
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ FTIR study of CO2 reduction on inorganic analogues of carbon monoxide dehydrogenase†
Ji-Eun
Lee
 a,
Akira
Yamaguchi
ab,
Hideshi
Ooka
a,
Tomohiro
Kazami
b,
Masahiro
Miyauchi
b,
Norio
Kitadai
cd and
Ryuhei
Nakamura
*ac
a,
Akira
Yamaguchi
ab,
Hideshi
Ooka
a,
Tomohiro
Kazami
b,
Masahiro
Miyauchi
b,
Norio
Kitadai
cd and
Ryuhei
Nakamura
*ac
aBiofunctional Catalyst Research Team, RIKEN Center for Sustainable Resource Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: ryuhei.nakamura@riken.jp
bDepartment of Materials Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8552, Japan
cEarth-Life Science Institute (ELSI), Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan
dSuper-cutting-edge Grand and Advanced Research (SUGAR) Program, Institute for Extra-cutting-edge Science and Technology Avant-garde Research (X-star), Japan Agency for Marine-Earth Science and Technology (JAMSTEC), 2-15 Natsushima-cho, Yokosuka 237-0061, Japan
First published on 2nd March 2021
Abstract
The CO2-to-CO reduction by carbon monoxide dehydrogenase (CODH) with a [NiFe4S4] cluster is considered to be the oldest pathway of biological carbon fixation and therefore may have been involved in the origin of life. Although previous studies have investigated CO2 reduction by Fe and Ni sulfides to identify the prebiotic origin of the [NiFe4S4] cluster, the reaction mechanism remains largely elusive. Herein, we applied in situ electrochemical ATR-FTIR spectroscopy to probe the reaction intermediates of greigite (Fe3S4) and violarite (FeNi2S4). Intermediate species assignable to surface-bound CO2 and formyl groups were found to be stabilized in the presence of Ni, lending insight into its role in enhancing the multistep CO2 reduction process.
Understanding how carbon dioxide (CO2) can be reduced to organic compounds is an important challenge, not only in terms of industrial applications, but also in terms of understanding the chemical processes underlying the biosphere. Of the six known pathways for biological carbon fixation, the Wood–Ljungdahl (W–L) pathway is arguably the simplest due to the absence of autocatalytic cycles and complex multi-carbon compounds.1 This simplicity, along with its phylogenetic diversity, has led previous studies to suggest the W–L pathway to be the most ancient form of biological carbon fixation which may have been present at the origin of life.2 Further, the simplicity of the W–L pathway implies that the underlying physicochemical concepts may be more readily applied towards artificial carbon capture and utilization compared to more complex pathways, such as the Calvin cycle, which is the most widespread carbon fixation pathway in the biosphere today.
Under anaerobic conditions, carbon fixation in the W–L pathway is initiated by the reduction of CO2 to CO by carbon monoxide dehydrogenase (CODH), which utilizes a highly conserved [NiFe4S4] cluster as the catalytic site (Scheme 1a).3 The generated CO can be combined with a methyl group (–CH3) to form a thioester, acetyl-CoA, which is a central metabolite of biological carbon metabolism (Scheme 1b).2,4 The first two-electron reduction of CO2 is thermodynamically uphill, and therefore, CODH affects the overall efficiency of carbon fixation.1a,5 Accordingly, although NiFe–CODH is highly sensitive to O2, it exhibits superb catalytic properties to generate CO at potentials near the thermodynamic equilibrium with nearly perfect selectivity.6 The origin of its high catalytic efficiency remains elusive, but it is likely attributable to the mechanisms by which CODH binds and activates CO2.3,7 Namely, previous crystallographic studies have shown that CODH interacts with the CO2 molecule in a multi-site conformation through both the Ni and Fe atoms within the [NiFe4S4] cluster (Scheme 1a), where a single Ni center associated with the iron–sulfur cluster specifically coordinates to the carbon atom of the CO2 molecule.3
