
User-defined, temporal presentation of bioactive molecules on hydrogel substrates using supramolecular coiled coil complexes†
M. Gregory
Grewal
 a,
Vincent P.
Gray
a,
Rachel A.
Letteri
a and
Christopher B.
Highley
*ab
a,
Vincent P.
Gray
a,
Rachel A.
Letteri
a and
Christopher B.
Highley
*ab
aDepartment of Chemical Engineering, University of Virginia, VA 22903, USA. E-mail: highley@virginia.edu
bDepartment of Biomedical Engineering, University of Virginia, VA 22903, USA
First published on 29th April 2021
Abstract
The ability to spatiotemporally control the presentation of relevant biomolecules in synthetic culture systems has gained significant attention as researchers strive to recapitulate the endogenous extracellular matrix (ECM) in vitro. With the biochemical composition of the ECM constantly in flux, the development of platforms that allow for user-defined control of bioactivity is desired. Here, we reversibly conjugate bioactive molecules to hydrogel-based substrates through supramolecular coiled coil complexes that form between complementary peptides. Our system employs a thiolated peptide for tethering to hydrogel surfaces (T-peptide) through a spatially-controlled photomediated click reaction. The complementary association peptide (A-peptide), containing the bioactive domain, forms a heterodimeric coiled coil complex with the T-peptide. Addition of a disruptor peptide (D-peptide) engineered specifically to target the A-peptide outcompetes the T-peptide for binding, and removes the A-peptide and the attached bioactive motif from the scaffold. We use this platform to demonstrate spatiotemporal control of biomolecule presentation within hydrogel systems in a repeatable process that can be extended to adhesive motifs for cell culture. NIH 3T3 fibroblasts seeded on hyaluronic acid hydrogels and polyethylene glycol-based fibrous substrates supramolecularly functionalized with an RGD motif demonstrated significant cell spreading over their nonfunctionalized counterparts. Upon displacement of the RGD motif, fibroblasts occupied less area and clustured on the substrates. Taken together, this platform enables facile user-defined incorporation and removal of biomolecules in a repeatable process for controlled presentation of bioactivity in engineered culture systems.
1. Introduction
The extracellular matrix (ECM) is intricate scaffolding that plays a central role in regulating cellular fates through multifaceted biophysical and biochemical processes.1–4 In efforts to recapitulate microenvironmental features of the ECM in vitro,5–7 the dynamic nature of the ECM must be considered, with the presentation of cell fate cues in flux during continual restructuring.4,8–10 To develop culture systems that influence cell migration, proliferation, and differentiation, approaches are needed to engineer the presentation of molecules involved in cell fate decisions.11–13 Hydrogel biomaterials are advantageous in vitro platforms as they can replicate tissue-specific mechanics and be modified with biomolecules through numerous established strategies.14–17The immobilization of biomolecules onto or within tissue culture substrates is important when engineering environments that mimic the ECM.18–21 One successful approach for incorporating bioactive molecules into scaffolds is photo-mediated thiol–ene click conjugation.22–26 Modifying hydrogel-forming polymers with norbornene groups enables spatial control over biomolecule presentation via photo-mediated thiol–ene click conjugation when used in conjunction with photomasks that selectively shield light.1,18,19,27 Controlling the localization of molecules on tissue culture scaffolds affords the ability to establish a spatial distribution of bioactive cues and gradients of signaling molecules to better recapitulate physiological environments and potentiate downstream cellular fates.22
While providing spatial control, a drawback of these covalent methods for conjugation of biomolecules to hydrogels is that the resulting materials do not capture the dynamic nature of in vivo cellular niches.11,12,28,29 Cells continually transduce signals provided by biochemical and biophysical cues in their microenvironment,30 and to achieve the dynamic characteristics of natural tissue in a biomaterial system, the ability to define the presentation of relevant signals, both spatially and temporally, is necessary.20 To this end, researchers have made significant strides developing techniques to dynamically introduce bioactive cues into hydrogel systems.31,32 For example, photo-mediated thiol–ene conjugation with subsequent photocleavage by means of o-nitrobenzyl-based ether linkers enabled reversible incorporation of bioactive compounds into hydrogel networks.11,28,33 Additionally, Grim et al. developed a method for repeatable biomolecule presentation via a reversible, light-mediated thiol–ene conjugation in conjunction with an engineered allyl-sulfide as a chain transfer agent.12,29 In a biologically inspired example, 3,4-dihydroxy-L-phenylalanine (DOPA), a catechol-containing amino acid present in mussels, facilitated reversible incorporation of biomolecules through dynamic-covalent esters formed between DOPA and phenylboronic acid.34 These methods demonstrate efficacy in reversible incorporation of biomolecules; however, they primarily leverage covalent bonds when immobilizing bioactive molecules – thus motivating exploration into reversibility driven by noncovalent interactions.
Supramolecular interactions offer approaches for dynamic incorporation of biomolecules into hydrogel scaffolds to capture the dynamic biochemical and biophysical features of cellular microenvironments.35–39 For example, host–guest pairs within hydrogels rapidly assemble, but can dissociate under externally applied forces.37,40,41 Boekhoven et al. achieved temporally controlled presentation of adhesive peptides within a hydrogel by appending the peptides to a naphthyl group for interaction with a β-cyclodextrin host immobilized to alginate.41 Subsequent addition of a bio-inert peptide attached to a higher affinity adamantane guest displaced the adhesive peptide and resulted in smaller 3T3 fibroblast cell areas.41 Additionally, oligonucleotides can be designed for reversible pairing through a process known as toehold-mediated strand displacement.42–44 Two complementary oligonucleotides pair, with one of the oligonucleotides designed with a longer ‘toehold’ region that can remain unpaired prior to introduction of a third, longer oligonucleotide designed to be fully complementary with the toehold-containing sequence. Adding the longer complementary strand displaces the shorter oligonucleotide due to the higher affinity interaction between the two longer oligonucleotides. This non-covalent interaction facilitates reversible and repeatable addition of biomolecules under short timescales through differences in association affinities on hydrogel scaffolds.42–44
We sought here to adopt concepts from each platform to develop a new method that affords reversible, dynamic incorporation of bioactive molecules into hydrogel networks with spatiotemporal control. We employ coiled coil-forming peptides that supramolecularly assemble in a specific manner in solution.45–47 Similar to toehold-mediated strand displacement with DNA, Gröger et al. showed coiled coil peptides can undergo a similar process.45 Introduction of a longer, higher affinity peptide to a lower affinity, toehold-containing coiled coil complex (dissociation constant, KD ∼10−8 M) displaced the shorter, lower affinity component and yielded a high affinity coiled coil (KD ∼10−9 M).45 These associations are similar in nature to other specific supramolecular assemblies, such as cyclodextrin-adamantane (KD ∼10−5 M)41 and cucurbituril host–guest systems (KD ∼10−11–10−12 M).36 We considered that the comparatively moderate affinities in the coiled coil system (KD ∼10−8–10−9 M)45 would allow for stable presentation of biomolecules over extended periods of time, with facile release potentiated via the addition of specific competitive molecules. Furthermore, while cyclodextrin and cucurbituril-based assemblies are reversible, the relatively straightforward synthesis and potential to reversibly trigger binding and release over multiple cycles under physiological conditions render coiled coil peptide platforms highly attractive for dynamic modulation of synthetic cellular microenvironments.
We hypothesized that coiled coil-forming peptides could be strategically designed to allow for both spatially-controlled conjugation via photo-mediated thiol–ene reactions and temporal control of biomolecule presentation via toehold-mediated strand displacement of coiled coil complexes. The ability to disrupt these associations and remove the biomolecules provides the desired constitutive “on/off” functionality – enabling facile reversible functionalization of in vitro culture systems. Herein, we describe the design, structural and thermodynamic characterization, and patterning of biomolecules using a coiled coil peptide-based system on both hyaluronic acid (HA) and fibrous polyethylene glycol (PEG) hydrogel surfaces. Using the patterned substrates, we demonstrate temporal attachment and release of biomolecules. To showcase the potential of this system in modulating bioactivity in engineered microenvironments, we build on previous work studying supramolecular assemblies in reversible modulation of cell adhesion and morphology in vitro.41,43 The reversible presentation of an adhesive sequence enables visual confirmation of changes occurring at the cellular level of in vitro models and may be of use in studies perturbing microenvironmental adhesion to ECM-derived peptide binding sequences to understand cell fate decisions.
Taken together, this coiled coil-forming peptide system represents a compelling platform for reversible, spatiotemporally controlled presentation of bioactive molecules. We note that this user-defined release process can be repeated over multiple cycles, lending itself to applications that require spatiotemporally controlled presentation of biomolecules that can be modulated through external cues as well as be reloaded for subsequent multi-stage release. In addition to the examples discussed here, this platform may be broadly applicable to understanding and controlling biomolecular composition in cellular microenvironments, for example to dynamically present growth factors and cytokines to modulate bioactivity in vitro.
