
Surfactant location and internal phase volume fraction dictate emulsion electrospun fiber morphology and modulate drug release and cell response
Pamela M.
Johnson
 a,
Kelsey E.
Knewtson
b,
Jacob G.
Hodge
a,
Justin M.
Lehtinen
b,
Anna S.
Trofimoff
b,
D. Joseph
Fritz
b and
Jennifer L.
Robinson
*ab
a,
Kelsey E.
Knewtson
b,
Jacob G.
Hodge
a,
Justin M.
Lehtinen
b,
Anna S.
Trofimoff
b,
D. Joseph
Fritz
b and
Jennifer L.
Robinson
*ab
aBioengineering Graduate Program, University of Kansas, USA. E-mail: jlrobinson@ku.edu; Tel: +1(785) 864-0298
bDepartment of Chemical and Petroleum Engineering, University of Kansas, USA
First published on 4th January 2021
Abstract
Emulsion electrospinning is a versatile technique used to create fibrous meshes for applications in drug delivery and tissue engineering. In this study, the effects of surfactant and increasing internal phase volume fraction on emulsion electrospun fiber morphology were investigated. The fiber diameter, surface topography, internal architecture, mesh hydrophobicity, and fiber volume fraction were all characterized and the resulting effects on model drug release and cell response were determined. Surfactant relocation to the fiber surface resulted in alterations to fiber surface topography and internal morphology, increased rate of water adsorption into the mesh, and reduced burst effects of drug release. Increasing the internal phase volume fraction within the emulsion resulted in minimal change to fiber diameter, surface morphology, fiber volume fraction, and rate of water adsorption illustrating the ability to increase drug loading without affecting fiber properties. Lastly, all meshes promoted cell adhesion and good viability with a trend of increased MTT absorbance from cells on the surfactant and emulsion fibers possibly suggesting that an increase in surface area via smaller fiber diameter and fiber volume fraction increases metabolic activity. Overall, these studies indicate that fiber morphology and mesh hydrophobicity can be tuned by controlling surfactant location within fibers and internal phase volume fraction. Modulating fiber properties within the emulsion electrospun mesh is important to achieve controlled drug release and cell response for tissue engineering applications.
Introduction
Electrospinning is a versatile technique capable of producing fibers in the nano- to micrometer diameter range that are used in applications such as membrane separations, nanogenerators, catalysis, and biomedical engineering. Within tissue engineering applications, electrospinning is used to recapitulate collagen fiber diameter and alignment and has been shown to provide native mechanical properties of many connective tissues.1,2 These fibers also provide spatial cell-instructive cues and a bridge for cell migration to promote new extracellular matrix generation and repair.3–5 Growth factors and signaling molecules can be incorporated into the polymer solution and electrospun while still maintaining their bioactivity. Further, fiber diameter, porosity, and polymer degradation rates can be modulated in the electrospinning fabrication process to dictate diffusion out of the fibers. As such, it is important to be able to tune the fiber diameter, surface topography, and internal fiber architecture for cell-fiber interactions and drug delivery applications.6–8Emulsion electrospinning is a process by which an emulsion, consisting of liquid droplets of one phase suspended in a second immiscible liquid phase and stabilized by a surfactant, is used to fabricate a fibrous mesh. Controlling components of emulsion chemistry such as internal phase droplet size, droplet stability, and surfactant chemistry can be used to template porosity and pore architecture of the resulting fibers. Emulsions have been used in electrospinning to template both porous and coaxial fiber architectures.9–15 Emulsion electrospun fibers can be fabricated with oil-in-water or water-in-oil (w/o) emulsions depending on the application and drug solubility.15–17 Electrospinning of emulsions has been used to encapsulate and maintain bioactivity of proteins with conformations that are dependent on a hydrophilic environment to reduce any denaturation that occurs in organic solvents traditionally used in electrospinning with poly(ester) solutions.18,19 Furthermore, emulsion electrospinning can be used as a tool to reduce burst release of drugs and achieve sustained release to maintain therapeutic drug levels over time. Xu et al. was one of the first to encapsulate a hydrophilic drug, doxorubicin, in a w/o emulsion with a hydrophobic polymer using emulsion electrospinning techniques.9,20 Results from this study showed that larger internal phase droplet sizes lead to discontinuous cores. Bazilevsky et al. modeled the influence of internal phase droplet size on resulting fiber internal architecture and illustrated electrospinning core shell fibers from a single nozzle.12 Yazgan et al. demonstrated that increasing surfactant concentration decreased internal phase droplet diameter linearly in w/o emulsions until the critical concentration was reached.11 The study also demonstrated that the ambient relative humidity plays a role in fiber internal architecture.
Fiber surface roughness can be modified during or after the electrospinning process. Focusing on modifications during the process, phase separation techniques are used to induce surface morphology on electrospun fibers during electrospinning.21 Vapor induced phase separation (VIPS) is a process through which ambient water vapor condenses on the surface and then penetrates the fibers, causing polymer-rich and polymer-poor regions and subsequent pores.22,23 Relative humidity is one way that VIPs can be used to create porous fibers and studies show that as the relative humidity was increased, fiber diameter, surface roughness, and internal architecture porosity also increased.11,24–26 Interestingly, increased porous architecture also correlated with an increased rate of drug release.11
Surfactants also play a critical role in the internal architecture and fiber morphology of electrospun meshes. Hu et al. demonstrated the effect of surfactant chemistry in electrospun scaffolds fabricated with poly(caprolactone) (PCL).27 The ability to modulate fiber diameter by altering intermolecular interactions and conductivity was demonstrated using samples fabricated with increasing concentrations of non-ionic, cationic and anionic surfactants. Also, it was shown that non-ionic surfactants can improve overall fiber morphology by reducing beading and branching due to reduced surface tension.16,28 It is important to note that fiber morphology improved as surfactant concentration increased, indicating that surface tension effects are likely occurring at the air-solvent electrospinning interface and surfactant relocated to this interface during the electrospinning process. Relocation of surfactant to the fiber surface has been previously shown and hypothesized to play a critical role in reducing drug burst release observed in samples lacking surfactant.20,29 However, to date, the mechanistic role of surfactant and internal phase volume fraction in emulsion electrospun fiber properties and resulting effects on drug release and cell behavior have not been fully explored and characterized.
Therefore, the aim of this paper is to determine the role of surfactant and aqueous phase volume fraction in a water immiscible solvent system on emulsion electrospun fiber properties and downstream effects on drug release and cell response. Specifically, the focus is on independently increasing drug encapsulation by increasing internal phase volume fraction while minimally altering fiber diameter, a large player in release kinetics, to rationally control release and provide fibrous networks with diameters for cell interaction. W/o emulsions comprised of a poly(caprolactone) (PCL) chloroform solution in the oil phase and the non-ionic surfactant Span 80 were used. The surfactant composition and concentration was kept constant and the effect of phase volume fractions on surfactant location was determined using contact angle measurements. The effect of fiber architecture on small molecule release as a function of surface roughness and internal fiber porosity was determined using the fluorophore Nile Red as a model hydrophobic drug. Lastly, the effect of surfactant location on protein adsorption and resulting human meniscal fibrochondrocyte (MFC) morphology, metabolic activity, and proliferation was assessed. Overall, these studies highlight the ability to tune compositional and processing parameters using emulsion electrospinning techniques to generate fibers with diameters, surface roughness, and internal architecture that modulate both drug release and cell response to the scaffold for tissue engineering applications.
Methods
Materials
Poly(caprolactone) (PCL) (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Mw) was purchased from CAPA lot #120625. Nile Red with a 99% purity was purchased from ACRO organics lot # A0395995. Premium fetal bovine serum (FBS) was purchased from Atlanta Biologicals lot # H19101. Sodium dodecyl sulfate (SDS) was purchased from Chem-Impex. Liberase was purchased from Roche. DAPI was purchased from Abcam. All other chemicals needed for emulsion electrospun mesh fabrication and cell culture and assessment were purchased from Sigma Aldrich and Gibco/Invitrogen, respectively.
000 Mw) was purchased from CAPA lot #120625. Nile Red with a 99% purity was purchased from ACRO organics lot # A0395995. Premium fetal bovine serum (FBS) was purchased from Atlanta Biologicals lot # H19101. Sodium dodecyl sulfate (SDS) was purchased from Chem-Impex. Liberase was purchased from Roche. DAPI was purchased from Abcam. All other chemicals needed for emulsion electrospun mesh fabrication and cell culture and assessment were purchased from Sigma Aldrich and Gibco/Invitrogen, respectively.
Electrospun mesh fabrication
The overall study schematic and electrospinning setup are shown in Fig. 1. Electrospun mesh was fabricated by loading either polymer or emulsion solution into a 5 mL glass syringe and extruded out of a 21-gauge blunted needle at a rate of 0.5 mL h−1 with a Harvard Apparatus pump. The tip of the needle was placed approximately 20 cm away from the collection plate. Constant voltage was applied to the needle tip at 15 kV with a gamma high voltage research power supply and two grounding wires were attached to the stationary copper collection plate. Samples were electrospun in an environment of 50 ± 5% relative humidity and 23 ± 2 °C temperature using PCL dissolved in chloroform at a concentration of 20% w/v. PCL only and PCL plus Span 80 without internal phase were fabricated as controls. The non-ionic surfactant, Span 80, was added at a concentration of 30% w/w to any sample with surfactant. To fabricate emulsions, an aqueous internal phase was added in 20 μL increments to achieve 2%, 4%, 8% w/o relative to the continuous phase. Emulsification was conducted using a FlackTek SpeedMixer DAC 150 FVZ-K for 30 seconds intervals at 2500 rpms. All mesh was dried for at least 48 hours in a vacuum oven prior to morphology characterization via SEM and cell studies. Samples used for contact angle measurements were dried at ambient conditions for at least a week prior to characterization and analysis.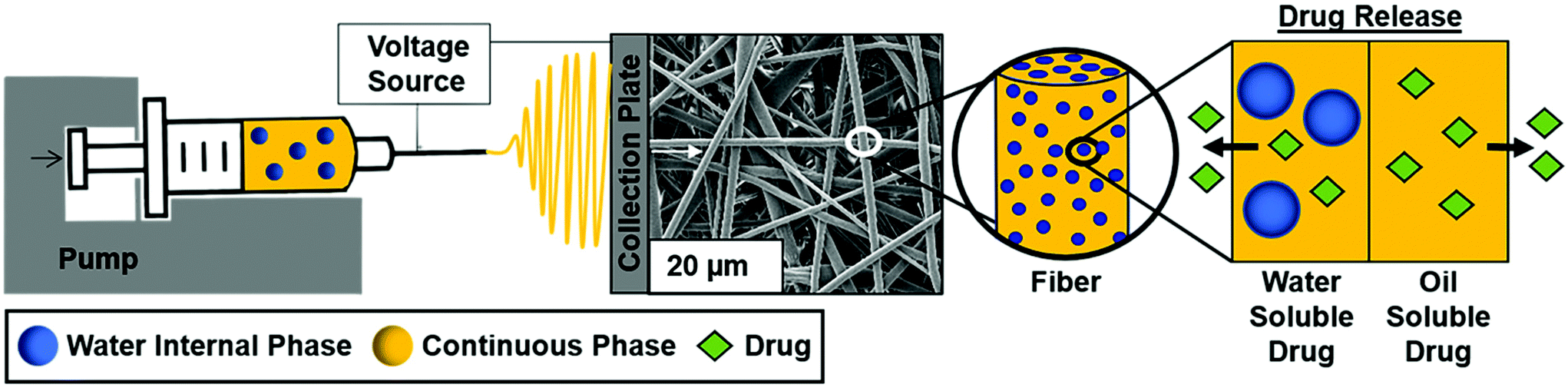 | ||
| Fig. 1 Emulsion electrospinning set-up showing loading of drug into fibers for controlled release. | ||
Fiber characterization
Scaffolds were analyzed with a Phenom Pro Desktop scanning electron microscope (SEM) to capture micrographs for fiber surface topography, diameter, volume fraction of fibers in mesh, and internal fiber architecture analysis. All samples were sputter coated with 8 nm of iridium and a 10 kV accelerating voltage was used with a backscatter detector.For PCL and Span 80 controls, internal fiber architecture was determined by fracturing the mesh in liquid nitrogen below the glass transition temperature of PCL (Tg = −61 °C)30 to preserve fiber structure. At least five fibers from each specimen for all samples were imaged. Fiber surface topography and morphology were determined by qualitative assessment of fibers. The fiber diameter and fiber volume fraction were determined from these specimens each from these electrospun samples of each composition by raster imaging at five different locations of each specimen based on studies by Szewczyk et al. and Qin et al.31,32 Each image was segmented and analyzed for fiber diameter and volume fraction of fibers using the best six segmentation algorithms in the software ImageJ and plugin DiameterJ. Histograms depicting distribution of fiber diameter were also created with DiameterJ.
Contact angle characterization
Water contact angle was conducted on all groups to assess the effect of mesh properties on wettability. Mesh fabricated with no surfactant, and surfactant with 0%, 2%, 4%, and 8% water internal phase were used to determine contact angle over time. Specimens were cut from the center of the mesh in approximately 1 cm × 2 cm rectangles and loaded with minimal wrinkles onto glass slides with double-sided tape. A Biolin Scientific Theta Lite 101 optical tensiometer was used to determine contact angle over a two-minute period at 30 frames per seconds. Using a glass syringe and a blunted needle, a droplet of ultra-pure water was placed on each of the dry mesh samples using a sessile drop technique. The contact angle was then analyzed using OneAttension software. For each run, time zero was determined as the time when the water droplet released from the needle tip. These samples were assessed in triplicate for a total of 9 specimens run per composition.Drug loading and release
Nile Red was used as a model hydrophobic, fluorescent drug to determine relative release over time from samples with surface roughness and a solid core (PCL control) compared to samples with a smooth surface with internal fiber architecture (Span 80). The electrospinning method described above was used to fabricate the samples with 0.1 mg mL−1 Nile Red. Specimens were prepared using an 8 mm histology punch. To determine the amount of Nile Red initially loaded into the electrospun mesh, samples of mesh were dissolved in chloroform and the relative fluorescence was determined using standard curves of known concentration of Nile Red in chloroform. These samples of 200 μL aliquots were removed and loaded into a black polystyrene 96-well plate. A Biotek Cytation 5 microplate reader was used to measure fluorescence with an excitation of 554 ± 20 nm and emission at 638 ± 20 nm. In order to determine the potential effects that PCL and Span 80 interaction with Nile Red may have on relative fluorescent levels, standards with solely PCL or PCL and Span 80 were run.To determine the amount of drug released over time, each sample was weighed to assess the dry mass and placed in 1 mL of ethanol. At time points of 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 10, 15, 30, 45, and 60 minutes, the full 1 mL ethanol was removed for sampling and replaced with 1 mL fresh ethanol. These 200 μL aliquot samples were removed from each time point sample and loaded into a black polystyrene 96-well plate. Sample data was normalized relative to weight of initial sample mass. Amount released was determined using a standard curve of known concentration of Nile Red in ethanol.
Preparation of mesh for protein adsorption and cell studies
Mesh specimens were prepared using an 8 mm biopsy punch and UV sterilized in a biosafety cabinet for 45 minutes. Punches were then treated with a wetting ladder to increase wettability. Specifically, 300 μL each of 70, 50, 30 and 0 v/v% ethanol in reverse osmosis water were added sequentially to each mesh, incubated at room temperature for 30 minutes, and then removed before the next solution was added. After removal of the final solution, meshes were allowed to dry for 30 minutes before the next solution was added.Fetal bovine serum protein adsorption
PCL only mesh was used for the no protein control. After completion of the successive dilutions of ethanol for wetting ladder, 300 μL of Dulbecco's modified Eagle medium (DMEM) (Gibco, 11995-065) with 40% v/v and 1% v/v penicillin/streptomycin (P/S) (Gibco, 15140-122) was added to each mesh. DMEM with 1% v/v P/S was used for no protein controls. After overnight incubation at 37 °C, the solution was removed and meshes were dried for 15 minutes. Meshes were washed once with 300 μL phosphate buffered saline (PBS) (Gibco, 10010-023) before 300 μL of complete DMEM containing 10% v/v premium FBS, 1% v/v P/S and 1% v/v L-glutamine (Gibco, 25030081) was added to each mesh. After incubation at 37 °C for 1 hours, the media was removed and meshes were washed 2× with 300 μL PBS. Meshes were then transferred to fresh wells containing 100 μL of 0.2% w/v sodium dodecyl sulfate (SDS) (Chem-Impex, 00270) and incubated at 37 °C, 100 rpm for 30 minutes. After incubation, meshes were removed from wells and 25 μL of each solution was transferred to a clear, flat bottomed 96 well plate for analysis. The Pierce BCA protein assay kit (Invitrogen, 23227) was used according to the manufacturer's protocol to analyze protein content of the SDS solution by absorbance at 562 nm using a Cytation 5 microplate reader (BioTek).Meniscal fibrochondrocyte (MFC) morphology, metabolic activity, and proliferation on emulsion electrospun meshes
Investigation of cell morphology, metabolic activity, and proliferation was conducted to determine initial cell–material interactions. Primary cells from the avascular region of the left, medial meniscus of a young, male donor were used to initially assess cell morphology, metabolic activity, and proliferation on the fibrous meshes. Meniscal tissue (516.9 mg) was resected from the body of the left medial meniscus of a 12-year old male with a parrot beak tear at Children's Mercy Hospital covered by IRB exemption STUDY00142627 (KU-Lawrence) and STUDY00000746 (Children's Mercy Hospital). Tissue was cut into small pieces (1–2 mm) using a scalpel and digested in DMEM containing 2% v/v P/S and 0.167 mg mL−1 of Liberase TM Research Grade (Roche, 05401127001) by adding 300 μL of this Liberase solution per 40 mg of tissue. Tissue was digested at 37 °C and 100 rpm for 3 h before being filtered through Swinnex Filter Holders (25 mm, Sigma-Aldrich, SX0002500) loaded with first 100 μm then 30 μm Spectra Mesh Nylon Filters (Spectrum Chemical Manufacturing, 888-13654 for 100 μM, 888-13681 for 30 μm) to remove undigested tissue. Cells were then cultured at 37 °C and 5% CO2 in T75 tissue culture treated flasks in complete DMEM until reaching 80–90% confluency, sub-cultured, and used at passage 3–5 for all studies. Electrospun mesh samples were prepared for cell studies by UV sterilization for 45 minutes followed by a wetting ladder of progressive dilutions of ethanol in water to ensure permeability of media solution throughout the mesh.To qualitatively assess cell morphology, MFC actin and nuclei were fluorescently tagged. MFCs were seeded at 20000 cells per mesh in 48 well plate (roughly 1 cm2, 3 specimens per group for each time point) on sterilized meshes that were weighed down with Teflon weights and incubated overnight in DMEM supplemented with 40 v/v% premium FBS and 1 v/v% P/S to promote protein adsorption for the requisite integrin-binding sites for cell adhesion. At day 1, 3, and 7, meshes were washed 1× with 300 μL PBS and transferred to a new well containing 300 μL PBS. PBS was removed and 300 μL 4 v/v% formaldehyde was added to each mesh, followed by a 15 minutes incubation at room temperature. Meshes were washed 2× with 300 μL PBS and then permeabilized by incubation in 300 μL 0.1% Triton X-100 at room temperature for 15 minutes. Meshes were washed 2× with 300 μL PBS before being stained with 200 μL Alexa Fluor 488 Phalloidin (1:400 PBS with 1% w/v BSA; Invitrogen, A12379). After incubation in the dark for 1 h at room temperature, meshes were washed 2× with 300 μL PBS. Nuclei were stained for 15 minutes at room temperature using DAPI (100 μM; Abcam, ab228549). Meshes were washed 2× with 300 μL of PBS before being imaged on a Zeiss Axio Observer A1 Inverted Fluorescence Microscope fitted with GFP and DAPI filters Zeiss Filter Sets 09 (for Alexa Fluor 488 Phalloidin) and 49 (for DAPI). Images were captured using an AxioCam MRc Color Camera. The light source was an X-Cite Series 120 Q. The fluorescence attenuator was set to 1, intensity was set to 40%, and exposure time was set to 40 miliseconds. Overlays were generated using the provided Zen Blue software. Brightness and contrast of images were enhanced equally across all compared images using Adobe Photoshop.
MFC metabolic activity and proliferation were assessed to determine the effects of mesh surface properties and diameter on cell behavior. Metabolic activity of cells on mesh was analyzed using the CyQUANT MTT cell viability assay (Invitrogen, V13154) at days 1, 3, and 7. Meshes were transferred to fresh wells containing 100 μL complete DMEM and 10 μL 12 mM MTT in PBS, and incubated for 4 h at 37 °C and 5% CO2. All media was then removed from each well and replaced with 100 μL DMSO and incubated at 37 °C and 100 rpm for 30 minutes to solubilize the dye. The solubilized dye solution was then transferred to clear, flat bottomed 96 well plates and the absorbance at 540 nm was read using a Cytation 5 Microplate Reader. Cell count and proliferation over time was estimated using the Quant-iT PicoGreen dsDNA assay kit (Invitrogen, P7589). At each time point (1, 3, and 7 day), meshes were washed 1× with 300 μL of PBS and transferred to a microcentrifuge tube containing 100 μL of 1× TE buffer with 0.2% Triton X-100 (IBI Scientific, IB07100). Meshes were subjected to 3× freeze/thaws with vortexing in between to lyse cells. Samples were stored at −80 °C until all being analyzed on the same day using the Quant-iT PicoGreen assay according to the manufacturer's protocol. Fluorescence was read using a Cytation 5 Microplate Reader with excitation set to 485 nm and emission set to 535 nm. The value of 7.7 ng DNA per cell was used to estimate cell number. For each assay and time point, a total of 15 specimens was assessed (3 samples of each mesh type + 5 specimens from each mesh sample).
Statistical analysis
All data was run in triplicate and data was expressed in mean ± standard deviation except for contact angle which was expressed in mean ± standard error of the mean to clearly demonstrate the trend in data. The ROUT method was used to check for outliers with Q = 0.1%; 1403, 16, 38, and 0 outliers were found for contact angle, fiber diameter, fraction of fibers, and relative drug loading, respectively. If outliers were determined, they were excluded from statistical analysis except the PicoGreen data. Fiber diameter and fraction of fiber were determined using one-way analysis of variance (ANOVA) with Levene's and Brown–Forsythe tests were used to determine the homogeneity of variance and a Games-Howell non-parametric post hoc test. Significance between Span 80 and no Span 80 samples loaded with Nile Red was determined using a t-test. For FBS adsorption, MTT, and PicoGreen analysis, the D'Agostino–Pearson omnibus normality test was used check the data for Gaussian distribution. The Brown–Forsythe test found that variance was not equal between samples. For these reasons, Brown–Forsythe and Welch ANOVA tests were run using Dunnett's T3 multiple comparisons test for FBS Adsorption. Two-way ANOVA was run using Bonferroni's multiple comparisons post hoc test for MTT and PicoGreen data. All analysis was performed using Prism and IBM SPSS software with significance indicated by * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001.Results and discussion
Fiber diameter, homogeneity, and fiber volume fraction
Fiber diameter decreased with the addition of surfactant and internal phase as shown visually in Fig. 2A–E. Distribution of fiber diameters was relatively normally distributed for all sample groups (Fig. 2F–J). The average fiber diameter decreased with the addition of surfactant from 4.30 ± 0.52 μm to 3.67 ± 0.21 μm. All samples were made with 30 wt% surfactant Span 80 to promote emulsification by reducing interfacial tension between to the two immiscible phases. Interestingly, these studies illustrated added benefits of Span 80 in controlling fiber properties that can be further capitalized on. Span 80 is a liquid at the temperature used in electrospinning. During the electrospinning process, the chloroform solvent evaporates in approximately less than 0.1 seconds allowing the Span 80 to have high mobility in the electrospun polymer jet.29,33 This decrease in fiber diameter may be due to Span 80 decreasing overall evaporation rate of the solvent or acting as a plasticizer for the polymer, allowing for greater stretching during the electrospinning process.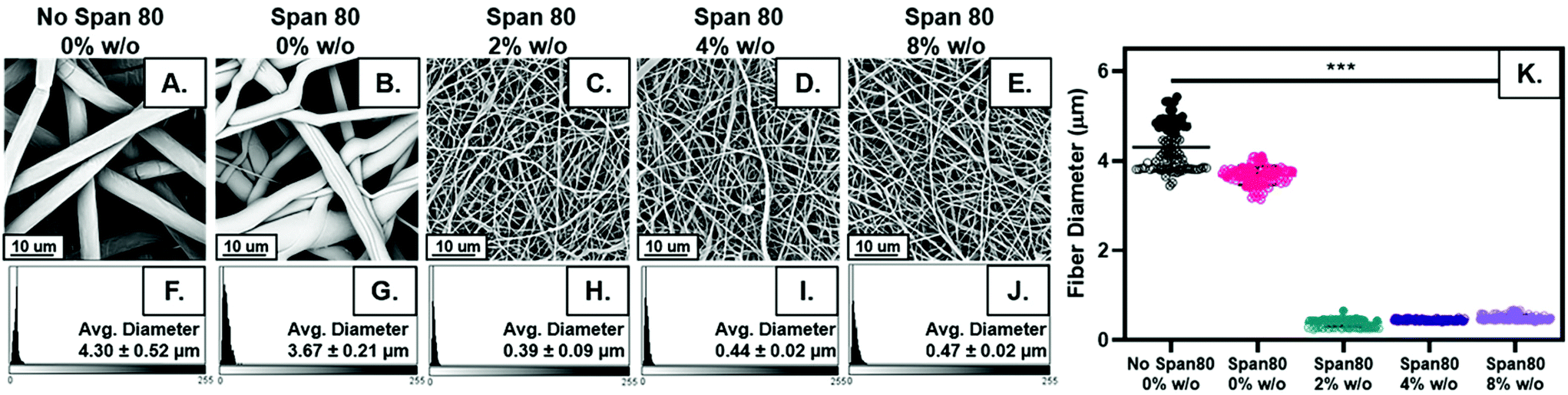 | ||
| Fig. 2 (A–E) SEM micrographs of electrospun fibers with increasing internal phase volume fraction and control. (F–J) Representative histograms of fiber diameter distribution for increasing internal phase volume fraction and control. (K) Dot plot of fiber diameter for each group. Data points from the same mesh are indicated by closed, open, or x shapes in the circle. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. | ||
The largest difference in fiber diameter can be seen with the addition of internal phase when comparing Fig. 2A–E, an effect likely due to changes in viscosity or conductivity with the addition of water in the emulsion which are hypothesized to influence fiber diameter.34 Increasing internal phase volume percentages of 2%, 4%, and 8% w/o slightly increased fiber diameter (0.39 ± 0.09 μm, 0.44 ± 0.02 μm, and 0.47 ± 0.02 μm) respectively. While emulsion fiber diameters are statistically significant when comparing all compositions, the difference between the diameters in samples with internal phase is quite small, a maximum increase of approximately 20% which fell within the error range (Fig. 2K). In fact, when neglecting the differences in homogeneity from control samples and analyzing these groups alone using a one-way ANOVA and a Bonferroni post hoc, the differences in fiber diameter are not statistically significant. These results indicate the ability to increase internal phase volume fraction, and the amount of drug loaded, with minimal change to fiber diameter.
Fiber surface topography and internal architecture
Fiber morphology was primarily cylindrical. Samples containing surfactant, including emulsions, demonstrated some adherence at junctions due to the slower evaporation of solvent when Span 80 was added to the system. Fig. 2B shows that fibers appear to be adhered at junctions in the sample with surfactant and no internal phase indicating that they likely collected wetter than in all other sample groups, an effect previously observed.29 Control PCL fibers without Span 80 exhibited surface roughness and a porous morphology. This is likely due to the fact that at high relative humidity, water has been shown to condense on the surface of fibers and create a porous morphology.24 These fibers had solid cross-sections due to polymer hydrophobicity, which prevented water from entering the center of the fibers resulting in water templating solely at the surface of the fibers. Fibers with Span 80 had smooth surface morphology and porous internal fiber architecture despite also being electrospun at high relative humidity as seen in Fig. 3A. As shown in Fig. 4, at high relative humidity, the surfactant relocated to the surface of the fiber and allowed water to penetrate the surface, absorb into the fiber, and template pores. Furthermore, this prevented water droplets from condensing on the surface of the fibers and remaining there while the solvent evaporated, creating a smooth fiber surface.24 It is important to understand how surfactant may be affecting internal architecture of the fiber, because porosity will alter the path for drug diffusion and release. Porous architectures may decrease the diffusive path of loaded drug compared to a solid architecture, because pores likely enhance fiber wetting. Furthermore, surface roughness has been shown to increase protein adsorption and dictate cell response including proliferation and differentiation.35–39 Therefore, controlling fiber architecture and surface roughness may be used as a tool to control drug release and cell response in tissue engineering applications. | ||
| Fig. 3 (A–E) SEM micrographs of the topography of electrospun fibers with increasing internal phase volume fraction and control. (F–G) Representative cross-sectional SEM micrographs of 0% w/o internal phase fibers with and without Span 80. | ||
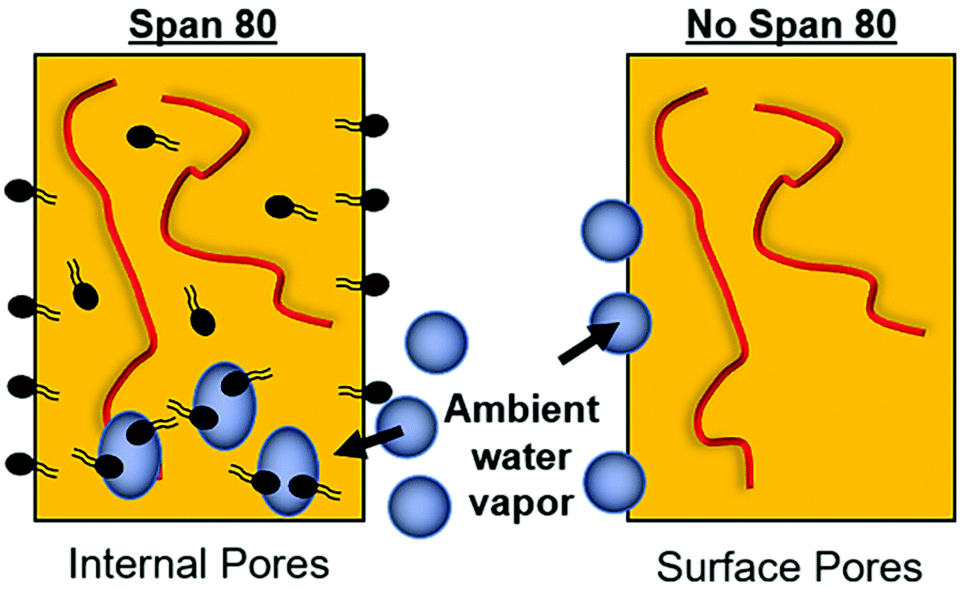 | ||
| Fig. 4 Ambient water interaction with fiber during electrospinning. In fibers with Span 80, water penetrates the core of the fiber. In fibers without Span 80, water droplets only condense on the fiber surface. | ||
Contact angle
The relocation of surfactant to the surface of the fiber was assessed via contact angle on the dry mesh over time. Controlling surfactant at the surface of the fiber is important in both controlling hydrophobicity of the overall mesh, and therefore wetting, for diffusional drug release and in modulating protein adsorption and subsequent cell adhesion to the integrin binding sites. Samples were electrospun at high relative humidity, thus making it thermodynamically favorable for surfactant to relocate at the hydrophobic solvent and polymer–air interface. As internal phase was increased in the system, it was hypothesized that more of the surfactant would be located at the oil–water interface within the fiber, instead of the oil–air interface at the surface of the fiber (Fig. 6).The contact angle of water on dry mesh was determined for all groups over two minutes. The PCL control meshes remained approximately constant at a contact angle of 130° due to the hydrophobicity of the polymer as seen in Fig. 5A and F. The samples with Span 80 but no aqueous internal phase, shown in Fig. 5B and G, exhibited a rapidly decreasing contact angle within the first 30 seconds as the water droplet absorbed into the scaffold. Fig. 5C–E and H–J depict the emulsion samples with surfactant and internal phase at 0 and 30 seconds, respectively. The droplets on these samples also absorbed into the scaffold within the first 30 seconds, but at a slower rate than the sample without internal phase. Fig. 5K shows the change in contact angle for all samples over time. The rapid decrease in contact angle when surfactant was present in the samples supports the hypothesis that surfactant is likely relocating to the surface of the fibers during electrospinning. In samples with internal phase, surfactant is at the oil–water interface and therefore lowering the concentration present on the surface of the fibers. This is supported by the slower absorption of water into the mesh relative to the sample with surfactant and no internal phase.
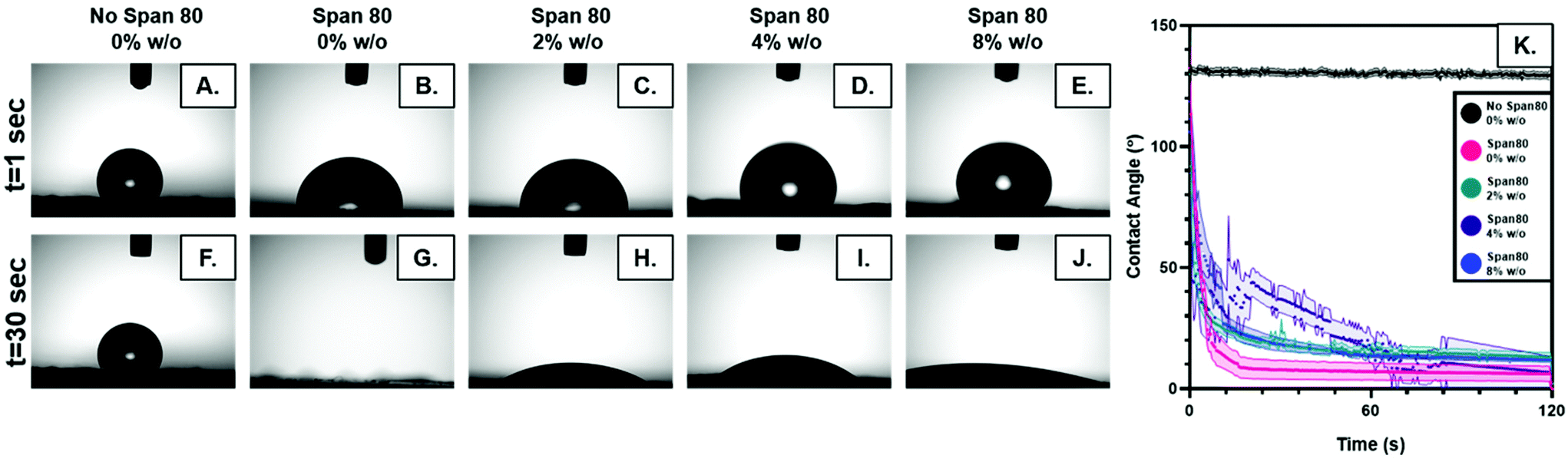 | ||
| Fig. 5 (A–E) Contact angle of water on electrospun mesh with increasing internal phase and no Span 80 control at 1 second. (F–J) Contact angle of water at 30 seconds. (K) Plot of contact angle for each group over time. | ||
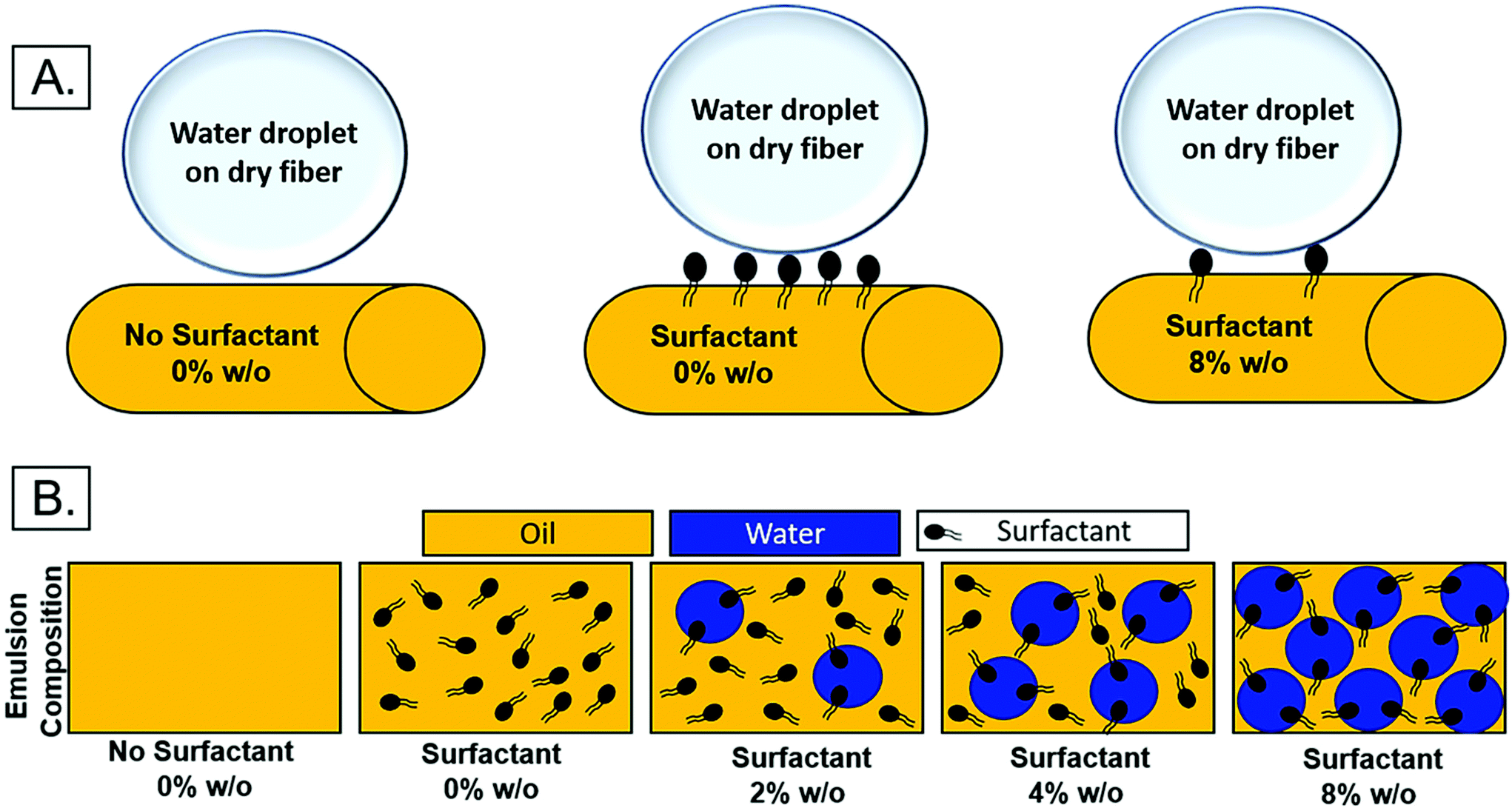 | ||
| Fig. 6 (A) Theoretical surfactant on the surface of electrospun fibers with increasing internal phase. (B.) Schematic of surfactant location within emulsion with increasing internal phase. | ||
Electrospun mesh hydrophobicity modulates protein adsorption, wettability, and cell–material interactions in tissue engineering scaffolds. Szewczyk et al. demonstrated the two main parameters that play a role in contact angle of the scaffold are surface roughness and fiber volume fraction.32 Surface roughness is the amount of porous topography on the fiber while the fiber volume fraction is the volume fraction of the mesh taken up by fiber (not air). Fiber volume fraction percentage was significantly different between samples with no Span 80 and samples with Span 80 and internal phase (Fig. 7). Significant differences were also seen between samples with 2% and 8% w/o. Fiber volume fraction is the opposite of air volume fraction within an electrospun mesh and is correlated to overall mesh hydrophobicity because of trapped air. Studies have shown that decreased fiber volume fraction is correlated with higher contact angle because more air is trapped in the scaffold.31,32,40
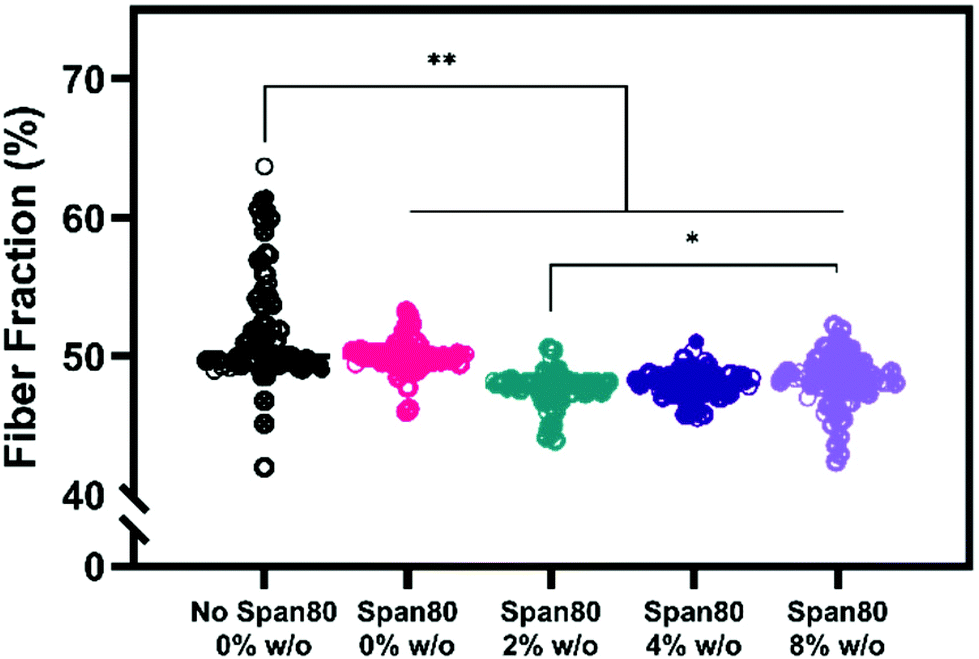 | ||
| Fig. 7 Fiber volume fraction for increasing internal phase and control. Data points from the same mesh are indicated by closed, open, or x shapes in the circle. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. | ||
While fiber volume fraction and surface roughness may have played a role in water absorption rate via contact angle, surfactant location is the more dominant parameter in our studies. Specifically, a decrease in fiber volume fraction did not result in higher contact angle supporting the hypothesis that surfactant movement to the fiber surface places a more significant role on wettability in this study. Fiber diameter, homogeneity, and fiber volume fraction are all critical material properties of electrospun meshes that dictate drug loading capacity, release rates, and cell response in drug delivery and tissue engineering applications. Controlling hydrophobicity of scaffolds and resulting liquid interactions for wetting is important for drug release and cell response.35–39,41,42 Furthermore, increasing internal phase volume fractions while controlling fiber diameter allows for increased loading capacity of aqueous drugs and greater control for cell response. To effectively use emulsions for drug loading, it is important to understand the role that surfactant is playing in decreasing fiber diameter and fiber volume fraction. These results are important because they indicate that increasing volume fractions of internal phase may be achieved with minimal or no changes to fiber diameter.
Drug loading and release
Nile Red was loaded into PCL and Span 80 control scaffolds as a preliminary model drug to determine release characteristics of the fibers as a function of surface roughness and internal fiber architecture. The loading capacity, a measure of the relative amount of drug per mass of the fibers, was not statistically different (P < 0.05), indicating that any differences in release rates were not a function of differences in drug loading (Fig. 8A). Drug release from PCL control fibers (rough surface, solid fiber core) occurred with a larger burst release and percentage of relative Nile Red released than samples with Span 80 (smooth surface, internal fiber porosity) (Fig. 8B). All samples demonstrated an initial burst release followed by a slower continuous release of drug over the time period of one hour. The smaller burst release of Nile Red from fibers with surfactant is likely due to two factors. First, Nile Red and Span 80 interact at the oil–water interface within the fiber, minimizing the amount of initial Nile red at the oil–air interface occurring in the mesh fabricated with no surfactant. Water likely penetrated the fibers because they were electrospun at high relative humidity and had porous internal architecture (Fig. 4F & G). Second, the PCL control fibers have increased surface area due to the surface roughness which may increase the release compared to the smooth Span 80 fibers with relatively lower surface area. It is important to understand how surfactant is playing a role in drug release because surfactant can be used as a tool to create porous internal architectures with potential to tune drug release rates. Furthermore, understanding the role that surfactants play in drug release is important in reducing burst release effects and incorporating aqueous drugs in emulsion electrospun fibers. The ability to rationally control fiber properties to reduce burst release and promote sustained release and reduce burst effects is beneficial in ensuring a therapeutic concentration of drug is maintained over longer periods of time.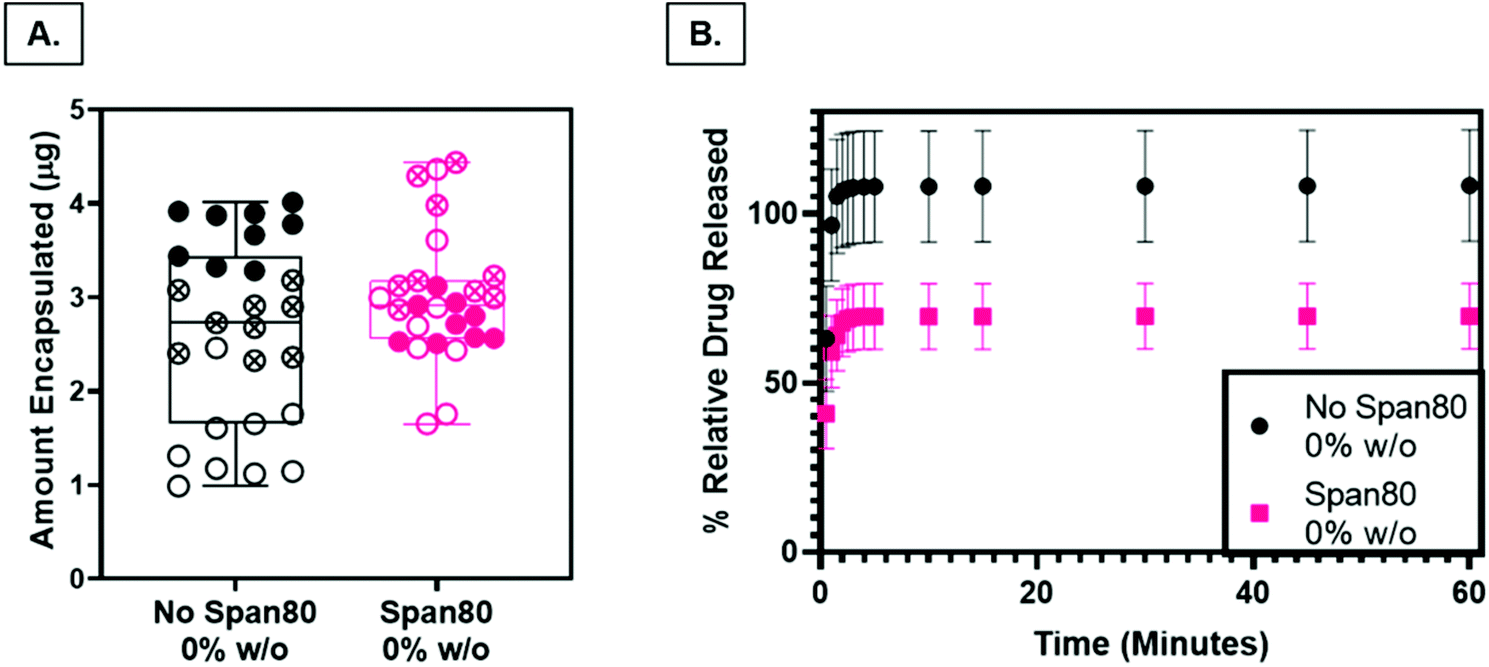 | ||
| Fig. 8 (A) Relative amount of Nile Red model drug loaded in fibers per weight of total specimen in samples with and without Span 80. * p ≤ 0.05. (B) Percent of relative Nile Red model drug released per total weight of specimen in samples with and without Span 80. Data points from the same mesh are indicated by closed, open, or x shapes in the circle. Data points from the same mesh are indicated by closed, open, or x shapes in the circle. | ||
Cell response
Electrospun fiber properties, such as fiber diameter and surface roughness, can induce cellular responses.35–38 Beyond the ability to modulate drug release over time, the emulsion electrospun fibers in this study were fabricated to support and enhance cell processes including metabolism and proliferation for tissue engineering applications. Human primary MFCs morphology, metabolic activity, and proliferation were assessed for initial cell response to the fibers for use as a tissue engineered scaffold for fibrous tissue repair. Of particular interest, evidence that Span 80 relocates to the fiber-environment interface prompted the need to determine how this amphiphilic compound may affect cell behavior. Cells on all mesh compositions experienced attachment and spreading as visualized by the spindle-shaped morphology (Fig. 9A). There were no visible differences in cell morphology on any of the meshes over the course of 7 days. The only significant difference in the total amount of FBS protein adsorbed onto the meshes after 1 day was between the 2 and 4% w/o emulsions compared to the 8% w/o emulsion (Fig. 9B) suggesting any difference in cell response was not due to differences in protein adsorption affecting cell adhesion. This is interesting as it was hypothesized that surfactant relocation to the surface would affect total protein adsorption, an effect that was not observed. Possible reasons for this difference in observed effect could be the wetting ladder treatment done to mimic the process for prepping for cell studies, the short time point, or the fact that specific proteins were not assessed. Previous findings illustrate fiber diameter and surface roughness altering cell morphology with an increase in cell elongation and area on the microfibers and those with increased surface roughness compared to smooth fibers and/or nanofibers.35,38 In this study, it is possible that MFCs do not follow this pattern or that more quantitative analysis should be conducted to directly assess this effect independent of surface chemistry.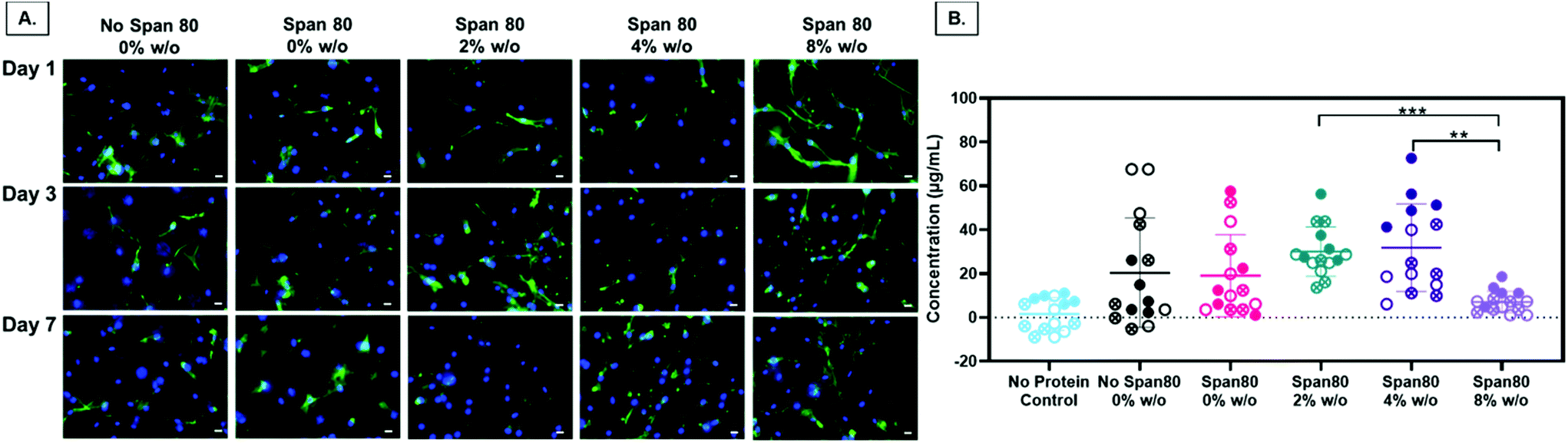 | ||
| Fig. 9 FBS adsorption and meniscal fibrochondrocyte (MFC) morphology on all fiber groups. MFC morphology on each mesh group at day 1, 3, and 7. (A) Alexa Fluor 488 Phalloidin stains the actin cytoskeleton green and nuclei stained blue with DAPI. (B) Total protein concentration after 24-hour FBS absorption on all groups with a PCL mesh no protein control. Data points from the same mesh are indicated by closed, open, or x shapes in the circle. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. Scale bar is 20 μm. | ||
Overall, there are two major findings after assessing initial cell metabolic activity and proliferation. First, PCL-based emulsion electrospun fibers with Span 80 promote cell metabolic activity and proliferation comparable to the no surfactant control. There is always the concern of surfactants in biomaterial systems leaching out and causing cell death by disrupting the cell plasma membrane. In all groups over the course of 3 days, viability was determined by live/dead imaging and there were no differences between the PCL control meshes and those with Span 80 (data not shown). Furthermore, meshes with Span 80 resulted in MFCs with increased metabolic activity at day 7 (Fig. 10A) and no difference in cell proliferation compared to the PCL control (Fig. 10B). The surfactant is likely anchored into the PCL fibers once the solvent evaporates off during the electrospinning process and any residual is removed during processing before cells are added such that none can diffuse out into the surrounding environment. These results suggest the ability to use Span 80 to control fiber morphology and surface properties without affecting cell viability.
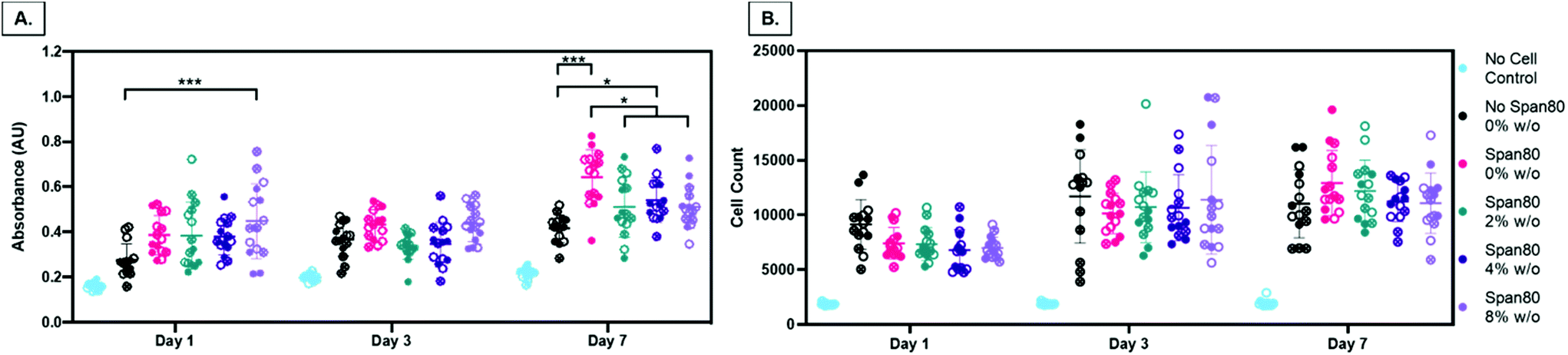 | ||
| Fig. 10 Meniscal fibrochondrocyte (MFC) metabolic activity and proliferation. (A) MTT absorbance at day 1, 3, and 7 illustrating relative metabolic activity for MFCs on all mesh groups and a PCL no cell control. (B) MFC cell counts at day 1, 3, and 7 using PicoGreen assay for quantifying double stranded DNA. Data points from the same mesh are indicated by closed, open, or x shapes in the circle. * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001. | ||
Second, in this study the change in fiber surface properties and diameter (μm to nm) had minimal effect on cell metabolic activity or proliferation using the assessment methods described here. Metabolic activity as measured by MTT absorbance at days 1, 3, and 7 is shown in Fig. 10A. At any of the time points, the only difference in metabolic activity was an increase in the 8% w/o emulsion meshes compared to the PCL control at day 1, PCL control compared to 4% w/o at day 7, and Span 80 compared to PCL control and 2% and 8% w/o emulsions at day 7. The metabolic activity from days 1 to 3, in the 4% and 8% w/o emulsion meshes increased. In comparing activity from days 3 to 7, Span 80, 2% and 4% w/o meshes resulted in an increase. Lastly the PCL, Span 80, 2% and 4% w/o meshes all exhibited an increase in MFC metabolic activity from days 1 to 7. Pabbruwe et al. illustrated constant MTT absorbance readings from MFCs in dextran bead microcarriers through 7 days suggesting this cell phenotype may not be highly metabolically active.43 However, the increased MTT absorbance from cells on the surfactant and emulsion fibers, an effect that is significant at Day 7, suggests that an increase in surface area via smaller fiber diameter and fiber volume fraction increases metabolic activity. This effect is being pursued further with more metabolically active cell types.
Proliferation was determined by changes in cell number over time via quantification of double stranded DNA and is illustrated in Fig. 10B. There was no difference in proliferation in comparing mesh composition at any of the time points. While there was no difference over the course of 7 days at the assessed time points for PCL, there were increases in the surfactant and 2% w/o groups from days 1 to 7 and increases in the 4% and 8% w/o emulsion meshes both from days 1 to 3 and days 1 to 7. Large rates of cell growth and resulting proliferation were not observed, even in the tissue culture polystyrene control wells (data not shown). This may be attributed to several factors. MFCs are mature, differentiated cells that are not highly proliferative and exhibit a slow doubling rate that decreases after 3–4 passages,44 unlike immortalized cell lines that maintain exponential growth through 20–30 passages. Also, the cells in this study were from primary harvest and at passage 3–5 depending on the study. Liang et al. showed population doubling of 0.4–0.5 per day through P3 and then a drop to 0.3 per day at P4.44 This explains the slow doubling seen in the cell counts as MFCs at these passage numbers are only experiencing a population doubling every 48–72 hours, compared to cells that double every 18–24 hours. Verdonk et al. saw minimal change in mass of DNA from MFCs in 2D culture through 7 days but then an exponential increase through 21 days suggesting these cells take time to adapt in vitro prior to doubling.45 In previous studies from our group, we observed good and comparable MFC viability on all mesh groups at 72 hours using the live/dead assay kit (data not shown) suggesting that the reduced MFC metabolic activity and proliferation is not due to a reduction in cell viability and any changes in viability are similar across groups. In support of this work, multiple other studies have shown the promising effect of MFCs on polyester-based fibrous electrospun scaffolds with incorporated biopolymer fibers for meniscus tissue repair but did not specifically look at cell metabolism or proliferation but rather gene expression of tissue specific genes.46–50 Preliminary data from our group using highly proliferative 3T3 mouse fibroblasts showed that all mesh groups promoted cell proliferation over 7 days. On day 7, a decrease in cell count on the Span 80 and all emulsions groups compared to the PCL control was observed suggesting an effect of the surfactant and/or the reduction in fiber diameter on cell proliferation. These early results further highlight the need for cell-specific characterization of the fiber properties in these emulsion electrospun meshes. Future work will focus on confirming our results with earlier passages of MFCs and more proliferative cell types such as mesenchymal progenitor cells and investigating transcriptional changes occurring in response to the scaffold properties.
Multiple studies have shown the ability to control cell proliferation and differentiation by modulating fiber surface roughness and diameter, with an increase in proliferation corresponding with increased roughness and smaller fibers.35–37 However, this was not observed in this study likely due to a number of factors including cell phenotype and the endpoint measurement techniques used. Other studies have shown contradictory effects as well. For example, myoblasts were shown to exhibit more significant increases in cell numbers and cell spreading on poly(lactic-co-glycolic acid) (PLGA) aligned fibers with up to 3 μm diameters compared to the nanofibers.51 Lastly, healing and regeneration is a time-dependent process and requires dynamic changes in the extracellular matrix to promote this process. Changes are typically seen over the course of weeks to months whereas this study was only conducted for one week.3 Thus, it will be informative to determine the role of modulating fiber properties including diameter and surface roughness on MFC behavior over time.
Conclusion
Emulsion electrospun meshes were fabricated and fiber diameter, surface roughness, internal fiber architecture, and surfactant location were modulated to control properties that dictate drug release and cell response. Excitingly, we have shown the ability to theoretically increase the amount of encapsulated drug via increasing volume fraction of the internal phase internal phase with minimal alteration of fiber diameter and homogeneity. Our data also shows that porous fibers created with surfactant can reduce burst drug release effects without detrimental effects to cells. Future work will continue studies on drug release and cell response by measuring drug release of hydrophilic and hydrophobic drugs loaded in emulsion electrospun fibers fabricated with increasing internal phase volume fraction. Further, subsequent effects of drug release kinetics on cell response will be investigated. Overall, this work demonstrates the ability to control fiber diameter, surface roughness, surfactant location, and internal fiber architecture using compositional parameters in emulsion electrospun meshes for applications in drug delivery and tissue engineering.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful to Katie Donnelly for help with contact angle sample preparation and Kara Hageman and Phil Elrod for aid with completing drug release studies. Further, the authors are grateful to Dr Donna Pacicca for the meniscal tissue from resection surgeries covered by IRB exemption STUDY00142627 (KU-Lawrence) and STUDY00000746 (Children's Mercy Hospital). This work was supported by startup funds from the University of Kansas (JR), NIH NIGMS P20GM103638 (KK and JR), NIH Biotechnology Training Grant (T32) Pharmaceutical Aspects of Drug Delivery (PJ), Howard Hughes Medical Institute through the James H. Gilliam Fellowships for Advanced Study program (PJ and JR), and University of Kansas Go Grant.References
- K. L. Moffat, A. S. Kwei, J. P. Spalazzi, S. B. Doty, W. N. Levine and H. H. Lu, Tissue Eng., Part A, 2009, 15, 115–126 CrossRef CAS.
- S. D. Subramony, B. R. Dargis, M. Castillo, E. U. Azeloglu, M. S. Tracey, A. Su and H. H. Lu, Biomaterials, 2013, 34, 1942–1953 CrossRef CAS.
- N. M. Lee, C. Erisken, T. Iskratsch, M. Sheetz, W. N. Levine and H. H. Lu, Biomaterials, 2017, 112, 303–312 CrossRef CAS.
- N. Lee, J. Robinson and H. Lu, Curr. Opin. Biotechnol., 2016, 40, 64–74 CrossRef CAS.
- G. C. Ingavle and J. K. Leach, Tissue Eng., Part B, 2014, 20, 277–293 CrossRef CAS.
- B. M. Baker, B. Trappmann, W. Y. Wang, M. S. Sakar, I. L. Kim, V. B. Shenoy, J. A. Burdick and C. S. Chen, Nat. Mater., 2015, 14, 1262–1268 CrossRef CAS.
- J. Zeng, X. Xu, X. Chen, Q. Liang, X. Bian, L. Yang and X. Jing, J. Controlled Release, 2003, 92, 227–231 CrossRef CAS.
- Y.-F. Goh, I. Shakir and R. Hussain, J. Mater. Sci., 2013, 48, 3027–3054 CrossRef CAS.
- X. Xu, X. Zhuang, X. Chen, X. Wang, L. Yang and X. Jing, Macromol. Rapid Commun., 2006, 27, 1637–1642 CrossRef CAS.
- J. Chen, P. Yang, C. Wang, S. Zhan, L. Zhang, Z. Huang, W. Li, C. Wang, Z. Jiang and C. Shao, Nanoscale Res. Lett., 2011, 6, 121 CrossRef.
- G. Yazgan, A. M. Popa, R. M. Rossi, K. Maniura-Weber, J. Puigmarti-Luis, D. Crespy and G. Fortunato, Polymer, 2015, 66, 268–276 CrossRef CAS.
- A. V. Bazilevsky, A. L. Yarin and C. M. Megaridis, Langmuir, 2007, 23, 2311–2314 CrossRef CAS.
- C. Wang, L. Wang and M. Wang, Mater. Lett., 2014, 124, 192–196 CrossRef CAS.
- L. Tian, M. P. Prabhakaran, X. Ding, D. Kai and S. Ramakrishna, J. Mater. Sci., 2012, 47, 3272–3281 CrossRef CAS.
- C. Zhang, F. Feng and H. Zhang, Trends Food Sci. Technol., 2018, 80, 175–186 CrossRef CAS.
- Y.-M. Lin and G. C. Rutledge, J. Membr. Sci., 2018, 563, 247–258 CrossRef CAS.
- N. Nikmaram, S. Roohinejad, S. Hashemi, M. Koubaa, F. J. Barba, A. Abbaspourrad and R. Greiner, RSC Adv., 2017, 7, 28951–28964 RSC.
- H. Frizzell, T. J. Ohlsen and K. A. Woodrow, Int. J. Pharm., 2017, 533, 99–110 CrossRef CAS.
- T. Briggs and T. L. Arinzeh, J. Biomed. Mater. Res., Part A, 2014, 102, 674–684 CrossRef.
- X. Xu, L. Yang, X. Xu, X. Wang, X. Chen, Q. Liang, J. Zeng and X. Jing, J. Controlled Release, 2005, 108, 33–42 CrossRef CAS.
- M. Srinivasarao, D. Collings, A. Philips and S. Patel, Science, 2001, 292, 79–83 CrossRef CAS.
- F. J. H. Abadi, M. A. Tehran, F. Zamani, M. Nematollahi, L. G. Mobarakeh and M. H. Nasr-Esfahani, Int. J. Polym. Mater. Polym. Biomater., 2014, 63, 57–64 CrossRef.
- P. Lu and Y. Xia, Langmuir, 2013, 29, 7070–7078 CrossRef CAS.
- B. Zaarour, L. Zhu, C. Huang and X. Jin, Nanoscale Res. Lett., 2018, 13, 285 CrossRef.
- B. Zaarour, L. Zhu, C. Huang and X. Y. Jin, RSC Adv., 2018, 8, 42353–42360 RSC.
- R. M. Nezarati, M. B. Eifert and E. Cosgriff-Hernandez, Tissue Eng., Part C, 2013, 19, 810–819 CrossRef CAS.
- J. Hu, M. P. Prabhakaran, X. Ding and S. Ramakrishna, J. Biomater. Sci., Polym. Ed., 2015, 26, 57–75 CrossRef CAS.
- T. Lin, H. Wang, H. Wang and X. Wang, Nanotechnology, 2004, 15, 1375–1381 CrossRef CAS.
- X. Li, Y. Su, S. Liu, L. Tan, X. Mo and S. Ramakrishna, Colloids Surf., B, 2010, 75, 418–424 CrossRef CAS.
- J. V. Koleske and R. D. Lundberg, J. Polym. Sci., Part A-2, 1969, 7, 795–807 CrossRef CAS.
- X.-H. Qin and D.-P. Xin, Fibers Polym., 2010, 11, 632–637 CrossRef CAS.
- P. Szewczyk, D. Ura, S. Metwally, J. Knapczyk-Korczak, M. Gajek, M. Marzec, A. Bernasik and U. Stachewicz, Polymers, 2018, 11, 34 CrossRef.
- S. Liao, R. Murugan, C. K. Chan and S. Ramakrishna, J. Mech. Behav. Biomed. Mater., 2008, 1, 252–260 CrossRef.
- J. C. Sy, A. S. Klemm and V. P. Shastri, Adv. Mater., 2009, 21, 1814–1819 CrossRef CAS.
- H. Chen, X. Huang, M. Zhang, F. Damanik, M. B. Baker, A. Leferink, H. Yuan, R. Truckenmuller, C. van Blitterswijk and L. Moroni, Acta Biomater., 2017, 59, 82–93 CrossRef CAS.
- L. Moroni, R. Licht, J. de Boer, J. R. de Wijn and C. A. van Blitterswijk, Biomaterials, 2006, 27, 4911–4922 CrossRef CAS.
- X. Wang, T. Lou, W. Zhao, G. Song, C. Li and G. Cui, J. Biomater. Appl., 2016, 30, 1545–1551 CrossRef CAS.
- T. L. Jenkins and D. Little, npj Regener. Med., 2019, 4, 15 CrossRef.
- K. M. Woo, V. J. Chen and P. X. Ma, J. Biomed. Mater. Res., Part A, 2003, 67, 531–537 CrossRef.
- C. P. Carroll and Y. L. Joo, Phys. Fluids, 2006, 18, 053102 CrossRef.
- S. T. Yohe, Y. L. Colson and M. W. Grinstaff, J. Am. Chem. Soc., 2012, 134, 2016–2019 CrossRef CAS.
- S. T. Yohe, J. D. Freedman, E. J. Falde, Y. L. Colson and M. W. Grinstaff, Adv. Funct. Mater., 2013, 23, 3628–3637 CrossRef CAS.
- M. B. Pabbruwe, K. Stewart and J. B. Chaudhuri, Biotechnol. Lett., 2005, 27, 1451–1455 CrossRef CAS.
- Y. Liang, E. Idrees, S. H. J. Andrews, K. Labib, A. Szojka, M. Kunze, A. D. Burbank, A. Mulet-Sierra, N. M. Jomha and A. B. Adesida, Sci. Rep., 2017, 7, 12148 CrossRef.
- P. C. Verdonk, R. G. Forsyth, J. Wang, K. F. Almqvist, R. Verdonk, E. M. Veys and G. Verbruggen, Osteoarthritis Cartilage, 2005, 13, 548–560 CrossRef CAS.
- J. Baek, M. K. Lotz and D. D. D'Lima, Tissue Eng., Part A, 2019, 25, 1577–1590 CrossRef CAS.
- J. Baek, S. Sovani, W. Choi, S. Jin, S. P. Grogan and D. D. D'Lima, Tissue Eng., Part A, 2018, 24, 81–93 CrossRef CAS.
- J. Baek, S. Sovani, N. E. Glembotski, J. Du, S. Jin, S. P. Grogan and D. D. D'Lima, Tissue Eng., Part A, 2016, 22, 436–448 CrossRef CAS.
- F. Qu, M. P. Pintauro, J. E. Haughan, E. A. Henning, J. L. Esterhai, T. P. Schaer, R. L. Mauck and M. B. Fisher, Biomaterials, 2015, 39, 85–94 CrossRef CAS.
- F. Qu, J. L. Holloway, J. L. Esterhai, J. A. Burdick and R. L. Mauck, Nat. Commun., 2017, 8, 1780 CrossRef.
- N. Narayanan, C. Jiang, C. Wang, G. Uzunalli, N. Whittern, D. Chen, O. G. Jones, S. Kuang and M. Deng, Front. Bioeng. Biotechnol., 2020, 8, 203 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |