
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast structural dynamics of in-cage isomerization of diiodomethane in solution†
Hanui
Kim
abc,
Jong Goo
Kim
c,
Tae Wu
Kim‡
 c,
Sang Jin
Lee
abc,
Shunsuke
Nozawa
de,
Shin-ichi
Adachi
de,
Kihwan
Yoon
f,
Joonghan
Kim
f and
Hyotcherl
Ihee
*abc
c,
Sang Jin
Lee
abc,
Shunsuke
Nozawa
de,
Shin-ichi
Adachi
de,
Kihwan
Yoon
f,
Joonghan
Kim
f and
Hyotcherl
Ihee
*abc
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: hyotcherl.ihee@kaist.ac.kr
bKI for the BioCentury, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
cCenter for Nanomaterials and Chemical Reactions, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
dPhoton Factory, Institute of Materials Structure Science, High Energy Accelerator Research Organization (KEK), Tsukuba, Ibaraki 305-0801, Japan
eDepartment of Materials Structure Science, School of High Energy Accelerator Science, The Graduate University for Advanced Studies, Tsukuba, Ibaraki 305-0801, Japan
fDepartment of Chemistry, The Catholic University of Korea, Bucheon 14662, Republic of Korea
First published on 7th December 2020
Abstract
Despite extensive studies on the isomer species formed by photodissociation of haloalkanes in solution, the molecular structure of the precursor of the isomer, which is often assumed to be a vibrationally hot isomer formed from the radical pair, and its in-cage isomerization mechanism remain elusive. Here, the structural dynamics of CH2I2 upon 267 nm photoexcitation in methanol were probed with femtosecond X-ray solution scattering at an X-ray free-electron laser. The determined molecular structure of the transiently formed species that converts to the CH2I–I isomer has the I–I distance of 4.17 Å, which is longer than that of the isomer (3.15 Å) by more than 1.0 Å and the mean-squared displacement of 0.45 Å2, which is about 100 times larger than those of typical regular chemical bonds. These unusual structural characteristics are consistent with either a vibrationally hot form of the CH2I–I isomer or the loosely-bound radical pair (CH2I˙⋯I˙).
Introduction
The solvent cage1,2 plays various roles in chemical dynamics by influencing the energy landscape of reacting species, altering the reaction rates, and even changing the reaction pathways.3–12 For example, the primary products of photodissociation, which are completely separated in the gas phase, collide with the surrounding solvent molecules and can be trapped in the solvent cage upon failure to escape the cage.1,2 A typical fate of the trapped photofragments is to undergo geminate recombination, a ubiquitous process in the solution phase, which generally leads the transient species to the initial ground state. In the case of haloalkanes, geminate recombination can yield an isomer species as well as the parent molecule.13–17 Since the formation of the isomer is rarely observed in the gas phase,18–21 the chemical dynamics of the isomer formation has been an important topic of many experimental investigations to understand the role of the solvent cage. In this regard, diiodomethane, CH2I2, is a prototypical example and its photodissociation is known to produce a unique isomer species, isodiiodomethane (CH2I–I),22–34 which has no analog in the gas phase (Fig. 1).19,35–37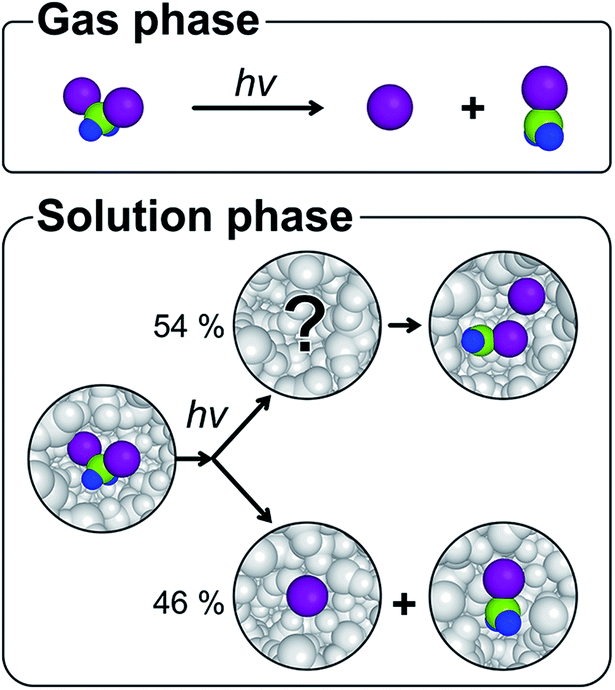 | ||
Fig. 1 The photodissociation dynamics of CH2I2 in the gas and solution phases. Whereas the products in the gas phase are CH2I˙ and I˙ radicals (CH2I˙ + I˙), the CH2I–I isomer is also observed in the solution phase. The green, blue and purple balls indicate carbon, hydrogen, and iodine atoms, respectively. Whereas the branching ratio into the CH2I–I isomer and CH2I˙ + I˙ of 54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :46 and the molecular structure of the CH2I–I isomer were determined in a previous TRXL study at a synchrotron, the exact structure of the precursor of the stable CH2I–I isomer remained to be determined and was probed with femtosecond TRXL in this study. :46 and the molecular structure of the CH2I–I isomer were determined in a previous TRXL study at a synchrotron, the exact structure of the precursor of the stable CH2I–I isomer remained to be determined and was probed with femtosecond TRXL in this study. | ||
The photodissociation dynamics of CH2I2 and the formation of CH2I–I isomer in solution have been mainly investigated with ultrafast transient absorption (TA) spectroscopy22–24,38,39 and transient resonance Raman (TRR) spectroscopy,25–28 which successfully identified the transient spectrum assigned to the isomer and the time scale of the isomer formation. As UV excitation of CH2I2 induces the n(I) → σ*(C–I) transition, one of the two C–I bonds in CH2I2 dissociates. The studies using TA spectroscopy uncovered that the excited molecule rapidly dissociates into CH2I˙ and I˙ fragments and recombines to form the CH2I–I isomer with a time constant of ∼5 ps.22–24 It is also known that a part of I˙ radicals escape the solvent cage and form CH2I˙ + I˙ within about 120 fs.38,39 Here CH2I˙ + I˙ denotes the CH2I˙ and I˙ photofragments that are completely separated and not in the same solvent cage. The TRR study suggested that the vibrationally hot CH2I–I isomer is initially formed upon UV excitation with the time scale of a few ps and then subsequently undergoes vibrational cooling on the 4–50 ps time scale.25–28 Based on their results, it was suggested that the solvation leads to the isomer via the interaction of the initially photolyzed CH2I˙ and I˙ radicals with the solvent cage. Nevertheless, the structural nature of the hot isomer, the precursor of the cold CH2I–I isomer, still remains unanswered. The detailed mechanism of the isomer formation and the structure of the hot isomer or the radical pair have been elusive and only speculated from indirect evidence. In this regard, the photodissociation dynamics of CH2I2 in solution was studied also by time-resolved X-ray liquidography (TRXL), also known as time-resolved X-ray solution scattering, which is an effective method for probing photoinduced structural changes of molecules in solution due to the structural sensitivity of X-ray scattering.7,40–42 The TRXL studies accurately identified the structure of the CH2I–I isomer,29,30 determined its fate at later times,31 and revealed that the photo-dissociated CH2I2 branches into the CH2I–I isomer and CH2I˙ + I˙ radical pathways with the 54:46 ratio.31 Nevertheless, since the temporal resolution of TRXL based on the synchrotron radiation was limited to ∼100 ps, the mechanism of isomer formation, which occurs at a few picosecond time scale, could not be investigated. With the advent of X-ray free electron lasers (XFELs),43–48 TRXL with femtosecond time resolution has become possible.49–56 In this article, we employed femtosecond TRXL on CH2I2 with 267 nm excitation to study the formation mechanism of the CH2I–I isomer with direct structural information. The structure of the isomer precursor is unveiled by the structural fitting analysis and also complemented by the quantum chemical calculations.
Experimental
The femtosecond TRXL experiment performed in this study is shown schematically in Fig. S1,† and the details of the experimental and analysis procedures are described in the ESI.† Briefly, a CH2I2 solution in methanol at the concentration of 50 mM was circulated through a sapphire nozzle with a 100 μm-thick aperture. A 100 fs laser pulse with the center wavelength of 267 nm generated by third-harmonic generation of the 800 nm femtosecond laser and with the fluence of 1 mJ mm−2 initiates the photodissociation of CH2I2 molecule. A time-delayed X-ray pulse with the center energy of 15 keV generated at the BL3 beamline of SACLA probes the progress of the reaction. The scattering patterns generated by X-ray pulses were measured with an area detector (Rayonix LX255-HS). Each scattering image was obtained by accumulating scattering intensities of 40 X-ray pulses, which provided a sufficient signal to noise ratio and suppressed the effect from jitter of the SASE process at SACLA. The difference scattering patterns were generated by subtracting the reference images which were acquired at −20 ps. The time-dependent changes were measured at various time delays ranging from −10 ps to 100 ps. By performing the singular value decomposition (SVD) analysis and kinetic analysis on our experimental data, two species-associated difference scattering curves (SADSs) and one time constant were obtained. The structural changes were extracted by the structural fitting analysis of the SADSs. The theoretical characterization of CH2I˙⋯I˙ in the solvent cage using CCSD(T) is described in the ESI.†Results and discussion
The experimental difference scattering curves were measured at various time delays from −10 ps to 100 ps, and those at selected time delays are shown in Fig. 2a. From the SVD analysis on the data and the subsequent kinetic analysis by solving the rate equations and convoluting with the instrument response function (IRF), as described in the ESI,† we obtained two SADSs shown in Fig. 2b and c and a single time constant of 6.2 ± 0.8 ps for the conversion from the first species to the second species with an IRF of 540 ± 90 fs full-width at half-maximum. We termed the first and second SADSs as SADS1 and SADS2, respectively. We first checked if SADS2 was consistent with the previous data, the curve at 150 ps measured at a synchrotron. A comparison confirms that they are identical within the experimental errors as shown in Fig. S5.† According to the previous TRXL study,31 23% of ground-state CH2I2 is initially photoexcited, and then 56.5% of the excited molecules directly relax back to the ground state, releasing a part of the photoexcitation energy as heat to surrounding solvent molecules. The remaining 43.5% of excited molecules form CH2I–I isomer and CH2I˙ + I˙ with the branching ratio of 54:46. When SADS2 is fitted with the fraction and I–I distance (RI–I) of CH2I–I isomer as fitting parameters, the best fit yielded 53.9 ± 5.2% and 3.15 ± 0.02 Å, which are consistent with 54% and 3.15 Å reported in a previous study,31 and the theoretical difference scattering curve gives an excellent agreement with SADS2 as shown in Fig. 2c. This result means that the CH2I–I isomer and CH2I˙ + I˙ are formed with a time constant of 6.2 ps via the conversion from the species generated at the onset of the reaction. Accordingly, the time constant must be responsible for the formation of either the CH2I–I isomer or CH2I˙ + I˙. Among these two possibilities, it is highly likely that the isomer is formed with the observed time constant of 6.2 ps because 6.2 ps agrees well with the values (5 ps in TA,22–24 4–50 ps time scales in TRR25–28) reported for the isomer formation in the previous spectroscopic studies.
 | ||
| Fig. 2 (a) Experimental difference scattering curves, qΔS(q), measured at various time delays from −1 ps to 100 ps (black) and calculated qΔS(q) (red) to fit the experimental curves. For clarity, data at selected time delays instead of all time delays are shown. (b and c) The structural fitting analysis of (b) SADS1 and (c) SADS2. The SADSs (black, solid line) are compared with the fitted curves without (top, red dashed line) and with (bottom, red solid line) introducing a DWF for the I–I distance. | ||
From the TA and TRR studies, it was suggested that the cold CH2I–I isomer is formed by the vibrational cooling of the hot CH2I–I isomer.22–28 However, since the spectroscopic signals at the optical frequencies are not directly related to the global molecular structures at the atomic level, the structure of the hot isomer, or the precursor of the cold isomer, remains to be determined. SADS1 contains the structural information necessary for this purpose, and to shed light on the identity of the precursor of the isomer, we performed the structural fitting analysis of SADS1. The detailed analysis protocol is described in our previous publications31 and also in the ESI.† To describe the relatively free movement of the weakly bound I atom, we incorporated a Debye–Waller factor (DWF),57,58 exp(−σ2q2/2), involving the mean-squared displacement (σ2) for RI–I into the equation for calculating the theoretical scattering pattern (see the ESI† for details). We note that for a typical analysis of TRXL data, DWF of 1 (that is, σ2 = 0) used for all interatomic distances suffices unless the interatomic distance has an unusually large σ2.
The fitting results summarized in Fig. 2b show that the theoretical SADS1 agrees well with the experimental SADS1 when a DWF is considered (Fig. 2b, red solid line). The best fit gives RI–I of 4.17 Å and σ2 of 0.45 Å2. The RI–I of 4.17 Å is longer than that of the isomer (3.15 Å) by more than 1.0 Å, and the σ2 value of 0.45 Å2 is about 100 times larger than that of typical regular chemical bonds (for example, 4.9 × 10−3 Å2 for RI–I in I3−).59 Such a long RI–I and an unusually large σ2 are consistent with a picture expected for the precursor of isomer as either the hot isomer or the loosely-bound radical pair in the cage. For a fair comparison, we performed the structural fitting analysis on SADS2 again with a DWF. As shown in Fig. 2c, the fit curve shows no noticeable change compared to the one calculated without using a DWF, and a typical small σ2 value (smaller than 1.2 × 10−3 Å2) was obtained, confirming that the CH2I–I isomer formed at later delays has a well-defined RI–I. To check the sensitivity of SADS1 to the structural parameters, we scanned the chi-square values as a function of both RI–I and σ2 of the precursor of isomer. The relative chi-square values, which are the chi-square values divided by the minimum chi-square value of SADS1, are displayed in Fig. 3a. Fig. 3a shows that the fit quality is comparable for a wide range of RI–I from 3.9 Å to 4.4 Å rather than a single well-defined value. For comparison, we performed the same analysis on SADS2 and obtained the relative chi-square values as a function of RI–I and σ2 of the cold isomer, which shows well-defined values contrary to the precursor of isomer (Fig. 3b). In Fig. 3a and b, the C–I–I angle was fixed at that of the optimized structure, 79.4° and 115° for the loosely-bound isomer precursor (Fig. 3a) and the CH2I–I isomer (Fig. 3b), respectively.
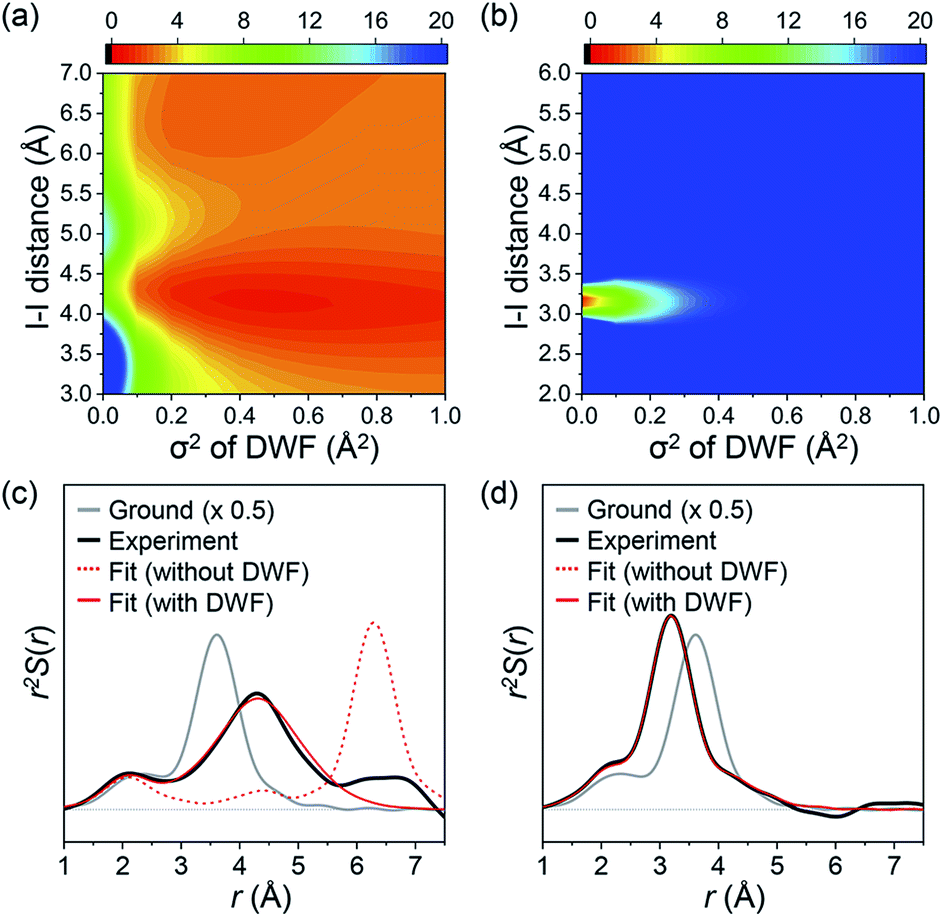 | ||
| Fig. 3 (a and b) Plots of the relative chi-square values of (a) SADS1 and (b) SADS2 as a function of the I–I distance (RI–I) and the mean-squared displacement (σ2) of (a) the loosely-bound isomer precursor and (b) the CH2I–I isomer. The loosely-bound isomer precursor has the minimum at RI–I = 4.2 Å and σ2 = 0.5 Å2 with large widths whereas the CH2I–I isomer has the minimum at RI–I = 3.2 Å and σ2 = 0.0 Å2 with sharp widths. (c and d) Two experimental species-associated RDFs, r2S(r), obtained by Fourier sine transformation of (c) SADS1 and (d) SADS2, after subtracting solvent and cage contributions. The RDF of the ground state (CH2I2) was added to the difference RDFs, to emphasize the contributions of the transient solute species. The experimental species-associated RDFs are compared with the RDF of the ground state CH2I2 (gray solid line, multiplied by 0.5) and the RDFs obtained from the structural fitting analysis of (c) SADS1 and (d) SADS2 with (red solid line) and without (red, dashed line) introducing a DWF for the I–I distance. | ||
A more intuitive picture of the structural changes that occur during the photodissociation and in-cage isomerization of CH2I2 can be obtained when we convert SADSs into the species-associated radial distribution functions (RDFs) in real space by Fourier sine transformation (Fig. 3c and d). To emphasize only the contributions of transient solute species, we subtracted the contributions from the solvent heating and solvent–solute cross term. We also added the RDF of the ground state (CH2I2) to the difference RDFs. Because all the solvent contributions are eliminated and the scattering from C and H atoms is much smaller compared to the scattering from I atoms, the RDFs shown in Fig. 3c and d mainly represent the interatomic distance between two iodine atoms and the contributions from C–I pairs are shown as a small shoulder at around 2.5 Å.
In the ground state, the peak corresponding to RI–I is at 3.58 Å as shown in the gray solid line in Fig. 3c and d. Right after the laser excitation, the RDF exhibits three distinct changes as shown in the black solid line in Fig. 3c. First, the amplitude of the peak becomes much smaller than that of the ground state since about half of the population goes into CH2I˙ + I˙, which has no I–I pair with a short enough RI–I visible in the RDF. Second, the I–I peak is shifted to a longer distance (4.17 Å), indicating that the C–I bond is dissociated and the isomer precursor exists as a loosely-bound state within the solvent cage. Third, the I–I peak becomes much broader than that of the ground state, meaning that the isomer precursor is loosely bound and thus does not have a well-defined distance. This is fully consistent with the conclusion inferred from the results of the structural fitting analysis of SADS1. It is shown in Fig. 3c that the theoretical RDF with the DWF (red solid line) with σ2 of 0.45 Å2 fits well the experimental RDF while the one without DWF (red dashed line) does not. At later time delays, the I–I peak is shifted to the distance of 3.15 Å, which is even shorter than that of the ground state, and the width of the peak is also narrowed again, as shown in Fig. 3d. This indicates that there exists a well-defined I–I bond assignable to the CH2I–I isomer formed at later time delays.
Finally, we analyzed all the experimental difference scattering curves measured at various time delays from 0.5 ps to 100 ps and all of them are well explained by the linear combinations of SADS1 and SADS2. The concentrations of each species at each time delay were determined from the coefficients of the corresponding SADSs as shown in Fig. 4a. The time-dependent changes of the concentration are then fitted with an exponential function and the time scale of the transition (6.2 ± 0.2 ps) agrees well with the one obtained from SVD analysis and also with the value reported in the previous spectroscopic studies.22–28 In Fig. 4b, we summarize the mechanism for the photoinduced in-cage isomerization of CH2I2. Upon photoexcitation of CH2I2, the dissociated iodine atom initially moves away from the carbon atom and the solvent molecules disturb the separation. Within our time resolution (∼540 fs), 46% of the dissociated iodine escapes from the solvent cage and generates CH2I˙ + I˙ radical species, while the remaining 54% is blocked by solvent molecules and forms the loosely-bound precursor of the isomer inside of the solvent cage. Subsequently, the isomer precursor transforms into a rigid CH2I–I isomer species with a time constant of 6.2 ps.
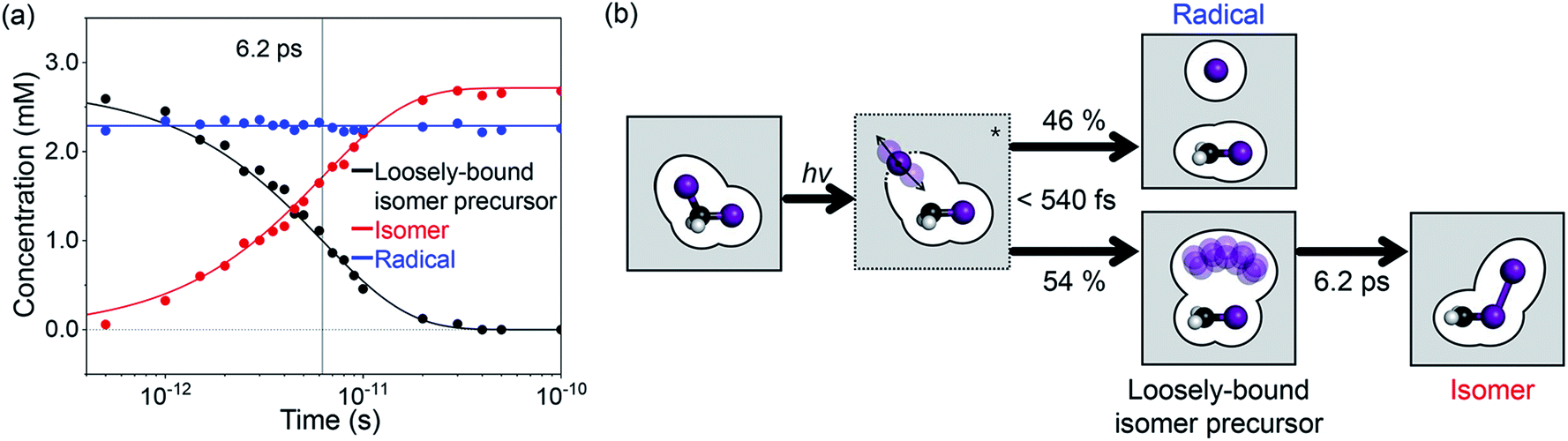 | ||
| Fig. 4 (a) Time-dependent concentrations of the three intermediate species and their transition kinetics. The black, red, and blue indicate the isomer precursor in the cage, the isomer (CH2I–I), and the radical (CH2I˙ + I˙), respectively. The lines are the concentrations obtained from the kinetics analysis. Dots represent the optimized concentrations calculated from the coefficients of SADSs obtained by fitting the experimental curve at each time point with a linear combination of SADSs. The vertical black line indicates the temporal position of the time constant of 6.2 ps. (b) Mechanism of photoinduced in-cage isomerization in CH2I2. Upon photoexcitation of CH2I2, the dissociated iodine atom initially moves away from the carbon atom and the solvent molecules disturb the separation. Within our time resolution (∼540 fs), 46% of the dissociated iodine escapes from the solvent cage (CH2I˙ + I˙), and 54% of the dissociated iodine is blocked by solvent molecules and forms the loosely-bound isomer precursor inside of the cage. The elongated RI–I (4.27 Å) and the Debye–Waller factor with a high mean-squared displacement (0.45 Å2) indicate that the isomer precursor in the cage is loosely bound and does not have a well-defined RI–I. Subsequently, it transforms into the CH2I–I isomer species with a time scale of 6.2 ps. | ||
Since the TRXL signal itself is not sensitive to the spin state, the structural aspect of the isomer precursor alone does not pin down the exact nature of the isomer precursor, that is whether it is a hot isomer or a radical pair. Nevertheless, we further considered the possibility that the isomer precursor is indeed a radical pair. As a matter of fact, while the concept of the radical pair has been frequently discussed in the literature, the structural aspect has never been determined experimentally and has rarely been addressed in theoretical calculation. The theoretical characterization of the weakly bound molecule is not trivial because of the difficulty in describing the weak interaction and a significant non-dynamic electron correlation effect of the singlet state molecule with a long bond length. Via performing CCSD(T)/AVTZ calculations, we found two molecular structures corresponding to radical pairs of the triplet state and they are consistent with the structure determined from our experiment (see the ESI† for details). The use of ab initio MD simulation with spin–orbit coupling in explicit solvent molecules would potentially be able to model the mechanism more accurately, but such an approach on a system with explicit solvent molecules is still challenging and beyond the scope of this work.
Conclusions
This work visualizes how the isomer is formed in the solvent cage via the loosely-bound isomer precursor, whose molecular structure remained to be determined, and provides structural information connecting chemical dynamics in the gas and liquid phases that were previously missing. This work also demonstrates that femtosecond TRXL offers a means of visualizing the entire process of photoinduced reactions in solution. Finally, we note that some other processes expected from the photodissociation dynamics of CH2I2 such as a rotational relaxation of the photofragments, the solvent cage rearrangement, and the escape dynamics of I˙ radical are not significant in our data, probably due to the limited time resolution. Nevertheless, by fully utilizing the recent advances of the femtosecond TRXL,52–56 future TRXL studies with improved time resolutions will surely reveal structural details of such processes and provide a comprehensive picture.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Institute for Basic Science (IBS-R004). We thank Junbeom Jo, Sungjun Park, Kyung Hwan Kim, Tokushi Sato, Ryo Fukaya, Kohei Ichiyanagi, and the beamline staff for the experiments performed at the BL3 of SACLA with the approval of the Japan Synchrotron Radiation Research Institute (proposal no. 2015B8054).Notes and references
- J. Franck and E. Rabinowitsch, Trans. Faraday Soc., 1934, 30, 120–130 RSC.
- S. J. Harris, D. Murdock, Y. Zhang, T. A. Oliver, M. P. Grubb, A. J. Orr-Ewing, G. M. Greetham, I. P. Clark, M. Towrie, S. E. Bradforth and M. N. Ashfold, Phys. Chem. Chem. Phys., 2013, 15, 6567–6582 RSC.
- I. A. Heisler and S. R. Meech, Science, 2010, 327, 857–860 CrossRef CAS.
- J. A. Kloepfer, V. H. Vilchiz, V. A. Lenchenkov and S. E. Bradforth, Chem. Phys. Lett., 1998, 298, 120–128 CrossRef CAS.
- D. A. Braden, E. E. Parrack and D. R. Tyler, Coord. Chem. Rev., 2001, 211, 279–294 CrossRef CAS.
- N. Huse, B. Bruner, M. Cowan, J. Dreyer, E. Nibbering, R. Miller and T. Elsaesser, Phys. Rev. Lett., 2005, 95, 147402 CrossRef CAS.
- K. H. Kim, J. H. Lee, J. Kim, S. Nozawa, T. Sato, A. Tomita, K. Ichiyanagi, H. Ki, J. Kim, S. Adachi and H. Ihee, Phys. Rev. Lett., 2013, 110, 165505 CrossRef.
- F. Liu, N. Luehr, H. J. Kulik and T. J. Martinez, J. Chem. Theory Comput., 2015, 11, 3131–3144 CrossRef CAS.
- S. Karashima, Y.-i. Yamamoto and T. Suzuki, J. Phys. Chem. Lett., 2019, 10, 4499–4504 CrossRef CAS.
- J. N. Miller and J. K. McCusker, Chem. Sci., 2020, 11, 5191–5204 RSC.
- C. Lee, C. H. Choi and T. Joo, Phys. Chem. Chem. Phys., 2020, 22, 1115–1121 RSC.
- H. Shimizu, K. H. Park, H. Otani, S. Aoyagi, T. Nishinaga, Y. Aso, D. Kim and M. Iyoda, Chem.–Eur. J., 2018, 24, 3793–3801 CrossRef CAS.
- G. Maier and H. P. Reisenauer, Angew. Chem., Int. Ed., 1986, 25, 819–822 CrossRef.
- X. Zheng and D. L. Phillips, Chem. Phys. Lett., 2000, 324, 175–182 CrossRef CAS.
- M. Wall, A. N. Tarnovsky, T. Pascher, V. Sundström and E. Åkesson, J. Phys. Chem. A, 2003, 107, 211–217 CrossRef CAS.
- C. P. Anderson, K. G. Spears, K. R. Wilson and R. J. Sension, J. Chem. Phys., 2013, 139, 194307 CrossRef.
- K.-C. Tang, J. Peng, K. G. Spears and R. J. Sension, J. Chem. Phys., 2010, 132, 141102 CrossRef.
- P. Y. Cheng, D. Zhong and A. H. Zewail, Chem. Phys. Lett., 1995, 237, 399–405 CrossRef CAS.
- V. A. Borin, S. M. Matveev, D. S. Budkina, P. Z. El-Khoury and A. N. Tarnovsky, Phys. Chem. Chem. Phys., 2016, 18, 28883–28892 RSC.
- J. Yang, X. Zhu, T. J. A. Wolf, Z. Li, J. P. F. Nunes, R. Coffee, J. P. Cryan, M. Gühr, K. Hegazy, T. F. Heinz, K. Jobe, R. Li, X. Shen, T. Veccione, S. Weathersby, K. J. Wilkin, C. Yoneda, Q. Zheng, T. J. Martinez, M. Centurion and X. Wang, Science, 2018, 361, 64–67 CrossRef CAS.
- Y. Liu, S. L. Horton, J. Yang, J. P. F. Nunes, X. Shen, T. J. A. Wolf, R. Forbes, C. Cheng, B. Moore, M. Centurion, K. Hegazy, R. Li, M.-F. Lin, A. Stolow, P. Hockett, T. Rozgonyi, P. Marquetand, X. Wang and T. Weinacht, Phys. Rev. X, 2020, 10, 021016 CAS.
- A. N. Tarnovsky, J. L. Alvarez, A. P. Yartsev, V. Sundstrom and E. Akesson, Chem. Phys. Lett., 1999, 312, 121–130 CrossRef CAS.
- A. N. Tarnovsky, V. Sundstrom, E. Akesson and T. Pascher, J. Phys. Chem. A, 2004, 108, 237–249 CrossRef CAS.
- K. Saitow, Y. Naitoh, K. Tominaga and K. Yoshihara, Chem.–Asian J., 2008, 3, 696–709 CrossRef CAS.
- W. M. Kwok, C. S. Ma, A. W. Parker, D. Phillips, M. Towrie, P. Matousek and D. L. Phillips, J. Chem. Phys., 2000, 113, 7471–7478 CrossRef CAS.
- X. M. Zheng and D. L. Phillips, J. Phys. Chem. A, 2000, 104, 6880–6886 CrossRef CAS.
- Y. L. Li, D. Q. Wang, K. H. Leung and D. L. Phillips, J. Phys. Chem. A, 2002, 106, 3463–3468 CrossRef CAS.
- X. Guan, X. Lin, W. M. Kwok, Y. Du, Y. L. Li, C. Zhao, D. Wang and D. L. Phillips, J. Phys. Chem. A, 2005, 109, 1247–1256 CrossRef CAS.
- J. Davidsson, J. Poulsen, M. Cammarata, P. Georgiou, R. Wouts, G. Katona, F. Jacobson, A. Plech, M. Wulff, G. Nyman and R. Neutze, Phys. Rev. Lett., 2005, 94, 245503 CrossRef.
- J. Vincent, M. Andersson, M. Eklund, A. B. Wohri, M. Odelius, E. Malmerberg, Q. Y. Kong, M. Wulff, R. Neutze and J. Davidsson, J. Chem. Phys., 2009, 130, 154502 CrossRef.
- S. Park, J. Choi, H. Ki, K. H. Kim, K. Y. Oang, H. Roh, J. Kim, S. Nozawa, T. Sato, S. I. Adachi, J. Kim and H. Ihee, J. Chem. Phys., 2019, 150, 224201 CrossRef.
- M. N. Glukhovtsev and R. D. Bach, Chem. Phys. Lett., 1997, 269, 145–150 CrossRef CAS.
- D. L. Phillips and W.-H. Fang, J. Org. Chem., 2001, 66, 5890–5896 CrossRef CAS.
- M. Odelius, M. Kadi, J. Davidsson and A. N. Tarnovsky, J. Chem. Phys., 2004, 121, 2208–2214 CrossRef CAS.
- M. Kawasaki, S. J. Lee and R. Bersohn, J. Chem. Phys., 1975, 63, 809–814 CrossRef CAS.
- P. M. Kroger, P. C. Demou and S. J. Riley, J. Chem. Phys., 1976, 65, 1823–1834 CrossRef CAS.
- U. Marvet, Q. Zhang, E. J. Brown and M. Dantus, J. Chem. Phys., 1998, 109, 4415–4427 CrossRef CAS.
- B. J. Schwartz, J. C. King, J. Z. Zhang and C. B. Harris, Chem. Phys. Lett., 1993, 203, 503–508 CrossRef CAS.
- K.-i. Saitow, Y. Naitoh, K. Tominaga and K. Yoshihara, Chem. Phys. Lett., 1996, 262, 621–626 CrossRef CAS.
- D. Rimmerman, D. Leshchev, D. J. Hsu, J. Hong, I. Kosheleva and L. X. Chen, J. Phys. Chem. Lett., 2017, 8, 4413–4418 CrossRef CAS.
- J. M. Budarz, M. P. Minitti, D. V. Cofer-Shabica, B. Stankus, A. Kirrander, J. B. Hastings and P. M. Weber, J. Phys. B: At., Mol. Opt. Phys., 2016, 49, 034001 CrossRef.
- T. C. Rossi, D. Grolimund, M. Nachtegaal, O. Cannelli, G. F. Mancini, C. Bacellar, D. Kinschel, J. R. Rouxel, N. Ohannessian, D. Pergolesi, T. Lippert and M. Chergui, Phys. Rev. B: Condens. Matter Mater. Phys., 2019, 100, 245207 CrossRef CAS.
- T. Ishikawa, H. Aoyagi, T. Asaka, Y. Asano, N. Azumi, T. Bizen, H. Ego, K. Fukami, T. Fukui, Y. Furukawa, S. Goto, H. Hanaki, T. Hara, T. Hasegawa, T. Hatsui, A. Higashiya, T. Hirono, N. Hosoda, M. Ishii, T. Inagaki, Y. Inubushi, T. Itoga, Y. Joti, M. Kago, T. Kameshima, H. Kimura, Y. Kirihara, A. Kiyomichi, T. Kobayashi, C. Kondo, T. Kudo, H. Maesaka, X. M. Maréchal, T. Masuda, S. Matsubara, T. Matsumoto, T. Matsushita, S. Matsui, M. Nagasono, N. Nariyama, H. Ohashi, T. Ohata, T. Ohshima, S. Ono, Y. Otake, C. Saji, T. Sakurai, T. Sato, K. Sawada, T. Seike, K. Shirasawa, T. Sugimoto, S. Suzuki, S. Takahashi, H. Takebe, K. Takeshita, K. Tamasaku, H. Tanaka, R. Tanaka, T. Tanaka, T. Togashi, K. Togawa, A. Tokuhisa, H. Tomizawa, K. Tono, S. Wu, M. Yabashi, M. Yamaga, A. Yamashita, K. Yanagida, C. Zhang, T. Shintake, H. Kitamura and N. Kumagai, Nat. Photonics, 2012, 6, 540–544 CrossRef CAS.
- T. Katayama, Y. Inubushi, Y. Obara, T. Sato, T. Togashi, K. Tono, T. Hatsui, T. Kameshima, A. Bhattacharya, Y. Ogi, N. Kurahashi, K. Misawa, T. Suzuki and M. Yabashi, Appl. Phys. Lett., 2013, 103, 131105 CrossRef.
- M. L. Shelby, P. J. Lestrange, N. E. Jackson, K. Haldrup, M. W. Mara, A. B. Stickrath, D. L. Zhu, H. T. Lemke, M. Chollet, B. M. Hoffman, X. S. Li and L. X. Chen, J. Am. Chem. Soc., 2016, 138, 8752–8764 CrossRef CAS.
- M. C. Langner, S. Roy, S. W. Huang, J. D. Koralek, Y. D. Chuang, G. L. Dakovski, J. J. Turner, J. S. Robinson, R. N. Coffee, M. P. Minitti, S. Seki, Y. Tokura and R. W. Schoenlein, Phys. Rev. Lett., 2017, 119, 107204 CrossRef CAS.
- Y. Obara, H. Ito, T. Ito, N. Kurahashi, S. Thurmer, H. Tanaka, T. Katayama, T. Togashi, S. Owada, Y. Yamamoto, S. Karashima, J. Nishitani, M. Yabashi, T. Suzuki and K. Misawa, Struct. Dyn., 2017, 4, 044033 CrossRef.
- P. Wernet, Philos. Trans. R. Soc., A, 2019, 377, 20170464 CrossRef CAS.
- K. H. Kim, J. G. Kim, S. Nozawa, T. Sato, K. Y. Oang, T. W. Kim, H. Ki, J. Jo, S. Park, C. Song, T. Sato, K. Ogawa, T. Togashi, K. Tono, M. Yabashi, T. Ishikawa, J. Kim, R. Ryoo, J. Kim, H. Ihee and S.-i. Adachi, Nature, 2015, 518, 385–389 CrossRef CAS.
- E. Biasin, T. B. van Driel, K. S. Kjaer, A. O. Dohn, M. Christensen, T. Harlang, P. Chabera, Y. Z. Liu, J. Uhlig, M. Papai, Z. Nemeth, R. Hartsock, W. Liang, J. X. Zhang, R. Alonso-Mori, M. Chollet, J. M. Glownia, S. Nelson, D. Sokaras, T. A. Assefa, A. Britz, A. Galler, W. Gawelda, C. Bressler, K. J. Gaffney, H. T. Lemke, K. B. Moller, M. M. Nielsen, V. Sundstrom, G. Vanko, K. Warnmark, S. E. Canton and K. Haldrup, Phys. Rev. Lett., 2016, 117, 013002 CrossRef.
- E. Biasin, T. Van Driel, G. Levi, M. Laursen, A. Dohn, A. Molkte, P. Vester, F. Hansen, K. Kjær, T. Harlang, R. Hartsock, M. Christensen, K. Gaffney, N. Henriksen, K. Møller, K. Haldrup and M. Nielsen, J. Synchrotron Radiat., 2018, 25, 306–315 CrossRef CAS.
- K. Haldrup, G. Levi, E. Biasin, P. Vester, M. G. Laursen, F. Beyer, K. S. Kjær, T. Brandt van Driel, T. Harlang, A. O. Dohn, R. J. Hartsock, S. Nelson, J. M. Glownia, H. T. Lemke, M. Christensen, K. J. Gaffney, N. E. Henriksen, K. B. Møller and M. M. Nielsen, Phys. Rev. Lett., 2019, 122, 063001 CrossRef CAS.
- K. S. Kjær, T. B. Van Driel, T. C. B. Harlang, K. Kunnus, E. Biasin, K. Ledbetter, R. W. Hartsock, M. E. Reinhard, S. Koroidov, L. Li, M. G. Laursen, F. B. Hansen, P. Vester, M. Christensen, K. Haldrup, M. M. Nielsen, A. O. Dohn, M. I. Pápai, K. B. Møller, P. Chabera, Y. Liu, H. Tatsuno, C. Timm, M. Jarenmark, J. Uhlig, V. Sundstöm, K. Wärnmark, P. Persson, Z. Németh, D. S. Szemes, É. Bajnóczi, G. Vankó, R. Alonso-Mori, J. M. Glownia, S. Nelson, M. Sikorski, D. Sokaras, S. E. Canton, H. T. Lemke and K. J. Gaffney, Chem. Sci., 2019, 10, 5749–5760 RSC.
- B. Stankus, H. Yong, N. Zotev, J. M. Ruddock, D. Bellshaw, T. J. Lane, M. Liang, S. Boutet, S. Carbajo, J. S. Robinson, W. Du, N. Goff, Y. Chang, J. E. Koglin, M. P. Minitti, A. Kirrander and P. M. Weber, Nat. Chem., 2019, 11, 716–721 CrossRef CAS.
- K. Kunnus, M. Vacher, T. C. B. Harlang, K. S. Kjaer, K. Haldrup, E. Biasin, T. B. van Driel, M. Papai, P. Chabera, Y. Z. Liu, H. Tatsuno, C. Timm, E. Kallman, M. Delcey, R. W. Hartsock, M. E. Reinhard, S. Koroidov, M. G. Laursen, F. B. Hansen, P. Vester, M. Christensen, L. Sandberg, Z. Nemeth, D. S. Szemes, E. Bajnoczi, R. Alonso-Mori, J. M. Glownia, S. Nelson, M. Sikorski, D. Sokaras, H. T. Lemke, S. Canton, K. B. Moller, M. M. Nielsen, G. Vank, K. Warnmark, V. Sundstrom, P. Persson, M. Lundberg, J. Uhlig and K. J. Gaffney, Nat. Commun., 2020, 11, 634 CrossRef CAS.
- J. G. Kim, S. Nozawa, H. Kim, E. H. Choi, T. Sato, T. W. Kim, K. H. Kim, H. Ki, J. Kim, M. Choi, Y. Lee, J. Heo, K. Y. Oang, K. Ichiyanagi, R. Fukaya, J. H. Lee, J. Park, I. Eom, S. H. Chun, S. Kim, M. Kim, T. Katayama, T. Togashi, S. Owada, M. Yabashi, S. J. Lee, S. Lee, C. W. Ahn, D.-S. Ahn, J. Moon, S. Choi, J. Kim, T. Joo, J. Kim, S.-i. Adachi and H. Ihee, Nature, 2020, 582, 520–524 CrossRef CAS.
- I. Hargittai and M. Hargittai, Stereochemical applications of gas-phase electron diffraction/Part A: the electron diffraction Technique, VCH, Weinheim, 1988 Search PubMed.
- H. Yong, J. M. Ruddock, B. Stankus, L. Ma, W. Du, N. Goff, Y. Chang, N. Zotev, D. Bellshaw, S. Boutet, S. Carbajo, J. E. Koglin, M. Liang, J. S. Robinson, A. Kirrander, M. P. Minitti and P. M. Weber, J. Chem. Phys., 2019, 151, 084301 CrossRef.
- H. Sakane, T. Mitsui, H. Tanida and I. Watanabe, J. Synchrotron Radiat., 2001, 8, 674–676 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05108j |
| ‡ Present address: Chemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, Illinois 60439, United States. |
| This journal is © The Royal Society of Chemistry 2021 |