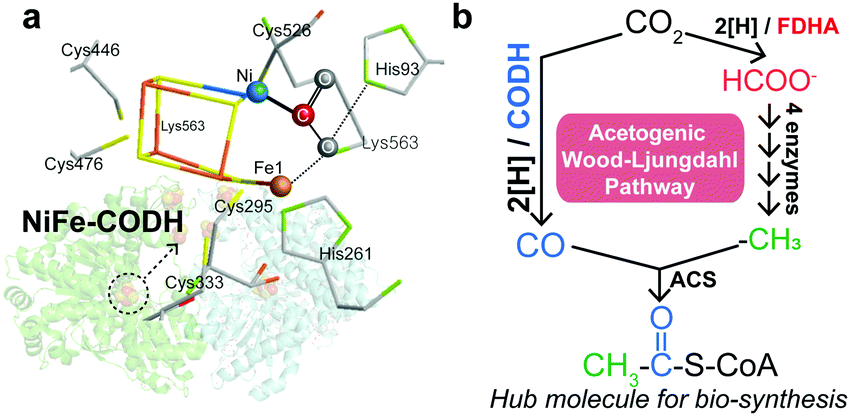 | ||
| Scheme 1 (a) The CO2 molecule interacts with the Ni, Fe, and histidine in the [NiFe4S4] cluster.3 (b) The acetogenic W–L pathway converts two molecules of CO2 into CO and formate, which can be combined to yield the acetyl group of acetyl-CoA.2b | ||
To identify the prebiotic origin of NiFe–CODH, several research groups have investigated the activity of Fe and Ni sulfides towards CO2 reduction under hydrothermal conditions.8 Recently, their electrocatalytic activity has attracted attention, due to their ability to catalyze the formation of C1 and multi-carbon compounds under conditions similar to deep-sea hydrothermal vents.9 However, the reaction mechanism remains largely unknown due to the lack of spectroscopic evidence particularly at the conditions where the reactions take place.10 Herein, we applied in situ electrochemical attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy to probe the reaction intermediates of NiFe sulfides during CO2 reduction. The formation of several surface-bound species was promoted in the presence of Ni, highlighting its possible role in enhancing the multistep CO2 reduction process.
As inorganic analogues of NiFe–CODH, greigite (Fe3S4) and violarite (FeNi2S4), which share the same crystal structure with the space symmetry group Fd3m (spinel), were synthesized following the method reported by Roldan et al. (ESI†).9d Ni-doped Fe sulfides were also synthesized on reduced-graphene oxide (rGO) nanosheets modified with polyaniline (PANI), as the amine groups may interact with NiFe sulfides in a similar way with the histidine coordination environment in the natural enzyme. Hereafter, these samples will be referred to as NiFeS–PANI. All peaks in the XRD patterns of the synthesized greigite and violarite were indexed to Fe3S4 and FeNi2S4, respectively, while the NiFeS–PANI was found to be a mixture of FeNi2S4, Fe3S4, and NiS2 (Fig. S1, ESI†). Energy-dispersive X-ray spectroscopy (EDX) imaging of NiFeS–PANI indicated that Ni, Fe, and S were uniformly distributed on the surface of polyaniline-coated rGO with a stoichiometric Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe ratio of 1:3 (Fig. S2–S4, ESI†). The electrostatic interaction of NiFeS with polyaniline was confirmed based on the shift of the IR bands of amine, benzenoid, and quinonoid rings upon the formation of the NiFeS nanoparticles (Fig. S5, ESI†). The interaction of the amine group of polyaniline with NiFeS was also indicated by X-ray photoelectron spectroscopy (XPS) analysis (Fig. S6 and S7, ESI†).
:Fe ratio of 1:3 (Fig. S2–S4, ESI†). The electrostatic interaction of NiFeS with polyaniline was confirmed based on the shift of the IR bands of amine, benzenoid, and quinonoid rings upon the formation of the NiFeS nanoparticles (Fig. S5, ESI†). The interaction of the amine group of polyaniline with NiFeS was also indicated by X-ray photoelectron spectroscopy (XPS) analysis (Fig. S6 and S7, ESI†).
The activity and selectivity of CO2 reduction on Fe3S4, FeNi2S4, and NiFeS–PANI was measured by performing electrolysis for 4 h at different potentials in CO2-saturated 0.1 M KHCO3 at 25 °C (Fig. 1). In this study, all potentials are referenced versus the reversible hydrogen electrode (RHE). The selectivity was evaluated based on faradaic efficiency (FE) measurements, which were reproducible across three independent sets of experiments (Fig. S8–S13, ESI†).
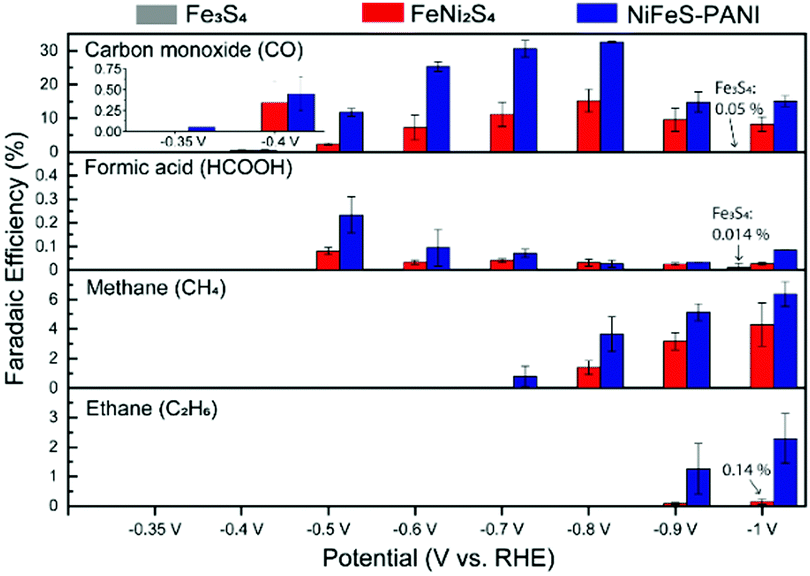 | ||
| Fig. 1 Performance and analysis of the CO2 electroreduction products for Fe3S4, FeNi2S4, and NiFeS–PANI nanostructures after 4 h of constant potential electrolysis. Calculated FEs are shown for the detected products at each potential. Error bars correspond to the standard error of the mean for at least three independent experiments. FE measurements for Fe3S4 were performed at −0.4, −0.7, and −1.0 V. | ||
In the case of Fe3S4, hydrogen was the dominant product, and essentially no CO2 reduction products were detected at all examined potentials (Fig. 1, grey bars: FEs for CO and HCOOH at −1.0 V are 0.05% and 0.014%, respectively). This finding is consistent with previous experiments showing the negligible activity of iron sulfides (FeS and Fe3S4) towards CO2 reduction.9a,b However, upon doping Ni into Fe3S4 (Fig. 1, red bars), CO was generated as a major product between −0.4 and −1.0 V, and further reduced products such as CH4 and C2H6 were also observed below −0.7 V. 13CO was detected when electrolysis was conducted in 13CO2-saturated 0.1 M KH12CO3, confirming that CO2 is the substrate for CO production (Fig. S14, ESI†). Introducing PANI into Ni-doped Fe sulfides further increased the CO2 reduction activity, in terms of both selectivity and overpotential. Namely, NiFeS–PANI reduced CO2 to CO from −0.35 V, which corresponds to an overpotential of approximately 250 mV (Fig. 1, blue bars). By applying a more negative potential, the FE for CO production increased to 30% at −0.8 V. The increase in selectivity may be due to the interaction of the PANI amine groups with CO2, as well as the enhanced hydrophobicity.9a,11 The partial current density of all products showed essentially the same potential dependence with the FE (Fig. S15–S17, ESI†). The time course of FEs and product concentrations are shown in Fig. S18 and S19 (ESI†). No liquid products other than formic acid were detected at a concentration higher than 0.1 mM in our experimental conditions.
To determine the mechanism by which Ni doping increases the efficiency of CO2 reduction on Fe sulfides, electrochemical ATR-FTIR spectroscopy was performed under in situ conditions with the catalysts coated on a single internal reflection prism (Ge) as the working electrode. The ATR-IR spectra were collected under the same conditions used for the aforementioned electrochemical CO2 reduction experiments (Fig. 1), except that H2O was replaced with D2O. The use of D2O enables a high signal-to-noise measurement in the spectral region from 1500 to 1700 cm−1, where CO2 related species such as adsorbed CO2 and HCO3−, exhibit vibrational bands.12 The ATR-FTIR spectra of Fe3S4, FeNi2S4, and NiFeS–PANI measured in CO2-saturated KDCO3 (0.1 M) are shown in Fig. 2. All three samples exhibited IR bands at 1363 cm−1 at potentials more negative than 0 V. This can be explained by the conversion of DCO3− to CO32−, considering that the pD at the electrode surface increases when cathodic reactions such as hydrogen evolution and/or CO2 reduction are occurring (pKa of CO32−/HCO3− is 10.5). The increase of local pD is consistent with the higher FE of CO relative to HCOOH at more negative potentials (Fig. 1).13 In control experiments using Ar-saturated KDCO3, no change in the IR bands of DCO3− and CO32− were observed (Fig. S20, ESI†).
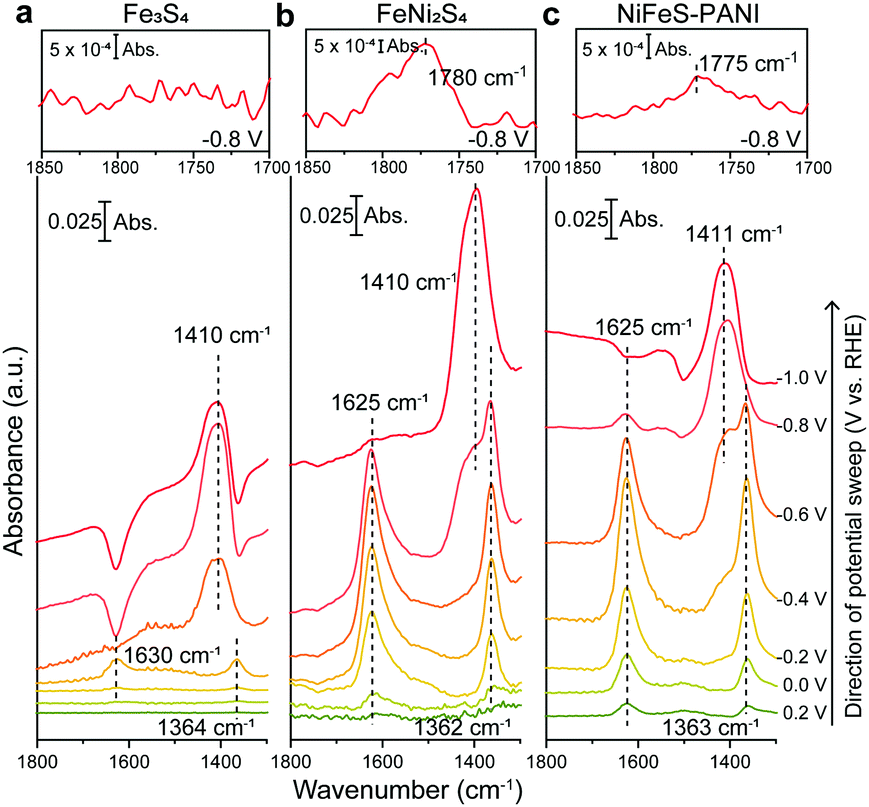 | ||
| Fig. 2 In situ ATR-FTIR spectra of (a) Fe3S4, (b) FeNi2S4, and (c) NiFeS–PANI were recorded during electrochemical CO2RR in CO2-saturated 0.1 M KDCO3. The insets in the upper panel of (a–c) show magnification of the 1700–1850 cm−1 region at −0.8 V. The spectra collected at +0.4 V vs. RHE were used as the reference. | ||
Upon further inspection of the IR spectra, a new band at 1625 cm−1 was observed upon the addition of Ni into Fe3S4 (Fig. 3a, black and red lines). Although this band was visible even in the case of Fe3S4 at −0.4 V, the intensity was an order of magnitude lower and also appeared across a smaller potential range. This band was also observed on NiFeS–PANI and exhibited a similar potential dependence (Fig. 3a, blue lines). Furthermore, exchanging 12CO2 with 13C-labeled CO2 resulted in a clear isotopic shift from 1625 to 1580 cm−1 on both FeNi2S4 and NiFeS–PANI (Fig. 3b and Fig. S21, ESI†). Therefore, the band at 1625 cm−1 is attributable to a reaction intermediate derived from the CO2 molecule.7a,12,14
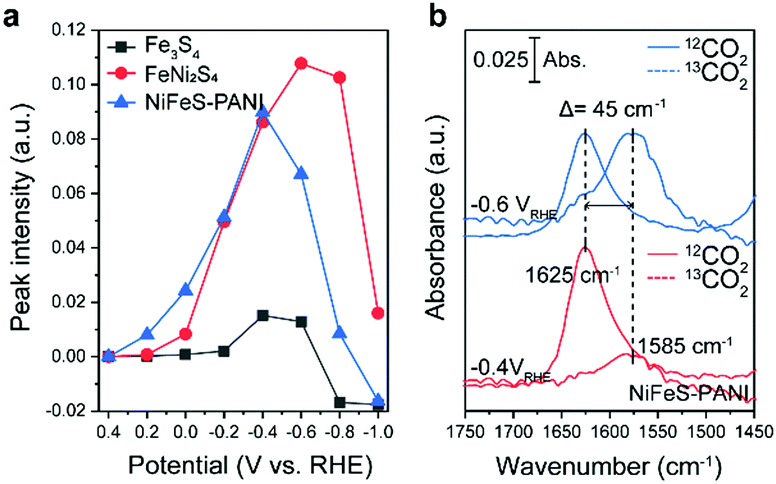 | ||
| Fig. 3 Analysis of the chemical species observed at 1625 cm−1. (a) The potential dependence of the absorption intensity at 1625 cm−1 relative to the baseline absorption value taken at 1700 cm−1. Adding Ni and PANI enhanced the formation of this species. (b) In situ ATR-FTIR spectra of NiFeS–PANI. An isotope shift of the species with 13C-labeled CO2 at 1625 cm−1 was observed at −0.4 and −0.6 V vs. RHE. | ||
The observed spectral position and isotopic shift value of 45 cm−1 are consistent with the asymmetric vibration of metal-associated CO2.7a,14,15 Considering that the intensity of the 1625 cm−1 band was largely increased upon Ni doping, a possible assignment to this species is adsorbed CO2 on the doped Ni site. Although bicarbonate also shows a vibrational band at a similar wavenumber,16 the intensity of the 1625 cm−1 band is independent of the bicarbonate bands at 1362 and 1410 cm−1, indicating that this band is not due to bicarbonate. The absence of the 1625 cm−1 band in the case of Fe3S4, despite clearly observable bicarbonate peaks, also supports that the 1625 cm−1 band is not due to bicarbonate.
In addition to the IR band at 1625 cm−1, another potential-dependent IR band was detected at 1780 cm−1 (inset of Fig. 2b and c). The formation of this band becomes evident at potentials more negative than −0.8 V and showed a clear isotopic shift to 1763 cm−1 upon exchanging 12CO2 with 13CO2 (Fig. S22, ESI†). This band is absent for Fe sulfide without Ni doping (inset of Fig. 2a). The observed band is located in the spectral region of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carbonyl, and close to that of surface-bound formyl, CHO, formed by electrochemical CO2 reduction.13,17 The possibility of assigning the band to the CO stretching of HCOOH can be ruled out because the deprotonated form is expected to become dominant under the reaction condition (pKa of HCOO−/HCOOH is 3.74).
O stretching of carbonyl, and close to that of surface-bound formyl, CHO, formed by electrochemical CO2 reduction.13,17 The possibility of assigning the band to the CO stretching of HCOOH can be ruled out because the deprotonated form is expected to become dominant under the reaction condition (pKa of HCOO−/HCOOH is 3.74).
Based on the electrochemical analysis and in situ ATR-FTIR observations, we propose a multistep reaction depicted in Fig. 4 as a possible mechanism for CO2 reduction on the surface of Ni-doped Fe sulfide. As seen from the evolution of the 1625 cm−1 band from +0.4 V, a surface-bound CO2 species forms on Ni-doped Fe sulfide prior to the initiation of CO2 reduction. The coverage of this species increases upon scanning the potential negatively, likely due to the enhanced nucleophilicity of the Ni site to coordinate to the carbon atom of CO2. The Fe site may act as an electrophilic center to interact with the oxygen atom of CO2.7a,18 The role of the Ni center to facilitate CO2 adsorption is consistent with recent DFT calculations by Posada-Pérez et al.,19 where the weak interaction of Fe3S4 with CO2 was attributed to the repulsion between the lone pair electrons of the oxygen atoms within the CO2 molecule and the spatially extended electronic clouds of the surface sulfur atoms.19 Posada-Pérez et al. also demonstrated that the partial substitution of Fe atoms by Ni strengthens the CO2 binding on Fe sulfides.19 Upon further scanning the potential negatively, the surface-bound CO2 species are reduced to CO, which is accompanied with the formation of a surface-bound formyl species (H–CO). This species has been proposed as an intermediate to facilitate the downstream reaction of CO to form CH4 and C2 compounds.13,17a,20 As a support for this hypothesis, when electrolysis was conducted in a CO saturated solution, CH4 and C2H6 productions were confirmed (Fig. S23, ESI†). Thus, the potential dependent formation of the carbonyl species, together with the concomitant production of CH4 and C2H6, suggests that surface-bound formyl species plays a role in facilitating the multi-electron reduction of CO on Ni-doped Fe sulfide.
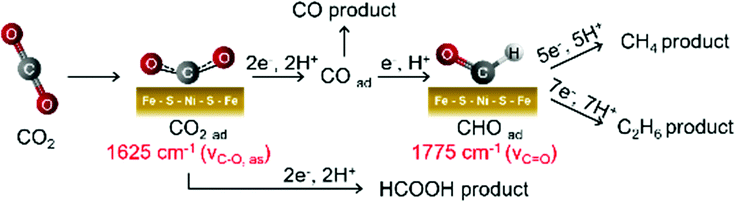 | ||
| Fig. 4 Proposed pathway for electrochemical CO2 reduction to CO, HCOOH, CH4, and C2H6 on Ni-doped Fe sulfide. | ||
In summary, we have identified the role of Ni during the electrochemical reduction of CO2 to CO by Fe sulfides. Ni enhances the formation of an intermediate assignable to surface-bound CO2, which leads to a substantial increase in the electrochemical selectivity. This intermediate is further reduced to surface-bound formyl species, which leads to products such as CH4 and C2H6. Recent studies have intensively argued the possibility that CO formation by NiFe-CODH is the oldest pathway of biological carbon fixation and was therefore involved in the origin of life.8a,b,9a–c,21 In this scenario, the reduction of CO2 to CO is proposed to proceed on the surfaces of metal sulfide minerals, either electrochemically15–17 using geochemically generated pH and temperature gradients as the driving force21,22 or hydrothermally, using H2 or metals as the electron source.8b Several Ni-containing Fe sulfides, such as violarite (FeNi2S4),9a pentlandite (Fe4.5Ni4.5S8),9e and awaruite (Ni3Fe),8b have been tested as possible prebiotic catalysts, but there is a lack of understanding of how CO2 can be converted to CO and multi-carbon compounds. The present study is the first to provide molecular level insight into the origin of the marked increase in CO2 reduction activity on iron sulfides by Ni doping. This will not only promote the development of biomimetic catalysts, but also yield a clue to identify prebiotic carbon fixation reactions.
J.-E. Lee acknowledges the support by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1A6A3A03012538). This work was partly supported by JSPS KAKENHI (grant number 18H05159) in scientific research on innovative areas “Innovations for Light Energy Conversion (I4LEC)” from MEXT, Japan.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. G. Peretó, A. M. Velasco, A. Becerra and A. Lazcano, Int. Microbiol., 1999, 2, 3 Search PubMed; (b) I. A. Berg, D. Kockelkorn, W. H. Ramos-Vera, R. F. Say, J. Zarzycki, M. Hügler, B. E. Alber and G. Fuchs, Nat. Rev. Microbiol., 2010, 8, 447 CrossRef CAS; (c) G. Fuchs, Annu. Rev. Microbiol., 2011, 65, 631 CrossRef CAS.
- (a) F. L. Sousa and W. F. Martin, Biochim. Biophys. Acta, 2014, 1837, 964 CrossRef CAS; (b) W. Nitschke and M. J. Russell, Philos. Trans. R. Soc. Lond., B, Biol. Sci., 2013, 368, 20120258 CrossRef.
- (a) J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461 CrossRef CAS; (b) J. Fesseler, J.-H. Jeoung and H. Dobbek, Angew. Chem., Int. Ed., 2015, 54, 8560 CrossRef CAS.
- P. S. Adam, G. Borrel and S. Gribaldo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E1166 CrossRef CAS.
- (a) S. W. Ragsdale, Crit. Rev. Biochem. Mol. Biol., 1991, 26, 261 CrossRef CAS; (b) A. Poehlein, S. Schmidt, A.-K. Kaster, M. Goenrich, J. Vollmers, A. Thürmer, J. Bertsch, K. Schuchmann, B. Voigt, M. Hecker, R. Daniel, R. K. Thauer, G. Gottschalk and V. Müller, PLoS One, 2012, 7, e33439 CrossRef.
- (a) W. Shin, S. H. Lee, J. W. Shin, S. P. Lee and Y. Kim, J. Am. Chem. Soc., 2003, 125, 14688 CrossRef CAS; (b) A. Parkin, J. Seravalli, K. A. Vincent, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2007, 129, 10328 CrossRef CAS.
- (a) C. Yoo, Y.-E. Kim and Y. Lee, Acc. Chem. Res., 2018, 51, 1144 CrossRef CAS; (b) J. B. Varley, H. A. Hansen, N. L. Ammitzbøll, L. C. Grabow, A. A. Peterson, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2013, 3, 2640 CrossRef CAS.
- (a) C. Huber and G. Wächtershäuser, Science, 1997, 276, 245 CrossRef CAS; (b) M. Preiner, K. Igarashi, K. B. Muchowska, M. Yu, S. J. Varma, K. Kleinermanns, M. K. Nobu, Y. Kamagata, H. Tüysüz, J. Moran and W. F. Martin, Nat. Ecol. Evol., 2020, 4, 534 CrossRef; (c) G. D. Cody, N. Z. Boctor, T. R. Filley, R. M. Hazen, J. H. Scott, A. Sharma and H. S. Yoder, Science, 2000, 289, 1337 CrossRef CAS.
- (a) A. Yamaguchi, M. Yamamoto, K. Takai, T. Ishii, K. Hashimoto and R. Nakamura, Electrochim. Acta, 2014, 141, 311 CrossRef CAS; (b) N. Kitadai, R. Nakamura, M. Yamamoto, K. Takai, Y. Li, A. Yamaguchi, A. Gilbert, Y. Ueno, N. Yoshida and Y. Oono, Sci. Adv., 2018, 4, eaao7265 CrossRef; (c) N. Kitadai, R. Nakamura, M. Yamamoto, K. Takai, N. Yoshida and Y. Oono, Sci. Adv., 2019, 5, eaav7848 CrossRef; (d) A. Roldan, N. Hollingsworth, A. Roffey, H. U. Islam, J. B. M. Goodall, C. R. A. Catlow, J. A. Darr, W. Bras, G. Sankar, K. B. Holt, G. Hogarth and N. H. de Leeuw, Chem. Commun., 2015, 51, 7501 RSC; (e) S. Piontek, K. J. Puring, D. Siegmund, M. Smialkowski, I. Sinev, D. Tetzlaff, B. Roldan Cuenya and U.-P. Apfel, Chem. Sci., 2019, 10, 1075 RSC; (f) K. Pellumbi, M. Smialkowski, D. Siegmund and U.-P. Apfel, Chem. – Eur. J., 2020, 26, 9938 CrossRef CAS.
- (a) K. R. Phillips, Y. Katayama, J. Hwang and Y. Shao-Horn, J. Phys. Chem. Lett., 2018, 9, 4407 CrossRef CAS; (b) Y. Deng, Y. Huang, D. Ren, A. D. Handoko, Z. W. Seh, P. Hirunsit and B. S. Yeo, ACS Appl. Mater. Interfaces, 2018, 10, 28572 CrossRef CAS.
- (a) H. Coskun, A. Aljabour, P. De Luna, D. Farka, T. Greunz, D. Stifter, M. Kus, X. Zheng, M. Liu, A. W. Hassel, W. Schöfberger, E. H. Sargent, N. S. Sariciftci and P. Stadler, Sci. Adv., 2017, 3, e1700686 CrossRef; (b) X. Wei, Z. Yin, K. Lyu, Z. Li, J. Gong, G. Wang, L. Xiao, J. Lu and L. Zhuang, ACS Catal., 2020, 10, 4103 CrossRef CAS.
- (a) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiely & Sons, Ins., 6th edn, 2008, ch. 1, p. 1 DOI:10.1002/9780470405888.ch1; (b) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiely & Sons, Ins., 6th edn, 2008, ch. 2, p. 275 DOI:10.1002/9780470405888.ch2.
- A. Wuttig, C. Liu, Q. Peng, M. Yaguchi, C. H. Hendon, K. Motobayashi, S. Ye, M. Osawa and Y. Surendranath, ACS Cent. Sci., 2016, 2, 522 CrossRef CAS.
- (a) C. Jegat, M. Fouassier, M. Tranquille, J. Mascetti, I. Tommasi, M. Aresta, F. Ingold and A. Dedieu, Inorg. Chem., 1993, 32, 1279 CrossRef CAS; (b) C. Jegat, M. Fouassier and J. Mascetti, Inorg. Chem., 1991, 30, 1521 CrossRef CAS.
- M. Aresta and C. F. Nobile, J. Chem. Soc., Dalton Trans., 1977, 708, 10.1039/DT9770000708.
- J. R. Bargar, J. D. Kubicki, R. Reitmeyer and J. A. Davis, Geochim. Cosmochim. Acta, 2005, 69, 1527 CrossRef CAS.
- (a) A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8 CrossRef CAS; (b) J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471 CrossRef CAS.
- C. Yoo and Y. Lee, Chem. Sci., 2017, 8, 600 RSC.
- S. Posada-Pérez, D. Santos-Carballal, U. Terranova, A. Roldan, F. Illas and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2018, 20, 20439 RSC.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251 CrossRef CAS.
- (a) H. Ooka, S. E. McGlynn and R. Nakamura, ChemElectroChem, 2019, 6, 1316 CrossRef CAS; (b) V. Sojo, A. Ohno, S. E. McGlynn, Y. M. A. Yamada and R. Nakamura, Life, 2019, 9, 16 CrossRef CAS; (c) M. J. Russell and W. Martin, Trends Biochem. Sci, 2004, 29, 358 CrossRef CAS.
- (a) R. Hudson, R. de Graaf, M. Strandoo Rodin, A. Ohno, N. Lane, S. E. McGlynn, Y. M. A. Yamada, R. Nakamura, L. M. Barge, D. Braun and V. Sojo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22873 CrossRef CAS; (b) R. Nakamura, T. Takashima, S. Kato, K. Takai, M. Yamamoto and K. Hashimoto, Angew. Chem., Int. Ed., 2010, 49, 7692 CrossRef CAS; (c) M. Yamamoto, R. Nakamura, T. Kasaya, H. Kumagai, K. Suzuki and K. Takai, Angew. Chem., Int. Ed., 2017, 56, 5725 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc07318k |
| This journal is © The Royal Society of Chemistry 2021 |