2. Materials and methods
2.1. Synthesis of norbornene-functionalized hyaluronic acid (NorHA)
NorHA was synthesized as previously described.18 Briefly, sodium hyaluronate (HA, Lifecore, 62 kDa) was dissolved in deionized (DI) water with Dowex 50 W × 8 ion-exchange resin (3 g resin per 1 g HA) for 2 h, and subsequently filtered, titrated to pH 7.02–7.05 with tert-butylammonium hydroxide (TBA, FisherSci) to yield HA-tert-butylammonium salt (HA-TBA). The final product was frozen at −80 °C, lyophilized, and stored under nitrogen. HA-TBA was then dissolved in anhydrous dimethyl sulfoxide (DMSO) and allowed to react with benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate coupling reagent (BOP, Sigma, 0.3 mol equivalents relative to carboxylic acids on HA), and 5-norbornene-2-methylamine (nor-amine, Sigma, 1 mol equivalent relative to carboxylic acids on HA) to functionalize HA with norbornene groups. After ∼2 h, the reaction was quenched with cold DI water, and the solution was transferred to a membrane (molecular weight cutoff: 6–8 kDa) and dialyzed against DI water for 5 d. Precipitate was removed by filtration, and the solution was re-dialyzed against DI water for 5 d prior to freezing at −80 °C, lyophilizing, purging with nitrogen, and storing at −20 °C until ready for use. The degree of modification was determined to be ∼25% by 1H nuclear magnetic resonance spectroscopy (1H NMR, 500 MHz Varian Inova 500).2.2. Peptide synthesis
All peptides used in this study, unless otherwise stated, were synthesized using a Liberty Blue (CEM) automated, microwave-assisted solid phase peptide synthesizer via Fmoc methods. Briefly, Rink amide resin (Advanced Chemtech, Rink Resin SS, 100–200 mesh, 1% DVB) was swollen with dimethylformamide (DMF, Aldrich, ACS reagent grade), and the immobilized Fmoc group removed with 20% (v/v) piperidine in dimethylformamide. Fmoc-protected amino acids (Advanced ChemTech, 0.2 M in DMF, 5 equivalents relative to theoretical available sites on the resin) and the coupling agents diisopropylcarbodiimide (DIC, Aldrich, 99%, 1 M in DMF) and Oxyma Pure (Advanced ChemTech, 1 M in DMF) were added to the reaction vessel and heated to 90 °C for 4 min. The Fmoc deprotection and coupling steps were repeated to build the peptide from the C-terminus to the N-terminus. For fluorescent peptides, 5(6)-carboxyfluorescein (Sigma Aldrich, ≥95%) was added last onto the N-terminus. The resultant peptides were cleaved from the resin with a cocktail of 92.5% trifluoroacetic acid (TFA, Aldrich, 99%), 2.5% triisopropylsilane (TIPS, Aldrich, 99%), 2.5% 2,2′-(ethylenedioxy) diethanethiol (DODT, Aldrich, 95%), and 2.5% DI water, and then isolated by precipitation into cold diethyl ether (Aldrich, ACS reagent, contains butylated hydroxytoluene as inhibitor) and centrifugation. After removal of ether under vacuum, the peptides were resuspended in DI water, frozen in liquid nitrogen, lyophilized, and stored at −20 °C as powders until ready for use. High performance liquid chromatography (HPLC) was used to determine peptide purity; since we noted no appreciable byproduct species, the peptides were used without further purification (Fig. S4†). Peptide primary structure was confirmed via electrospray ionization mass spectrometry (ESI MS, Fig. S2, 3 and Table S1†). Secondary structures were determined by circular dichroism (CD) spectroscopy (Fig. S5†).2.3. Isothermal titration calorimetry
Experiments were performed using a standard volume affinity isothermal titration calorimeter (TA Instruments, New Castle, DE) with peptide solutions prepared in either 1× phosphate buffered saline (PBS) or NIH 3T3 fibroblast culture medium at indicated concentrations. Peptide solutions were adjusted to pH 7.4 using NaOH or HCl and then degassed for 10 min at 25 °C. Titrations consisted of an initial 2 μL injection, followed by 24 or 49 injections (10 μL each) of one peptide solution (150–200 μM) into 1.3 mL of a second peptide solution (10–20 μM). Following an initial delay of 200 s, injections were separated by 200 s. Experiments were performed at 25 °C with the stirring speed set to 125 rpm and the cooling rate set to medium. The reference cell was filled with 1.3 mL of degassed, deionized water. The thermograms were analyzed using NanoAnalyze software (TA Instruments) and heats of binding (in kJ mol−1) were obtained by integrating the area under each injection peak in the baseline-subtracted thermograms, then dividing by moles of each injected volume. When possible, the resultant curves were then fit to either the independent (one site) or multiple sites binding models to obtain KD values. Heats of dilution from blank injections – either peptide (150–200 μM) into 1× PBS/fibroblast medium or 1× PBS/fibroblast medium into peptide (10–20 μM) – were subtracted from experimental heats to yield the blank-corrected data.48 In all analyses, we neglected the heats from the initial 2 μL injection.2.4. Fabrication of NorHA hydrogels
Prior to formation of NorHA hydrogels, glass coverslips (22 × 22 mm) were functionalized with 3-(mercaptopropyl) trimethoxysilane (MTS, Sigma Aldrich, 95%) to present pendant sulfhydryl groups as follows. Briefly, glass coverslips were plasma treated (Harrick Plasma) for 3 min, and MTS was added dropwise to plasma treated surface prior to being baked at 100 °C for 1 h, and 120 °C for 10 min in an exhausted oven. The coverslips were washed sequentially in dichloromethane (DCM), 70% ethanol in water, and DI water, then stored under inert atmosphere until ready for use. NorHA hydrogels were synthesized from a solution consisting of 5% (w/v) NorHA, 1 mM lithium phenyl-2,4,6-trimethyl benzoylphosphinate (LAP) photoinitiator to induce thiyl radicals, and dithiothreitol (DTT) for crosslinking ([thiol]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[norbornene] = 0.6) in PBS. For each hydrogel, 50 μL of the NorHA solution was pipetted onto a thiol-functionalized glass coverslip (22 × 22 mm), sandwiched with an 18 × 18 mm coverslip, and crosslinked by irradiation for 2 min at 365 nm (10 mW cm−2, Omnicure) to covalently stabilize the gel network. NorHA hydrogels to be used in spatial patterning experiments were incubated in a 1% (w/v) bovine serum albumin (BSA) solution in PBS for 30 min to limit nonspecific binding prior to subsequent experiments; NorHA hydrogels to be uniformly patterned were incubated solely in PBS prior to use in experiments.
:[norbornene] = 0.6) in PBS. For each hydrogel, 50 μL of the NorHA solution was pipetted onto a thiol-functionalized glass coverslip (22 × 22 mm), sandwiched with an 18 × 18 mm coverslip, and crosslinked by irradiation for 2 min at 365 nm (10 mW cm−2, Omnicure) to covalently stabilize the gel network. NorHA hydrogels to be used in spatial patterning experiments were incubated in a 1% (w/v) bovine serum albumin (BSA) solution in PBS for 30 min to limit nonspecific binding prior to subsequent experiments; NorHA hydrogels to be uniformly patterned were incubated solely in PBS prior to use in experiments.
2.5. Preparation of norbornene-functionalized polyethylene glycol (PEG-NB) electrospun fibers
The electrospinning protocol was adapted from Sharma and coworkers24 and all fibers were collected on thiolated coverslips – identical to those used for preparing the 2D NorHA hydrogels. Solutions consisting of 8-arm PEG-NB (10% w/v, ∼20 kDa, JenKem Technology, USA), polyethylene oxide (5% w/v, ∼400 kDa, carrier polymer), DTT ([thiol]:[norbornene] = 0.6), and 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (I2959, 0.05% w/v) were mixed for at least 24 h in PBS. Electrospinning was conducted on a custom setup with the following parameters: 16-gauge needle; ∼15 cm between the needle and collection surface; 0.8 mL h−1 flow rate; 10–14 kV positive voltage applied to the needle; and 6 kV negative voltage applied to the collection surface. Fibers were collected for at least 10 min and crosslinked for 15 min (365 nm, 10 mW cm−2, Omnicure) under nitrogen. Fibers were then incubated in a 1% (w/v) BSA/PBS solution if they were to be spatially patterned or in PBS alone if they were to be uniformly patterned prior to subsequent experimentation.
2.6. Photoligation of peptides to hydrogels and fibers
NorHA hydrogels and PEG-NB fibers were fabricated with a 0.6 thiol:norbornene ratio to avail norbornene groups for photo-patterned attachment of thiolated peptides after crosslinking. For fluorescent-based experiments (Fig. 2 and 3), solutions of thiolated peptides (20 μM, T-peptide or thiolated fluorophore), BSA (1% w/v), and LAP (1 mM) in PBS were added dropwise to the surface of the hydrogels/fibers, covered with photomasks (CAD/Art Services), and irradiated (365 nm, 10 mW cm−2) for 2 min. For cell-based experiments (Fig. 4 and 5), a 100 μM solution of the T-peptide with 1 mM LAP in PBS was added dropwise to the surface of the hydrogels/fibers and irradiated with light (365 nm, 10 mW cm−2) for 2 min. Following radical-induced thiol–ene coupling of the peptides to the hydrogel/fiber surfaces, samples were washed 3× in PBS for at least 30 min per wash to remove unreacted peptide and stored at room temperature until further use. Hydrogels/fibers with covalently tethered fluorophores were imaged directly after the wash steps, while other samples were used in coiled coil experiments as described below.
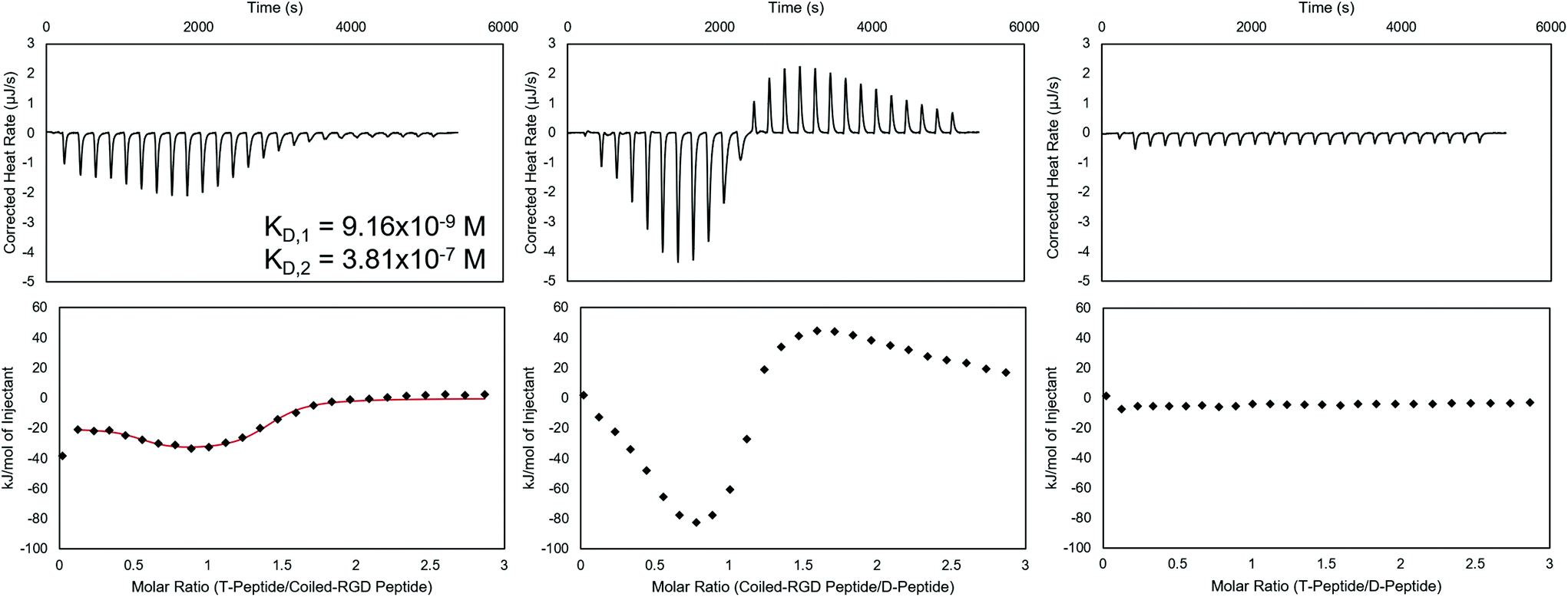 | ||
Fig. 1
Isothermal titration calorimetry of T-peptide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :coiled-RGD peptide, coiled-RGD peptide:D-peptide, and T-peptide:D-peptide interactions in NIH 3T3 fibroblast medium. (Top) Baseline-subtracted ITC thermograms, integrated to yield the heats of interaction in kJ mol−1 (bottom plots). If possible, the integrated plots were fit to models (shown as a red line) that provide parameters for the interaction in solution. The T-peptide:coiled-RGD peptide complex exhibits high affinities, as evidenced by the KD values on the order of 10−7–10−9 M. The larger exothermic heats of interaction measured for the coiled-RGD peptide:D-peptide complex as compared to the T-peptide:coiled-RGD peptide complex demonstrate the greater strength of these interactions. No appreciable heats of interaction were observed for non-complementary T-peptide:D-peptide pairs. :coiled-RGD peptide, coiled-RGD peptide:D-peptide, and T-peptide:D-peptide interactions in NIH 3T3 fibroblast medium. (Top) Baseline-subtracted ITC thermograms, integrated to yield the heats of interaction in kJ mol−1 (bottom plots). If possible, the integrated plots were fit to models (shown as a red line) that provide parameters for the interaction in solution. The T-peptide:coiled-RGD peptide complex exhibits high affinities, as evidenced by the KD values on the order of 10−7–10−9 M. The larger exothermic heats of interaction measured for the coiled-RGD peptide:D-peptide complex as compared to the T-peptide:coiled-RGD peptide complex demonstrate the greater strength of these interactions. No appreciable heats of interaction were observed for non-complementary T-peptide:D-peptide pairs. | ||
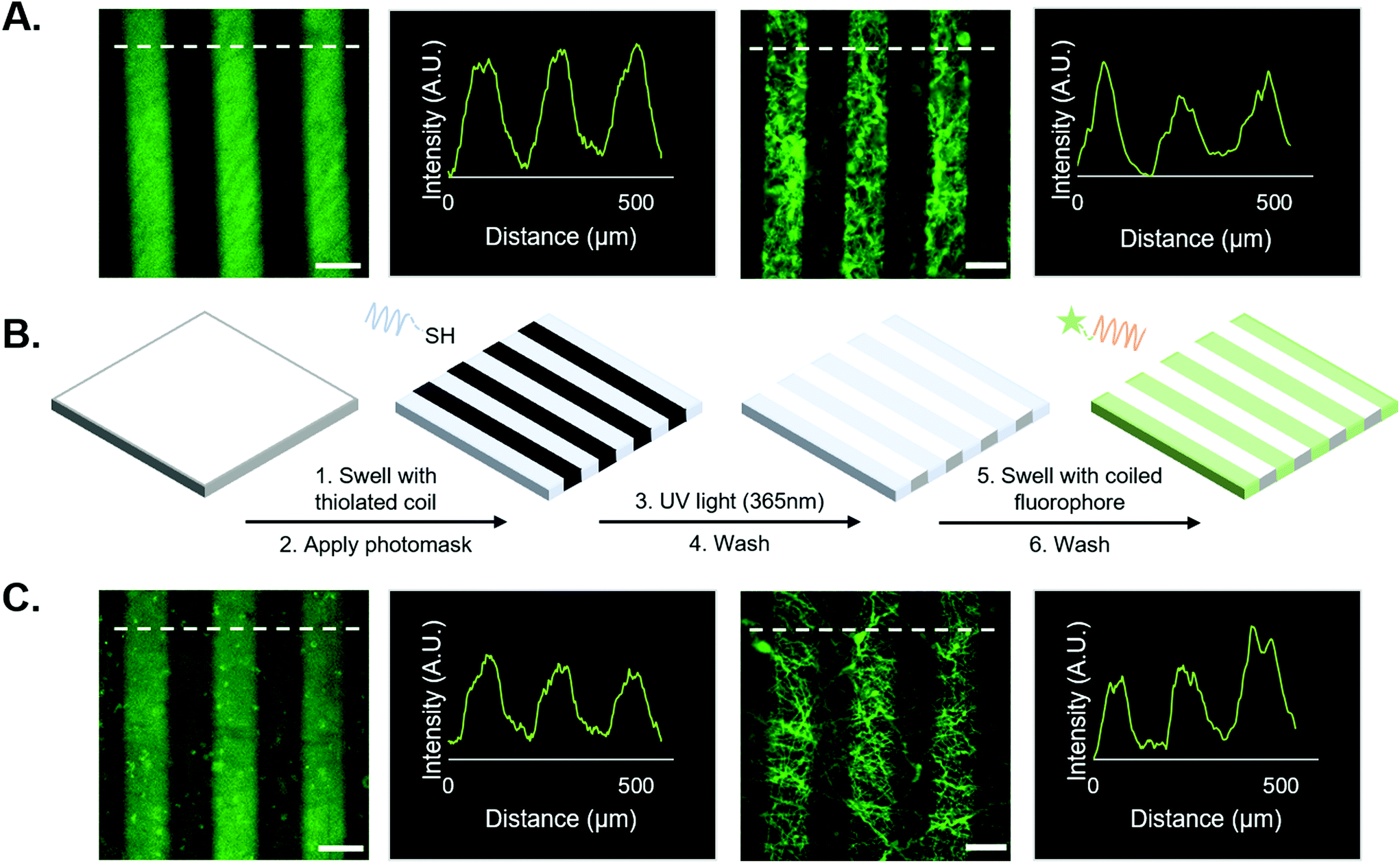 | ||
| Fig. 2 Supramolecular vs. covalent immobilization of FAM on NorHA hydrogels and PEG-NB fibers. (A) Covalent system (from left to right): representative micrographs of covalently photopatterned FAM on a NorHA hydrogel; representative intensity line profile; representative micrograph of covalently bound FAM on PEG-NB fibers; representative intensity profile. (B) Schematic of the supramolecular patterning process for FAM utilizing our coiled-coil system: swelling of substrate – either 2D hydrogel or fibers –with thiolated T-peptide, application of photomask and irradiation with 365 nm light; substrate is then washed and swelled with the complementary FAM-tagged A-peptide; finally, substrate is then washed again to remove unbound peptide. (C) Coiled system (from left to right): representative micrograph of FAM bound by coiled-coil system on a NorHA hydrogel; representative intensity profile; representative micrograph of FAM bound by coiled-coil system on PEG-NB fibers; representative intensity profile. Scale bars = 100 μm. Dashed white lines indicate sample location of intensity profiles plotted for each micrograph. | ||
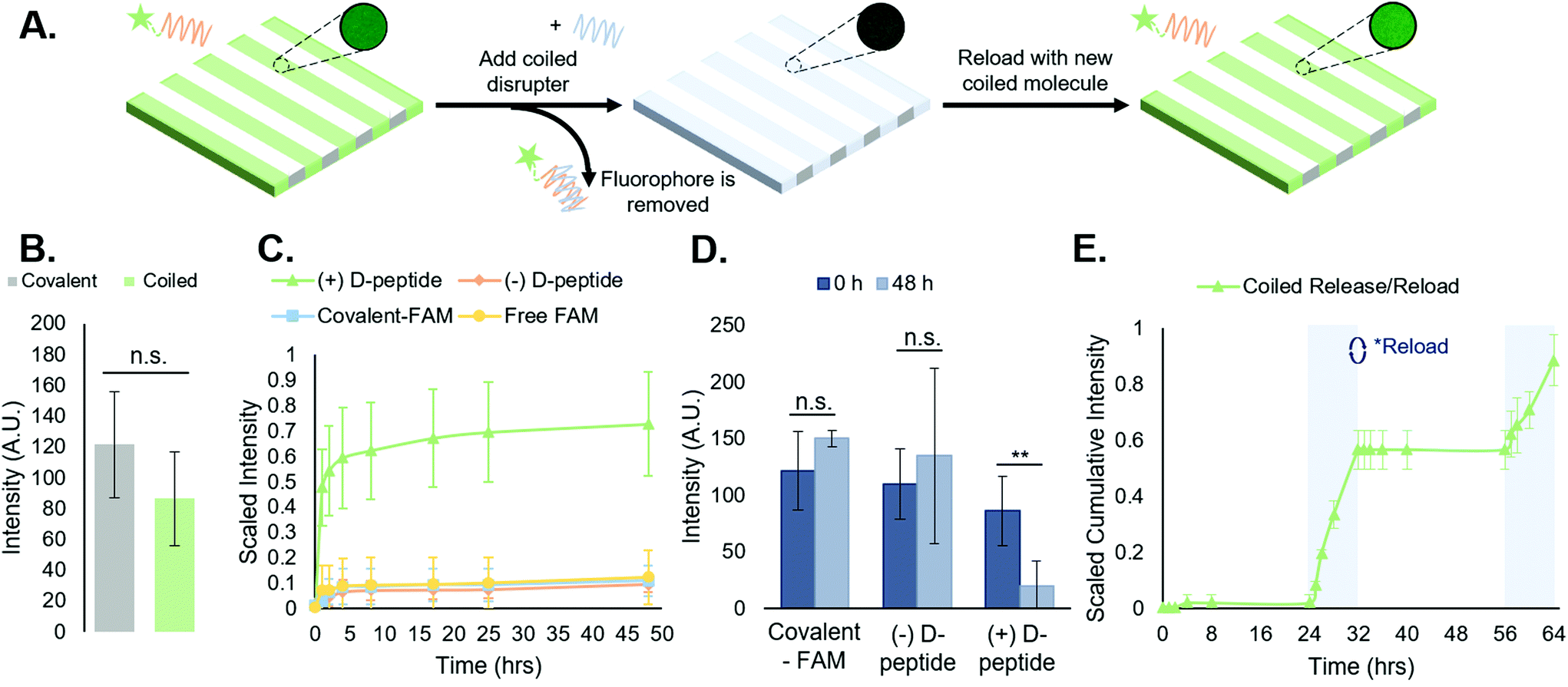 | ||
| Fig. 3 Reversible, repeatable addition of biomolecules to peptide-functionalized hydrogels. (A) Schematic of FAM-tagged A-peptide release from substrates via toehold-mediated strand displacement upon addition of the complementary D-peptide, with inset images showing intensity- and exposure-controlled resultant changes in stripe fluorescence, with the ability to reload the vacant T-peptide sites in order to repeat the process. (B) Quantitative comparison of the average intensities of FAM by covalent and coiled-coil conjugations indicates no significant difference in stripe intensity between both platforms. (C) Cumulative release curves of the coiled coil disruption compared to the various controls. This indicates a coiled coil complex forming that is stable, with reversibility being disrupter-dependent. (D) Average stripe intensity at 0 h and 48 h time points, based on fluorescent micrographs, indicating a statistically significant difference (**p < 0.01) stripe intensity at 48 h for only the ‘(+) D-peptide’ test group. (E) Repeated loading and release of FAM-tagged peptides from hydrogels shown by holding the hydrogels in PBS for 24 hours prior to inducing a D-peptide-dependent release for 8 h. Gels were then reloaded with A-peptide and the process was repeated. Profile shown is cumulative release from both disruption cycles. This further reinforces the notion that the system is stable over time in buffer and disruption can occur more than once via a release and reload protocol. Error bars represent standard deviation. | ||
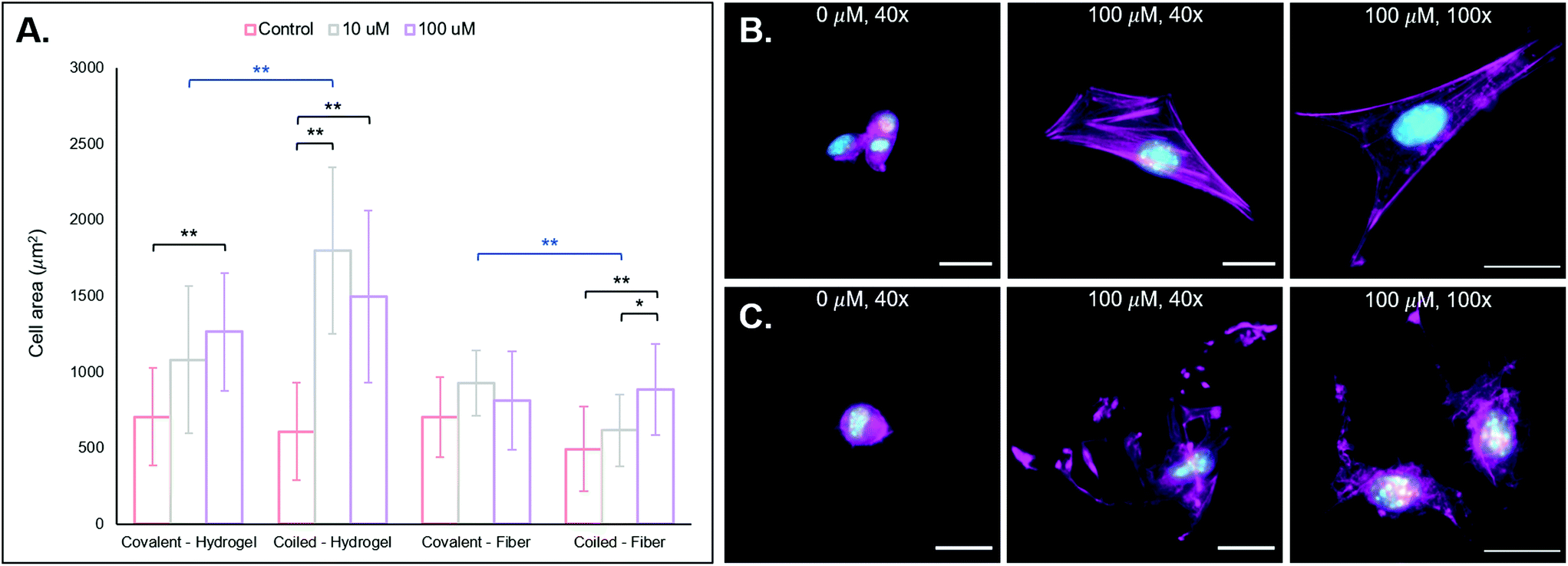 | ||
| Fig. 4 3T3 fibroblast behavior on substrates with presentation of RGD through the coiled-coil platform. (a) Quantifications of cell area for each of the groups with comparisons to their covalently conjugated analogs. (b) NorHA 2D hydrogel (from left to right): 0 μm RGD, 100 μm RGD, and 100 μm RGD hydrogel zoomed in to better visualize cell structure. (c) PEG-NB hydrogel fibers (from left to right): 0 μm RGD, 100 μm RGD, and 100 μm RGD fibers zoomed in to better view cell structure. It is clear qualitatively from the images, and quantitatively from the cell area comparisons, that generally the addition of the coiled RGD peptide to the substrates improves bioactivity, and thus cell area increases. Furthermore, it is important to note that the F-actin stress fibers are more organized for cells on NorHA scaffolds than those on PEG-NB fibers. Scale bars = 25 μm, **p < 0.01, *p < 0.05. Error bars represent standard deviation. | ||
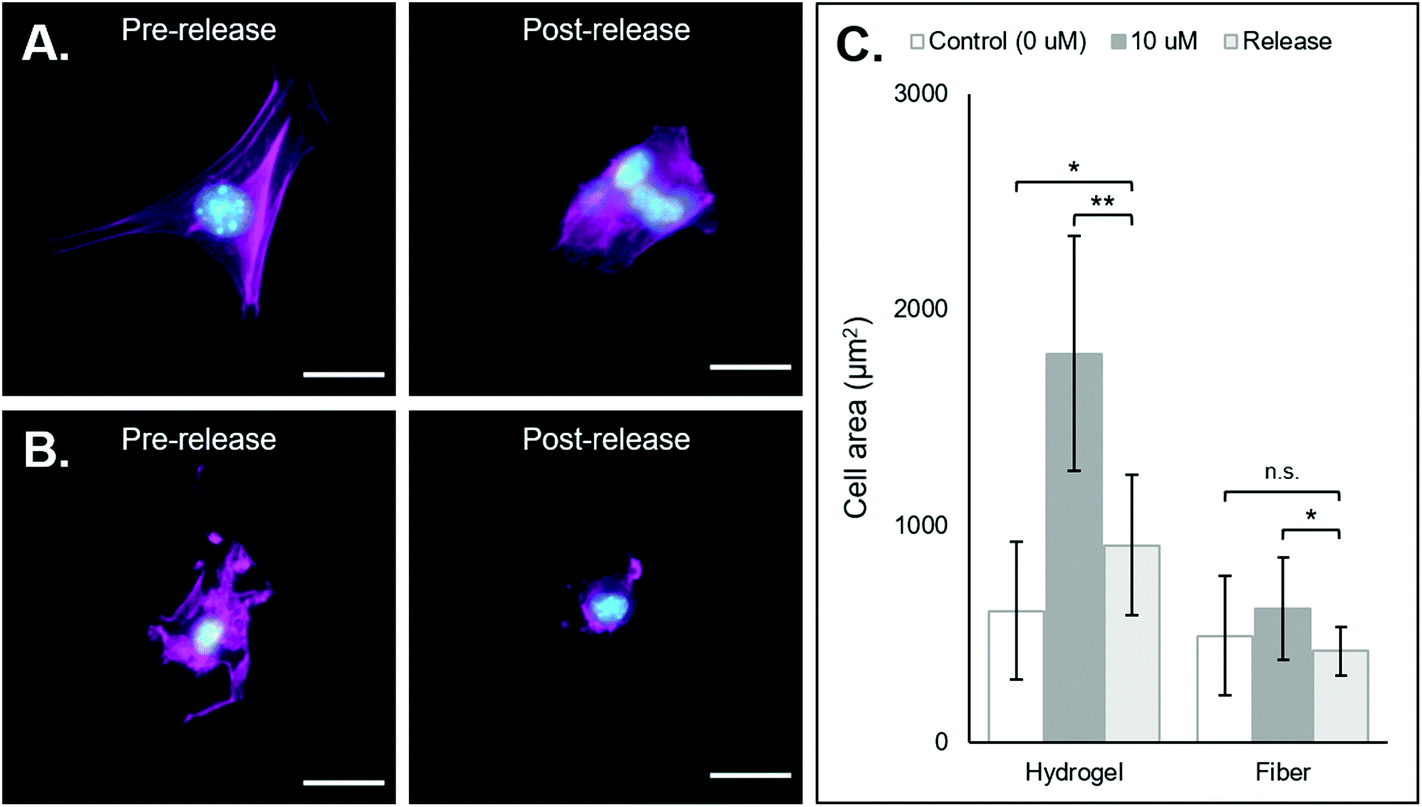 | ||
| Fig. 5 3T3 morphology changes following removal of coiled-RGD peptide via the addition of the competing D-peptide. (A) NorHA hydrogel (from left to right): representative fluorescent micrograph of a fibroblast seeded on a NorHA hydrogel functionalized with 10 μm of the coiled-RGD motif; representative fluorescent micrograph of fibroblasts on a NorHA hydrogel originally functionalized with 10 μm of the coiled-RGD motif and treated with 100 μm of the D-peptide. Cells exhibited fewer extensions and appeared to aggregate. (B) PEG-NB fibers (left to right): representative fluorescent micrograph of a fibroblast seeded on PEG-NB fibers functionalized with 10 μm of the coiled-RGD motif; representative fluorescent micrograph of a fibroblast on PEG-NB fibers originally functionalized with 10 μm of the coiled-RGD motif and treated with 100 μm of the D-peptide. Cells exhibited fewer extensions and covered less surface area. (C) Quantification of cell area across groups in release experimentation. Statistics solely compared cell area after treatment with D-peptide to the control and 10 μm coiled-RGD groups prior to treatment with D-peptide. Scale bars = 25 μm, n.s. = no significance, *p < 0.05, **p < 0.01, error bars represent standard deviation. | ||
2.7. Formation of coiled coil complexes and subsequent peptide release
To induce coiled coil peptide complex formation on NorHA hydrogels and PEG-NB fibers, the scaffolds with tethered T-peptide were swollen with a 20 μM solution of the complementary A-peptide (2 mL per well) for 3 min prior to washing 3× with PBS for at least 30 min per wash to remove unbound peptide. To release the A-peptide, D-peptide was introduced into the system (3 mL per well, 20 μM for fluorescence experiments) at multiple time points. During the disruption process, the higher affinity D-peptide binds A-peptides, disrupting the A-peptide:T-peptide coiled coil and removing the A-peptides from the surface. Solution (1 mL) was removed at predetermined timepoints, and remaining 2 mL were aspirated off and replaced with fresh D-peptide solution. Aliquots collected at each time point were stored at 4 °C until analysis.
To introduce an Arg-Gly-Asp (RGD) cell adhesion motif into hydrogels and fibers for cell culture, 100 μM of the T-peptide was tethered to the substrates by the radical-induced thiol–ene click reaction as described above. Subsequently, following the same protocol as above, solutions of either 0 μM, 10 μM, or 100 μM of a complementary A-peptide containing an RGD sequence (GYG![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif)
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) SPG(KIAALKE)4) were added to supramolecularly attach the adhesion motif to the surface. For disruption of this complex and removal of RGD from the system, 100 μM solutions of the D-peptide were used. For covalent RGD immobilization, a thiolated version of the RGD peptide (GCGYGSPG, Genscript) was added to the surface at the designated RGD concentrations for photo-mediated thiol–ene attachment.
SPG(KIAALKE)4) were added to supramolecularly attach the adhesion motif to the surface. For disruption of this complex and removal of RGD from the system, 100 μM solutions of the D-peptide were used. For covalent RGD immobilization, a thiolated version of the RGD peptide (GCGYGSPG, Genscript) was added to the surface at the designated RGD concentrations for photo-mediated thiol–ene attachment.
2.8. Determination of peptide release
Kinetics of peptide release from NorHA hydrogels were indirectly assessed using plate reader measurements of fluorophore intensity in the supernatant at time points during disruption. Briefly, the A-peptide was synthesized as described above with 5(6)-carboxyfluorescein (FAM) included on the N-terminus during peptide synthesis, and the fluorescence of the disruption solution at each time point was determined via a BioTek Synergy 4 fluorescence spectrophotometer (excitation: 495 nm; emission: 518 nm). Three hydrogels were assessed for each experimental group.Peptide release was further assessed visually using fluorescence microscopy (Leica DMi8 Widefield) during disruption. At each time point, fluorescent images (20×, dry) were taken of each NorHA hydrogel and the average intensity of photopatterned stripes was determined via ImageJ pixel intensity analysis. Three stripes per hydrogel were measured across three hydrogels for each experimental group.
2.9. Cell culture
NIH 3T3 fibroblasts (kindly provided by Dr Steven Caliari at the University of Virginia) were used for all cell experiments (passages 4–8). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) fortified with 10% (v/v) calf bovine serum (ATCC) and 1× antibiotic-antimycotic (Gibco). Prior to seeding cells on 2D NorHA hydrogels or PEG-NB fibers, hydrogels and fibers were sterilized with germicidal light for 2 h and swelled with culture medium for at least 30 min. Cells suspended in culture medium were seeded at a density of 5 × 104 cells per hydrogel or fiber sample and allowed 24 h to adhere to the surface. Cells were then fixed for subsequent analysis, as described below.For release experiments, hydrogel/fibrous scaffolds were similarly seeded at a density of 5 × 104 cells per scaffold, and cells were allowed to adhere for 24 h. Following the 24 h window, the medium was removed and replaced with culture medium containing D-peptide (2 mL, supplemented with 100 μM of the D-peptide) to induce release of the A-peptide. The D-peptide-containing medium was exchanged a total of two times, with exchanges at 1 h intervals, to facilitate displacement of coiled-RGD peptide. An incubation time of 1 h was allowed after the second treatment for a cumulative 3 h window. Following this release cycle, cells were fixed and treated for subsequent analysis.
2.10. Cell staining
For analysis of cell experiments, fibroblasts were fixed in a 10% (v/v) solution of neutral buffered formalin for 15 min before permeabilization with a 0.1% (v/v) Triton X-100/PBS solution for 10 min. Samples were then blocked by incubation in a 3% (w/v) BSA solution for at least 1 h to prevent nonspecific binding. F-actin was visualized by staining with Alexa Fluor-488-phalloidin (Thermofisher, 1:600 dilution) for at least 1 h and nuclei were visualized by staining with DAPI (ThermoFisher, 1:1 × 104 dilution) for 1 min. Samples were washed once with PBS, once with 0.1% (v/v) TWEEN-20 in PBS, and again in PBS after the staining steps to remove unbound fluorophore. All samples were protected from light and stored at 4 °C until imaging.
2.11. Imaging and image analysis
All imaging was conducted on a Leica DMi8 Widefield microscope. Coverslips with NorHA hydrogels or PEG-NB fibers were placed on microscope slides, sandwiched with a 25 × 25 mm coverslip, and inverted for imaging. Imaging settings (exposure time and light intensity) were held constant for all imaging where fluorescence intensities were compared across multiple samples. For imaging of hydrogels containing fluorescent peptides, three distinct photopatterned stripes per scaffold from three scaffolds were imaged for analysis. Images acquired with the 20× dry objective were used for intensity comparisons. To evaluate pattern fidelity, we plotted the normalized intensity line profiles across 3 stripes on each sample; all intensity profiles were normalized to the lowest intensity value corresponding to each representative image.For cellular experiments, at least three distinct areas per scaffold for three hydrogel and three fibrous scaffolds were imaged for cell spread area analyses. A 40× dry objective was used for cell area measurements, while a 100× oil immersion objective was used to visualize F-actin formation.
2.12. Statistical analyses
For quantitative comparisons between two experimental groups, independent t-tests were used; for comparisons with more than two experimental groups, a one-way ANOVA was leveraged in conjunction with a Tukey HSD post-hoc test with an α value of 0.95 indicating statistical significance.3. Results and discussion
Hydrogels are advantageous for use in synthetic ECM-mimetic materials due to their high water content – similar to natural tissue – and tailorability to specific applications.4,14 Moreover, many tissue-specific ECMs have fibrous components, and fibers are thus attractive in vitro models of physiological milieus.4,14,49,50 Towards introducing spatiotemporally controlled signals within these model environments, we investigated the ability of supramolecular coiled coil complexes to facilitate dynamic presentation of molecular adhesion motifs on or within both 2D NorHA hydrogels and fibrous PEG-NB hydrogels.Hyaluronic acid is a hydrophilic, non-sulfated glycosaminoglycan that is ubiquitous in natural ECM, and thus intrinsically biocompatible.16,17 PEG is a hydrophilic, biocompatible synthetic polymer used widely for biomedical applications, including for solubilization of therapeutics and as components of ECM-mimetic hydrogels.14,29 Both HA and PEG are amenable to chemical modification either on the side chains or at the chain ends.14 We installed norbornene moieties on both HA and PEG (NorHA and PEG-NB, respectively) to enable efficient, spatially controlled photo-mediated thiol–ene click reactions for addition of thiolated cross-linkers and biomolecules.18,24,51,52 The resulting NorHA hydrogels and PEG-NB fibers were crosslinked using dithiothreitol (DTT) as a crosslinker, adjusting the stoichiometry to leave residual norbornene groups available for post-crosslinking addition of thiolated peptides.1,18,24
Coiled coil-forming peptides were designed as shown in Scheme 1 based on a previously described complementary glutamic acid/lysine (E/K)-rich peptide pairs that form heterodimeric coiled coils45,47,53 and undergo toehold-mediated strand displacement.45 We sought to modify the sequences with cysteine residues to facilitate thiol–ene conjugation to NorHA and PEG-NB and demonstrate transfer of fluorophore or adhesive motif-tagged complementary peptides. For immobilization to the hydrogel surfaces, a tethered peptide (T-peptide) was designed with a cysteine for conjugation,54 a glycine spacer, and three repeating heptads of EIAALEK as the glutamic acid (E)-rich coiled coil-forming motif (I = isoleucine, A = alanine, L = leucine).45,47 The complementary association peptide (A-peptide) was designed with four repeating, lysine (K)-rich complementary KIAALKE heptads.45,47 The extra heptad repeat provides a toehold motif for triggered removal of the A peptide in the presence of the higher affinity disruptor peptide (D-peptide) having four complementary repeating E-rich EIAALEK heptads.45 We hypothesized that this difference in affinities would facilitate removal of A-peptides from the hydrogels by disrupting the A-peptide:T-peptide coiled coils upon introduction of the D-peptide in solution. We further extend this platform for dynamic incorporation of adhesive ligands (here, the fibronectin-derived RGD motif) for use in cell culture systems. To accomplish this, we modified the A-peptide with an RGD sequence (“coiled-RGD”) at the N-terminus.
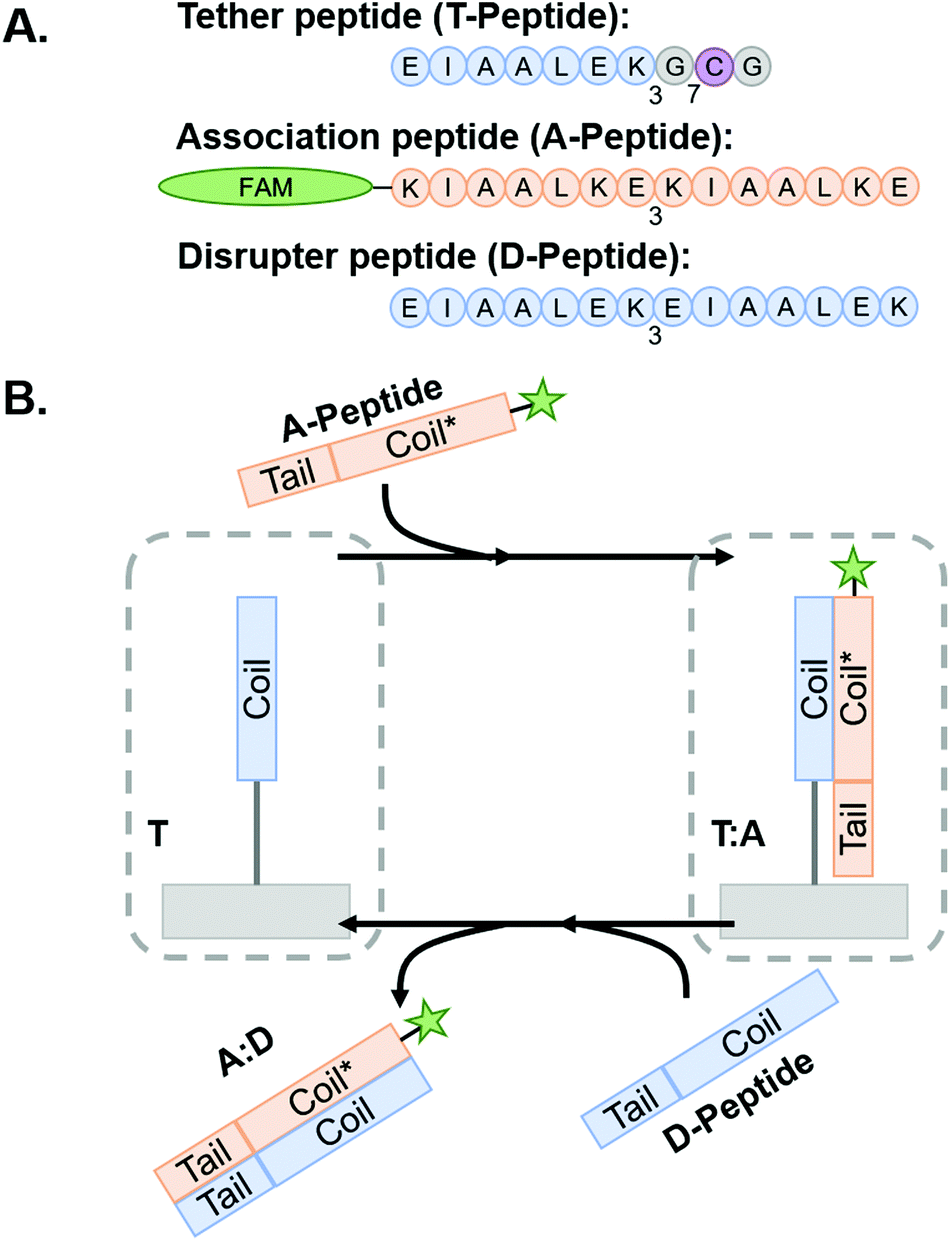 | ||
| Scheme 1
Coiled coil peptides and process schematic of peptide association and subsequent removal via toehold-mediated strand displacement. (A) Representative peptides used in this study. Blue regions indicate E-rich coiled coil-forming heptads, orange regions indicate complementary k-rich heptads, and yellow regions indicate the toehold motifs. (B) Tethered peptides are covalently conjugated to NorHA/PEG-NB surfaces prior to incubation with A-peptide to form T:A coiled-coil complex. The system is then incubated with D-peptide to interrupt the complex and form the A:D coiled-coil – thus removing the FAM-tagged A-peptide from the hydrogel and leaving behind a vacant T-peptide. Figure inspired by Gröger et al.45 | ||
3.1. Thermodynamic characterization of coiled coil peptide interactions using isothermal titration calorimetry (ITC)
Prior to applying these peptides to NorHA hydrogels and PEG-NB fibers for reversible biomolecule attachment, their interactions when forming complexes were characterized using ITC. ITC is capable of assessing thermodynamic properties of associations in solution.48,55 The coiled coil forming peptide pairs shown in Scheme 1 were analyzed in either NIH 3T3 fibroblast medium, PBS, or both, and representative baseline-subtracted thermograms and integrated data are shown in Fig. 1 for the complexes used in the cell culture studies – namely T-peptide:coiled-RGD peptide and coiled-RGD peptide:D-peptide, as well as a control experiment showing no interactions between non-complementary T-peptide and D-peptide pairs. Other replicates in medium, as well as PBS trials, are included in the ESI (Fig. S6–8† for medium and Fig. S9–13† for PBS).
Analysis of the ITC data in culture medium indicates that the T-peptide:coiled-RGD peptide forms two distinct, independent sites of interaction with strong affinities (KD,1 ∼10−7–10−9 M, KD,2 ∼10−6–10−7 M, represented in Fig. 1). This result is consistent with two-stage binding processes reported for coiled coils formed from peptides with mismatched lengths.45,56 Conversely, ITC of the same two peptides without the RGD moiety (i.e., T-peptide:A-peptide association) showed only one binding site (KD ∼10−6, Fig. S9†). Therefore, the multiple binding sites are likely encouraged due to the presence of RGD causing a greater mismatch in peptide lengths. As noted by the KD values, both sites of the T-peptide:coiled-RGD peptide complex exhibit strong binding affinities, which is advantageous for stable presentation of biomolecules.
Interestingly, the coiled-RGD peptide:D-peptide trace also seems to exhibit two-stage binding; however, the presence of both exothermic and endothermic heats of interaction prevents a model from fitting the data. This two-stage binding is intriguing as the coiled-RGD peptide and D-peptide both contain 4 coiled coil-forming heptad repeats. Therefore, the additional RGD residues yield mismatched lengths which may explain the multi-stage model. Moreover, these endothermic peaks may be indicative of higher order structures forming in solution, as has been reported for peptides that undergo self-complementary assembly.57 Nevertheless, the larger magnitude of the heats of interaction from the coiled-RGD peptide:D-peptide interaction (∼−80 kJ mol−1, Fig. 1) compared to those of the T-peptide:coiled-RGD peptide (∼−30 kJ mol−1, Fig. 1) indicate that the coiled-RGD peptide:D-peptide complex is thermodynamically favored over the T-peptide:coiled-RGD peptide complex. Therefore, we conclude that the coiled-RGD peptide will preferentially interact with the D-peptide in the presence of the T-peptide – facilitating reversibility in our system.
The T-peptide:D-peptide analysis indicates no discernible interactions between the two peptides in solution, with heats of interaction of essentially 0 kJ mol−1 after correcting for the heats of dilution of T peptide into media and media into D-peptide. These results indicate that, as expected, the D-peptide does not interact with the T-peptide, and the D-peptide should displace the coiled-RGD peptide from the T-peptide:coiled-RGD peptide coiled coil due to the differences in their strengths of interaction – comparable to the results presented by Gröger et al.45 using similar peptides to form coiled coils in solution.
These ITC experiments were also conducted in the presence of PBS to investigate how the absence of serum affects peptide complex affinities. The resultant ITC thermograms and integrated analyses (Fig. S9–13†) indicate only marginally different heats of interaction when PBS is used rather than cell culture medium.
3.2. Spatial patterning of FAM-tagged peptides onto NorHA hydrogels and PEG-NB hydrogel fibers
First, we tested the hypothesis that supramolecular coiled coil-mediated immobilization would yield similar patterns as covalent immobilization of fluorophores onto hydrogels. In these experiments, 100 μm-wide stripes were generated using a photomask and standard lithographic techniques.1,10,18 Covalently bound fluorophores were introduced by patterning a thiolated FAM onto NorHA/PEG-NB surfaces (see Fig. 2A). For supramolecular patterning, unlabeled, thiolated T-peptide was first photopatterned onto NorHA and PEG-NB substrates, followed by incubation with a solution of the FAM-tagged complementary A-peptide. NorHA and PEG-NB scaffolds were washed with PBS to remove any unbound fluorophore prior to imaging (refer to Fig. 2B for a schematic of the process, and Fig. 2C for representative images). The resulting micrographs (Fig. 2) demonstrate that the NorHA and PEG-NB systems can be modified with fluorophore to generate stripe patterns either by covalent or supramolecular methods. The covalent systems typically yielded more uniform intensity profiles with higher peaks when compared to their coiled coil counterparts (Fig. 2A and C). Qualitatively, this can be visualized by the non-patterned regions of the coiled coil systems exhibiting more fluorescent signal than the covalent non-patterned regions. It is possible that steric hindrance near the surface limited uniform conjugation of FAM in the coiled coil system and/or that non-specific interactions between the hydrogel and peptides were greater when using coiled structures compared to the shorter peptides used in covalent conjugation.Prior to the release studies, we compared the stripe intensities of the covalent and coiled coil systems to determine the system's net intensity under identical light conditions – analogous to a ‘loading capacity’ of prospective biomolecules. Analysis of stripe intensities from fluorescent micrographs indicated that although the average intensity of the coiled coil system was marginally lower than the covalent system, an independent t-test suggests there is no statistical significance (Fig. 3B). Therefore, our platform for supramolecular addition of biomolecules generates patterned stripes similarly to its covalent ligation analog at the experimental (20 μM) concentrations, allowing equivalent conjugation of fluorophore based on our semi-quantitative analysis.
3.3. Supramolecular immobilization of FAM via coiled coil peptide complexes is reversible and repeatable
Following experiments demonstrating spatially controlled supramolecular addition of FAM-tagged peptides to hydrogels using the coiled coil system, we sought to evaluate its efficacy as a reversible, repeatable process towards temporally controlled presentation of biomolecules on and in cell culture substrates.We measured FAM concentration in solution as a function of time to determine the stability of the supramolecular coiled coil structure in buffer over time and monitor the toehold-mediated disruption of the complexes by adding the disrupter peptide (Fig. 3A). All release studies were performed on NorHA hydrogels due to the ability to precisely and reproducibly form substrates with consistent surface area across samples, enabling a closer comparison across all groups. First, we examined the release of the FAM-tagged A-peptide in the presence of the D-peptide (hereafter “(+) D-peptide”). Plate reader fluorescence measurements of supernatant samples indicated a large cumulative release in the (+) D-peptide test group, suggesting that release is dependent on the addition of the D-peptide to disrupt the T:A coiled coil. To support this conclusion, we compared the (+) D-peptide test group to various controls. These controls consisted of a coiled coil system that was not subjected to D-peptide treatment (hereafter “(−) D-peptide”), a covalently bound FAM (hereafter “covalent-FAM”), and the FAM molecule in solution (“free-FAM”). The (−) D-peptide group was designed to evaluate supramolecular complex stability in buffer for extended periods of time (∼48 h), and the free-FAM control was designed to determine the extent of non-specific interactions between the fluorophore in solution with the T-peptide-modified substrates. Using standard plate reader fluorescence measurements of supernatant samples, we measured FAM release profiles. The results indicated large cumulative release from the coiled coil group in comparison to controls that exhibited minimal, if any, release of fluorescent molecules (Fig. 3C). Furthermore, the supramolecular coiled coils persist over days in the absence of the D-peptide, indicating that this system can stably, but reversibly, incorporate biomolecules onto norbornene-modified hydrogels. The free-FAM control contained no stripes to quantify in subsequent analysis, suggesting no discernable interactions between the free molecule and the T-peptide surface.
To further quantify the release of the FAM-tagged A-peptide via addition of the disrupter, we looked at the average stripe intensities of the ‘(+) D-peptide’ test group compared to the (−) D-peptide and ‘covalent-FAM’ controls at the first and last time points to compare the decrease in intensity upon FAM release. There were no significant differences between the stripe intensities at the first and last timepoints (0 h and 48 h) for the controls (Fig. 3D). However, the (+) D-peptide test group showed a statistically significant reduction in intensity after D-peptide treatment (**p < 0.01). Together, these data confirm the hypotheses that: (1) coiled coil peptides enable supramolecular immobilization on hydrogels; and (2) introduction of the D-peptide facilitates removal of supramolecularly immobilized molecules by toehold-mediated strand displacement.
We further investigated how different concentrations of D-peptide affected removal of the FAM-tagged A-peptide (Fig. S14†). Increasing concentrations of D-peptide generally enhanced and accelerated removal of the FAM-tagged A-peptide from the hydrogel surfaces; however, we began to observe diminishing increases in removal at higher concentrations of D-peptide. We postulate that at these higher concentrations, kinetics of peptide displacement might limit reversibility in the system over availability of D-peptide, with steric hindrances imposed by NorHA surfaces possibly limiting reversibility as well. Thus, increasing the concentration of D-peptide has the most significant effects on increasing A-peptide removal at lower concentrations of D-peptide, with only marginal improvement at higher concentrations of the competitive disrupter. Additionally, removal of A-peptide from T:A coiled coils in the presence of 3T3 fibroblast medium was investigated as a precedent for subsequent cell studies. Shown in Fig. S14,† removal in culture medium is similar to removal in PBS. Combining this observation with ITC measurements (Fig. S6–8†), we conclude that the specific and dynamic properties of the coiled-coil system extend to protein-rich environments, such as cell media.
Toehold-mediated removal of the A-peptide by addition of the complementary, higher affinity D-peptide leaves T-peptide sites vacant on the surface of NorHA hydrogels for binding other bioactive molecules. As a proof-of-concept, after release of the initially bound FAM-tagged A-peptide, we incubated the same scaffolds with a fresh solution of FAM-tagged A-peptide and rinsed the substrates with PBS to remove unbound peptide. After a 24 h incubation in PBS, D-peptide was reintroduced to cue a second removal of FAM-tagged peptide to demonstrate the repeatability of the process (Fig. 3E). The second release profile (t = 56–64 h) resembled the first (t = 24–32 h), albeit with a slightly smaller magnitude of cumulative release. These results suggest that this is a reversible and repeatable process, but we can also postulate that the D-peptide does not completely remove the A-peptide (Fig. 3D, where the ‘( + ) D-peptide’ group does not return to a 0-intensity value following disruption). This may be due to interactions with the NorHA surface that sterically interfere with removal, interactions with material in the gels that prevent reaching 100% removal, or equilibrium of toehold-mediated strand displacement.
3.4. Supramolecular, coiled coil-mediated immobilization of RGD confers bioactivity that supports 3T3 fibroblast adhesion to NorHA hydrogels and PEG-NB fibers with comparable morphologies to traditional covalent ligation
After using FAM-tagged A-peptides as a proof-of-concept to establish the viability of coiled coils for immobilization of biomolecules onto NorHA hydrogels and PEG-NB fibers, we next incorporated the cell-adhesive RGD-containing peptide sequence GYGSPG into the A-peptide at the N-terminus to impart cell adhesive properties.1 Here, photopatterned T-peptide hydrogels bound RGD-functionalized A-peptide (“coiled-RGD”). A thiolated version of the RGD peptide (G![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) GYGSPG) was also covalently bound to NorHA and PEG-NB substrates for comparisons against our coiled coil system. To test cell behavior on scaffolds displaying the RGD motif, we cultured NIH 3T3 fibroblasts on NorHA hydrogels and PEG-NB fibers previously modified with different RGD concentrations (0, 10, 100 μM RGD via coiled coil or covalent conjugation). After incubation for 24 h, the samples were fixed and stained for fluorescent visualization of F-actin and cell nuclei, and cell area quantified. See Fig. 4A for cell area quantification and 3B/C for fluorescent micrographs of NorHA gels and PEG-NB fibers, respectively.
GYGSPG) was also covalently bound to NorHA and PEG-NB substrates for comparisons against our coiled coil system. To test cell behavior on scaffolds displaying the RGD motif, we cultured NIH 3T3 fibroblasts on NorHA hydrogels and PEG-NB fibers previously modified with different RGD concentrations (0, 10, 100 μM RGD via coiled coil or covalent conjugation). After incubation for 24 h, the samples were fixed and stained for fluorescent visualization of F-actin and cell nuclei, and cell area quantified. See Fig. 4A for cell area quantification and 3B/C for fluorescent micrographs of NorHA gels and PEG-NB fibers, respectively.
We also observe generally larger cell areas for the 10 μM and 100 μM RGD groups with our coiled coil system compared to the covalent groups, with a statistically significant difference between the 10 μM supramolecular and covalent groups (**p < 0.01). We hypothesize that this is likely due to the non-covalent, supramolecular characteristics of our coiled coil system that potentially allows cellular remodeling of RGD ligands within its environment. A covalent conjugation permanently immobilizes a pendant ligand on a substrate, allowing cells to exert traction forces.62,63 These traction forces are ubiquitous and implicated in dynamic tissue processes (e.g. contraction),62–64 and they may induce remodeling of the supramolecular coiled coil system. Since the conjugated T-peptide provides open and dynamic sites for coiled coil complexes to form, we postulate that cells may be able to reorganize the physical locations of the A-peptide motifs on the NorHA hydrogel surface through traction forces – thus providing a dynamic surface that leads to increased cell area. Dynamic interactions with 2D surfaces and 3D hydrogel matrices are known to play important roles in cell fate processes,65–67 and supramolecular functionalization of materials may allow for ongoing, cell-mediated changes in surface properties.
In considering both materials systems, we observe that the supramolecular addition of the coiled RGD ligand promotes 3T3 fibroblast adhesion and spreading on both NorHA hydrogels and PEG-NB fibers, and it presents a generalizable method for temporally-controlled functionalization of hydrogel substrates for cell adhesion in further studies.
3.5. The addition of the D-peptide induces removal of the coiled adhesive motif which actuates changes in cell morphology
After demonstrating that cells can adhere and spread on substrates supramolecularly functionalized with coiled-RGD, we sought to investigate the effects of its removal via the addition of the competing D-peptide on cell adherence and morphology. Both the NorHA hydrogel and PEG-NB hydrogel fiber systems exhibited changes in cell adherence that could be observed qualitatively upon the addition of the D-peptide (Fig. 5A and B). Quantitatively, we saw significant changes in cell areas in both systems, with cell areas on the NorHA hydrogels decreasing by roughly 50% and cell areas on the PEG-NB fibers decreasing to the same sizes as seen on unmodified PEG-NB fibers (Fig. 5C).Nonetheless, overall changes in cell adhesion and morphology were temporally controlled via the addition of the D-peptide to displace the coiled-RGD in both NorHA and PEG-NB systems. This demonstrates the potential for user-controlled perturbation of cellular microenvironments. Taken together with previous results, this coiled coil system offers both spatial and temporal control over patterning of ligands that can affect cell behaviors.
4. Conclusions and outlook
Coiled coil peptides offer a versatile system for engineering spatial and temporal signals into hydrogel environments. Biofunctionality of a peptide can readily be altered through standard peptide synthesis techniques, and thiol groups in cysteine residues allow their incorporation via light-controlled reactions amenable to spatial patterning, as well as by other bioconjugation reactions, such as Michael additions. The supramolecular coiled coil interaction also presents a reversible platform that allows for the repeated introduction and removal of bioactivity within in vitro hydrogel and hydrogel fiber culture systems. Proof-of-concept experiments showing reversible immobilization of FAM demonstrated comparable efficacy in functionalizing photoreactive biomaterials with high spatial control using coiled coil complexes compared to covalent photoligations. We confirmed the stability of the supramolecular coiled coil association over time, with release being dependent on toehold-mediated strand displacement by the D-peptide – a process that can be repeated after subsequent reloading with A-peptide. We then developed an extension of the A-peptide that included a cell-adhesive RGD motif on the N-terminus. Using this coiled-RGD for cell studies illustrated the ability to culture fibroblasts on materials functionalized with RGD via this coiled coil system. The concentration of the coiled-RGD peptide bound to the T-peptide affected the cell spread area on both NorHA hydrogels and PEG-NB fibers. Finally, removal of the coiled-RGD via introduction of the D-peptide caused a statistically significant decrease in cell spread area – on both hydrogels and fibers – indicating that the reversal of RGD presentation has a direct impact on fibroblast morphology. On the basis of these observations, future work should allow for the investigation of how dynamism in cell culture environments affects downstream cell behaviors. User-defined perturbations to these culture environments will allow for incremental advancements based on discrete changes to the microenvironment. Future work will also consider differences in cell behaviors on hydrogels and hydrogel-based fibers, as well as differences between hydrogel backbone materials in this system. We believe this platform might be applied to many other areas of research that desire user-controlled addition and subsequent temporal release of bioactive compounds that can be reloaded for multiple release cycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to Dr Steven R. Caliari at the University of Virginia for providing the NIH 3T3 fibroblasts as well as usage of the BioTek Synergy 4 spectrophotometer. Furthermore, the authors would like to thank Dr Nicholas E. Sherman for usage of circular dichroism spectroscopy and for conducting electrospray ionization mass spectrometry in the Biomolecular Analysis Facility which is supported by the University of Virginia School of Medicine.This work was supported by the University of Virginia and the National Institutes of Health through UVA Biotechnology Training Program NIGMS 5T32 GM008715 and GM136615. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- R. J. Wade, E. J. Bassin, W. M. Gramlich and J. A. Burdick, Adv. Mater., 2015, 27, 1356–1362 CrossRef CAS PubMed.
- B. M. Baker, B. Trappmann, W. Y. Wang, M. S. Sakar, I. L. Kim, V. B. Shenoy, J. A. Burdick and C. S. Chen, Nat. Mater., 2015, 14, 1262–1268 CrossRef CAS PubMed.
- C. D. Davidson, W. Y. Wang, I. Zaimi, D. K. P. Jayco and B. M. Baker, Sci. Rep., 2019, 9, 12 CrossRef PubMed.
- B. M. Baker and C. S. Chen, J. Cell Sci., 2012, 125, 3015–3024 CAS.
- R. J. Wade and J. A. Burdick, Mater. Today, 2012, 15, 454–459 CrossRef CAS.
- A. Velasco-Hogan, J. Xu and M. A. Meyers, Adv. Mater., 2018, 30(52), 1800940 CrossRef PubMed.
- B. Grigoryan, S. J. Paulsen, D. C. Corbett, D. W. Sazer, C. L. Fortin, A. J. Zaita, P. T. Greenfield, N. J. Calafat, J. P. Gounley, A. H. Ta, F. Johansson, A. Randles, J. E. Rosenkrantz, J. D. Louis-rosenberg, P. A. Galie, K. R. Stevens and J. S. Miller, Science, 2019, 364, 458–464 CrossRef CAS PubMed.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- R. J. Wade, E. J. Bassin, C. B. Rodell and J. A. Burdick, Nat. Commun., 2015, 6, 6639 CrossRef CAS PubMed.
- C. A. Deforest and K. S. Anseth, Angew. Chem., Int. Ed., 2012, 51, 1816–1819 CrossRef CAS PubMed.
- J. C. Grim, I. A. Marozas and K. S. Anseth, J. Controlled Release, 2015, 219, 95–106 CrossRef CAS PubMed.
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1138 CrossRef CAS PubMed.
- S. R. Caliari and J. A. Burdick, Nat. Methods, 2016, 13, 405–414 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- C. B. Highley, G. D. Prestwich and J. A. Burdick, Curr. Opin. Biotechnol., 2016, 40, 35–40 CrossRef CAS PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Healthcare Mater., 2011, 23, H41–H56 CrossRef CAS PubMed.
- W. M. Gramlich, I. L. Kim and J. A. Burdick, Biomaterials, 2013, 34, 9803–9811 CrossRef CAS PubMed.
- E. Hui, K. I. Gimeno, G. Guan and S. R. Caliari, Biomacromolecules, 2019, 20, 4126–4134 CrossRef CAS PubMed.
- J. A. Burdick and W. L. Murphy, Nat. Commun., 2012, 3, 1269 CrossRef PubMed.
- C. D. Davidson, D. K. P. Jayco, D. L. Matera, S. J. DePalma, H. L. Hiraki, W. Y. Wang and B. M. Baker, Acta Biomater., 2020, 105, 78–86 CrossRef CAS PubMed.
- S. A. Fisher, R. Y. Tam, A. Fokina, M. M. Mahmoodi, M. D. Distefano and M. S. Shoichet, Biomaterials, 2018, 178, 751–766 CrossRef CAS PubMed.
- R. Y. Tam, L. J. Smith and M. S. Shoichet, Acc. Chem. Res., 2017, 50, 703–713 CrossRef CAS PubMed.
- S. Sharma, M. Floren, Y. Ding, K. R. Stenmark, W. Tan and S. J. Bryant, Biomaterials, 2017, 143, 17–28 CrossRef CAS PubMed.
- J. J. Roberts and S. J. Bryant, Biomaterials, 2013, 34, 9969–9979 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- O. I. Kalaoglu-Altan, B. Verbraeken, K. Lava, T. N. Gevrek, R. Sanyal, T. Dargaville, K. De Clerck, R. Hoogenboom and A. Sanyal, ACS Macro Lett., 2016, 5, 676–681 CrossRef CAS.
- C. A. Deforest and D. A. Tirrell, Nat. Mater., 2015, 14, 523–531 CrossRef CAS PubMed.
- J. C. Grim, T. E. Brown, B. A. Aguado, D. A. Chapnick, A. L. Viert, X. Liu and K. S. Anseth, ACS Cent. Sci., 2018, 4, 909–916 CrossRef CAS PubMed.
- G. Weng, U. S. Bhalla and R. Iyengar, Science, 1999, 284, 92–97 CrossRef CAS PubMed.
- C. Xie, W. Sun, H. Lu, A. Kretzschmann, J. Liu, M. Wagner, H. J. Butt, X. Deng and S. Wu, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- F. M. Fumasi, N. Stephanopoulos and J. L. Holloway, J. Appl. Polym. Sci., 2020, 137, 1–16 CrossRef PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- L. Liu, X. Tian, Y. Ma, Y. Duan, X. Zhao and G. Pan, Angew. Chem., Int. Ed., 2018, 57, 7878–7882 CrossRef CAS PubMed.
- J. L. Mann, A. C. Yu, G. Agmon and E. A. Appel, Biomater. Sci., 2018, 6, 10–37 RSC.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- C. B. Highley, C. B. Rodell and J. A. Burdick, Adv. Mater., 2015, 27, 5075–5079 CrossRef CAS PubMed.
- E. A. Appel, X. J. Loh, S. T. Jones, F. Biedermann, C. A. Dreiss and O. A. Scherman, J. Am. Chem. Soc., 2012, 134(28), 11767–11773 CrossRef CAS PubMed.
- J. D. Tang, C. Mura and K. J. Lampe, J. Am. Chem. Soc., 2019, 141, 4886–4899 CrossRef CAS PubMed.
- C. B. Highley, C. B. Rodell, I. L. Kim, R. J. Wade and J. A. Burdick, J. Mater. Chem. B, 2015, 2, 8110–8115 RSC.
- J. Boekhoven, C. M. Rubert Pérez, S. Sur, A. Worthy and S. I. Stupp, Angew. Chem., Int. Ed., 2013, 52, 12077–12080 CrossRef CAS PubMed.
- S. L. McNamara, Y. Brudno, A. B. Miller, H. O. Ham, M. Aizenberg, E. L. Chaikof and D. J. Mooney, ACS Biomater. Sci. Eng., 2020, 6(4), 2159–2166 CrossRef CAS PubMed.
- R. Freeman, N. Stephanopoulos, Z. Álvarez, J. A. Lewis, S. Sur, C. M. Serrano, J. Boekhoven, S. S. Lee and S. I. Stupp, Nat. Commun., 2017, 8, 15982 CrossRef CAS PubMed.
- R. Freeman, M. Han, Z. Alvarez, J. A. Lewis, J. R. Wester, N. Stephanopoulos, M. T. McClendon, C. Lynsky, J. M. Godbe, H. Sangji, E. Luijten and S. I. Stupp, Science, 2018, 813, 808–813 CrossRef PubMed.
- K. Gröger, G. Gavins and O. Seitz, Angew. Chem., Int. Ed., 2017, 201705339, 14217–14221 CrossRef PubMed.
- A. N. Lupas and M. Gruber, Adv. Protein Chem., 2005, 70, 37–78 CrossRef CAS PubMed.
- J. R. Litowski and R. S. Hodges, J. Biol. Chem., 2002, 277, 37272–37279 CrossRef CAS PubMed.
- W. R. Archer and M. D. Schulz, Soft Matter, 2020, 16, 8760–8774 RSC.
- R. J. Wade and J. A. Burdick, Nano Today, 2014, 9, 722–742 CrossRef CAS.
- C. D. Davidson, D. K. P. Jayco, W. Y. Wang, A. Shikanov and B. M. Baker, J. Biomech. Eng., 2020, 142, 1–9 CrossRef PubMed.
- M. Iglesias-Echevarria, L. Durante, R. Johnson, M. Rafuse, Y. Ding, W. Bonani, D. Maniglio and W. Tan, Biomater. Sci., 2019, 7, 3640–3651 RSC.
- A. M. Kloxin, C. J. Kloxin, C. N. Bowman and K. S. Anseth, Adv. Mater., 2010, 22, 3484–3494 CrossRef CAS PubMed.
- B. Apostolovic and H. A. Klok, Biomacromolecules, 2008, 9, 3173–3180 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- M. Kabiri and L. D. Unsworth, Biomacromolecules, 2014, 15, 3463–3473 CrossRef CAS PubMed.
- G. De Crescenzo, J. R. Litowski, R. S. Hodges and M. D. O'Connor-McCourt, Biochemistry, 2003, 42, 1754–1763 CrossRef CAS PubMed.
- M. Kabiri, I. Bushnak, M. T. McDermot and L. D. Unsworth, Biomacromolecules, 2013, 14, 3943–3950 CrossRef CAS PubMed.
- C. Gaudet, W. A. Marganski, S. Kim, C. T. Brown, V. Gunderia, M. Dembo and J. Y. Wong, Biophys. J., 2003, 85, 3329–3335 CrossRef CAS PubMed.
- J. A. Rowley and D. J. Mooney, J. Biomed. Mater. Res., 2002, 60, 217–223 CrossRef CAS PubMed.
- R. Oria, T. Wiegand, J. Escribano, A. Elosegui-Artola, J. J. Uriarte, C. Moreno-Pulido, I. Platzman, P. Delcanale, L. Albertazzi, D. Navajas, X. Trepat, J. M. García-Aznar, E. A. Cavalcanti-Adam and P. Roca-Cusachs, Nature, 2017, 552, 219–224 CrossRef CAS PubMed.
- B. L. Ekerdt, R. A. Segalman and D. V. Schaffer, Biotechnol. J., 2013, 8, 1411–1423 CrossRef CAS PubMed.
- W. R. Legant, J. S. Miller, B. L. Blakely, D. M. Cohen, G. M. Genin and C. S. Chen, Nat. Methods, 2010, 7, 969–971 CrossRef CAS PubMed.
- W. R. Legant, A. Pathak, M. T. Yang, V. S. Deshpande, R. M. McMeeking and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10097–10102 CrossRef CAS PubMed.
- J. M. Viola, C. M. Porter, A. Gupta, M. Alibekova, L. S. Prahl and A. J. Hughes, Adv. Mater., 2020, 2002195, e2002195 CrossRef PubMed.
- S. Khetan, M. Guvendiren, W. R. Legant, D. M. Cohen, C. S. Chen and J. A. Burdick, Nat. Mater., 2013, 12, 458–465 CrossRef CAS PubMed.
- J. N. Roberts, J. K. Sahoo, L. E. McNamara, K. V. Burgess, J. Yang, E. V. Alakpa, H. J. Anderson, J. Hay, L.-A. Turner, S. J. Yarwood, M. Zelzer, R. O. C. Ore, R. V. Ulijn and M. J. Dalby, ACS Nano, 2016, 10, 6667–6679 CrossRef CAS PubMed.
- W. Li, Z. Yan, J. Ren and X. Qu, Chem. Soc. Rev., 2018, 47, 8639–8684 RSC.
- C. Loebel, R. L. Mauck and J. A. Burdick, Nat. Mater., 2019, 18, 883–891 CrossRef CAS PubMed.
- J. Dumbleton, P. Agarwal, H. Huang, N. Hogrebe, R. Han, K. J. Gooch and X. He, Cell. Mol. Bioeng., 2016, 9, 277–288 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm00016k |
| This journal is © The Royal Society of Chemistry 2021 |