex 52. This switch in regioselectivity was rationalised on the basis of a different mechanism of activation. It was suggested that the electrocyclic (oxidative) coupling of CO2 with the π-bound phenylacetylene occurred. Finally, when the alkyne was changed to the parent acetylene, double CO2 insertion products were additionally observed, cis-[Fe{C(H)C(CO2H)C(O)O}(dmpe)2] and cis-[Fe{C(H)C(H)C(O)O}(dmpe)2], in a ratio of approximately 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Similar observations were also found for the corresponding complexes containing bis(diethylphosphino)ethane as a co-ligand.77
:1. Similar observations were also found for the corresponding complexes containing bis(diethylphosphino)ethane as a co-ligand.77
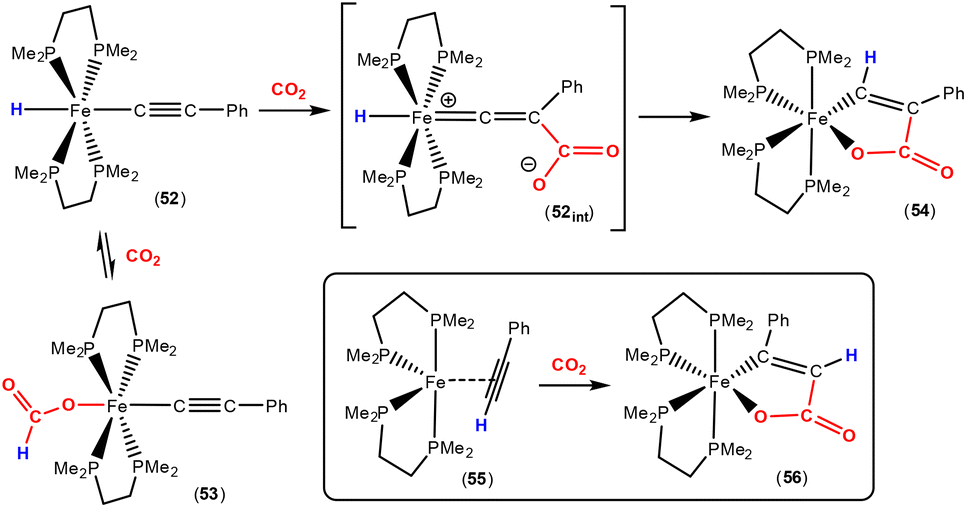 | ||
| Scheme 14 Ferralactone regioisomers via CO2 fixation in iron acetylido/acetylene complexes. | ||
Similar oxidative coupling transformations, where alkenes are utilised as the AB coupling unit, have been observed across a range of other transition metals. Selected examples are highlighted in Scheme 15. Early examples by the groups of Cohen and Bercaw described the coupling of CO2 and coordinated alkene in titanocene at −78 °C to form stable, bright red titanalactone 58 (Scheme 15, top). This metallalactone functional group was confirmed by infrared spectroscopy where a vibration band for the CO stretch at 1653 cm−1 was observed.78 The use of 13C isotope labelling confirmed the 13C–13C bond in the lactone fragment formed with a coupling constant of 52 Hz between the β-methylene carbon and carbonyl carbon of [(Cp*)2Ti{O13C(O)–13CH2CH2}]. Attempts to reproduce the same reactivity at 0 °C led to unidentified products.
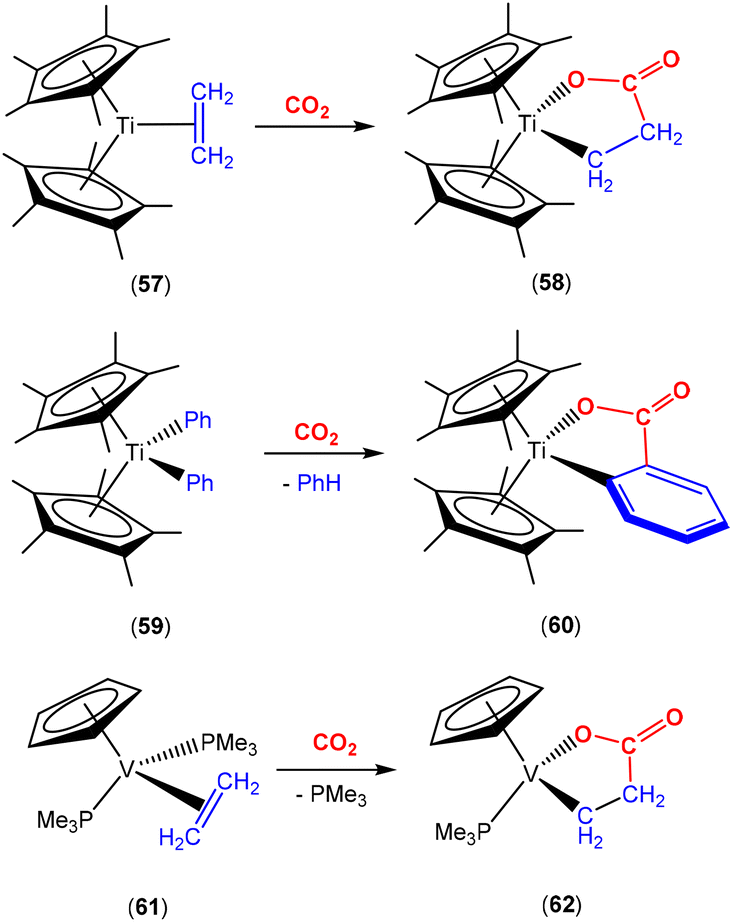 | ||
| Scheme 15 Selected examples of CO2 and alkyl/alkene coupling on early transition metal complexes. | ||
Interestingly, the two-atom bridge can also be part of an aromatic system such as the phenyl unit in the example of 59 (Scheme 15, middle), where the carbon of the CO2 molecule adds to the β-carbon in the phenyl unit.79 More recently, Miqueu and co-workers characterised the short-lived n2-benzyne titanacene intermediate formed from complex 59 by UV-photospectroscopy and DTF calculations.80 The insertion of CO2 into the 2-atom bridge (CαCβ) of this short-lived intermediate explains the formation of metallalactone 60 (Scheme 15, middle). Furthermore, a related example was reported by O'Hare where the two Cp* ligands were replaced with a fused version of these ligands, i.e. η8-permethylpentalene (Pn*).81 Titanium dialkyl complexes of the type [Ti(Pn*)R2] (where R = Me, CH2Ph) were isolated and exposed to CO2 to give the products of double insertion, [Ti(Pn*)(κ2-O,O-O2CR)2], where CO2 inserted into the M–R bond. In these cases, coordinated carboxylate groups were the resulting products.
The reactivity of vanadium complex 61 (Scheme 15 bottom) with CO2 to form metallalactones was studied by the Teuben group.82 Despite the presence of β-hydrogen atoms, the degree of planarity of the lactone ring increases their distance to the metal centre and increases its stability against β-hydride elimination.
Although palladalactones, such as complexes 63 and 64 (Fig. 2), are accepted intermediates in the mechanism of the palladium catalysed formation of acrylates from CO2 and alkenes, they have not been isolated from stoichiometric investigations. Nonetheless, they have been studied by computational methods83 and prepared by alternative synthetic methods by the groups of Yamamoto84 and Limbach.85 Other metallalactones form via the reaction of an alkene molecule with CO2 using the precursors [Zr(Cp)2(C2H4)(PMe3)]86 and [Rh(bpy)(C2H4)Cl],87 respectively.
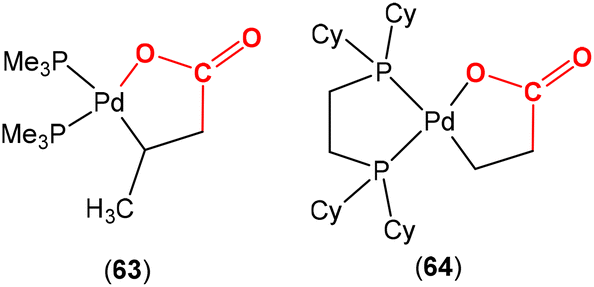 | ||
| Fig. 2 Examples of palladalactones prepared by alternative synthetic methods. | ||
Pursuing the catalytic synthesis of acrylic acid, the Carmona and Galindo groups developed a series of molybdenum and tungsten complexes, which inserted CO2 into the 2-atom bridge of a coordinated alkene.88–90 In these cases, species containing coordinated CO2 or intermediate metallalactone could neither be detected nor isolated. Nonetheless, these are postulated as intermediates within the transformations. Attempts to liberate the acrylate from stable dimeric complexes and regenerate the starting complexes were not successful and unfortunately a catalytic cycle was not realised.88,89 More recently, in 2019 Carmona and co-workers further outlined the synthesis of related complexes of the same metals where both CO2 and alkenes were simultaneously coordinated to the metal centre.90 Crystal structures of these complexes were reported. In these cases, however, attempts to react CO2 with the alkene were unsuccessful and no acrylate formation was observed.
Recently, Iwasawa and co-workers reported the reactivity of Ru(0) complex 65 (Scheme 16), which contained a tetradentate phosphorus ligand (L4).91 This complex was active for the catalytic conversion of ethene and CO2 into acrylate salts as shown in the scheme. In addition to the isolation of the main ruthenalactone product of CO2 activation involving the two-atom bridge bond (CC) (complex 66), small quantities of the corresponding carbonato complex [Ru(L4)(κ2-O,O-CO3)] were also observed. This suggested that some disproportionation of CO2 into [CO3]2− and CO occurred, similarly to a number of other investigations highlighted herein. Indeed, further heating of the mixtures gave greater conversions to the carbonato complex. Alongside, two additional complexes were obtained. These were the carbonyl complex, [Ru(L4)(CO)], and hydridoacrylato complex 67. The formation of the carbonyl complex confirmed the disproportionation-type reactivity. Complex 67 formed as a result of β-hydride elimination. Whilst the selectivity to generate complex 67 was poor, it was found that treatment with a base in the presence of further ethene gave the acrylate salts and regenerated complex 65, thus providing a catalytic means of synthesising acrylate salts from CO2. At this stage the catalytic activity is limited. Nevertheless, it highlights the potential for the synthesis of CO2 derived acrylates with zerovalent ruthenium complexes. This system was explored in further mechanistic detail, which revealed a second competitive pathway for the formation of acrylate salts.92
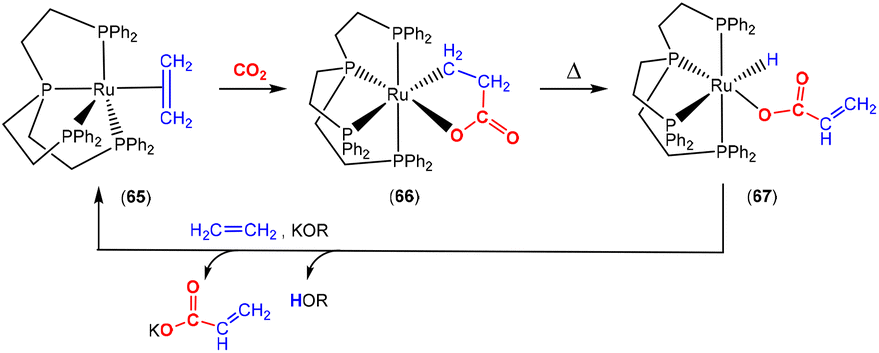 | ||
| Scheme 16 Investigations leading to the first example of the ruthenium-catalysed synthesis of acrylate salts from ethene and CO2. | ||
Finally, Hopmann and co-workers reported some computational investigations into the mechanisms of carboxylation reactions involving M–C(sp3) and M–C(sp2) species, where M = Cu, Rh, Pd.93,94 These revealed that depending on the type of ligand, steric effects and the nature of the metal, inner-sphere or outer sphere mechanisms involving cyclic or acyclic transition states were possible.
Type 2 activation
One other common strategy for the activation of CO2 is for the outer B atom of the AB unit to contain a nucleophilic functional group. In this way, it attacks the electrophilic carbon and a five-membered ring is formed, as outlined in Scheme 9 above. There are a wide variety of functional groups that have been utilised for this mode of activation. Selected examples are given in Scheme 17.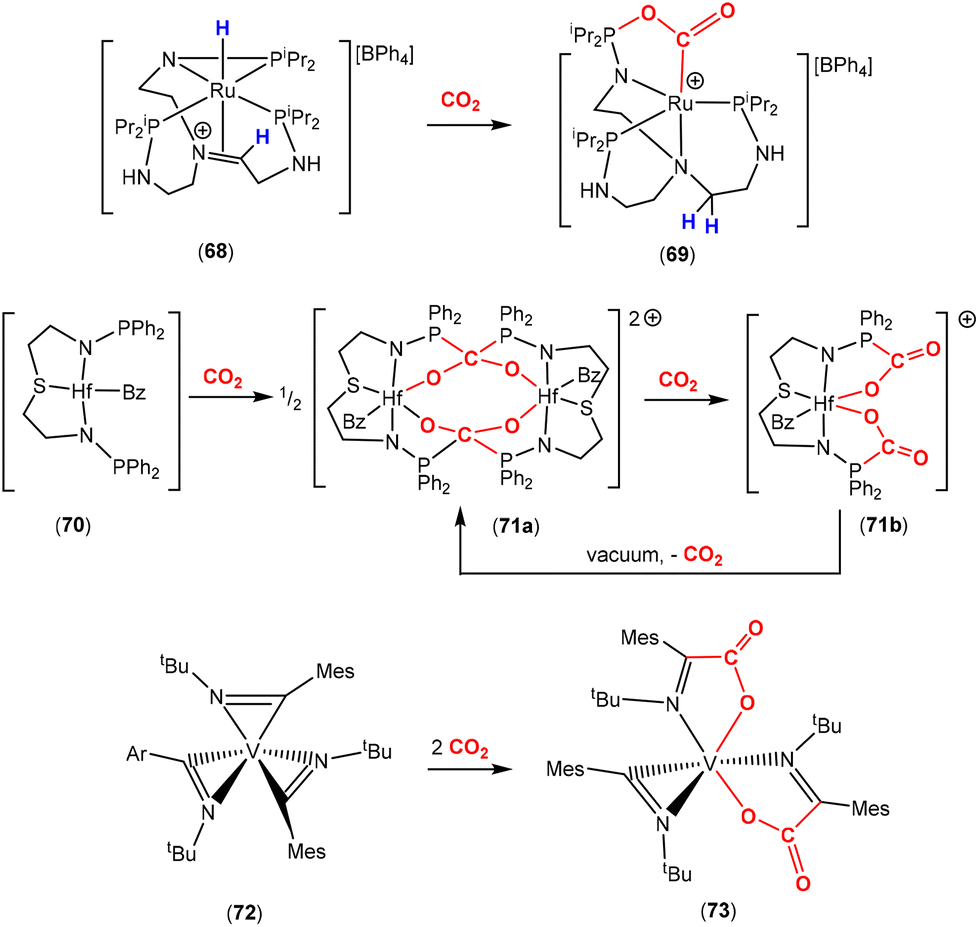 | ||
| Scheme 17 Selected examples of the type 2 activation of CO2. | ||
An example of a multidentate ligand system that was explored by the Stephan group for CO2 activation was shown in Scheme 17, top.95 They reported the transformation of complex 68, which contained a ligand derived from [N{(CH2)2N(H)PiPr2}3] via a deprotonation step followed by a halide exchange step. The complex contains the κ2-N,P coordination mode of one of the “arms” of the ligand. In this case, a MLC process takes place via a hydride migration step and its incorporation into the ligand structure. This permits a molecule of CO2 to interact with the metal centre and leads to its activation by the Lewis basic phosphorus donor. The transformation leads to the formation of the five-membered ring [RuNPOC(O)] structure in the new complex, 69, as shown in the scheme. In the case of a related neutral complex, it was found that an alternative activation pathway occurred across a N–P bond where it was broken and a five-membered ring [RuNC(O)OP] structure formed instead.
Other chelating ligands containing phosphinoamides within their framework were also utilised by Stephan and co-workers to promote CO2 activation by early transition metal complexes (Scheme 17, middle).96 Hafnium complex 70 exhibited reactivity with CO2 by promoting its insertion between the two-atom bridge involving the N–PPh2 units. Slow and controlled addition of CO2 led to the precipitation of dinuclear complex 71a, as single crystals. Complex 71a formed as the result of the activation of two CO2 molecules across two metal complexes (1:1). Crystallographic analysis revealed that activation by the N–PPh2 units led to a doubly bridged P2C(O–Hf)2 motif. When this compound was placed in solution, it converted into a mononuclear complex, which was then further transformed in the presence of CO2 into complex 71b. This contained two activated CO2 molecules per hafnium centre featuring two, [Hf–NPC(O)O], five-membered rings. Placing 71b under reduced pressure led to the second equivalent of CO2 being reversibly lost. On the other hand, when an isopropyl phosphine-based ligand was used instead of a phenyl-based phosphine, the more basic phosphine centres meant that once double CO2 activation had occurred, they could not be removed under reduced pressure.
An interesting example of activation involving η2-bound iminoacyl units was reported by Floriani and co-workers (Scheme 17, bottom).97 They reported the synthesis of vanadium complex 72, which contained three η2-bound iminoacyl units, via the insertion of tertbutylisocyanide into the three V–C bonds of [V(Mes)3(THF)]. Two of the three “V{CN(R)}” units acted as two atom bridging units for the irreversible activation of CO2 leading to complex 73. In this case, the more nucleophilic part of the ligand involved the carbon of the iminoacyl group leading to the product of insertion into the V–C bonds.
More recently, the Anwander group reported the reversible binding of CO2 to multiple sites on a set of cerium complexes including [Ce(Me2pz)4]2 (Scheme 18; Me2pz = 3,5-dimethylpyrazolyl).98 In this case, the AB unit corresponds to the N–N unit of the pyrazolyl ligands and the addition of CO2 across this two-atom bridge. This allowed for complexes such as 74 to be synthesised and for the development of materials able to adsorb up to 20 wt% of CO2.99 The insertion was reversible, acting as a “CO2 sponge”, and could be exploited for the catalytic formation of cyclic carbonates from epoxides.
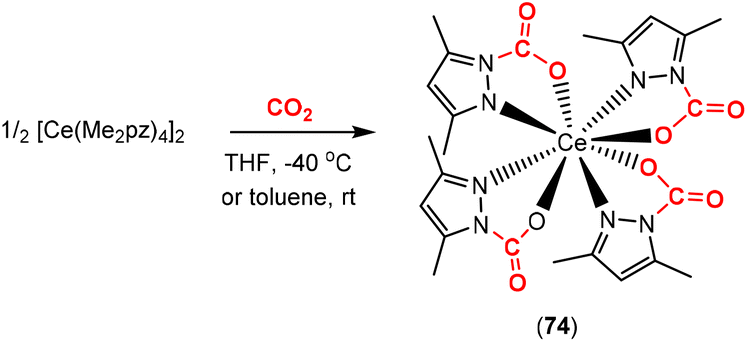 | ||
| Scheme 18 Multiple type 2 CO2 activations on the cerium–pyrazole complex. | ||
Carmona and co-workers described the use of an iridium complex containing a pyridylidene ligand in addition to a hydrotris(pyrazolyl)borate co-ligand (Scheme 19). Exposure to CO2 led to the formation of a carbamate complex (75). In this case, the AB unit corresponds to C–N (within the pyridylidene ligand) and the nitrogen atom acts as the nucleophile where the final product is the result of CO2 activation involving a 2-atom bridge (CN) between the metal and the ligand.
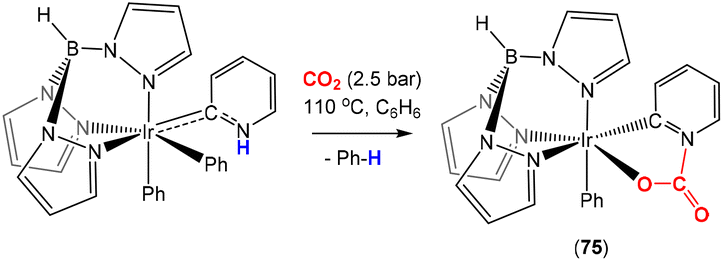 | ||
| Scheme 19 In situ generation of the Ir–pyridylidene complex and its reactivity towards CO2 fixation via 2-atom bridge type 2 activation. | ||
The Mann research group reported a cyclometallated iridium(III) complex where the AB atom bridge corresponded to a hydrazino (H2N–NH2) ligand (Scheme 20). They showed that the coordinated hydrazine could slowly, yet, irreversibly bind CO2 in an efficient way, forming carbazate complex 77.100 After N-carboxylation of one of the hydrazino units and proton transfer to the second ligand, a metal-coordinated ammonium carbamate ion pair formed. This lost a single hydrazino ligand as [H2NNH3][X], followed by immediate chelation of the bidentate carbazate moiety to form the neutral iridium(III) carbazate species 77.
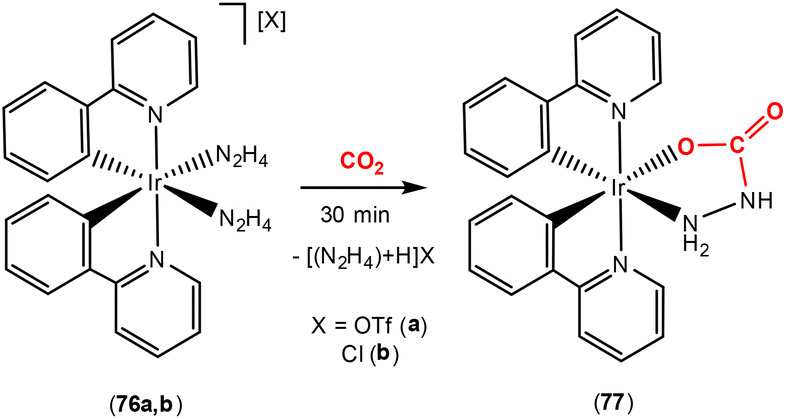 | ||
| Scheme 20 Type 2 activation of CO2 using a hydrazido scaffold. | ||
Very recently, Cossairt and co-workers reported further developments on systems containing nitrogen based nucleophiles for the activation of CO2 (Scheme 21).101 In this case, they utilised protic N-heterocyclic carbene units within a multidentate ligand scaffold. The carbene carbon and adjacent nitrogen acted as the two-atom bridge during activation. Within the transformation, deprotonation of the N–H group on the NHC led to a basic and nucleophilic site, which attacked the CO2 molecule. Coordination of oxygen to the metal facilitated activation of the CO2 molecule and led to a system of Type 2. The authors intentionally designed the system with the concept of ligand cooperativity in mind by introducing protic sites in close proximity to the metal centre. Here, a multidentate electron-rich ligand was used to permit the formation of thermodynamically stable and long-lived catalytic species. The dependency of the complex's activity in converting CO2 into formate was studied in relation to the quantity and type of base used. Additionally, labelled 13CO2 and X-ray crystallography allowed the identification of the relevant active intermediates such as complexes 78 and 79, where L was a coordinating solvent such as THF.
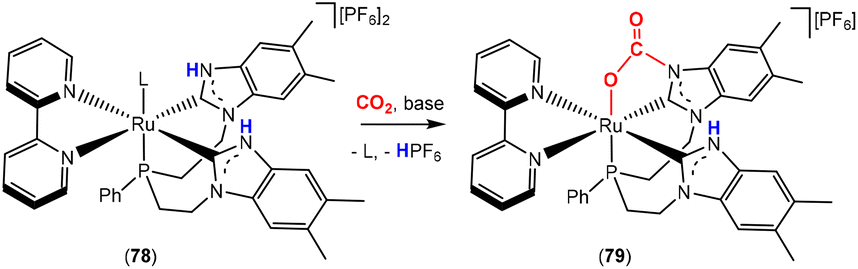 | ||
| Scheme 21 Activation of CO2 by ligand cooperation on benzimidazole-based NHC complexes (L = weakly coordinating solvent ligand). | ||
Type 3 activation
One of the archetypical ligand motifs for the activation of CO2 is those of the type shown in Scheme 22. These examples involve a pincer motif with a polar N–H functional group. Initial examples of systems of this type were reported by the groups of Hazari, Crabtree and Bernskoetter.64,102 The effect of this two atom bridge (E–H) unit was studied via computational methods. As shown in the scheme, a CO2 molecule was found to insert into the iridium–hydride bond of complex 80. Insertion of CO2 into such a bond can occur without any MLC. However, in this case, the presence of the N–H unit facilitates the transformation. This would fit into type 3 reactivity when considering that the N–H unit acts as a scaffold holding activated CO2 in place for attack at the electrophilic carbon centre, e.g. by a hydride. It should be noted, however, that the mechanism for this transformation is not implied by this classification. The impact of hydrogen bonding, and the eventual formation of a six-membered ring, on activation was significant. The calculations showed that the activation energy of insertion with the involvement of a hydrogen bond [N–H⋯OC(O)] was significantly lower, cf. 52.1 kJ mol−1 (with) and 71.3 kJ mol−1 (without). Highly stable complex 80 was found to be one of the most active catalysts for the transformation of CO2 into formate salts, achieving TON and TOF values of 348000 and 18780 h−1, respectively.
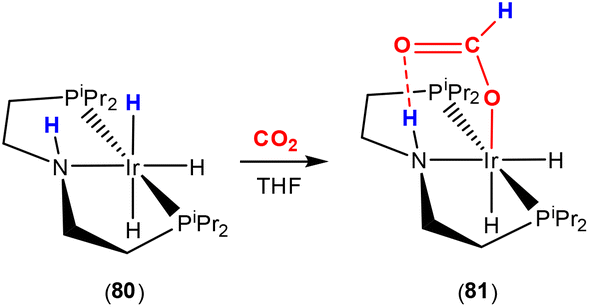 | ||
| Scheme 22 Type 3 activation of CO2 involving ligand cooperation of an N–H bridge. Insertion reaction assisted by secondary sphere interactions. | ||
Activation of CO2 in systems containing a three-atom bridge
This section focuses on systems that present a three-atom spacer between activated CO2 and the transition metal centre. As shown in Scheme 23, in a typical mononuclear system, activation involving a three-atom bridge leads to the formation of a six-membered ring with the metal centre.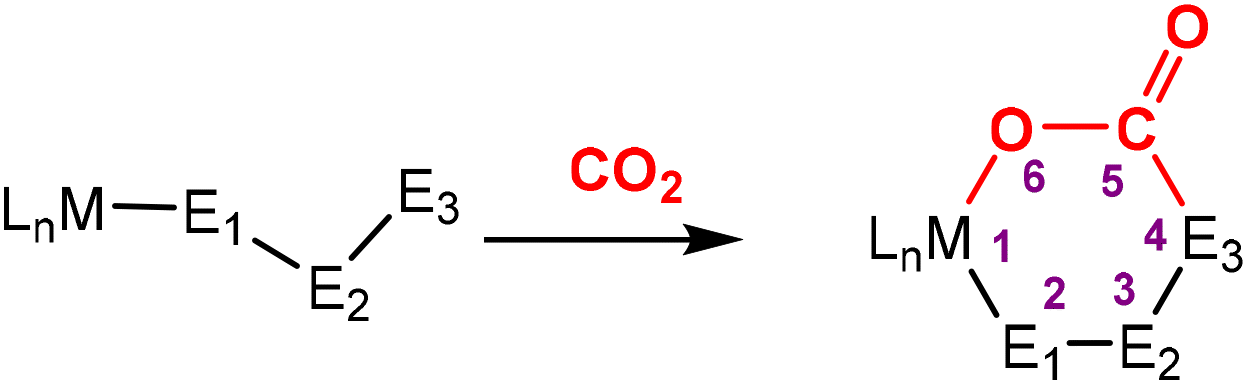 | ||
| Scheme 23 Schematic representation of the activation of CO2 utilising a three-atom bridging unit (in some examples, an interaction between M and E3 is possible). | ||
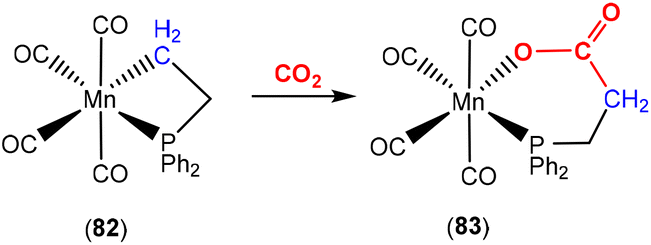
An early example of CO2 activation involving a three-atom bridge was reported by Behr and co-workers (Scheme 24).103 They reported manganese carbonyl complex 82, which contained a strained four-membered phosphametallacycle. Reaction with CO2 led to new complex 83, which formed via a formal insertion into the manganese–carbon bond. The resulting product was a ring expanded six-membered phospha-metallalactone complex.
 | ||
| Scheme 24 Utilising the ring strain of a phospha-metallacycle to activate CO2. | ||
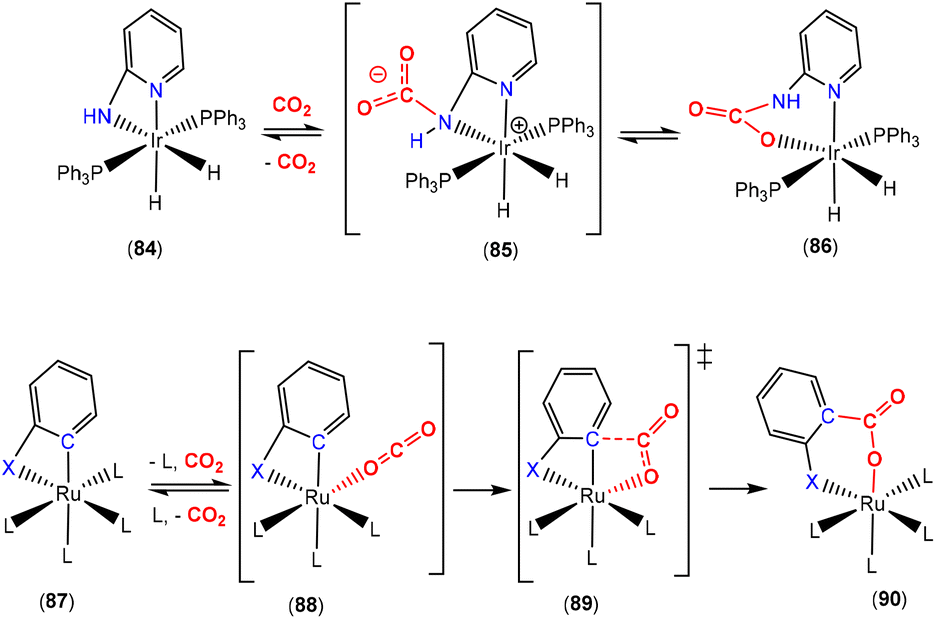
The Crabtree and Hazari groups demonstrated that a deprotonated 2-amino-pyridine (2-NH2py) unit could be utilised as a three-atom bridge for CO2 activation (Scheme 25, top).104 They observed this reactivity whilst investigating the [IrH3(κ2-N,N-2-NH2py)(PPh3)2] complex with CO2. They expected the CO2 molecule to insert into one of the Ir–H bonds in accordance with the reactivity of other Ir(III)–hydride complexes. Instead, the complex underwent an initial dehydrogenation step leading to the corresponding amido (κ2-N,N-NHpy) complex 84 in the scheme. Complex 84 was found to react with CO2via a formal insertion into the iridium–amido bond to form the cyclic carbamate species, [Ir{κ2-O,N-2-OC(O)NHpy}H2(PPh3)2] (86). In this study, the researchers synthesised a family of compounds involving the substitution of the amine functional group with methyl and phenyl groups in addition to changing the pyridine to pyrimidine. They also investigated the impact of exchanging the PPh3 ligands for PCy3. It was found that the rate of CO2 insertion was faster with a more nucleophilic amide. An investigation into the mechanism using DFT suggested that insertion occurred following an initial nucleophilic attack on the CO2 molecule by the nitrogen lone pair of the amide to form the N-bound carbamato intermediate 85, which subsequently underwent rearrangement to 86. CO2 insertion was also found to be reversible in the presence of H2, although some free 2-NH2py was also observed. This reactivity involves different resonance forms of the pyridine unit. More well-defined examples demonstrating dearomatisation of the pyridine units are outlined in a later section (vide infra).
 | ||
| Scheme 25 Two examples highlighting various aromatic three atom scaffolds, which are utilised to activate CO2. Species 85 was predicted as an intermediate by DFT for the top reaction. For the bottom computational investigation, one of the two pathways is shown, X = NH, CH2, O; L = PMe3, PH3 (see text for details). | ||
A related study was carried out by Vadivelu and co-workers who investigated a series of ruthenium complexes containing substituted-phenyl ligands that were cyclometallated (Scheme 25, bottom).105 Their computational study revealed two possible pathways for CO2 activation leading to either insertion of the CO2 unit into the Ru–C bond or insertion into the Ru–N bond (where X = NH). The first pathway (as shown in the scheme) occurs in the cases where X = CH2 and O. This is found where phosphine dissociation is energetically feasible. The generation of an empty coordination site allows for CO2 to coordinate and undergo a 1,2-migratory insertion into the ruthenium–carbon bond via transition state 89 from 88. Complex 89 then undergoes a final transformation to form complex 90 where the resultant product is that of CO2 insertion between the metal and the three-atom bridge. In the case where X = NH, the dissociation of a phosphine ligand was found to be too high energy and so the second pathway was determined to be operational. This pathway was found to be similar to that observed by the Crabtree and Hazari groups (Scheme 25, top) where the CO2 molecule reacted at the nucleophilic nitrogen centre.104 This was also consistent with other investigations.106,107
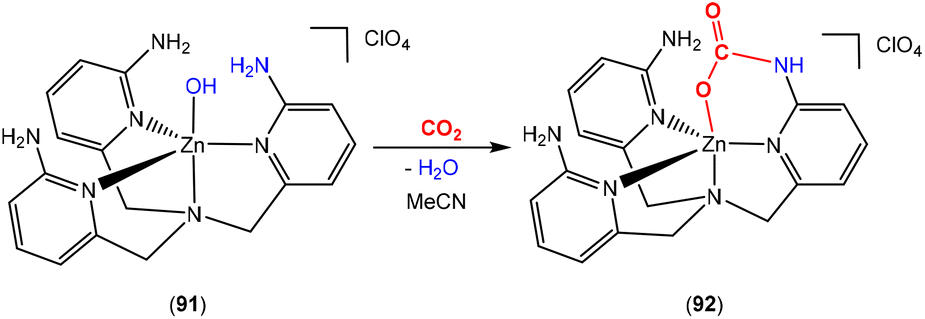
The Masuda group reported an interesting zinc system containing a tetradentate ligand, tris(6-amino-2-pyridylmethyl)amine (Scheme 26).108 In this case, zinc hydroxide complex 91 was found to react with CO2 to form the corresponding carbamate complex 92. This transformation was found to proceed via an intermediate carbonate species, [ZnOC(O)OH], formed from the reaction of CO2 with the initial ZnOH complex. This then converts into the carbamate complex following intramolecular attack of one of the three amine groups of the multidentate ligand. Overall, 92 is the product resulting from the addition of CO2 to the 3-atom bridge of one “arm” of the ligand, i.e. a six-membered ring involving the atoms [ZnNCNC(O)O]. This transformation mimics the transformation found in biotin-dependent carboxylase.
 | ||
| Scheme 26 Type 3 activation of CO2 with a zinc coordination complex. | ||
A key functional group that is utilised as a three-atom bridge is an allylic unit. Metal complexes containing allylic species tend to promote the insertion of CO2 to form carboxylates, which coordinate to the metal centre with a κ2-O,O coordination mode. Different plausible mechanisms were proposed that highlighted the double bond functioning as a nucleophile. Johansson and Wendt studied the insertion of CO2 into the allyl unit of palladium pincer complex 93 (Scheme 27).109 The insertion of CO2 (4 atm) into the allyl moiety in C6D6 at room temperature was much faster than that of the analogous Pd–CH3 complex. Thus, the three-atom allyl unit serves to promote insertion into the palladium–carbon bond via the process shown in the scheme. They proposed a six-membered cyclic transition state for the carboxylation of the palladium–allyl species as shown. This rearranges to give what appears to be direct insertion of CO2 into the palladium–carbon bond in product 94.
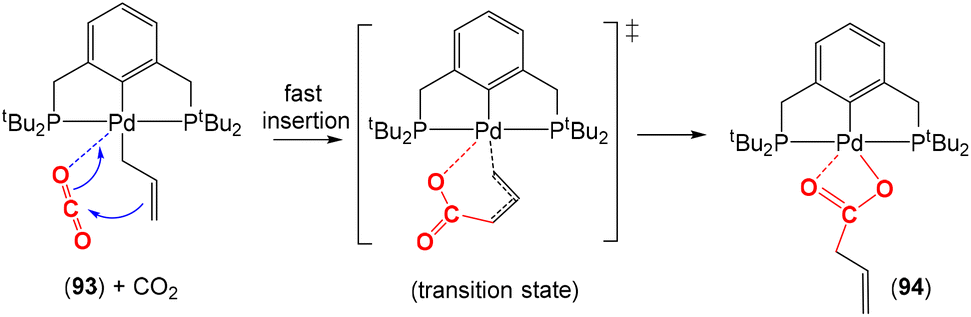 | ||
| Scheme 27 Proposed mechanism for the fast insertion of CO2 into a Pd–allyl complex containing the PCP pincer co-ligand. | ||
Additional computational and experimental studies, which also corroborated that such carboxylation of metal–allylic systems occurred directly on the terminal carbon of the allyl ligand, were reported.94,110,111 The intermediate product resembles a six-membered metallalactone and is related to the addition of CO2 to a three-atom bridge.111 The coordination mode of the allylic species also plays a role. While sigma η1-allylic species in monomeric palladium complexes lead to the formation of a carboxylate, the η3 coordination mode does not react with CO2. In the particular case of dimeric Pd(I) with bridging allyl ligands, only those containing two allyl bridging ligands exhibit reactivity towards CO2. Additionally, the presence of other ancillary ligands influences the reaction mechanism for the carboxylation reaction where more electron-rich ligands facilitate CO2 insertion.93
In summary, out of the various modes of CO2 activation, via transition metal complexes, type 2 activation involving a two-atom bridge is one mode that is predominant. The formation of cyclic structures involving the metal (metallalactones) plays a vital role in the activation process and these species are typically thermodynamically stable. The coupling of CO2 with either alkenes or alkynes seems to be a facile process, although the isolation of complexes containing both functional groups coordinated simultaneously at the metal centre is rare. In particular, the work by Field and co-workers76 shows that the activation of CO2 and its fixation on an alkyne moiety may occur through different mechanisms that depend on how the alkyne group is bound to the metal centre. The different mechanisms will lead to the formation of different regio-isomers. This can be viewed as evidence of the cooperation of the alkyne ligand in the activation process by changing the steric and electron density at the metal centre. The activation of CO2 on complexes containing alkenes played an important role in the development of catalytic systems for the synthesis of acrylic acid and acrylates; this needs further progress to achieve practical applications. Typically, coordinatively unsaturated early or late transition complexes in low oxidation state are employed for the activation of CO2. The use of electron rich multidentate ligands was widely explored in different ways: (i) by increasing the electron density on the metal centre to facilitate the interaction with CO2, (ii) by introducing nucleophilic sites on the multidentate ligand framework, which directly facilitates CO2 activation, and (iii) by introducing functional groups on the ligand scaffold that are able to promote hydrogen-bonding towards the activated CO2 molecule. More importantly, this latter use of chelate ligands is crucial when coupled with transition metal hydrides units for the reduction of CO2 to formate ions. This combination successfully led to catalytic systems.
Two main strategies were utilised for the activation of CO2 involving three-atom bridges, are defined in Scheme 23. These are: (i) the use of strained 4-membered rings containing the metal centre, where CO2 insertion expands the ring to a thermodynamically more favoured 6-membered system, and (ii) the use of multidentate ligands. In both cases, the ligands are designed to have a nucleophilic site, able to cooperate with the metal centre by attacking the electrophilic carbon of the CO2 molecule, and be directly involved in CO2 fixation by forming heterolactones or carbamate functional groups.
Dearomatisation–aromatisation systems (Fig. 1D)
Amongst the many transition metal complexes used in catalytic synthesis and bond activations, pincer type metal complexes were extensively investigated (Fig. 3). This is due to their exceptional stability, diverse reactivity, high catalytic activities and selectivity.112–114 Furthermore, this class of ligand system is readily tuned and modified, furnishing great control over the intended reactivity. The mer-κ3-E,E′,E coordination motif not only offers chemical stability for the complex, but can also be functionalised with a range of different heteroatoms, though in the context of this review, the pyridine central unit is ubiquitous. These ligand systems can facilitate the activation of substrates such as CO2 by weakening the inert CO bonds. The chemical literature contains a whole host of examples of activation across a wide range of substrates with this unique ligand platform. The desired selectivity, reactivity and stability can be designed for specific catalytic applications by tuning the steric/electronic environment around the metal center.115 Moreover, the pincer motif opens the possibility for MLC where the ligand contains an aromatic unit at its centre. As demonstrated in this section, this central unit possesses the ability to undergo a dearomatisation–aromatisation process that can be used to activate small molecules. These systems provide examples of two-atom and three-atom bridging units for the activation of CO2. Thus, these examples could have been included in those sections above. We have chosen to consider them separately due to the rather unique nature of these activation pathways.
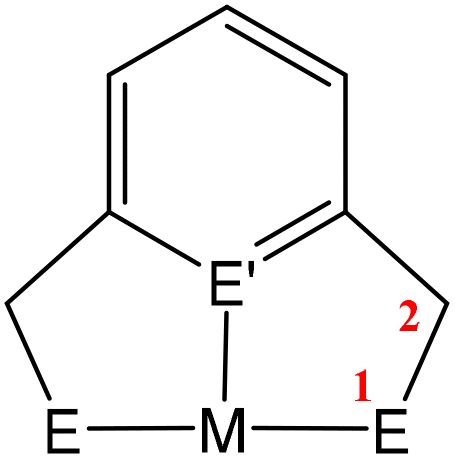 | ||
| Fig. 3 Generalised pincer complex containing a central aromatic unit; as demonstrated below, more elaborate related systems were developed (E and E′ represent different donor atoms containing various substituents; other ligands at the metal centre are omitted for clarity). | ||
Milstein and co-workers were the first to show that this ligand motif could be utilised in the context of MLC. They demonstrated this reactivity across a range of ligands, as shown in Scheme 28. They showed that when using complexes based on lutidine (L1–L4), phenanthroline (L5) and acridine (L6) ligand scaffolds, a unique cooperation between metal and ligand took place. In these cases, a dearomatisation–aromatisation pathway for a pyridine unit occurs (Scheme 28).116 Treatment of pyridine-based pincer complexes with a base can lead to deprotonation of the methylene unit of one of the ligand “arms” as shown in the scheme. Deprotonation results in a dearomatised form of the complex. The net result is a change in the nature of the central nitrogen donor, which goes from an L-type in the pyridine form to an X-type in the dearomatised form. This ligand transformation is facilitated by the acidity of the methylene protons, stabilisation of the dearomatised ligand by the coordinated metal and the relatively low resonance energy of the pyridine ring. The important advantage of the formed pyridyl dearomatised complex is that it can then undergo further reactivity with a wide range of substrates (containing both polar and non-polar bonds) via low energy pathways. For example, the energy difference between the dearomatised and aromatised forms in the case of H2 activation is between 1 and 4 kcal mol−1.117 It should be noted that during the process, the oxidation state of the metal centre remains constant.118
 | ||
| Scheme 28 A selection of pincer type ligands shown to exhibit dearomatisation–aromatisation processes involving a pyridine unit. The selectivity shown for H–E activation occurs when E is an element that is more electronegative than H (e.g. OR, NR2, CR3). | ||
This fascinating mode of MLC was applied in the activation of a broad range of E–H bonds, including those in alcohols,119,120 amines, nitriles,121,122 boranes,123 dihydrogen,124 and dioxygen125 as well as in the activation of Csp2–H and Csp3–H bonds.126,127 As the synergy between the metal and the ligand has a great influence on bond activation, several pincer complexes based on different metal centres such as Ru,128,129 Rh,130 Ir,131 Re,132 Fe,133,134 Co,135 Ni136 and Mn137 were employed. The Milstein group discovered this mode of activation when they were trying to prepare a Ru(0) complex via dehydrochlorination of Ru(II) complex 95, shown in Scheme 29. An abbreviation notation highlighting the donor atoms coordinating to the metal centre is typically utilised. For instance, the RPNN system indicates a phosphorus-based ligand arm, a central pyridine and a nitrogen-based ligand arm, respectively. The superscripts are sometimes used to indicate substituents on the donor atoms. The reaction of tBuPNN complex 95 with a base resulted in deprotonation at the phosphine methylene arm instead of hydride loss from the metal centre leading to the formation of stable dearomatised complex 96, where the oxidation state of the metal centre was maintained.128 DFT calculations on the related PNP system (L1, R = tBu) confirmed that the dearomatised Ru(II) complex was slightly lower in energy than the corresponding aromatised Ru(0) complex (ΔG = −2.2 kcal mol−1).138 DFT was also used to confirm that deprotonation on the phosphine arm was more favourable.139
 | ||
| Scheme 29 Initial discovery of dearomatisation by Milstein and co-workers; proton is removed from the ligand rather than the metal centre. | ||
For the aim of utilising CO2 as a C1 building block, the activation of this inert substrate using transition metal complexes was explored in detail. In 2012, the Milstein128 and Sanford140 groups independently investigated the reactivity of CO2 with dearomatised pincer complexes at ambient temperatures (Scheme 30). In these cases, the CO2 molecule was found to undergo a 1,3-addition between the metal centre and the sp2 hybridised nucleophilic carbon centre adjacent to the dearomatised unit. This led to new products containing new C–C and M–O bonds (Scheme 30). In this case, the P–C unit within the ligand acts as a two-atom bridge for the activation of CO2.
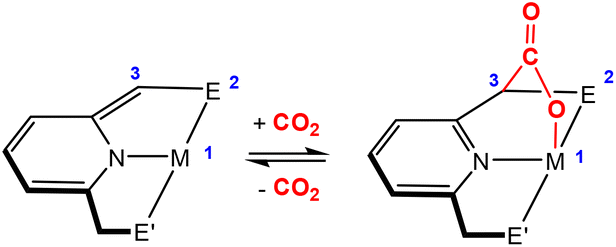 | ||
| Scheme 30 Generalised scheme highlighting the potential for reversible CO2 activation via MLC involving dearomatisation–aromatisation. This is a 1,3-addition across a two-atom bridge. | ||
In the first example involving CO2, the Milstein group reported a ruthenium complex containing a PNPtBu pincer ligand, 97 (Scheme 31), which was deprotonated using a base to form the dearomatised complex [Ru(PNPtBu)(H)(CO)], 98.128 This complex subsequently undergoes reaction with CO2 to form the 1,3-addition product 99. CO2 addition was found to be reversible and complex 98 was reformed when a benzene solution of 99 was placed under vacuum. It should be noted, however, that complex 99 was stable against the loss of CO2 in the solid state. The reaction of CO2 with complex 98 was easily followed by NMR spectroscopy with two new signals in the 31P{1H} NMR spectrum [107.0 and 114.0 ppm (2JPP = 251.0 Hz)] for the two chemically inequivalent phosphorous atoms. Moreover, a doublet resonance at 172.6 ppm (2JCP = 9.2 Hz) in the 13C{1H} NMR spectrum confirmed the presence of the newly formed carboxylate group. Infrared spectroscopy was also informative with a band at 1628 cm−1 again confirming the formation of a carboxylate species. The crystal structure of 99 unambiguously confirmed the reactivity of CO2 with 98 and confirmed 1,3-addition across the ruthenium and carbon centres. The structure revealed a distorted octahedral Ru(II) complex in which the CO ligand was located trans to the pyridine ring. The 1,3-addition occurs within this ruthenium complex in preference to other theoretically possible binding or activation modes of CO2. It might have been envisaged that CO2 would insert into the ruthenium–hydride bond instead. DFT calculations agreed with the experimental results and confirmed that 1,3-addition was the most favourable mode of activation in these complexes.
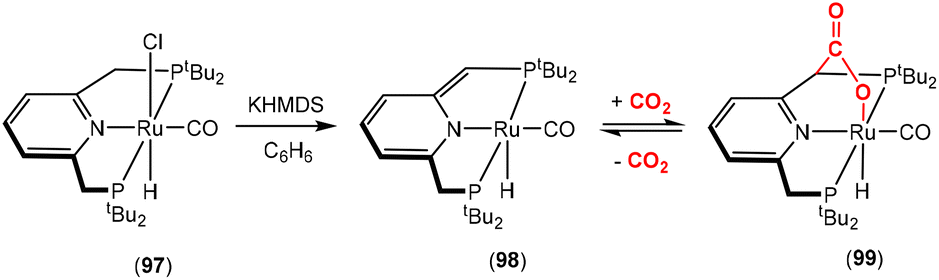 | ||
| Scheme 31 The first example of the use of dearomatisation MLC for the activation of CO2. | ||
For the second example, Sanford and co-workers showed that a PNN pincer ligand could be employed for the activation of CO2 in a similar manner (Scheme 32).140 Rather than containing two phosphorus “arms” for the ligand, the Sanford ligand contained one phosphorus and one nitrogen “arm” on either side of the central pyridyl moiety. The reaction of complex 96 with 1 atm CO2 at room temperature led to the 1,3-addition product, 100, where C–C bond formation between CO2 and the carbon occurred on the side of the phosphine “arm” (as found for the prior dearomatisation step). Complex 100 was found to exhibit analogous coordination to that found in the case of Milstein's complex, 99, above. However, by heating a solution of complex 96 at 70 °C for 15 min (or leaving the solution overnight at room temperature), the species rearranged to the more thermodynamically stable product 101. In this case, the CO2 unit transferred to the methylene carbon centre on the amine “arm”. Carbon-13 labelling studies demonstrated that the CO2 ligand in 100 could be exchanged with 13CO2, thus coordination at this position was confirmed as being reversible. In contrast, the same 13CO2 exchange did not occur in complex 101. Furthermore, complex 96 was not observed when 101 was placed under vacuum. This confirmed that addition of CO2 on the amine “arm” was irreversible at room temperature.
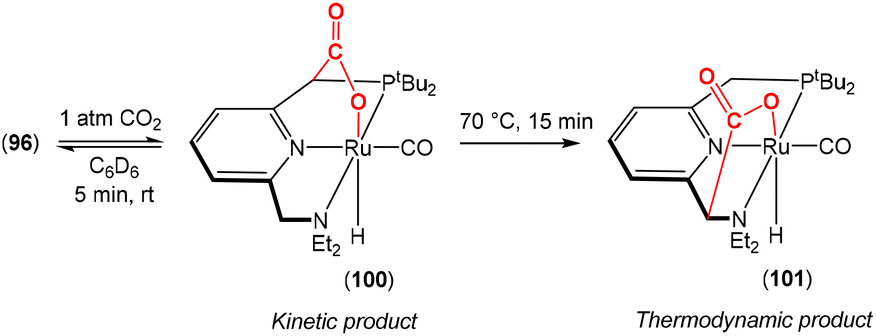 | ||
| Scheme 32 Reactivity of CO2 with dearomatised complex 96 and the formation of complexes 100 and 101. | ||
This unique mode of CO2 activation was also demonstrated with other pincer complexes with a number of different metal centres including Fe,141 Re,142 and Ir.131 More recently, CO2 activation was studied using a manganese pincer complex143 (Scheme 33) employing two different modes of MLC. Manganese complex 102, as shown in Scheme 33 (top), features a PNN′ ligand where the N′ component is a tertiary amine group. In the presence of a base the ligand is deprotonated in the same way as that observed in the previous complexes where dearomatisation of the pyridine unit occurs to form the corresponding complex 103. Complex 103 further reacts with CO2via 1,3-addition to form complex 104, which features new Mn–O and C–C bonds, as in the previous cases. If, however, the tertiary amine group is changed to a secondary amine then different reactivity is observed. This is demonstrated in complex 105 (Scheme 33; bottom). Upon addition of a base to 105, deprotonation of the amine functional group occurs rather than a dearomatisation step. The resulting complex 106 contains an amido functional group and the pyridine unit remains in the aromatic form. Thus, upon addition of CO2 to the complex, 1,2-activation occurs to form new Mn–O and C–N bonds in complex 107 which contains a carbamate functional group. This demonstrates how the reactivity can be tuned.
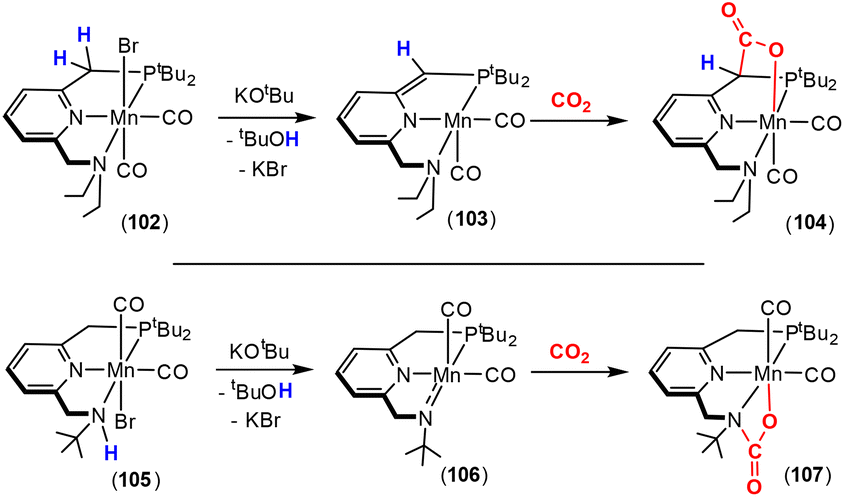 | ||
| Scheme 33 Activation of CO2 by Mn–pincer complexes with two differing modes of activation. | ||
Activation of CO2 using a dearomatisation–aromatisation approach was also studied by Vogt and co-workers.142 They demonstrated that the pincer motif was not a prerequisite for MLC. The group showed that rhenium(I) triscarbonyl complexes containing both 2-aminomethyl pyridine and 2-iminomethylpyridine could be utilised as precursors leading to MLC reactivity. In their work, the treatment of 2-aminomethyl pyridine complex 108 (Scheme 34) with excess base (LiHMDS; HMDS = hexamethyldisilazide) resulted in double deprotonation of the complex to afford the anionic amido complex 110a. Whilst this complex was not isolated, it was characterised in situ. Two resonance structures are feasible, the dearomatised form (110a) and the aromatised pyridine form (110b). Spectroscopic data were consistent with the existence of 110a as the major component. Complex 110a could also be synthesised via an alternative route. The 2-iminomethylpyridine complex 109 could be reduced in the presence of potassium metal to form complex 110a. As with the pincer-based examples above, dearomatised complex 110a was found to be highly reactive with CO2 and underwent 1,3-addition. The nucleophilicity of the rhenium centre, the Lewis acidity of the counter cation (Li+ or K+), together with MLC via dearomatisation/aromatisation, were all factors leading to the formation of new complex 111. Addition was also found to be reversible and 111 readily underwent exchange with labelled 13CO2.
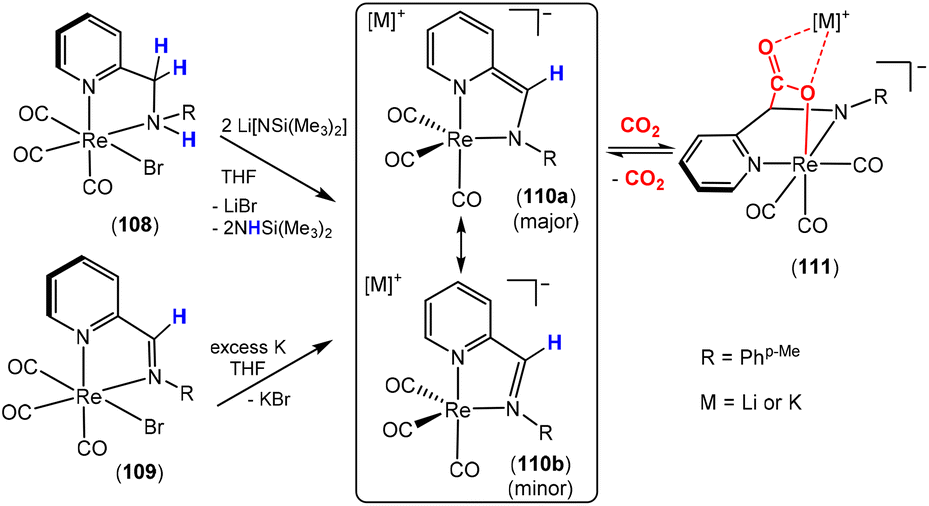 | ||
| Scheme 34 Rhenium–triscarbonyl complexes and their reactivity with CO2. | ||
Ozerov and co-workers also demonstrated that the central pyridine unit was not a requirement for opening up pathways for dearomatisation/aromatisation MLC.144 They synthesised rhenium(V) oxo complexes of the type [Re(O)Cl2(PCP)], 112 (Scheme 35). Treatment of complex 112 with a base led to the dearomatised complex, 113. The reactivity of this complex was explored with small molecules. Typically, for systems where the MLC pathways exist, the activation of H2 is predominant. In this case, however, no reactivity with this molecule was observed. It is believed that the reactivity is not favoured since the product of activation would place a hydride ligand on the metal centre trans to an oxo ligand as well as the fact that rhenium has a low tendency to form dihydrogen complexes; a key step for H2 splitting. It was reasoned that the activation of polar bonds could be feasible on the other hand. Indeed, it was found that dearomatised complex 113 readily reacted with CO2, via 1,3-addition at ambient temperatures. In this case the product of activation placed the less trans-influencing oxygen donor (of the activated CO2) trans to the oxo ligand (Scheme 35). The addition of CO2 was confirmed by NMR and IR spectroscopy. X-ray crystallographic studies were carried out on 114. However, the crystals showed disorder around the rhenium carboxylate region. Thus, the corresponding iodide complexes, 115 and 116, were prepared and fully characterised. The crystal structure of 116 revealed a distorted octahedral complex and confirmed the formation of a new C–C bond at the benzylic position of the pincer ligand.
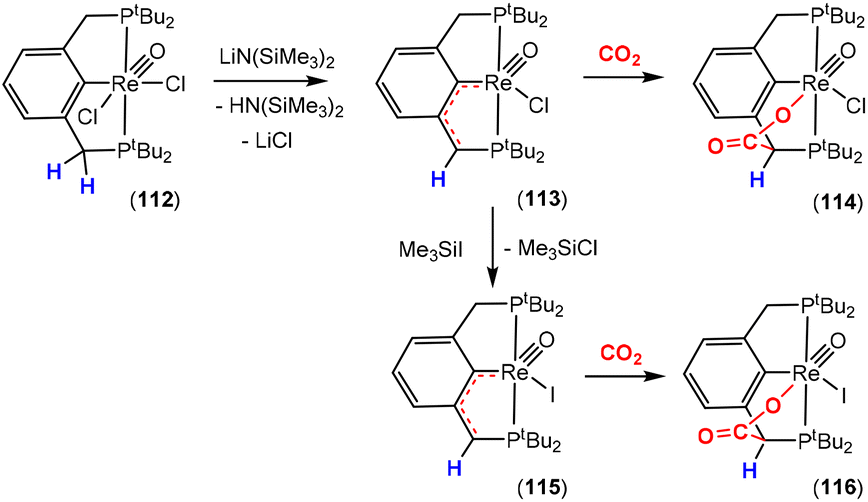 | ||
| Scheme 35 Reactivity of the [ReOCl2(PCP)] complex, deprotonation reaction and the reactivity with CO2. | ||
It was also shown that pincer complexes containing PNP ligands could exhibit other CO2 activation pathways. A particularly interesting example, which underwent some unexpected transformations (Scheme 36), was reported by Milstein and co-workers.145 They were exploring the reactivity of a dearomatised nickel–hydride complex (117) with CO2. It was found that the addition of CO2 gas resulted in the formation of the aromatised product where the hydride species shifted to the ligand and a molecule of CO2 coordinated to the nickel centre with a rare η1-C coordination mode (complex 118). Whilst this new complex could be isolated, it underwent further transformation in the presence of CO2. When heated, full conversion of 118 was observed to form two new species, 119 and 120. Complex 119 formed as a result of C–P bond cleavage and the addition of a CO2 unit to the resulting carbon where the resulting PiPr2 fragment was transferred to the metal centre and oxidised to form P(O)iPr2. The source of the oxygen atom appears to originate from a CO2 unit thereby forming CO, which is the ligand found in the second complex, 120 (Scheme 36). Whilst this transformation leads to destruction of the pincer ligand, it highlights some interesting reactivity leading to the conversion of CO2 to CO. Disproportionation of the CO2 molecule, as a result of a MLC step, was also observed in some of the other systems presented herein.
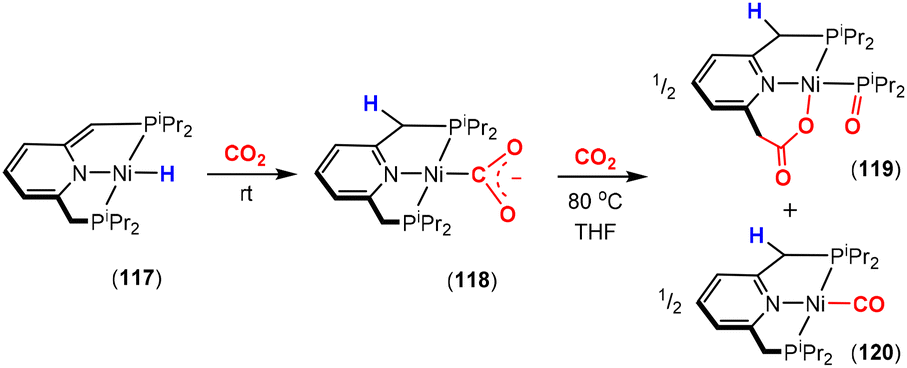 | ||
| Scheme 36 Reactivity of nickel–hydride pincer type complexes with CO2 and subsequent transformation. | ||
CO2 reductive cleavage using MLC was investigated using iridium PNP complexes.146 In this case, the activation of CO2 led to an interesting set of transformations resulting in the formation of a CO ligand and H2O as a by-product. These transformations are relevant to the reverse gas water shift reaction and could be a possible sustainable pathway that could open up the industrial scale use of CO2 as a CO surrogate. In this system, the reactivity focused on the transformations involving iridium(III) dihydride complex 121, which features a PNPtBu pincer ligand (Scheme 37). Deprotonation of this complex results in a mixture of two iridium–hydride complexes, 122 and 123, which differ in the position of one of the hydrogen atoms. The pyridine unit in 122 exists in the dearomatised form where there are two hydride ligands cis to each other at the iridium(III) centre. For 123, the pyridine unit is aromatised and there is one hydride at the metal centre [Ir(I)]. These two species were found to interconvert within the reaction mixture; however, this interconversion ceased in the absence of a proton transfer agent. The mixture of 122 and 123 was found to react with CO2 at room temperature over the course of 18 hours to form a new dearomatised complex, 124 (Scheme 37, top reaction). In this case, the CO2 molecule was converted into a CO ligand and the second oxygen from this molecule was converted into H2O, where the precursor complexes 122 and 123 were the sources of the two hydrogens.
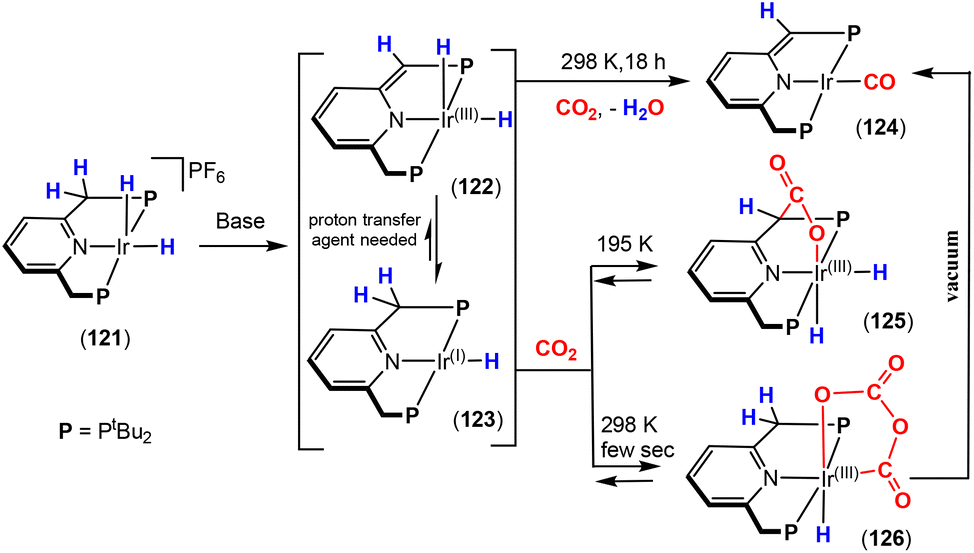 | ||
| Scheme 37 Reactivity of complexes 122 and 123, formed by deprotonation of 121, followed by CO2 under various conditions. Transformation of CO2 into CO and water. | ||
In order to explore this reactivity further, different reaction conditions were investigated to identify possible intermediates and understand the mechanism of CO2 cleavage. As also shown in Scheme 37, the same mixture of complexes 122 and 123 was reacted with CO2 at low temperature in toluene (195 K). Under these conditions, the 1,3-addition product was confirmed and complex 125 was characterised by spectroscopic methods. On the other hand, when the reaction with CO2 was carried out at ambient temperature in higher concentrations of this gas and spectroscopic data were obtained immediately following addition, intermediate species 126 was observed. This intermediate contained a new κ2-C,O-[C2O4]2− ligand where the reductive coupling of two CO2 molecules had occurred. However, under both conditions it was found that the reaction with CO2 was reversible and could interconvert depending on the reaction conditions. For example, complex 124 was formed after complex 126 was placed under vacuum.
Based on their experimental results, the detected intermediates, and computational studies, the Milstein group proposed a series of mechanisms for the transformations involving complexes 124 and 126 (Scheme 38). Complex 125 is formed via an analogous 1,3-addition to those observed in previous examples. Complexes 124 and 126 are formed via the same initial intermediate Aapical, where the carbon of the CO2 molecule is attached to the nucleophilic iridium(I) centre [η1-C-(CO2)] at the apical site. With the CO2 ligand connected at this position, one of the oxygen atoms is positioned in proximity to the methylene unit. This undergoes deprotonation by the nucleophilic oxygen (facilitated by MLC dearomatisation) to form intermediate B. This species undergoes subsequent loss of water to form carbonyl complex 124. In the next transformation, Aapical isomerises to form Abasal. In the presence of a second equivalent of CO2, one of the oxygen atoms of the η1-C coordinated CO2 ligand attacks the electrophilic carbon of free CO2 to form complex 126.
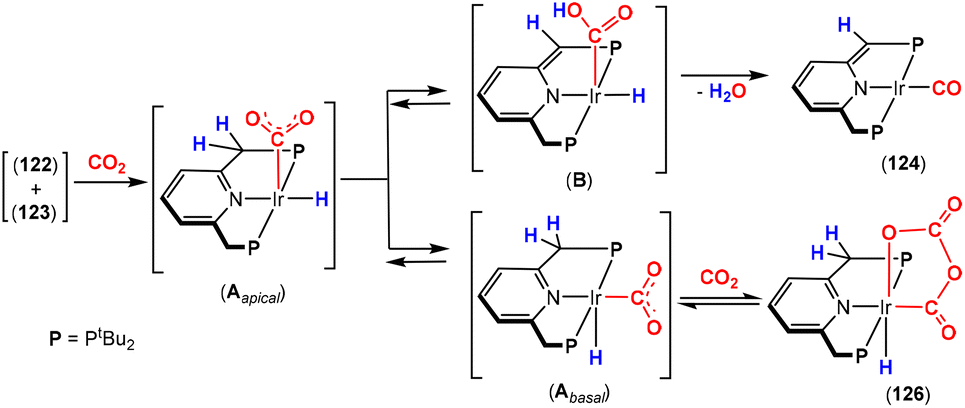 | ||
| Scheme 38 Proposed intermediates leading to the formation of complexes 124 and 126, supported by DFT calculations. | ||
The Milstein group applied their findings to the reductive cleavage of CO2 using MLC for the preparation of benzaldehyde using CO2 as the C1 source and carbonyl surrogate.130 This is exemplified in the stepwise cycle shown in Scheme 39. Whilst not catalytic, this is an important development since there is a wealth of chemistry involving CO as a molecular building block. The transformation of CO2 into CO at transition metal centres offers significant potential for utilising CO2 as a C1 source. Following the same MLC steps as shown in the previous examples, rhodium(I)–hydride pincer complex 127 reacted with CO2via a reductive cleavage pathway to form the dearomatised complex rhodium(I)–carbonyl complex 128 and water. A short-lived formate species [η1-OC(O)H] was identified spectroscopically at low temperatures and crystalised at low temperatures (sufficient for obtaining a crystal structure). The 1H NMR spectrum showed a signal at 8.6 ppm (as a doublet of triplets, 3JRhH = 2.8 Hz, 4JPH = 1.4 Hz) corresponding to the formate proton. The corresponding 13C NMR spectrum confirmed this functional group as a broad signal at 168.1 ppm. This, however, rapidly converted into complex 128 in the mixture. Additionally, it was found that another species was present within the reaction mixture consistent with [Rh(PNP)(CO)][OH], which would result from the reaction of 128 with one equivalent of water and the pincer ligand in the aromatised form.
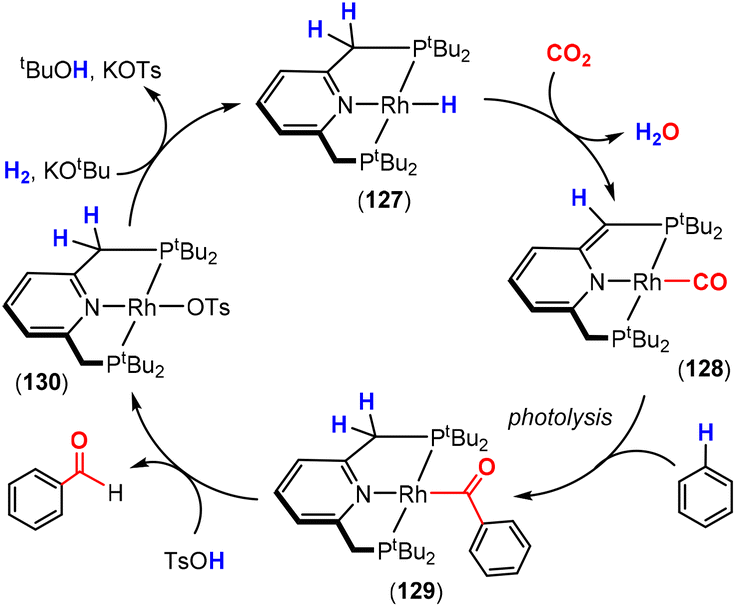 | ||
| Scheme 39 Carbonylation of benzene using a Rh(I) pincer complex via MLC. | ||
The route to 128 from CO2 offers significant promise. Unfortunately, complex 128 was found to be quite unreactive. Nevertheless, photolysis of the complex in the presence of benzene led benzoyl complex 129. This is a formal product of C–H activation of the solvent, transfer of the hydride to the pincer ligand (re-aromatisation) and a 1,1-migratory insertion of the carbonyl ligand into the resulting rhodium–phenyl species. None of the intermediate species of this part of the transformation were observed. One of the challenges of this reaction was the competing decarbonylation transformation, which involved the elimination of CO gas into the headspace of the reaction vessel under the UV light conditions. Furthermore, a number of conditions were trialled in order to find a methodology for releasing the benzoyl fragment. It was found that a strong organic acid such as p-toluenesulfonic acid (TsOH) was required. This led to the formation of benzaldehyde and tosyl complex 130. Through the use of an internal standard, it was found that the stoichiometric conversion to benzaldehyde was approximately 50%. Further treatment of this complex with a base (KOtBu) and H2 resulted in the reformation of the main active rhodium–hydride complex 127. This was only achieved following the removal of the volatiles, including benzaldehyde. Whilst a full catalytic cycle for this transformation was not realised, this example serves to inspire the potential of MLC for utilising CO2 as a surrogate for CO.
Other transition metal complexes with pincer ligands containing symmetrical N-heterocyclic carbene (NHC) donors were reported. These include NHCs based on a lutidine central unit, including CNN147,148 and CNC149 systems. These also showed interesting reactivity in the activation of small molecules via the dearomatisation–aromatisation process. In terms of the activation of CO2 by such complexes, differing reactivities were observed depending on the pincer ligand as well as the metal centre. For example, ruthenium complex 131, containing a bis (NHC) pincer ligand, undergoes a reaction with a base to form the dearomatised complex 132 (Scheme 40) where deprotonation occurs at one of the methylene units of the pincer ligand. The dearomatised complex subsequently undergoes the 1,4-addition of CO2 in a similar fashion to the previously described examples to form complex 133. This mode of activation differs by the size of the ring, which is formed upon formation of the CO2 adduct. In this case, a six-membered ring was generated. This can be viewed as an activation involving a three-atom bridge. Pidko and co-workers demonstrated that complex 131 was an active catalyst in the hydrogenation of CO2 into formates.150 Whilst there was a greater propensity for the activation of H2 and CO2 in these CNC complexes, the catalytic performances of the PNP complexes highlighted above (such as complex 98, Scheme 31) were found to be higher. Furthermore, it was found that the ratio of H2 to CO2 utilised in these reactions was important for determining the catalyst's activity. Further investigations revealed that stable complex 133 was found to be catalytically inactive within the catalytic cycle.
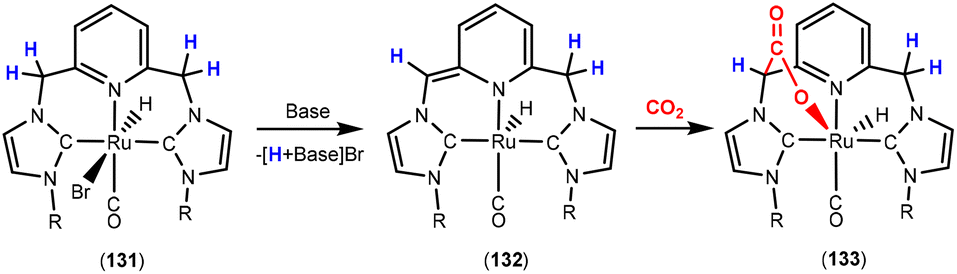 | ||
| Scheme 40 Dearomatisation reactivity involving ruthenium complexes containing NHC-based pincer ligands and subsequent MLC activation of CO2via 1,4-addition. | ||
The Casado and Polo groups further reported on the reactivity of a series of cationic rhodium complexes bearing pincer carbene CNC ligands, as outlined in Scheme 41.149,151 Deprotonation of complex 134 was achieved with a strong base, resulting in the formation of dearomatised neutral complex 135. This complex was found to react with CO2via MLC activation. In contrast to the mode of activation of complex 132 (Scheme 40), the reaction with CO2 resulted in the formation of a new C–C bond between CO2 and the activated CH arm of the CNC ligand to form zwitterionic complex 136. In this case, the 1,4-addition product was not observed with no Rh–O bond being formed. This is likely due to the preference for Rh(I) to remain square planar and the larger distance between the site of attachment of the CO2 unit and the metal centre. Interestingly, complex 136 further reacted with protic amines to form 137, which is the re-aromatised product with a carbamate ion as the counterion that demonstrates promise for further transformation.
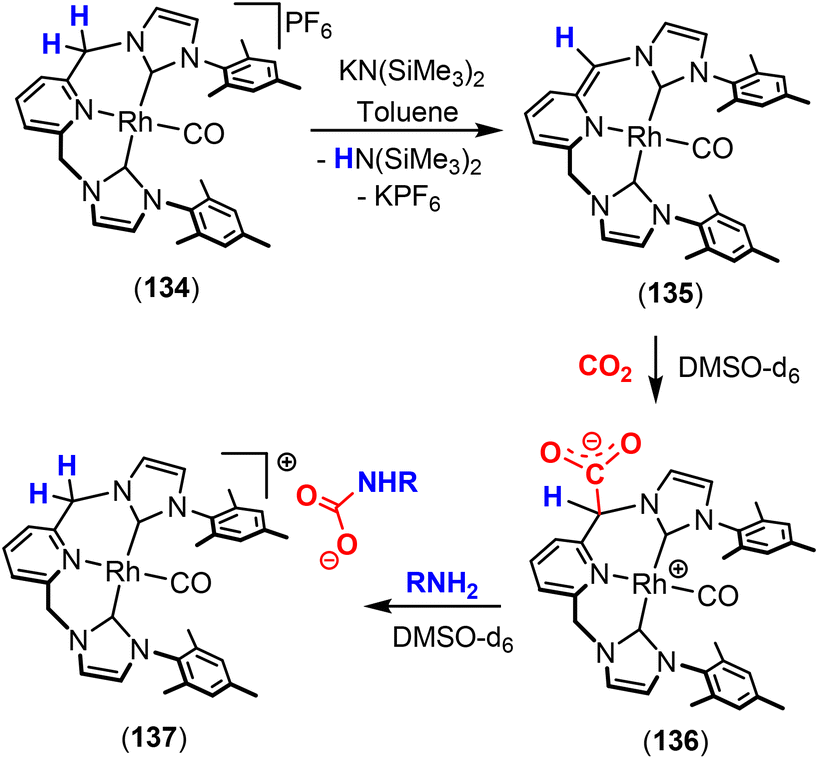 | ||
| Scheme 41 Preparation of a neutral rhodium complex during a dearomatisation step and further reactivity with CO2 and amines. | ||
This rare type of CO2 activation with dearomatised complexes was also observed in the case of the doubly deprotonated anionic nickel complex 139, as shown in Scheme 42.136 This complex was formed by a second deprotonation step on the previously dearomatised complex 138 with methyllithium. In these transformations, the Milstein group showed that CO2 addition occurred directly on the carbon atom of the pincer motif to form complex 140 and that the Ni(II) centre was not involved in the coordination of the CO2 unit. This is an example where 1,3-addition is not observed in contrast to the examples containing a PNP pincer ligand outlined above (vide supra).
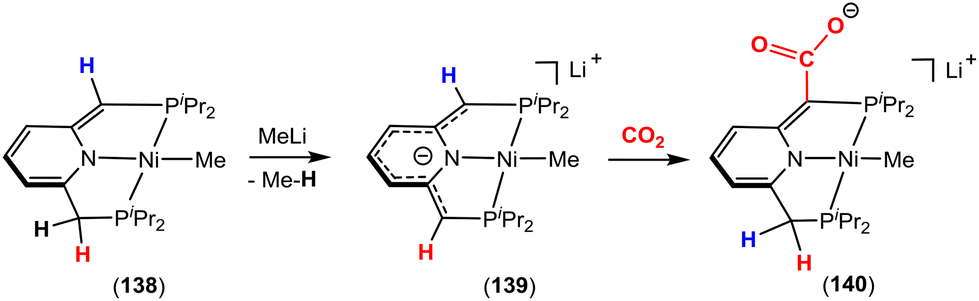 | ||
| Scheme 42 Double deprotonation of a Ni–PNP pincer complex and further reactivity with CO2. | ||
In summary, MLC activation involving dearomatisation–aromatisation opens new prospects towards the design of new transition metal complexes that can be utilised for the clean and efficient transformation of a wide range of substrates, including CO2, to high added-value chemicals. As discussed in this section, MLC activation of CO2 demonstrated different reactivities and afforded intermediates that would be difficult to obtain without the dearomatisation/aromatisation steps. The addition of CO2 to the dearomatised complexes leads to the formation of 1,3-addition (or 1,4-addition) products as key intermediates obtained under relatively mild conditions. In many cases these additions are reversible. However, the current literature seems to indicate that the dearomatisation/aromatisation sequence for the catalytic transformation of CO2 demonstrates a lack of efficient activity. This is due to the high stability of the 1,3-addition products. As such, it seems that 1,3-addition leads to deactivation of the metal complexes, many of which are thereby not catalytically active. In some reports, it is possible to avoid this deactivation and improve the catalytic performance by modifying the reaction conditions, for example, in the catalytic hydrogenation of CO2 to formates by using lutidine–Ru–CNC pincer complexes and optimising the H2/CO2 ratio. The use of CO2 for the carbonylation of organic substrates in place of toxic and petroleum derived carbon monoxide would be a very promising approach from a sustainability perspective. The potential for this was demonstrated in the example involving the carbonylation of benzene. This example shows that MLC based on dearomatisation/aromatisation could be a useful tool for the splitting of CO2 and formation of catalytically active carbonyl complexes. However, further development is needed to realise this potential and achieve efficient catalytic processes that can transform CO2 into high-added value chemicals.
Bimetallic activation
During the writing of this manuscript, an excellent perspective review was published by Mankad and co-workers in this journal.152 The perspective provides a comprehensive overview of cooperative bimetallic chemistry involving the activation of CO2 (in addition to NO2). Thus, aspects are only covered briefly here to highlight the utilisation of two metals in the cooperative activation of CO2 and link with those systems covered in the above sections. The reader is referred to Mankad's and other excellent reviews153–156 for more comprehensive reviews on this topic.As an extension of the one atom bridge between the transition metal and cooperating element (i.e. ME; where E = CR2, SiR2, NR, O), this section describes examples where E is another metal fragment. This leads to a situation where the second metal is acting as a metalloligand (L, X or Z type) and provides an opportunity to tune the properties of the first metal. Heterobimetallic cooperative activation is akin to frustrated Lewis pair (FLP) bond activation where electronic frustration between bulky Lewis acid and Lewis base main group moieties mediates CO2 activation.157 When compared to FLP or the MLC methods we outlined above, the presence of a second redox active centre offers the potential to facilitate electron transfer to the substrate and is a key advantage for the selective reduction of CO2.
The dipole in the M–M′ bond determines the regiochemistry of CO2 activation (Fig. 4). The strength of the bonds formed, i.e. M–C and M–O, is also an important parameter.158 For example, the electronegativity difference between a transition metal and p-block element creates a polarised bonding situation in which p-block elements can function as a Lewis acidic Z-type σ-acceptor ligand to a nucleophilic metal centre. More typically, the M–M′ bond is supported by bridging ligands such as hydrides, oxo units, carbonyls and halide ligands, although there are some examples of unsupported M–M′ bonds or those supported by elaborate ligand scaffolds.159
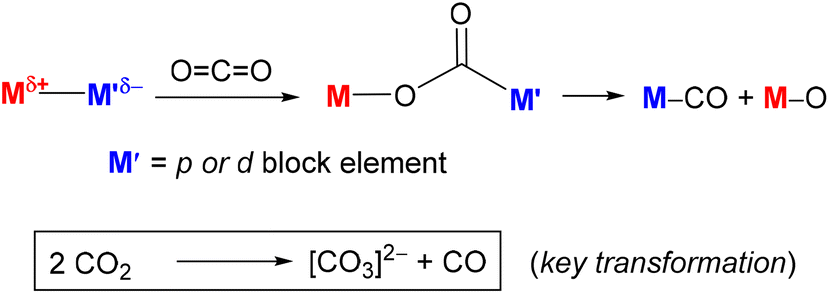 | ||
| Fig. 4 Schematic illustration of cooperation between two metals where one of the oxygen atoms of the CO2 molecule is activated by the electropositive centre and the electrophilic carbon centre is activated by the nucleophilic metal centre. Most often bimetallic cooperation results in the disproportionation of CO2. | ||
Bimetallic architectures provide new avenues for bond activation by working in cooperation with each other. This field has flourished from just a handful of examples to many well-defined structures and intriguing CO2 activation modes in recent years.160 However, improved rational design, understanding of bonding and detailed mechanistic insights are needed to fully exploit the potential of CO2 activation. At times, each metal can act independently in the bimetallic unit, thus hindering the desired cooperativity mode of activation. This section briefly outlines selected literature examples wherein cooperation between bimetallic sites results in interesting CO2 activation and transformations. We focus on enzymatic mimics first and then move on to other relevant examples involving M–M′ cooperation.
The active sites present within metalloenzymes have inspired the design of bimetallic complexes for multisite activation of the small molecules utilised by nature. For example, carbon monoxide dehydrogenases (CODHs) contain bimetallic active sites [e.g. the Fe–Ni system (141) highlighted in Fig. 5] and can lead to reversible 2-electron transformations involving the reduction of CO2 to CO and the oxidation of CO to CO2. Lee and co-workers recently reported a paramagnetic iron–nickel heterobimetallic complex that could be considered as a synthetic structural model of the CODH active site.161 They reported the synthesis of carboxylate complex 142 in which both the nickel and iron centres contained the amido-pincer ligand [N{(C6H3Me)P(iPr)2}2] (PNP). The coordination environment of the activated CO2 molecule was determined to be μ2–κ1C:κ2O,O′, bridging the two metal centres. Upon activation, new Ni–C and Fe–O bonds are formed, consistent with the polarity difference between the metal centres (cf. electronegativities, Fe: 1.83 and Ni: 1.91) and the strength of the new bonds formed.162 Activation of the CO2 molecule brings about a change in hybridisation of the carbon centre as shown in the crystal structure where the O–C–O angle is found to be 116.5°. This is also reflected in the various bond lengths {Ni–C 1.858(1) Å, Fe–O 2.204(1) and 2.066(1) Å, C–O 1.269(2) and 1.289(2) Å}. The binding mode represents the extreme limit of a 2-electron reduced species coordinated at the nickel centre (i.e. Ni(II)–[CO2]2−). This is consistent with the very low stretching frequency of the carbon–oxygen bonds in the IR spectrum [ν(CO) = 1510 cm−1]. Preliminary studies suggested that C–O bond cleavage was possible upon addition of HBF4·Et2O to generate [Ni(PNP)(CO)][BF4] and [Ni(PNP){C(O)OH}] in addition to other unidentified products. Later Lu and co-workers reported a new example of an iron–nickel heterobimetallic complex as a functional model of the C-cluster of the CODH active site.163
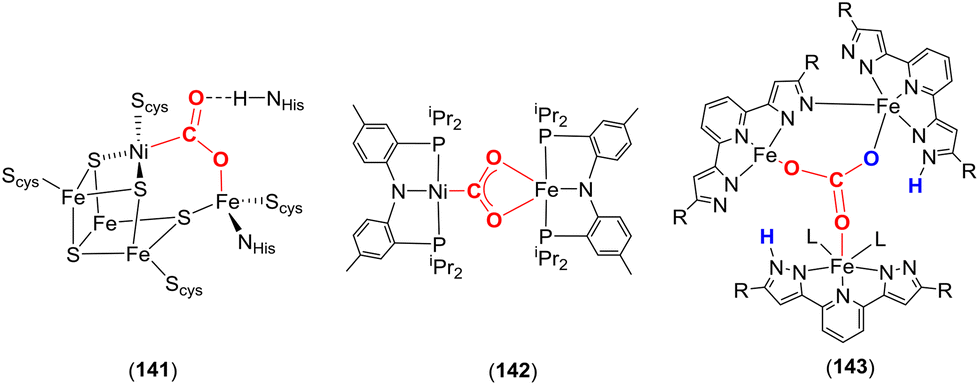 | ||
| Fig. 5 Carbon monoxide dehydrogenase C-cluster active site (141) found in the enzyme and its biomimetic model (142). Trinuclear iron complex 143 is the functional model of carbonic anhydrase. | ||
The active site in carbonic anhydrase contains a tetrahedral redox inactive Zn(II) centre stabilised by three histidine residues and hydroxide. This catalyses the conversion of CO2 and water into carbonates. In 2018, Caulton and co-workers reported a carbonic anhydrase functional mimic based on the trimetallic complex 143 (Fig. 5), which involved an interesting proton shuttling cooperative mechanism.164 Complex 143 was prepared by the reaction of [Fe(NNN)(DMAP)3] with CO2/H2O in CH3CN (where NNN = bis-pyrazolatopyridine, DMAP = dimethylaminopyridine). The trinuclear complex contains a [CO3]2− core, which originates from the CO2 molecule and oxygen atom from H2O. Two of the basic pendant nitrogen moieties at the β-position of the pyrazolate groups act as bases resulting in the double deprotonation of H2O. It is postulated that the transformation proceeds via an oxo intermediate of the type [Fe(O)(NNN + H)(DMAP)2]. All three oxygen atoms from the resulting bridging carbonate unit are each bound to an iron centre providing an overall coordination mode of μ3:η1:η1:η1. Structural characterisation revealed lengths of 1.985(3), 1.992(3) and 2.022(3) Å for the Fe–O bonds and 1.294(5), 1.285(5) and 1.262(4) Å for the C–O bonds. The flexibility in bonding within the “[Fe(NNN)]” core coupled with the oxophilicity of the iron centres results in the cooperative capture and conversion of CO2 and H2O into [CO3]2− and two protons. A similar pattern of reactivity involving a nucleophilic MO species with CO2 leading to a κ2-O,O-CO3 species was highlighted above (vide supra).
In recent times, there has been a renaissance in the study of the synthesis and reactivity of heterobimetallic complexes featuring highly polarised Mδ+–Mδ− bonds in the context of CO2 activation. The early work of Bergman and co-workers laid the foundation for this research area where the reaction of [(Cp)2Zr(μ-NtBu)Ir(Cp*)] with CO2 gave [(Cp)2Zr(μ-NtBu){μ-OC(O)}Ir(Cp*)]. Here, the CO2 molecule is attacked by the nucleophilic iridium centre and stabilised by the formation of a new Zr–O bond.165 Other relevant examples of bimetallic complexes such as [(Cp)2(CO)2M–ZrCl(Cp)2] (where M = Fe, Ru) were prepared and characterised. Their reactions with CO2 lead to complexes of the type [(Cp)2(CO)2M(CO2)ZrCl(Cp)2] where the CO2 ligand contains a μ,κ1-C(M),κ2-O,O(Zr).166,167
In one of the more recent examples of CO2 activation using heterobimetallic complexes, the Mankad group reported the selective formation of CO during their investigations into the hydroboration of CO2 using HBpin (pin = C6H12O2) in the presence of heterobimetallic complexes [(NHC)Cu–MLn] {MLn = [FeCp(CO)2] (144), [WCp(CO)3] (145), [MoCp(CO)3] (146)} (10 mol%) (Scheme 43).168 The cooperation of the second metal (M) led to facile deoxygenation of the initially formed hydroboration product, HC(O)OBpin. This offered a differing selectivity to that typically found in such reactions. Furthermore, the selectivity ratio of CO:HCO2Bpin was dependent on the nature of MLn. Mechanistic investigations suggest that during the initial step the bimetallic complex dissociates into two distinct monometallic entities by reaction with HBpin forming half an equivalent of hydride bridged dimer [(NHC)CuH]2 and [LnM–Bpin] (147–149). At first, the [(NHC)CuH]2 complex mediates CO2 hydroboration with HBpin to form HC(O)OBpin. In a subsequent step, the decarbonylation of HC(O)OBpin to CO, O(Bpin)2 and [M]–H was achieved by the cooperative action of the second metal centre [LnM–Bpin]. The tandem hydroboration of CO2 and decarbonylation of formate species HCO2Bpin depends on the relative nucleophilicity of the [M] anion.
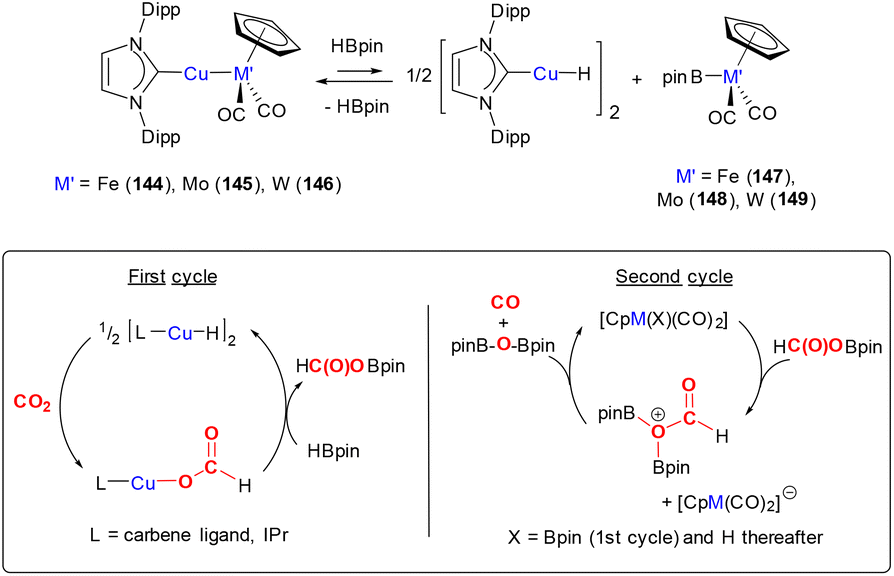 | ||
| Scheme 43 Mechanism for the 2-electron reduction of CO2 to CO catalysed by heterobimetallic complexes [(NHC)Cu–MLn] {MLn = [FeCp(CO)2], [WCp(CO)3], [MoCp(CO)3]}. | ||
A very recent example was reported by Tilley and co-workers involving the reductive disproportionation of CO2 into [CO3]2− and CO (Scheme 44).169 In this case the diamagnetic nickel(I) bimetallic complex, [Ni(μ-CH3)(IPr)]2 [IPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene] was shown to react with CO2 (1 atm) in C6D6 at 22 °C. After 15 min, two new bimetallic complexes were formed. These were Ni(II) carbonate [Ni(CH3)2(μ–κ2-O,O-CO3)(IPr)] (150), with 50% conversion as determined by NMR, and Ni(0) bridged carbonyl product [Ni(IPr){(μ-CO)(μ–η2η2-CO2)}Ni(IPr)] (151) with 32% conversion. The loss of the methyl groups in 151 was not fully clear, although the presence of methane was observed in the 1H NMR spectrum of the reaction mixture. A low temperature NMR experiment revealed that 150 existed as two isomers. A crystal structure of the isomer where the methyl groups had an anti-relationship to each other (i.e. the second isomer of the structure shown in Scheme 44) was obtained. This confirmed the coordination of the carbonate between the nickel centres. The C–O bond length involving the oxygen bridged between the two nickel centres was found to be 1.331(1) Å, whilst the other C–O bonds were shorter, 1.264(1) and 1.265(1) Å, respectively. Interestingly, the Ni–O–Ni angle was almost linear at 174.95(8)°. The crystal structure for complex 151 was previously reported by Sadighi and co-workers.170 They investigated the reactivity of the analogous binuclear Ni(0) complex, [Ni(IPr)]2 (where one of the aryl rings of the NHC ligand bridges to the other nickel centre via a η6 coordination mode) with CO2. In this original report, they suggested the disproportionation of CO2 into CO and [CO3]2−. The isolation of both 150 and 151 by Tilley and co-workers supported this postulation. The structure of 151 is rather interesting since it is a novel coordination mode for CO2 that has not been observed in any subsequent systems since the original report in 2007. It features a bent CO2 moiety bridged between the two nickel centres with two η2-C,O binding modes. The two C–O bond lengths were found to be 1.257(2) and 1.2552(19) Å. The O–C–O angle was 133.43(15)°. These bond metrics reflect a pronounced bond activation with respect to the free CO2 molecule (cf. 1.16 Å and 180°). The disproportionation reactivity (i.e. CO2 to CO and [CO3]2−) found in these systems shares similarities with other modes of CO2 bond cleavage pathways discussed in the previous sections (e.g. in the one-atom bridge section, see Scheme 8, and dearomatisation–aromatisation section, see Scheme 36).
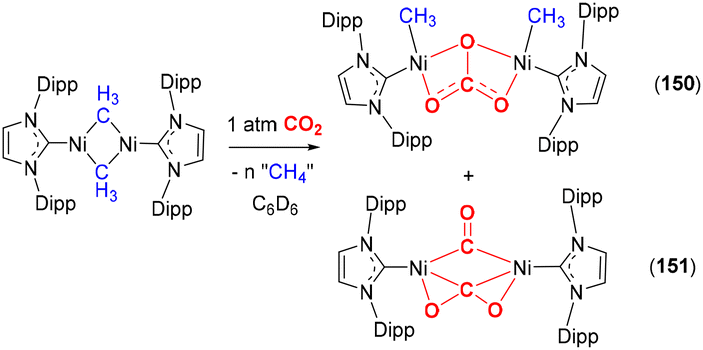 | ||
| Scheme 44 Disproportionation transformations of the CO2 moiety facilitated by a Ni(I) binuclear system. | ||
The utilisation of metalloligands where the second metal is a main group metal, or a Z-type Lewis acid function, has also become a research area of interest. Such complexes were demonstrated to be highly beneficial in CO2 activations. Whilst the metalloligand centre is not a directly involved in activation, its influence on the position trans to it on the other metal centre is significant. The cooperation between the two centres leads to novel catalytic systems. For instance, Takaya, Iwasawa and co-workers developed an interesting rigid and planar 6,6′′-bis(phosphino)-terpyridine ligand containing N3P2 donors that was utilised to support both Pd and E centres, where E = the group 13 elements, Al, Ga and In (Fig. 6).171 The Lewis acidic main group 13 element withdraws electron density from the metal via a σ-acceptor interaction to influence bond activation. The structure of the ligand scaffold holds the two centres in proximity, thereby facilitating the Pd–E interactions. Using this C2 symmetric ligand scaffold, a family of palladium–metalloligand complexes were synthesised (e.g.152a–c) and their catalytic activities were examined. Amongst them, the Pd–Al complex (152a) was found to be an excellent catalyst (0.1 mol%, 25 °C, 1 mol% cesium pivalate, 1 h) for the selective hydrosilylation of CO2 to silyl formate with the highest TOF of 19300 h−1 under mild conditions. Facile tuning of the Pd–group 13 element interaction through Z-type bonding is proposed to be the key for high catalytic performance.
 | ||
| Fig. 6 Example of complexes containing a transition metal and metalloligand reported by Takaya, Iwasawa and co-workers. | ||
Due to the strong Pd–E interactions in the 152 complexes and accessibility of the metalloligand site for CO2, other modes of reactivity including disproportionation (vide supra) of this molecule across the Pd–E bond to form an “OC–Pd–OE” species was not observed. A rational mechanism for the high catalytic activity of 152a is yet to be unravelled. However, it is likely that the use of Lewis acidic group 13 metalloligands can either stabilise carboxylate bridges such as Al–OC(O)–Pd through the formation of the Alδ+–Pdδ− zwitterionic intermediate or exert a high trans influence on the Al–Pd–OC(O)H intermediate, which is important for the selectivity of the transformation.172
In the same year, the Lu research group introduced a “double decker” multidentate ligand of the type [N{o-C6H4(NCH2PiPr2)}3]3− to support bimetallic structures.173–175 This was utilised to support M–E complexes [M = Ni, Co, Fe; E = Al, Ga, In] for the purposes of CO2 insertion into M–H bonds where the hydride was trans to the E component. In one such example involving cobalt and gallium, the complex was found to be an efficient catalyst for the formation of formate in the presence of a base. Turnover numbers of 19200 were obtained with a TOF of 27000 h−1. Of note, in the absence of H2, the complexes underwent disproportionation where two equivalents of CO2 were converted into [CO3]2− and CO, which remained as a ligand at the metal centre.176
Bourissou and co-workers illustrated cooperation between the platinum and aluminium centres in [{(Mes)2PC(CHPh)Al(tBu)2}Pt(PPh3)].177 This complex possessed an ambiphilic Pt → Al Z-type interaction featuring a T-shaped platinum centre. It reacted with CO2 to yield a six membered metallacycle containing a PtPCAlOC ring where cooperation between the platinum and aluminium centres stabilised the initially formed η1-CO2 adduct through a Pt–C(O)O–Al intermediate. More recently, the Aldridge and Goicoechea groups prepared a series of coinage metal–aluminium heterobimetallic complexes containing yet more highly polarised Alδ+–Mδ− covalent bonds and studied their reactivity with CO2.178,179 In one such example, the reaction with CO2 (1 atm) resulted in its insertion into the Al–Au bond to form the gold(I) metalla-carboxylate Al(μ-O2C)Au, where the μ-κ1-C,κ2-O,O bridge was supported by cooperative Au–η1-C and Lewis acidic Al–κ2-O,O bonds.180 Similarly, a number of other heterobimetallic complexes containing Cu–Al,181 Ir–Al,182 and Fe–Al183 bonds were reported. The examples discussed above highlight the cooperation between nucleophilic and Lewis acidic metal centres.
In summary, this section highlighted the applications of the bimetallic activation of CO2. There are, however, only a handful of examples of their catalytic application. There are some examples that demonstrate the disproportionation reactivity of two equivalents of CO2, converting it into [CO3]2− and coordinated CO. This draws several parallels with the monometallic counterparts outlined above where M–CO and M–O species are observed.
Conclusions and future prospects
The various sections above highlighted a range of different strategies that were employed for the activation of CO2 across two sites in cooperation with each other. In the earlier sections, this was as a result of activation across a metal and ligand. In the last section, this cooperation occurred between two metal centres or a metal in combination with a metalloid. Whilst there have been significant developments and achievements for cooperative methodologies involving the stoichiometric activation of CO2, the development of efficient catalytic systems remains a challenge.From the examples presented herein, two overall key transformations stand out where one of the strong double bonds in CO2 is cleaved (Scheme 45). The first is a reductive cleavage of CO2, effectively splitting it across a LnM–E species and leading to a stable LnM–CO compound and a “oxo” species bound to the E component. This latter component can be either an early transition metal or a main group species, which forms strong bonds to oxygen (e.g. Si, P, Al or Ga). The formation of these species serves as a thermodynamic sink for reductive cleavage. The second involves a disproportionation of the CO2 molecule into carbonate and “oxo”, which is facilitated again by an LnM–E moiety. In this scenario, the carbonate anion is formed and is either coordinated to the metal or non-coordinated as a free ion.
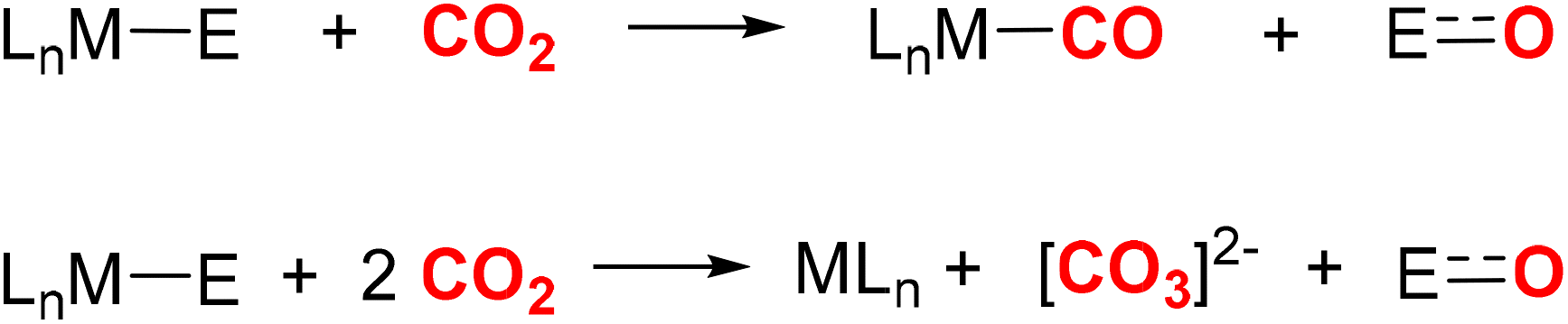 | ||
| Scheme 45 Key transformations involving the transformation of CO2 utilising MLC strategies, where M = transition metal and in this case E = either transition metal or a main group species. The carbonate is shown as a free anion. There are, however, cases where this is bound to the metal centre. | ||
With these in mind, in order to develop catalytic systems via MLC strategies, tandem methodologies will need to be developed. One such approach would be to use CO, generated by deoxygenation of CO2, within a multicomponent catalytic system where CO2 is utilised as a CO surrogate. This would find application in current industrial scale reactions such as carbonylation, carboxylation and hydroformylation. Current systems do exist, for example, the Pd-catalysed Hiyama–Denmark cross coupling for the synthesis of pharmaceutically important diaryl ketones.184 This methodology also helps to avoid the direct use of the toxic C1 building block CO and other ineffective CO resources. Further developments are needed to make such processes sustainable, however, to avoid the need for stoichiometric oxide scavengers such as silicon. Although not truly catalytic, the Milstein example highlighted in Scheme 39 (complex 127) where one of the oxygen atoms from CO2 is converted into water suggests real promise in this area. Secondly, the disproportionation of CO2 into carbonate [CO3]2− and CO is a common transformation found across a broad range of examples in many of the cooperative methodologies discussed herein. It could be envisaged that the carbonates could be converted into functionalised products by treatment with homogeneous hydrogenation catalysts. In this regard, examples of the hydrogenation of inorganic carbonates (K2CO3, Na2CO3) are still quite rare with only a few catalysts known to perform this task. For example, iridium–NHC complexes that produce formates from carbonates and renewable H2 sources are known.185 Alternatively, carbonates could also be functionalised by treatment with amines followed by hydrogenation in one pot for the N-methylation of amines, R2NH.186 Future work should focus on the rational development of practical catalysts under mild conditions based on a mechanistic understanding of CO2 transformations. To achieve this, a comprehensive understanding of the transformation of CO2 within the coordination sphere of transition metal complexes is needed. We hope to have demonstrated that cooperation between a metal and ligand or between two metals plays an important role in the activation of CO2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Welsh Government for funding this work. The project is funded by a Sêr Cymru II Capacity Builder Accelerator Award (80761-USW208). The Sêr Cymru II scheme is part-funded by the European Regional Development Fund through the Welsh Government.Notes and references
- National Oceanic and Atmospheric Administration, Global monitoring Laboratory https://keelingcurve.ucsd.edu/ (accessed 11 May 2022).
- IEA, Energy Efficiency 2021, IEA, Paris, 2021, https://www.iea.org/reports/energy-efficiency-2021 Search PubMed.
- A. Kumar, P. Daw and D. Milstein, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- T. Schaub, Chem. – Eur. J., 2021, 27, 1865–1869 CrossRef CAS PubMed.
- A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002–4033 RSC.
- Emissions – Global Energy & CO2 Status Report 2019 – Analysis - IEA, https://www.iea.org/reports/global-energy-co2-status-report-2019.
- C. Le Quéré, R. M. Andrew, P. Friedlingstein, S. Sitch, J. Hauck, J. Pongratz, P. A. Pickers, J. I. Korsbakken, G. P. Peters, J. G. Canadell, A. Arneth, V. K. Arora, L. Barbero, A. Bastos, L. Bopp, F. Chevallier, L. P. Chini, P. Ciais, S. C. Doney, T. Gkritzalis, D. S. Goll, I. Harris, V. Haverd, F. M. Hoffman, M. Hoppema, R. A. Houghton, G. Hurtt, T. Ilyina, A. K. Jain, T. Johannessen, C. D. Jones, E. Kato, R. F. Keeling, K. K. Goldewijk, P. Landschützer, N. Lefèvre, S. Lienert, Z. Liu, D. Lombardozzi, N. Metzl, D. R. Munro, J. E. M. S. Nabel, S. Nakaoka, C. Neill, A. Olsen, T. Ono, P. Patra, A. Peregon, W. Peters, P. Peylin, B. Pfeil, D. Pierrot, B. Poulter, G. Rehder, L. Resplandy, E. Robertson, M. Rocher, C. Rödenbeck, U. Schuster, J. Schwinger, R. Séférian, I. Skjelvan, T. Steinhoff, A. Sutton, P. P. Tans, H. Tian, B. Tilbrook, F. N. Tubiello, I. T. van der Laan-Luijkx, G. R. van der Werf, N. Viovy, A. P. Walker, A. J. Wiltshire, R. Wright, S. Zaehle and B. Zheng, Earth Syst. Sci. Data, 2018, 10, 2141–2194 CrossRef.
- Electricity – Global Energy & CO2 Status Report 2019 – Analysis - IEA, https://www.iea.org/reports/global-energy-co2-status-report-2019/electricity.
- https://iea.blob.core.windows.net/assets/181b48b4-323f-454d-96fb-0bb1889d96a9/CCUS_in_clean_energy_transitions.pdf .
- S. Dabral and T. Schaub, Adv. Synth. Catal., 2019, 361, 223–246 CrossRef CAS.
- M. D. Burkart, N. Hazari, C. L. Tway and E. L. Zeitler, ACS Catal., 2019, 9, 7937–7956 CrossRef CAS.
- H.-Q. Liang, T. Beweries, R. Francke and M. Beller, Angew. Chem., Int. Ed., 2022, 61, e202200723 CAS.
- Y. R. Luo, Bond dissociation energies, in CRC Handbook of Chemistry and Physics, CRC Press, 90th edn, 2010 Search PubMed.
- D. R. Lide, Bond lengths and angles in gas-phase molecules, in CRC Handbook of Chemistry and Physics, CRC Press, 84th edn, 2004 Search PubMed.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed.
- C. Das, J. Grover, Tannu, A. Das, D. Maiti, A. Dutta and G. K. Lahiri, Dalton Trans., 2022, 51, 8160–8168 RSC.
- W. H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- Y. N. Li, R. Ma, L. N. He and Z. F. Diao, Catal. Sci. Technol., 2014, 4, 1498–1512 RSC.
- S. T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Chem. Soc. Rev., 2021, 50, 4259–4298 RSC.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- A. Paparo and J. Okuda, Coord. Chem. Rev., 2017, 334, 136–149 CrossRef CAS.
- H. Li, J. Zhao, L. Luo, J. Du and J. Zeng, Acc. Chem. Res., 2021, 54, 1454–1464 CrossRef CAS PubMed.
- M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS PubMed.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, 2005 Search PubMed.
- B. Chatterjee, W. C. Chang, S. Jena and C. Werlé, ACS Catal., 2020, 10, 14024–14055 CrossRef CAS.
- J. Takaya, Chem. Sci., 2021, 12, 1964–1981 RSC.
- M. T. Whited, Dalton Trans., 2021, 50, 16443–16450 RSC.
- N. Hazari and J. E. Heimann, Inorg. Chem., 2017, 56, 13655–13678 CrossRef CAS PubMed.
- Q. Chen, C. Shen, G. Zhu, X. Zhang, C.-L. Lv, B. Zeng, S. Wang, J. Li, W. Fan and L. He, ACS Catal., 2021, 11, 9390–9396 CrossRef CAS.
- F. Schneck, J. Ahrens, M. Finger, A. C. Stückl, C. Würtele, D. Schwarzer and S. Schneider, Nat. Commun., 2018, 9, 1161 CrossRef PubMed.
- P. Amara, J.-M. Mouesca, A. Volbeda and J. C. Fontecilla-Camps, Inorg. Chem., 2011, 50, 1868–1878 CrossRef CAS PubMed.
- E. Maatta, Metal-Ligand Multiple Bonds, 1989, vol. 8 Search PubMed.
- R. A. Manzano and R. D. Young, Coord. Chem. Rev., 2021, 449, 214215 CrossRef CAS.
- H. G. Raubenheimer, Dalton Trans., 2014, 43, 16959–16973 RSC.
- S. Dey, M. Rawat and T. K. Hollis, Carbene-Based Pincer Ligands, Elsevier Inc., 3rd edn, 2021 Search PubMed.
- M. T. Whited, Beilstein J. Org. Chem., 2012, 8, 1554–1563 CrossRef CAS PubMed.
- M. T. Whited and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 5874–5875 CrossRef CAS PubMed.
- N. J. Brookes, A. Ariafard, R. Stranger and B. F. Yates, J. Am. Chem. Soc., 2009, 131, 5800–5808 CrossRef CAS PubMed.
- E. A. Lapierre, W. E. Piers and C. Gendy, Organometallics, 2018, 37, 3394–3398 CrossRef CAS.
- E. A. LaPierre, W. E. Piers and C. Gendy, Dalton Trans., 2018, 47, 16789–16797 RSC.
- D. J. Mindiola and G. L. Hillhouse, J. Am. Chem. Soc., 2002, 124, 9976–9977 CrossRef CAS PubMed.
- J. S. Price and D. J. H. Emslie, Organometallics, 2020, 39, 4618–4628 CrossRef CAS.
- M. T. Whited, J. Zhang, S. Ma, B. D. Nguyen and D. E. Janzen, Dalton Trans., 2017, 46, 14757–14761 RSC.
- M. T. Whited, J. Zhang, T. M. Donnell, V. H. Eng, P. O. Peterson, M. J. Trenerry, D. E. Janzen and B. L. H. Taylor, Organometallics, 2019, 38, 4420–4432 CrossRef CAS.
- M. T. Whited, J. Zhang, A. M. Conley, S. Ma, D. E. Janzen and D. Kohen, Angew. Chem., Int. Ed., 2021, 60, 1615–1619 CrossRef CAS PubMed.
- For additional discussions on this ligand system see: Metal/Organosilicon Complexes; Structure, Reactivity and Considerations for Catalysis, ed. M. T. Whited. and B. L. H. Taylor, Comments Inorg. Chem., 2020, vol. 40, pp. 217–276 Search PubMed.
- T. D. Lohrey, E. A. Cortes, J. I. Fostvedt, A. K. Oanta, A. Jain, R. G. Bergman and J. Arnold, Inorg. Chem., 2020, 59, 11096–11107 CrossRef CAS PubMed.
- D. S. Morris, C. Weetman, J. T. C. Wennmacher, M. Cokoja, M. Drees, F. E. Kühn and J. B. Love, Catal. Sci. Technol., 2017, 7, 2838–2845 RSC.
- M. Y. Wang, N. Wang, X. F. Liu, C. Qiao and L. N. He, Green Chem., 2018, 20, 1564–1570 RSC.
- I. Knopf, T. Ono, M. Temprado, D. Tofan and C. C. Cummins, Chem. Sci., 2014, 5, 1772–1776 RSC.
- J. S. Silvia and C. C. Cummins, Chem. Sci., 2011, 2, 1474–1479 RSC.
- J. S. Silvia and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 2169–2171 CrossRef CAS PubMed.
- D. S. Glueck, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1989, 111, 2719–2721 CrossRef CAS.
- D. S. Glueck, J. Wu, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1991, 113, 2041–2054 CrossRef CAS.
- M. Kinauer, M. Diefenbach, H. Bamberger, S. Demeshko, E. J. Reijerse, C. Volkmann, C. Würtele, J. Van Slageren, B. De Bruin, M. C. Holthausen and S. Schneider, Chem. Sci., 2018, 9, 4325–4332 RSC.
- D. J. Mindiola, R. Waterman, V. M. Iluc, T. R. Cundari and G. L. Hillhouse, Inorg. Chem., 2014, 53, 13227–13238 CrossRef CAS PubMed.
- S. J. Goodner, A. Grünwald, F. W. Heinemann and D. Munz, Aust. J. Chem., 2019, 72, 900–903 CrossRef CAS.
- E. N. Lapsheva, T. Cheisson, C. Álvarez Lamsfus, P. J. Carroll, M. R. Gau, L. Maron and E. J. Schelter, Chem. Commun., 2020, 56, 4781–4784 RSC.
- S. Chakraborty, O. Blacque and H. Berke, Dalton Trans., 2015, 44, 6560–6570 RSC.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed.
- C. L. Boyd, E. Clot, A. E. Guiducci and P. Mountford, Organometallics, 2005, 24, 2347–2367 CrossRef CAS.
- A. E. Guiducci, A. R. Cowley, M. E. G. Skinner and P. Mountford, J. Chem. Soc., Dalton Trans., 2001, 1392–1394 RSC.
- P. A. Zhizhko, N. S. Bushkov, A. V. Pichugov and D. N. Zarubin, Coord. Chem. Rev., 2021, 448, 214112 CrossRef CAS.
- J. I. Fostvedt, L. N. Grant, B. M. Kriegel, A. H. Obenhuber, T. D. Lohrey, R. G. Bergman and J. Arnold, Chem. Sci., 2020, 11, 11613–11632 RSC.
- H. Hoberg, D. Schaefer, G. Burkhart, C. Krüger and M. J. Romão, J. Organomet. Chem., 1984, 266, 203–224 CrossRef CAS.
- Y. Inoue, Y. Itoh and H. Hashimoto, Chem. Lett., 1977, 6, 855–856 CrossRef.
- G. Burkhart and H. Hoberg, Angew. Chem., Int. Ed. Engl., 1982, 21, 76 CrossRef.
- H. Hoberg, K. Jenni, K. Angermund and C. Krüger, Angew. Chem., Int. Ed. Engl., 1987, 26, 153–155 CrossRef.
- H. Hoberg and D. Schaefer, J. Organomet. Chem., 1982, 236, C28–C30 CrossRef CAS.
- S. M. Rummelt, H. Zhong, I. Korobkov and P. J. Chirik, J. Am. Chem. Soc., 2018, 140, 11589–11593 CrossRef CAS PubMed.
- T. T. Adamson, S. P. Kelley and W. H. Bernskoetter, Organometallics, 2020, 39, 3562–3571 CrossRef CAS.
- P. M. Jurd, H. L. Li, M. Bhadbhade, J. D. Watson and L. D. Field, J. Organomet. Chem., 2022, 961, 122252 CrossRef CAS.
- P. M. Jurd, H. L. Li, M. Bhadbhade, S. J. Dalgarno, R. D. McIntosh and L. D. Field, Organometallics, 2020, 39, 1580–1589 CrossRef CAS.
- S. A. Cohen and J. E. Bercaw, Organometallics, 1985, 4, 1006–1014 CrossRef CAS.
- I. S. Kolomnikov, T. S. Loveeva, V. V. Gorbachevskaya, G. G. Aleksandrov, Y. T. Struckhov and M. E. Vol'pin, J. Chem. Soc. D, 1971, 972–973 RSC.
- K. Miqueu, S. Labat, E. Daiann Sosa Carrizo and J.-M. Sotiropoulos, Eur. J. Inorg. Chem., 2018, 2018, 2717–2729 CrossRef CAS.
- R. T. Cooper, F. M. Chadwick, A. E. Ashley and D. O'Hare, Chem. Commun., 2015, 51, 11856–11859 RSC.
- B. Hessen, A. Meetsma, F. Van Bolhuis, J. H. Teuben, G. Helgesson and S. Jagner, Organometallics, 1990, 9, 1925–1936 CrossRef CAS.
- Y. Li, Z. Liu, R. Cheng and B. Liu, ChemCatChem, 2018, 10, 1420–1430 CrossRef CAS.
- K. Nagayama, F. Kawataka, M. Sakamoto, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 1999, 72, 573–580 CrossRef CAS.
- S. C. E. Stieber, N. Huguet, T. Kageyama, I. Jevtovikj, P. Ariyananda, A. Gordillo, S. A. Schunk, F. Rominger, P. Hofmann and M. Limbach, Chem. Commun., 2015, 51, 10907–10909 RSC.
- H. G. Alt and C. E. Denner, J. Organomet. Chem., 1990, 390, 53–60 CrossRef CAS.
- M. Aresta and E. Quaranta, J. Organomet. Chem., 1993, 463, 215–221 CrossRef CAS.
- A. Galindo, A. Pastor, P. J. Perez and E. Carmona, Organometallics, 1993, 12, 4443–4451 CrossRef CAS.
- C. Collazo, M. del Mar Conejo, A. Pastor and A. Galindo, Inorg. Chim. Acta, 1998, 272, 125–130 CrossRef CAS.
- M. Álvarez, A. Galindo, P. J. Pérez and E. Carmona, Chem. Sci., 2019, 10, 8541–8546 RSC.
- T. Ito, K. Takahashi and N. Iwasawa, Organometallics, 2019, 38, 205–209 CrossRef CAS.
- K. Takahashi, Y. Hirataka, T. Ito and N. Iwasawa, Organometallics, 2020, 39, 1561–1572 CrossRef CAS.
- D. García-López, L. Pavlovic and K. H. Hopmann, Organometallics, 2020, 39, 1339–1347 CrossRef.
- M. Obst, L. Pavlovic and K. H. Hopmann, J. Organomet. Chem., 2018, 864, 115–127 CrossRef CAS.
- M. J. Sgro, F. Dahcheh and D. W. Stephan, Organometallics, 2014, 33, 578–586 CrossRef CAS.
- M. J. Sgro and D. W. Stephan, Chem. Commun., 2013, 49, 2610–2612 RSC.
- J. Ruiz, M. Vivanco, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Organometallics, 1993, 12, 1811–1822 CrossRef CAS.
- U. Bayer, D. Werner, C. Maichle-Mössmer and R. Anwander, Angew. Chem., Int. Ed., 2020, 59, 5830–5836 CrossRef CAS PubMed.
- U. Bayer, Y. Liang and R. Anwander, Inorg. Chem., 2020, 59, 14605–14614 CrossRef CAS PubMed.
- K. R. Schwartz and K. R. Mann, Inorg. Chem., 2011, 50, 12477–12485 CrossRef CAS PubMed.
- M. C. Johnson, D. Rogers, W. Kaminsky and B. M. Cossairt, Inorg. Chem., 2021, 60, 5996–6003 CrossRef CAS PubMed.
- Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2015, 6, 4291–4299 RSC.
- A. Behr, U. Kanne and G. Thelen, J. Organomet. Chem., 1984, 269, c1–c3 CrossRef CAS.
- G. E. Dobereiner, J. Wu, M. G. Manas, N. D. Schley, M. K. Takase, R. H. Crabtree, N. Hazari, F. Maseras and A. Nova, Inorg. Chem., 2012, 51, 9683–9693 CrossRef CAS PubMed.
- P. Vadivelu and C. H. Suresh, Inorg. Chem., 2015, 54, 502–512 CrossRef CAS PubMed.
- J. F. Hartwig, R. G. Bergman and R. A. Andersen, J. Am. Chem. Soc., 1991, 113, 6499–6508 CrossRef CAS.
- P. Vadivelu and C. H. Suresh, Dalton Trans., 2015, 44, 16847–16853 RSC.
- S. Yamaguchi, T. Takahashi, A. Wada, Y. Funahashi, T. Ozawa, K. Jitsukawa and H. Masuda, Chem. Lett., 2007, 36, 842–843 CrossRef CAS.
- R. Johansson and O. F. Wendt, Dalton Trans., 2007, 488–492 RSC.
- J. Wu, J. C. Green, N. Hazari, D. P. Hruszkewycz, C. D. Incarvito and T. J. Schmeier, Organometallics, 2010, 29, 6369–6376 CrossRef CAS PubMed.
- D. P. Hruszkewycz, J. Wu, J. C. Green, N. Hazari and T. J. Schmeier, Organometallics, 2012, 31, 470–485 CrossRef CAS.
- D. Gelman and R. Romm, Top. Organomet. Chem., 2013, 40, 289–318 CrossRef CAS.
- L. Piccirilli, D. L. J. Pinheiro and M. Nielsen, Catalysts, 2020, 10, 773 CrossRef CAS.
- H. Li, T. P. Gonçalves, D. Lupp and K. W. Huang, ACS Catal., 2019, 9, 1619–1629 CrossRef CAS.
- D. Morales-Morales, Pincer compounds: Chemistry and Applications, 2018 Search PubMed.
- C. Gunanathan and D. Milstein, Organomet. Chem, 2011, 55–84 CAS.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- P. Hu, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2014, 4, 2649–2652 CrossRef CAS.
- M. Montag, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2012, 134, 10325–10328 CrossRef CAS PubMed.
- J. O. Bauer, G. Leitus, Y. Ben-David and D. Milstein, ACS Catal., 2016, 6, 8415–8419, DOI:10.1021/acscatal.6b02946.
- A. Nerush, M. Vogt, U. Gellrich, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6985–6997 CrossRef CAS PubMed.
- M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004–17018 CrossRef CAS PubMed.
- A. Anaby, B. Butschke, Y. Ben-David, L. J. W. Shimon, G. Leitus, M. Feller and D. Milstein, Organometallics, 2014, 33, 3716–3726 CrossRef CAS.
- E. Ben-Ari, G. Leitus, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2006, 128, 15390–15391 CrossRef CAS PubMed.
- M. Feller, E. Ben-Ari, Y. Diskin-Posner, R. Carmieli, L. Weiner and D. Milstein, J. Am. Chem. Soc., 2015, 137, 4634–4637 CrossRef CAS PubMed.
- L. Schwartsburd, M. A. Iron, L. Konstantinovski, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2010, 29, 3817–3827 CrossRef CAS.
- R. Langer, I. Fuchs, M. Vogt, E. Balaraman, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2013, 19, 3407–3414 CrossRef CAS PubMed.
- M. Vogt, M. Gargir, M. A. Iron, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2012, 18, 9194–9197 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- A. Anaby, M. Feller, Y. Ben-David, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2016, 138, 9941–9950 CrossRef CAS PubMed.
- M. Feller, E. Ben-Ari, Y. Diskin-Posner and D. Milstein, J. Coord. Chem., 2018, 71, 1679–1689 CrossRef CAS.
- M. Vogt, A. Nerush, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. Sci., 2014, 5, 2043–2051 RSC.
- R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 2120–2124 CrossRef CAS PubMed.
- T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed.
- A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS.
- M. Vogt, O. Rivada-Wheelaghan, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, Organometallics, 2013, 32, 300–308 CrossRef CAS.
- N. A. Espinosa-Jalapa, A. Nerush, L. J. W. Shimon, G. Leitus, L. Avram, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2017, 23, 5934–5938 CrossRef CAS PubMed.
- E. Fogler, M. A. Iron, J. Zhang, Y. Ben-David, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon and D. Milstein, Inorg. Chem., 2013, 52, 11469–11479 CrossRef CAS PubMed.
- X. Yang and M. B. Hall, J. Am. Chem. Soc., 2010, 132, 120–130 CrossRef CAS PubMed.
- C. A. Huff, J. W. Kampf and M. S. Sanford, Organometallics, 2012, 31, 4643–4645 CrossRef CAS.
- O. Rivada-Wheelaghan, A. Dauth, G. Leitus, Y. Diskin-Posner and D. Milstein, Inorg. Chem., 2015, 54, 4526–4538 CrossRef CAS PubMed.
- R. Stichauer, A. Helmers, J. Bremer, M. Rohdenburg, A. Wark, E. Lork and M. Vogt, Organometallics, 2017, 36, 839–848 CrossRef CAS.
- A. Kumar, P. Daw, N. A. Espinosa-Jalapa, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Dalton Trans., 2019, 48, 14580–14584 RSC.
- A. J. Kosanovich, C. H. Komatsu, N. Bhuvanesh, L. M. Pérez and O. V. Ozerov, Chem. – Eur. J., 2018, 24, 13754–13757 CrossRef CAS PubMed.
- D. Oren, Y. Diskin-Posner, L. Avram, M. Feller and D. Milstein, Organometallics, 2018, 37, 2217–2221 CrossRef CAS PubMed.
- M. Feller, U. Gellrich, A. Anaby, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6445–6454 CrossRef CAS PubMed.
- I. Ortega-Lepe, A. Rossin, P. Sánchez, L. L. Santos, N. Rendón, E. Álvarez, J. López-Serrano and A. Suárez, Inorg. Chem., 2021, 60, 18490–18502 CrossRef CAS PubMed.
- Y. Sun, C. Koehler, R. Tan, V. T. Annibale and D. Song, Chem. Commun., 2011, 47, 8349–8351 RSC.
- P. Hermosilla, P. García-Orduña, F. J. Lahoz, V. Polo and M. A. Casado, Organometallics, 2021, 40, 3720–3732 CrossRef CAS.
- G. A. Filonenko, D. Smykowski, B. M. Szyja, G. Li, J. Szczygieł, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2015, 5, 1145–1154 CrossRef CAS.
- P. Hermosilla, P. García-Orduña, P. J. Sanz Miguel, V. Polo and M. A. Casado, Inorg. Chem., 2022, 61, 7120–7129 CrossRef CAS PubMed.
- S. Sinhababu, Y. Lakliang and N. P. Mankad, Dalton Trans., 2022, 51, 6129–6147 RSC.
- J. Campos, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS.
- N. P. Mankad, Chem. – Eur. J., 2016, 22, 5822–5829 CrossRef CAS PubMed.
- R. J. Somerville and J. Campos, Eur. J. Inorg. Chem., 2021, 2021, 3488–3498 CrossRef CAS PubMed.
- M. Navarro and J. Campos, Bimetallic frustrated Lewis pairs, Elsevier Inc., 1st edn, 2021, vol. 75 Search PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 20002–20014 CrossRef CAS PubMed.
- K. L. Mears, C. R. Stennett, E. K. Taskinen, C. E. Knapp, C. J. Carmalt, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2020, 142, 19874–19878 CrossRef CAS PubMed.
- For an example see: C. M. Thomas, Comments Inorg. Chem., 2011, 32, 14–38 CrossRef CAS.
- R. M. Charles and T. P. Brewster, Coord. Chem. Rev., 2021, 433, 213765 CrossRef CAS PubMed.
- C. Yoo and Y. Lee, Chem. Sci., 2017, 8, 600–605 RSC.
- M. Weller, T. Overton, J. Rourke and F. Armstrong, Inorganic Chemistry, 7th Edition, 2018, Oxford University Press, ISBN: 9780198768128 Search PubMed.
- J. R. Prat, C. A. Gaggioli, R. C. Cammarota, E. Bill, L. Gagliardi and C. C. Lu, Inorg. Chem., 2020, 59, 14251–14262 CrossRef CAS PubMed.
- B. J. Cook, A. V. Polezhaev, C. H. Chen, M. Pink and K. G. Caulton, Inorg. Chim. Acta, 2018, 483, 510–515 CrossRef CAS.
- T. A. Hanna, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 11363–11364 CrossRef CAS.
- J. R. Pinkes, B. D. Steffey, J. C. Vites and A. R. Cutler, Organometallics, 1994, 13, 21–23 CrossRef CAS.
- H. Memmler, U. Kauper, L. H. Gade, I. J. Scowen and M. McPartlin, Chem. Commun., 1996, 1751–1752 RSC.
- S. Bagherzadeh and N. P. Mankad, J. Am. Chem. Soc., 2015, 137, 10898–10901 CrossRef CAS PubMed.
- R. J. Witzke and T. D. Tilley, Organometallics, 2022, 41, 1565–1571 CrossRef CAS.
- C. H. Lee, D. S. Laitar, P. Mueller and J. P. Sadighi, J. Am. Chem. Soc., 2007, 129, 13802–13803 CrossRef CAS PubMed.
- J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 6074–6077 CrossRef CAS PubMed.
- K. M. Paskaruk, B. E. Cowie and D. J. H. Emslie, Coordination Chemistry of Lewis Acidic Ligands, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr., vol. 1, Elsevier, 2021; pp 717–805 Search PubMed.
- R. C. Cammarota, M. V. Vollmer, J. Xie, J. Ye, J. C. Linehan, S. A. Burgess, A. M. Appel, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2017, 139, 14244–14250 CrossRef CAS PubMed.
- J. Ye, R. C. Cammarota, J. Xie, M. V. Vollmer, D. G. Truhlar, C. J. Cramer, C. C. Lu and L. Gagliardi, ACS Catal., 2018, 8, 4955–4968 CrossRef CAS.
- M. V. Vollmer, J. Ye, J. C. Linehan, B. J. Graziano, A. Preston, E. S. Wiedner and C. C. Lu, ACS Catal., 2020, 10, 2459–2470 CrossRef CAS.
- M. V. Vollmer, R. C. Cammarota and C. C. Lu, Eur. J. Inorg. Chem., 2019, 2019, 2140–2145 CrossRef CAS.
- M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917–4926 CrossRef CAS PubMed.
- J. Hicks, A. Mansikkamäki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237–241 CrossRef CAS PubMed.
- C. McManus, J. Hicks, X. Cui, L. Zhao, G. Frenking, J. M. Goicoechea and S. Aldridge, Chem. Sci., 2021, 12, 13458–13468 RSC.
- D. Sorbelli, L. Belpassi and P. Belanzoni, J. Am. Chem. Soc., 2021, 143, 14433–14437 CrossRef CAS PubMed.
- H. Y. Liu, R. J. Schwamm, M. S. Hill, M. F. Mahon, C. L. McMullin and N. A. Rajabi, Angew. Chem., Int. Ed., 2021, 60, 14390–14393 CrossRef CAS PubMed.
- L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844–4856 CrossRef CAS PubMed.
- S. Sinhababu, M. R. Radzhabov, J. Telser and N. P. Mankad, J. Am. Chem. Soc., 2022, 144, 3210–3221 CrossRef CAS PubMed.
- Z. Lian, D. U. Nielsen, A. T. Lindhardt, K. Daasbjerg and T. Skrydstrup, Nat. Commun., 2016, 9, 1–7 Search PubMed.
- Y.-J. Cheong, K. Sung, S. Park, J. Jung and H.-Y. Jang, ACS Sustainable Chem. Eng., 2020, 8, 6972–6978 CrossRef CAS.
- J. R. Cabrero-Antonino, R. Adam, K. Junge and M. Beller, Catal. Sci. Technol., 2016, 6, 7956–7966 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |