
Effect of backbone and end-group regioisomerism on thermomechanical properties of vanillin-based polyurethane networks†
Adithya
Rangamani
 and
Christopher A.
Alabi
*
and
Christopher A.
Alabi
*
Robert Frederick Smith School of Chemical and Biomolecular Engineering, Olin Hall, Cornell University, Ithaca, NY 14853, USA. E-mail: caa238@cornell.edu
First published on 29th December 2020
Abstract
Monomer composition, cross-link density, and cross-link distribution are the primary determinants of material properties in thermosetting networks. Here, we investigate the effect of regioisomerism and composition in polymeric networks via a recently developed scalable sequence-defined polyurethane macromer (SD-PUM) platform. The iterative, support-free SD-PUM platform facilitates the assembly of macromers with tunable backbones and pendant groups at the gram-scale. A series of SD-PUMs made with different end-groups and vanillin positional isomers were synthesized and cross-linked into thermosetting networks via multivalent thiol cross-linkers. The results show that the thermomechanical properties of the ensuing networks are sensitive to the choice of regioisomer embedded in the SD-PUM backbone as well as the type of macromer end-group. When a cross-linker with higher functionality is employed, regioisomeric differences result in more pronounced changes in material properties. This work showcases the robustness of the SD-PUM platform and its use of backbone and end-group regioisomers to modulate material properties.
Introduction
Thermosetting networks are widely used due to their excellent thermomechanical properties and stability that results from the presence of permanent covalent cross-links.1 These networks are made from cross-linking polyfunctional monomers (e.g., phenolic and urea-formaldehyde resins) or reactive polymer or macromer chains (e.g., unsaturated polyesters) via permanent covalent bonds to give insoluble solid materials with high modulus, solvent resistance, high-temperature stability, and low creep.2–4 Polyurethanes are one of the largest class of cross-linked polymers and have found extensive use in a wide variety of applications, including adhesives, sealants, automobile parts, sheeting, footwear, and much more.5 Epoxy resins are an example of cross-linked networks via small molecules (diglycidyl ether of bisphenol A with a multifunctional cross-linker) and are widely used as structural adhesives in coatings, optical communication devices, packaging, and others.6 Studies have shown that the molecular architecture of the monomers and cross-linkers used in monomer-based resins, in addition to their functionality, can strongly affect the mechanical properties of the resulting network.7One of the modes by which monomer and cross-linker architecture affect material properties is by influencing cross-link density. Cross-link density and distribution can vary depending on the functionality and geometry of the monomer and cross-linker and are one of the primary determinants of material property. Since most cross-linking reactions are kinetically controlled (e.g., condensation and radical-based reactions), kinetically trapped states are formed and driven in part by the structure of the monomer. Thus, network topology is highly dependent on monomer structure, and this effect is more prevalent in monomers with rigid aromatic groups.
Propagation of high network connectivity demands specific geometric requirements from the monomer. Molecular structure dictates the network reaction kinetics, loop formation, and strongly influences the network topology. A number of studies have investigated how molecular structure affects network connectivity and material properties. The review by Dusek et al.8 lists monomers with multiple geometric differences, including star-like and hyperbranched molecules with various degrees of freedom and length between functional groups and their effect on material properties. The effect of conformation, regioisomerism, stereoisomerism on physical properties has also been explored in bisphenol,9,10 benzoxazine,11–13 and azobenzene14,15 based systems. Others have investigated the effect of changes in ring substitutions on the physical properties of the ensuing network.16–21 Riad et al.6 investigated the effect of ortho-, meta- and para- substitutions in epoxy-amine networks and found a significant change (para- > meta- > ortho-) in the glass transition temperatures (Tg) of the networks due to minor structural changes in the monomer structure. These afore-mentioned studies are based on the use of structural isomers of multifunctional small molecule monomers.
Unlike small molecules, modulation of the spacing, regioselectivity, and sterics of a multifunctional unimolecular oligomer (i.e., a macromer) is non-trivial and often requires extensive synthetic manipulations. Due to these synthetic challenges, studies into how macromer structure affects network connectivity and material properties are lacking. Addressing this question requires studying network formation with well-defined and easily accessible functional macromers. Sequence-defined macromers fill a void that exists between precise small molecule monomers and large homo- and copolymers. When assembled from functional monomers, these well-defined unimolecular macromers can serve as versatile building blocks for a wide range of polymeric materials and networks.22,23 However, simple gram-scale synthesis is required to understand how the structure and composition of a macromer can affect network topology and material properties.
Recently, our research group developed a robust platform for the synthesis of sequence-defined polyurethane macromers (SD-PUMs)24 using a sustainable feedstock, vanillin, and high-yielding reactions. The SD-PUMs have modifiable pendant groups and a rigid aromatic vanillin-based backbone. The initial report by Hoff et al.24 shows the importance of sequence on the properties of cross-linked networks. This work inspired further investigations, carried out in this study, where we investigate the effect of regioisomeric backbone and end-group substitutions on network topology and material properties. In this study, we show that the SD-PUM platform can be used to create and tune network properties by varying backbone structure and end-group composition. To study how changes in backbone and end-group structure affect network topology and properties, four macromers (two pairs of regioisomers) were synthesized and cross-linked using multivalent thiols. The thermal and mechanical properties of the ensuing networks were measured and compared. The results reported herein illustrate how subtle changes in backbone and end-group structure, via the use of positional isomers, significantly impacts network performance.
Results and discussion
Synthesis of SD-PUMs
The SD-PUM synthetic platform involves the use of an inexpensive biorenewable compound, vanillin, to create SD-PUMs on the gram-scale (Fig. 1). Vanillin is converted to a divanillin carbonate monomer with two reactive sites: the aldehyde that undergoes a reductive amination reaction to generate a secondary amine and a carbonate that is reacted with the secondary amine to form a carbamate. The synthetic process involves sequential reductive amination and carbamation reactions, followed by a simple aqueous workup after each step (Fig. 1). Performing the assembly and purification steps in this manner enables the production of precision SD-PUMs at scale with only a round-bottom flask, a separatory funnel for stepwise purification, and a rotovap for solvent removal.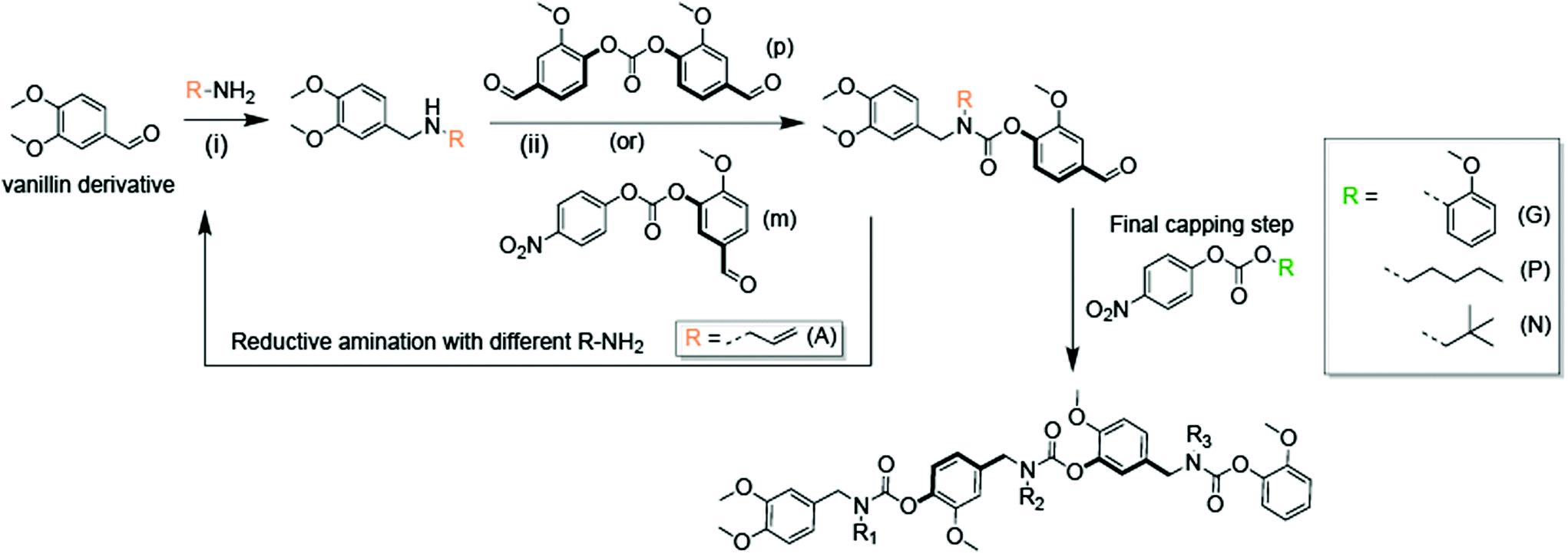 | ||
| Fig. 1 General synthesis scheme for sequence-defined polyurethane macromers. (i) MeOH, NaBH4, room temperature, (ii) triethylamine, CH3CN, 50 °C,. (A) – allyl, (G) – guaiacaol, (P) – pentyl, (N) – neopentyl. | ||
In this study, allylamine is used as the pendant group for all macromers. Macromers with 1,4-backbone substitutions were made using divanillin carbonate for the carbamate formation reaction with a secondary amine. However, for the macromers with a 1,3-backbone substitution, a 4-nitrophenyl activated carbonate of isovanillin was used. This was done due to the high pKa of isovanillin (∼8.8), which makes it a poor leaving group relative to vanillin (pKa ∼ 7.4) or nitrophenol (pKa ∼ 7.2). The final capped macromers were purified using column chromatography. The individual reaction steps had recovered yields of 70–85%. The recovered yields of the purified macromers were in the 16–22% range at the gram-scale. Each oligomer is named according to its monomeric units, where “A” represents an allyl pendant group, “m” represents an isovanillin backbone unit and “p” represents a vanillin backbone unit. The end-groups used to cap the macromers were guaiacol (G), pentanol (P), and neopentanol (N). The four macromers that were studied were AmAG, ApAG, ApAN, and ApAP (Fig. 2A). The AmAG and ApAG macromers are regioisomers in their backbone, while ApAN and ApAP are regioisomers at their end-group. The progression of the SD-PUM synthesis was followed by 1H and 13C NMR spectroscopy and liquid chromatography-mass spectrometry (LC-MS) (ESI Fig. 1–4†). The final 1H NMR spectra of the macromers after purification via column chromatography is shown in Fig. 2B. The full spectra of all macromers with complete integration can be found in ESI Fig. 5–8.†
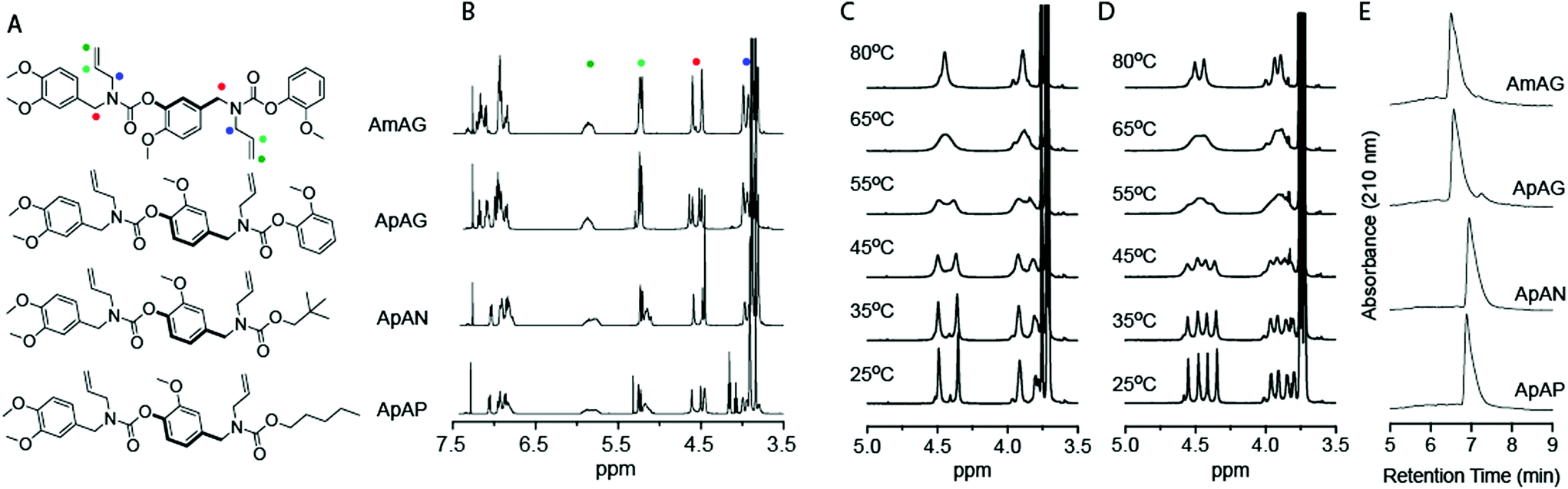 | ||
| Fig. 2 (A) Chemical structure of macromers evaluated in this study; (B) 1H NMR spectra of macromers; VT NMR of (C) AmAG and (D) ApAG; (E) retention time of the macromers on a C4 reverse-phase column. | ||
Structural differences of regioisomeric SD-PUMs
The 1H NMR spectra of the AmAG and ApAG regioisomers shared similar spectral features with notable differences in the splitting patterns of the benzylic protons centered around 4.6 ppm and the allylic protons at 4 ppm (Fig. 2B and ESI Fig. 5 and 6†). This hinted at differences in conformation or electronic environment. To probe the solution conformational dynamics of each macromer, a two-dimensional rotating frame Overhauser effect spectroscopy (1H−1H ROESY) experiment was conducted for both macromers at 0 and 200 ms mixing times (ESI Fig. 9 and 10†). The presence of off-diagonal exchange peaks in the 200 ms spectra confirmed conformational exchange between the different positions due to rotation around the C(O)–N bond. The AmAG macromer only had two diagonal peaks at the benzylic protons, whereas ApAG had four diagonal peaks at the benzylic protons at 0 ms. At 200 ms, the AmAG macromer had a single set of exchange peaks while the ApAG macromer had two sets of exchange peaks (ESI Fig. 9B and 10B†). The rate constants for conformational exchange were calculated using EXSYcalc, and both macromers had similar rate constants (k1 = 8.23 s−1 for AmAG and k1 = 8.56, 8.42 s−1 for ApAG). Further studies via heteronuclear single quantum coherence spectroscopy (1H–13C HSQC) (ESI Fig. 11 and 12†) confirmed the presence of eight methylene protons (four benzylic and four allylic) for both macromers. The four benzylic protons in ApAG were separated both in the 13C and 1H spectra, indicating that the two benzyl groups in the macromer were in different environments. However, the benzylic protons in AmAG were separated only in the 13C spectra suggesting that the two benzyl positions in the macromer were in identical chemical environments. This observation speaks to how a single positional change in the macromer backbone can alter the molecular electronic environment.Variable temperature NMR (VT-NMR) was also used to confirm the structural dynamics of the macromers (Fig. 2C and D). As the temperature was increased from 25 to 80 °C, the two benzylic proton peaks at 4.6 ppm coalesced into a broad peak due to the rapid exchange between conformations brought about by rotation around the C(O)–N bond. The coalescence occurred at 65 °C for both macromers (Fig. 2C and D). The presence of two distinct peaks at 4.6 ppm at 80 °C in the ApAG spectra concurs with the conclusions from HSQC spectra, which suggests that the two benzyl groups in ApAG are in different environments. Similarly, the presence of a single peak at 4.6 ppm at 80 °C in the AmAG spectra confirms the identical environments for the two benzylic positions.
The retention times of the macromers on a C4 reverse-phase column is shown in Fig. 2E. The retention time is a measure of relative hydrophobicity, and the data shows that the macromers with the aliphatic end-groups have higher retention times (hence, more hydrophobic) than those with aromatic end-groups. The data also shows that the regioisomers, in both cases, have similar retention times.
Effect of end-group composition and regioisomerism on macromer physical properties
To study the effect of regioisomerism on the physical properties of the macromers, viscosity measurements were performed using a parallel plate rheometer at 45 °C with a constant shear rate of 0.1 s−1 over 60 seconds. The measured viscosities were compared to motor oil and honey in order to provide a physical frame of reference using common household items. The viscosities of the SD-PUMs with rigid aromatic guaiacol end groups are comparable to honey, with AmAG and ApAG having viscosities of 8.29 and 5.35 Pa.s, respectively (Fig. 3). The viscosities of SD-PUMs with aliphatic end groups are comparable to motor oil, with ApAN and ApAP macromers having viscosities of 1.56 and 0.84 Pa.s, respectively. The viscosity clearly decreases with increasing end-group rotational flexibility, i.e., going from a rigid guaiacol cap in AmAG and ApAG to aliphatic end caps in ApAN and ApAP. Flexible chains in the molecular structure increase the available free volume in the fluid, which results in enhanced mobility and diminished resistance to flow. ApAN, due to its bulky and rigid neopentyl end-group, had a higher viscosity (almost 2-fold) than ApAP as expected. Interestingly, the subtle change in the backbone structure from a 1,3 substitution in AmAG to a 1,4 substitution in ApAG led to a ∼40% decrease in the viscosity (Fig. 3).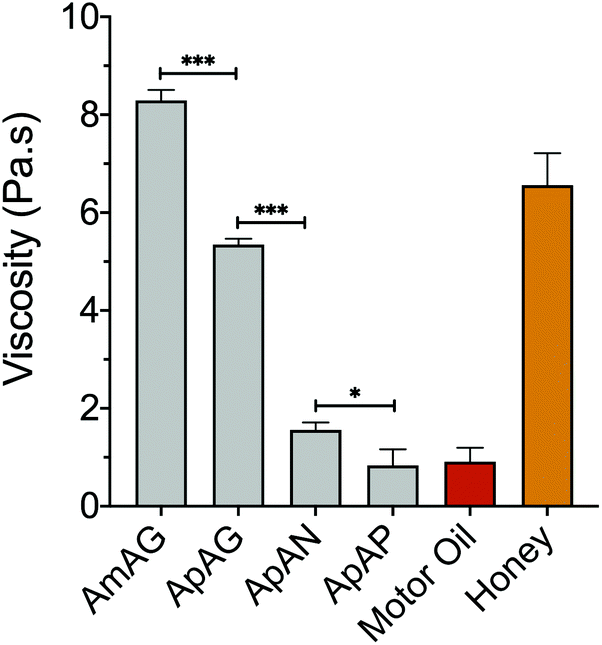 | ||
| Fig. 3 Viscosity measurements made with a parallel plate rheometer at a temperature of 45 °C and a constant shear rate of 0.1 s−1. ***, p-value < 0.0001, *, p-value < 0.05. | ||
Preparation and characterization of SD-PUM networks
To understand the effect of composition and regioisomerism on the thermal and mechanical properties of thermosetting networks, the allyl groups in the SD-PUMs were cross-linked with a tetravalent and trivalent thiol via thiol–ene reactions. The two cross-linkers were selected to study the effect of cross-linker valency on material properties. The size of the cross-linkers were kept small, such that their chain flexibility would not mask the effect of SD-PUM structure. Networks were prepared via a thiol–ene photopolymerization reaction between the allyl groups of the SD-PUM and the thiol groups on the cross-linker. The four SD-PUMs and two cross-linkers were used to prepare eight unique thioether linked networks.The networks were prepared by solvent casting a mixture of the SD-PUM, multivalent cross-linker, and a photoinitiator in acetonitrile into a rectangular PTFE mold (25 mm × 20 mm × 1 mm deep). Cross-linking was initiated with UV light (365 nm, 20 mW cm−2), and the networks were cured for 60 minutes. Following irradiation and gelation, each film was removed from the mold and swelled in acetonitrile for 48 h to remove unreacted macromers, cross-linker, and photoinitiator from the solid network. The residual acetonitrile solvent was removed, and the film was cut into ∼5 mm wide rectangular strips, as seen in Fig. 4B, and dried in a vacuum oven for 12 h at 100 °C.
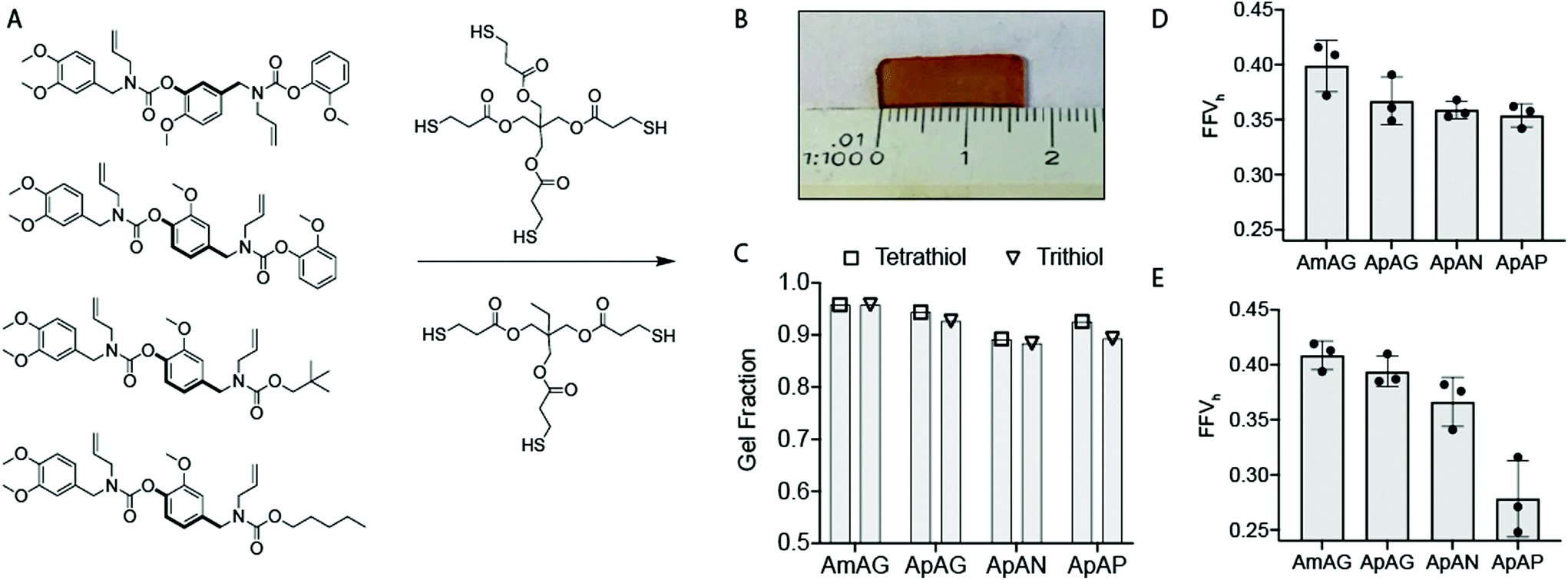 | ||
| Fig. 4 (A) Macromer cross-linking via a photo-initiated thiol–ene reaction with a tetrathiol and trithiol. (B) Image of crosslinked film (C) Gel fraction for each of the crosslinked networks after 60 minutes of irradiation. Fractional free volume of static holes (FFVh) of macromers crosslinked with the (D) tetrathiol, and (E) trithiol cross-linker. | ||
To examine the extent of cross-linking across all networks, the gel fractions of the networks were determined gravimetrically by subtracting the mass of the residual mixture from the total mass of the starting materials (not including the initiator). The gel fraction results in Fig. 4C indicate that the extent of cross-linking is similar (>90%) for all eight networks. Using the fluorescence signal of the SD-PUMs (ESI Fig. 13†), fluorescence measurements were acquired to measure the amount of residual SD-PUM in the recovered mixture by generating calibration curves for each macromer. The residual mixture gave no fluorescence emission, which suggests that all macromers were incorporated in their respective networks.
We also examined the initial rate of cross-linking across all networks using a model kinetics experiment with a monothiol such that –ene conversion could be measured via1H NMR. Thiol–ene reaction kinetics were monitored by reacting the SD-PUMs with 3-mercaptopropionic acid (3-MPA) under the same reaction conditions as the network cross-linking reaction. The reaction conversion was observed at various time points by monitoring the disappearance of the allyl olefin peaks at 5.9 and 5.2 ppm via1H NMR spectroscopy (ESI Fig. 14†), and the conversion was plotted as a function of time (ESI Fig. 15†). The kinetics data shows similar reaction kinetics across all SD-PUMs. This, along with the gel fraction data in Fig. 4C, indicates that the rates and extent of cross-linking are not affected by the SD-PUM structure. It follows then that any differences in the network properties can be attributed to monomer structure and composition.
The fractional free volume,25–30 an empirical parameter that characterizes static hole volume in polymers in the glassy state, was measured for all eight cross-linked networks at −10 °C. This static hole volume is the unoccupied space, in excess of the occupied volume (V0), that is created due to the orientation and arrangement of the molecules in the frozen network. Beyond Tg, molecular vibrations are large enough to create enough free volume, in excess of the static hole volume and V0, to allow for large-scale segmental motions of polymer chains, which allows the material to flow. In the glassy state, higher static hole volumes are associated with polymer networks that are unable to pack efficiently due to rigid or bulky groups with constrained centers or shapes. Here, the fractional free volume of static holes (FFVh), is the ratio of the estimated specific static hole volume (SSHV), also commonly referred to as the specific free volume (SFV), and the experimentally measured specific volume (V) (eqn (3)).
| SSHV = V − V0 | (1) |
| V0 = 1.3 × Vvdw | (2) |
| FFVh = SSHV/V = 1 − (1.3 × Vvdw)/V | (3) |
The SSHV was calculated using the Bondi group contribution method27 in eqn (1) and (2), where V0 is the occupied volume of the constituent moieties that comprise the network and Vvdw is the van der Waals volume, i.e., the volume occupied by the molecule that is impenetrable by other molecules. The coefficient of 1.3 represents the average packing density of polymers at 0 K and is an approximation adopted from Van Krevelen26 and Privalko31 due to the lack of data for these exact networks. The experimentally measured specific volume is the inverse of the density, which was obtained by measuring the mass and volume of the rectangular strips of films (3 replicates measured at −10 °C). The van der Waals volume of the network was estimated numerically using values from Park et al.30 as well as MarvinSketch (ESI Fig. 16 and 17†). The two estimates were similar, and thus volume estimates using MarvinSketch were used to calculate FFVh (Fig. 4D and E). One limitation of the FFVh parameter as calculated is that the packing density embedded within it does not take into account different spatial arrangements due to the regioisomeric SD-PUMs. The positron annihilation lifetime spectroscopy measurement (PALS)32–34 is a unique probe for characterizing the local hole volume in networks and could also be used to measure FFVh, though it has been reported that densely cross-linked networks such as epoxy networks tend to have small hole volumes that push the limit of the PALS method.35
The data Fig. 4D and E show the AmAG SD-PUM with the highest FFVh in both tetrathiol and trithiol cross-linked networks. In the tetrathiol networks, ApAG, ApAN, and ApAP gave similar FFVh values while the trithiol network showed ApAP with a significantly lower FFVh than its regioisomer, ApAN. As indicated earlier, high FFVh values are associated with polymer networks that are unable to pack efficiently due to rigid or bulky groups with constrained centers or shapes. This suggests that materials with high FFVh values have a high barrier to bond rotation, which is required for polymer unwinding and segmental motion of chains. Thus, our results suggest that the AmAG network is geometrically constrained in the glassy state relative to the other SD-PUM networks, and this should translate to higher Tg. Furthermore, our results imply that the ApAP trithiol linked network, given its much lower FFVh, should have a low Tg relative to the other SD-PUM networks.
Effect of end-group composition and regioisomerism on thermal and mechanical properties
The Tg of the cross-linked networks were evaluated by differential scanning calorimetry (DSC) (Fig. 5A and B). Overall, the data showed higher Tg's for networks made with the tetrathiol cross-linker (Fig. 5A) relative to those made with the trithiol cross-linker (Fig. 5B). We also see a general trend of decreasing Tg with increasing end-group flexibility as we go from AmAG and ApAG with rigid aromatic end-groups to ApAN and ApAP with flexible aliphatic end-groups. A predictable regioisomeric effect was observed; ApAN networks with the bulky neopentyl group had a higher Tg relative to the ApAP network composed of a linear and flexible pentyl end-group. This observation is consistent with the conclusions from the FFVh data. Furthermore, the data showed that the AmAG networks, made with both tetrathiol and trithiol, had the highest Tg values (Fig. 5A and B). Again, this observation is consistent with the conclusions from the FFVh data. In addition to DSC measurements, Tg was also determined from DMA studies via tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ plots (Fig. 6). Similar trends seen in the DSC data were also recorded in the tanδ plots. For the tetrathiol cross-linked networks, the Tg's for AmAG, ApAG, ApAN, and ApAP were 35.4 °C, 24.1 °C, 22.6 °C, and 6.1 °C, respectively. Similarly, the Tg's for the trithiol cross-linked networks were 22.1 °C, 21.3 °C, 11.2 °C, and −4.1 °C in the same order. Again, Tg decreases with increasing end-group flexibility. The difference in the Tg of the networks made with AmAG relative to ApAG could be due to the higher molecular rigidity or constrained shape of the AmAG macromer within the network. This effect is more pronounced in the tetrathiol cross-linked networks than in the trithiol networks. We posit that the additional connectivity in the tetrathiol amplifies the geometric constraints of the macromers in the networks.
δ plots (Fig. 6). Similar trends seen in the DSC data were also recorded in the tanδ plots. For the tetrathiol cross-linked networks, the Tg's for AmAG, ApAG, ApAN, and ApAP were 35.4 °C, 24.1 °C, 22.6 °C, and 6.1 °C, respectively. Similarly, the Tg's for the trithiol cross-linked networks were 22.1 °C, 21.3 °C, 11.2 °C, and −4.1 °C in the same order. Again, Tg decreases with increasing end-group flexibility. The difference in the Tg of the networks made with AmAG relative to ApAG could be due to the higher molecular rigidity or constrained shape of the AmAG macromer within the network. This effect is more pronounced in the tetrathiol cross-linked networks than in the trithiol networks. We posit that the additional connectivity in the tetrathiol amplifies the geometric constraints of the macromers in the networks.
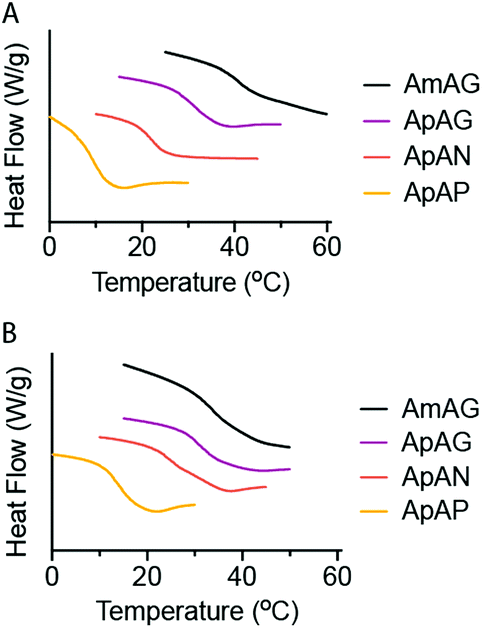 | ||
| Fig. 5 Differential scanning calorimetry of the (A) tetrathiol and, (B) trithiol cross-linked networks. | ||
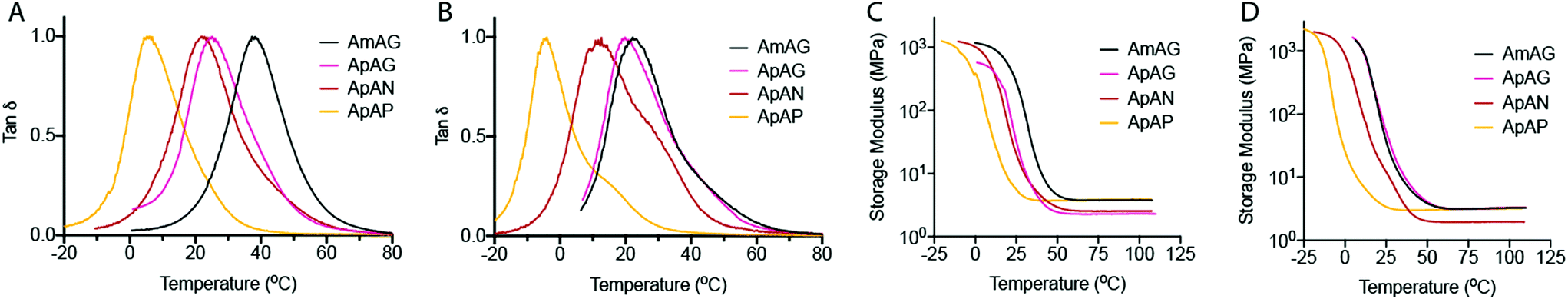 | ||
| Fig. 6 Thermal and mechanical properties of sequence defined networks (60 min. UV-cure). (A–D) Dynamic mechanical analysis of cross-linked networks. The tanδ curves of networks cross-linked with (A) tetrathiol and (B) trithiol. Plots of storage modulus (log scale) as a function of temperature for (C) tetrathiol cross-linked networks and (D) trithiol cross-linked networks. | ||
The storage modulus, determined via DMA measurements as a function of temperature, is shown in Fig. 6C and D. Above Tg, all networks had similar storage moduli values ranging between 2–4 MPa in the rubbery regime. The rubbery modulus for the ApAG tetrathiol cross-linked network was ∼45% lower than that of its backbone regioisomer, AmAG. However, the rubbery modulus of the ApAN network was ∼40% lower than its end group regioisomer, ApAP. The latter is observed with both the tetrathiol and trithiol cross-linked networks (Fig. 6C and D). At room temperature, the storage modulus largely follows similar trends to the Tg and viscosity measurements.
The FFVh values of the networks are correlated with the measured Tg. Networks with higher Tg values comprise chains that are composed of structurally “constrained” moieties, as observed in the viscosity data (Fig. 3). This molecular constraint results in the accumulation of larger static hole volumes in the glassy state, hence high FFVh. Polymeric networks that are unable to pack efficiently due to these molecular constraints, for the same reason, have a high barrier to bond rotation, which is required for polymer unwinding and segmental motion of chains. Thus, networks with poor packing efficiencies due to geometric constraints have a high FFVh and high Tg relative to others. Overall, our results, when paired with the gel fraction and kinetics data, support the conclusion that the observed differences in thermomechanical properties are related to the impact of SD-PUM structure (i.e. regioisomerism).
Conclusions
This study sought to investigate the effect of regioisomerism and composition on material properties via a recently developed scalable SD-PUM platform. This was achieved by designing two sets of regioisomeric SD-PUMs with different backbone and end-groups. Analysis of the synthesized SD-PUMs via NMR and viscosity measurements showed changes in physical properties owing to their structural differences. Following cross-linking with multivalent thiols, the thermomechanical properties of the networks were obtained experimentally. Here, we observed that the change in backbone connectivity influenced the Tg to a greater extent when cross-linking was performed with a tetrathiol relative to a trithiol. The difference in the thermomechanical properties was determined to be solely due to composition and structure by ruling out other factors such as the extent and rate of cross-linking. With the use of regioisomers, this study clearly illustrates how subtle changes in backbone and end-group structure significantly impacts network performance. We discovered that a subtle change from 1,3 to 1,4 connectivity leads to a decrease in viscosity as well as attenuated thermomechanical properties of the ensuing networks. This response in material properties to the subtle change in backbone connectivity could be used to alter the properties of commercial resins, e.g., tuning the mechanical, self-healing, and adhesive properties of phenylene diamine-based epoxy resins. Armed with the versatility of the SD-PUM platform, the next step beyond backbone conformation and terminal group composition is to combine these elements along with sequence to generate multifunctional macromers with multiple handles that can be used to further tune material properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Center for Sustainable Polymers under award number CHE1413862. This work made use of the Cornell University NMR Facility, which is supported, in part, by the NSF under award number CHE1531632. This work also made use of DSC and DMA in the Cornell Center for Materials Research (CCMR) Shared Facilities, which is supported by the NSF DMR award DMR-1719875. The authors thank Dr Ivan Keresztes and Anthony Condo for 2D NMR data collection and analysis. The authors also thank Dr Emily Hoff for her critical assessment of the manuscript.References
- Q. Guo, Thermosets: Structure, Properties, and Applications, Elsevier, 2nd edn, 2018 Search PubMed, Hardcover ISBN: 9780081010211, eBook ISBN: 9780081010280.
- A. Srinivasan and S. Bandyopadhyay, Advances in Polymer Materials and Technology, CRC Press, 1st edn, 2016 Search PubMed, ISBN 9781498718813.
- J. Pascault, H. Sautereau, J. Verdu and R. J. J. Williams, Thermosetting Polymers, CRC Press, 1st edn, 2002 Search PubMed, ISBN 9780824706708.
- H. Dodiuk and S. Goodman, Handbook of Thermoset Plastics, Elsevier, 2014, DOI:10.1016/C2011-0-09694-1.
- S. Thomas, J. Datta, J. Haponiuk and A. Reghunadhan, Polyurethane Polymers: Blends and Interpenetrating Polymer Networks, Elsevier, 2017 Search PubMed, Hardcover ISBN: 9780128040393, eBook ISBN: 9780128040850.
- K. B. Riad, R. Schmidt, A. A. Arnold, R. Wuthrich and P. M. Wood-Adams, Polymer, 2016, 104, 89–93 CrossRef.
- K. Dusek, Polym. Bull., 2007, 58, 321–338 CrossRef CAS.
- K. Dusek and M. Duskova-Smcrkova, Prog. Polym. Sci., 2000, 25(9), 1215–1260 CrossRef CAS.
- S. T. Knox, A. Wright, C. Cameron and J. P. A. Fairclough, Macromolecules, 2019, 52(18), 6861–6867 CrossRef CAS.
- J. Liu and I. Hatsuo, Macromolecules, 2014, 47(16), 5682–5690 CrossRef CAS.
- S. N. Kolanadiyal, M. Minami and T. Endo, Macromolecules, 2017, 50(9), 3476–3488 CrossRef.
- K. Zhang, Y. Liu, C. J. Evans and S. Yang, Macromol. Rapid Commun., 2020, 41(5), 1900625 CrossRef CAS.
- J. Liu, T. Agag and H. Ishida, Polymer, 2010, 51(24), 5688–5694 CrossRef CAS.
- A. A. Blevins and G. J. Blanchard, J. Phys. Chem. B, 2004, 108(16), 4962–4968 CrossRef CAS.
- P. Bortolus and S. Monti, J. Phys. Chem. A, 1979, 83(6), 648–652 CrossRef CAS.
- X. Q. Liu, M. Jikei and M. A. Kakimoto, Macromolecules, 2001, 34(10), 3146–3154 CrossRef CAS.
- M. Ghosh, Polyimides: fundamentals and applications, CRC Press, 1996 Search PubMed, ISBN 9780824794668.
- T. J. Dingemans, E. Mendes, J. J. Hinkley, E. S. Weiser and T. L. StClair, Macromolecules, 2008, 41(7), 2474–2483 CrossRef CAS.
- C. J. Campo, T. Anastasiou and K. E. Uhrich, Polym. Bull., 1999, 42, 61–68 CrossRef CAS.
- D. A. Boyles and J. T. Reams, J. Appl. Polym. Sci., 2011, 121(2), 756–63 CrossRef.
- Q. Wang, H. Xie, C. Yang, G. Zhong and H. Zhang, Liq. Cryst., 2010, 37(10), 1339–1346 CrossRef.
- J. C. Barnes, D. J. Ehrlich, A. X. Gao, F. A. Leibfarth, Y. Jiang, E. Zhou, T. F. Jamison and J. A. Johnson, Nat. Chem., 2015, 7(10), 810–815 CrossRef CAS.
- A. S. Knight, E. Y. Zhou, M. B. Francis and R. N. Zuckermann, Adv. Mater., 2015, 27(38), 5665–5691 CrossRef CAS.
- E. A. Hoff, G. X. De Hoe, C. M. Mulvaney, M. A. Hillmyer and C. A. Alabi, J. Am. Chem. Soc., 2020, 142(14), 6729–6736 CrossRef CAS.
- W. M. Lee, Polym. Eng. Sci., 1980, 20(1), 65–69 CrossRef CAS.
- D. W. van Krevelen Klaas te Nijenhuis, Properties of Polymers, Elsevier, 4th edn, 2009 Search PubMed, Hardcover ISBN: 9780080548197, eBook ISBN: 9780080915104.
- A. V. Bondi, J. Phys. Chem. A, 1964, 68(3), 441–451 CrossRef CAS.
- N. R. Horn, J. Membr. Sci., 2016, 518, 289–294 CrossRef CAS.
- R. Recio, A. E. Lozano, P. Prádanos, A. Marcos, F. Tejerina and A. Hernández, J. Appl. Polym. Sci., 2008, 107(2), 1039–1046 CrossRef CAS.
- J. Y. Park and D. R. Paul, J. Membr. Sci., 1997, 125(1), 23–39 CrossRef CAS.
- V. P. Privalko, Polym. J., 1975, 7(2), 202–206 CrossRef CAS.
- G. Dlubek, J. Pionteck, V. Bondarenko, G. Pompe, C. Taesler, K. Petters and R. Krause-Rehberg, Macromolecules, 2002, 35(16), 6313–6323 CrossRef CAS.
- Y. C. Jean, Microchem. J., 1990, 42(1), 72–102 CrossRef CAS.
- H. J. Zhang, S. Sellaiyan, K. Sako, A. Uedono, Y. Taniguchi and K. Hayashi, Polym. J., 2020, 190, 122225 Search PubMed.
- G. Dlubek, E. M. Hassan, R. Krause-Rehberg and J. Pionteck, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 73(3), 031803 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01578d |
| This journal is © The Royal Society of Chemistry 2021 |