
Cationic amphiphilic alternating copolymers with tunable morphology†
Jingling
Zhang
 a,
Xiaoxi
Yu
b,
Bingqian
Zheng
b,
Jiachun
Shen
b,
Surita R.
Bhatia
b and
Nicole S.
Sampson
*b
a,
Xiaoxi
Yu
b,
Bingqian
Zheng
b,
Jiachun
Shen
b,
Surita R.
Bhatia
b and
Nicole S.
Sampson
*b
aDepartment of Materials Science and Chemical Engineering, Stony Brook University, Stony Brook, New York 11794-2275, USA
bDepartment of Chemistry, Stony Brook University, Stony Brook, New York 11794-3400, USA. E-mail: nicole.sampson@stonybrook.edu
First published on 29th July 2020
Abstract
A series of ionic amphiphilic alternating copolymers were characterized via SAXS, TEM and DLS to help understand factors that could potentially affect self-assembly, including the degree of polymerization, the length of hydrophobic spacers between ionic units, the distance between charged groups and polymer backbone, solvent environment and counterions.
 Nicole S. Sampson | Nicole S. Sampson received her education and training as a chemist and chemical biologist from Harvey Mudd College (BS), the University of California—Berkeley (PhD), and Harvard University (postdoctoral fellow). She is currently Distinguished Professor of the Chemistry and Dean of the College of Arts and Sciences at Stony Brook University. She is a Fellow of the American Chemical Society. Her research interests include the design of chemical probes of mammalian fertilization and exploiting metabolic pathways that enable survival of Mycobacterium tuberculosis for drug discovery. Her work with polymers in fertilization led to the development of new methodology for the synthesis of alternating copolymers by ring-opening metathesis polymerization from 1-substituted cyclobutenes and unstrained cycloalkenes. |
Amphiphilic polymers contain both hydrophobic and hydrophilic components and are of great interest not only because of their potential applications in the fields of drug delivery, catalysis, coatings, and cosmetics,1–5 but also because studying their chemistry provides insight into the mechanism of formation of the higher-order structures of amphiphilic biomacromolecules such as DNA, RNA, proteins, and polysaccharides.6 Such insight can in turn facilitate the development of new therapeutics. For example, the sophisticated, well-defined higher-order structures of proteins, which are amphiphilic biomacromolecules with hydrophobic and ionizable hydrophilic components, arise from finely tuned electrostatic interactions among hydrophilic units that are precisely located along the chains. Mimicking the interactions involved in the formation of such structures in living organisms can be expected to lead to the establishment of new methods for developing medicines, especially in the fields of drug delivery and gene therapy.7–10
Self-assembly, the spontaneous and reversible organization of molecular units into ordered structures in a process driven by noncovalent interactions, is often seen in amphiphiles. This process has been extensively studied not only because it is crucial for understanding many important biological structures but also because it offers strategies for organizing matter on a large scale.11 Self-assembly of amphiphiles happens because the energies of the interactions of their hydrophobic and hydrophilic components with the solvent surface differ.12–16 When the repeat units consist of charged hydrophilic components, self-assembly of polymers can be complicated by a variety of electrostatic phenomena that do not occur in charge neutral systems.17 Ionic amphiphilic systems can be affected by electrostatic repulsion, osmotic swelling, and counterion condensation, as well as by external factors such as changes in solution pH and the presence of salts that screen charges.18 Therefore, it is essential to understand the mechanisms of self-assembly and the variables that affect the formation, solubility, and stability of self-assembled aggregates.
Assemblies of amphiphilic synthetic polymers with various architectures can be carefully constructed. Amphiphilic block copolymers have been widely studied, and they can self-assemble into a diverse array of morphologies, depending on the solvent and the structure of the polymer chain.1–3,5,18–22 Ionic amphiphilic block copolymers have attracted attention because they combine highly tunable structures with the ability to respond to external stimuli such as changes in solvent pH or ionic strength.23–26 Sternhagen et al. has reported a unique method to precisely tailor the structure of small spherical micelles formed by ionic block copolymers by controlling the sequence and position of the ionic monomers.27 Amphiphilic dendritic polymers with various morphologies have also drawn attention.28–31 In addition, homopolymers comprising monomers that contain both hydrophobic and hydrophilic moieties have been synthesized and shown to form micelles or inverse micelles depending on the solvent environment.32
In contrast, there have been only a few studies focused on amphiphilic alternating copolymers.33–36 Examples include the amphiphilic alternating polyesters designed by Wang et al., who used ring-opening polymerization to form micelles with hydrophobic cores and thermosensitive shells in aqueous solution.37 In addition, amphiphilic copolymers consisting of alternating sodium maleate and dodecyl vinyl ether units form unicore or multicore flower micelles depending on the degree of polymerization (DP).38 Furthermore, investigators have carried out molecular dynamics simulations on amphiphilic copolymers and discovered that the aggregates readily adopt compact spherical conformations in which hydrophobic chains cluster at the globular core and hydrophilic groups envelop the core.36 However, the factors that affect self-assembly of amphiphilic alternating copolymers have not been systematically studied.
In previously reported work, our group used alternating ring-opening metathesis polymerization (AROMP) to synthesize quaternary ammonium (QA)-bearing cationic amphiphilic alternating polymers. After expanding the monomer scope of the method, we were able to obtain long, linear alternating copolymers with varied spacing and a diverse array of heteroatomic functionality.39–42 Of particular interest was an AROMP system involving bicyclo[4.2.0]oct-1(8)-ene-8-carboxamides and cycloalkenes, which opened up new opportunity for studies of the morphology of amphiphilic alternating copolymers.
Herein, we report the synthesis of a panel of QA-bearing cationic amphiphilic alternating copolymers via AROMP followed by post-polymerization functionalization to investigate how their morphology was affected by (1) DP, (2) the length of the alkyl side chains (3) the length of the spacer between the cationic units, (4) the solvent, and (5) the counterion (Fig. 1).
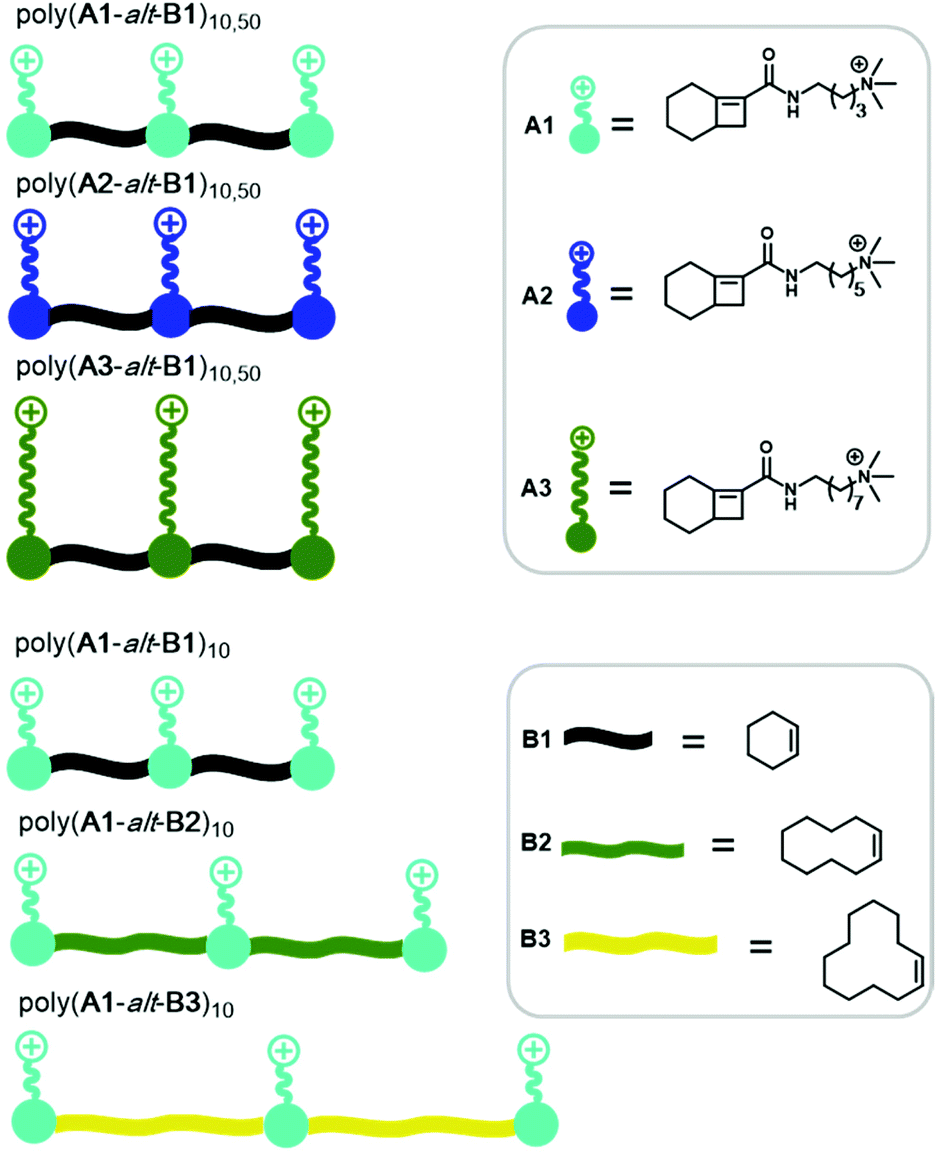 | ||
| Fig. 1 Structures of monomers (before ring-opening) and polymers synthesized and characterized in this work. | ||
The route used to synthesize cationic amphiphilic alternating polymers poly(A-alt-B)n is presented in Scheme 1. First, Cl-containing alternating polymers poly(A′-alt-B)n were synthesized via AROMP of monomers A′ and B with the 3rd generation Grubbs catalyst (3-BrPyr)2Cl2(H2IMes)Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh (G3), and the molecular weights of these intermediate polymers were determined by means of gel permeation chromatography (GPC) (Table 1). All the AROMP reactions reached 100% conversion and yielded linear alternating copolymers with acceptable dispersities and the correct chain lengths according to NMR spectra and GPC. Then the desired cationic amphiphilic copolymers poly(A-alt-B)n were obtained by means of post-polymerization functionalization of the intermediate polymers. Again, conversion was 100%, as indicated by the disappearance of the signal for H1 proton from the 1H NMR spectra of the reaction mixtures. In a control reaction (see General methods and Synthesis in ESI† for control experiment details), aqueous trimethylamine was confirmed unable to alter polymer backbone of this work at 50 °C for 24 h. This result indicates that the backbone of poly(A′-alt-B)n can be expected to remain unchanged by treatment with aqueous trimethylamine and that the only reaction was replacement of the Cl atom with the trimethyl amine moiety to yield poly(A-alt-B)n. The liberated Cl atom remained in the system in the form of a Cl− counterion. Excess trimethylamine and salts were removed by dialysis, and the copolymers were characterized by NMR spectroscopy, which showed them to be free of impurities. Because of the difficulty of obtaining molecular weight data for poly(A-alt-B)n by means of aqueous GPC, the length of each poly(A-alt-B)n was represented by the length of the corresponding intermediate polymer poly(A′-alt-B)n. Integration values for the terminal phenyl groups in the 1H NMR spectra of poly(A-alt-B)n (7.0–7.5 ppm) were also examined to ensure that the cationic copolymers had the expected lengths (see Fig. S12–S27†).
CHPh (G3), and the molecular weights of these intermediate polymers were determined by means of gel permeation chromatography (GPC) (Table 1). All the AROMP reactions reached 100% conversion and yielded linear alternating copolymers with acceptable dispersities and the correct chain lengths according to NMR spectra and GPC. Then the desired cationic amphiphilic copolymers poly(A-alt-B)n were obtained by means of post-polymerization functionalization of the intermediate polymers. Again, conversion was 100%, as indicated by the disappearance of the signal for H1 proton from the 1H NMR spectra of the reaction mixtures. In a control reaction (see General methods and Synthesis in ESI† for control experiment details), aqueous trimethylamine was confirmed unable to alter polymer backbone of this work at 50 °C for 24 h. This result indicates that the backbone of poly(A′-alt-B)n can be expected to remain unchanged by treatment with aqueous trimethylamine and that the only reaction was replacement of the Cl atom with the trimethyl amine moiety to yield poly(A-alt-B)n. The liberated Cl atom remained in the system in the form of a Cl− counterion. Excess trimethylamine and salts were removed by dialysis, and the copolymers were characterized by NMR spectroscopy, which showed them to be free of impurities. Because of the difficulty of obtaining molecular weight data for poly(A-alt-B)n by means of aqueous GPC, the length of each poly(A-alt-B)n was represented by the length of the corresponding intermediate polymer poly(A′-alt-B)n. Integration values for the terminal phenyl groups in the 1H NMR spectra of poly(A-alt-B)n (7.0–7.5 ppm) were also examined to ensure that the cationic copolymers had the expected lengths (see Fig. S12–S27†).
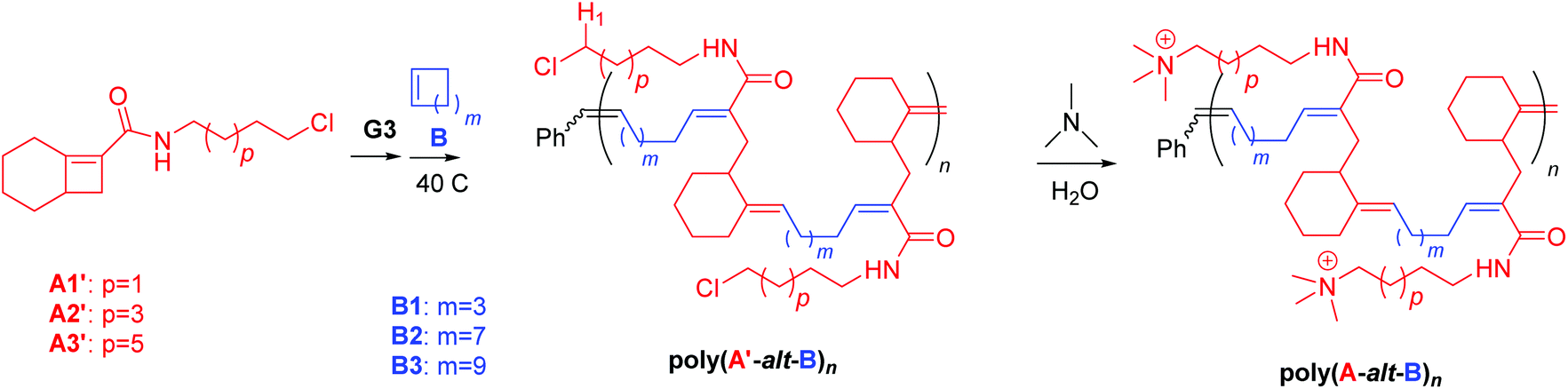 | ||
| Scheme 1 Synthesis of intermediate copolymers poly(A′s-alt-Bs)nvia AROMP and cationic copolymers poly(As-alt-Bs)nvia post-polymerization functionalization. | ||
| A′ | B | [A′]/[B]/[G3]b | Conv.c (%) |
M
n, theo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d (kDa) d (kDa) |
M
n, mease (kDa) |
M
w, measf (kDa) |
Đ M | Reaction time (h) | |
|---|---|---|---|---|---|---|---|---|---|
|
a All preparative polymerization experiments were performed twice. Representative data from a single polymerization experiment are presented in this table.
b G3 is the 3rd-generation Grubbs catalyst (3-BrPyr)2Cl2(H2IMes)RuCHPh.
c Conversion was determined by monitoring the 1H NMR spectrum of the reaction mixture for the disappearance of the amide resonance of monomer A′.
d Theoretical number-average molecular weight calculated from the monomer/catalyst feed ratio.
e Number-average molecular weight determined by GPC with refractive index detection.
f Weight-average molecular weight.
|
|||||||||
| 1 | A1′ | B1 | 10:10:1 |
100 | 3.2 | 3.8 | 4.9 | 1.3 | 2 |
| 2 | A1′ | B1 | 50:50:1 |
100 | 16.2 | 13.0 | 19.5 | 1.5 | 6 |
| 3 | A1′ | B2 | 10:10:1 |
100 | 3.8 | 5.0 | 6.8 | 1.4 | 1 |
| 4 | A1′ | B3 | 10:10:1 |
100 | 4.1 | 5.0 | 7.0 | 1.4 | 6 |
| 5 | A2′ | B1 | 10:10:1 |
100 | 3.5 | 4.0 | 5.6 | 1.4 | 2 |
| 6 | A2′ | B1 | 50:50:1 |
100 | 17.6 | 19.0 | 28.0 | 1.5 | 6 |
| 7 | A3′ | B1 | 10:10:1 |
100 | 3.8 | 5.0 | 7.4 | 1.5 | 3 |
| 8 | A3′ | B1 | 50:50:1 |
100 | 19.0 | 22.0 | 32.0 | 1.5 | 12 |
The effects of DP, spacing between charged units, and side-chain length on the structure of the cationic alternating copolymers were studied by means of small-angle X-ray scattering (SAXS) analysis of samples dissolved in deionized water and in 100 mM aqueous NaCl. In addition, counterions with various charges were used to determine the effect of the counterion on copolymer self-assembly. The structures of the copolymers are shown in Fig. 1. Two independent replicates of each copolymer sample were analyzed by SAXS, and the results appeared to be repeatable; therefore, data from one replicate per sample are reported in Table 1. Sample concentrations of 0.5 and 1.0 wt% in the deionized water were tested, and the structural results were similar for the two concentrations. Therefore, to achieve the best resolution, the data for the 1.0 wt% samples were used for the SAXS analysis. Data points with large uncertainties due to low signal intensities in the region of high scattering vector (q) were eliminated from the analysis. Detailed SAXS analysis was performed over a q range of 0.01–0.5 Å−1, which can provide information about structural features in the 12.56–628 Å range.
The SAXS curves of assemblies of poly(A1-alt-B1)10, poly(A1-alt-B2)10, and poly(A1-alt-B3)10 were investigated to study the influence of the spacing between the charged units (Fig. 2). A major peak appeared in the q region of 0.02–0.04 Å−1, indicating large structures with a characteristic size of 16–31 nm (d = 2π/qpeak). There was a minor peak in the q region of 0.10–0.12 Å−1, attributable to secondary structures with average sizes of 5–6 nm. As the spacing along the backbone between the cationic units was increased from 6 carbons to 12 carbons, both the major peak and the minor peak shifted to lower q, suggesting that on average, the lengths of the major and minor structures of the copolymer assemblies increased. On the basis of these results, the sets of SAXS data were fitted to a core–shell ellipsoid model with a hard sphere structure factor (Fig. 2). The model includes a major equatorial radius and a minor polar radius. The hard sphere structure factor provides information about the volume fraction occupied by the spheres and monodisperse spherical particles interacting through excluded-volume interactions. The results (with errors on fitting parameters) based on goodness-of-fit are reported in Table 2 (entries 1–3).
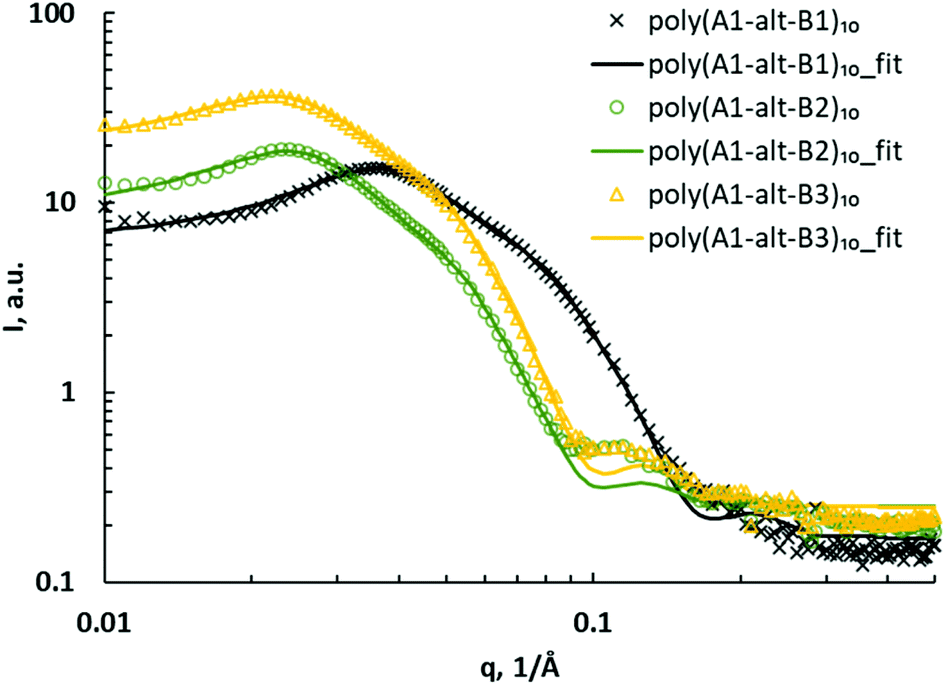 | ||
| Fig. 2 SAXS data for poly(A1-alt-Bs)10 assemblies (symbols) and corresponding fits to a core–shell ellipsoid model with a hard sphere structure factor (curves). | ||
| Polymers |
R
eqb (nm) |
R
pc (nm) |
t
shelld (nm) |
φ | D (nm) | |
|---|---|---|---|---|---|---|
| a The results (with errors on fitting parameters) are based on goodness-of-fit. b Equatorial radius. c Polar radius. d Shell thickness. e Volume fraction. f Diameter of ellipsoid, in equatorial direction, calculated as two times the sum of the equatorial radius and shell thickness. | ||||||
| 1 | (A1-alt-B1)10 | 2.5 ± 0.2 | 4.8 ± 0.1 | 4.1 ± 0.3 | 0.1 ± 0.007 | 17.9 |
| 2 | (A1-alt-B2)10 | 4.1 ± 0.3 | 8.2 ± 0.2 | 5.5 ± 0.5 | 0.1 ± 0.008 | 27.4 |
| 3 | (A1-alt-B3)10 | 4.2 ± 0.2 | 7.9 ± 0.08 | 6.3 ± 0.3 | 0.1 ± 0.004 | 28.2 |
| 4 | (A2-alt-B1)10 | 1.8 ± 0.3 | 4.5 ± 0.2 | 2.4 ± 0.08 | 0.08 ± 0.02 | 14.9 |
| 5 | (A3-alt-B1)10 | 2.9 ± 0.2 | 5.2 ± 0.1 | 3.9 ± 0.3 | 0.1 ± 0.006 | 18.1 |
| 6 | (A1-alt-B1)50 | 2.0 ± 0.2 | 4.4 ± 0.1 | 3.3 ± 0.3 | 0.2 ± 0.009 | 15.3 |
| 7 | (A2-alt-B1)50 | 2.1 ± 0.3 | 5.3 ± 0.2 | 4.1 ± 0.05 | 0.1 ± 0.01 | 19.0 |
| 8 | (A3-alt-B1)50 | 2.1 ± 0.4 | 6.9 ± 0.3 | 4.7 ± 0.09 | 0.2 ± 0.02 | 23.2 |
The ellipsoidal poly(A1-alt-Bs)10 assemblies were not very elongated: the polar radius was approximately half the equatorial radius, and the average diameter was in the 18–28 nm range. The SAXS results confirmed that as the spacing between the charged units increased, the overall diameter of the ellipsoids increased and the shell thickness slightly increased. The core–shell aspect reflects the fact that the ellipsoidal assemblies of the 10-mers consisted of two components —a core and a shell—each with a different scattering length density. The average sizes of the copolymer assemblies in deionized water were measured by means of dynamic light scattering (DLS) at a scattering angle of 173°, which was chosen to minimize the effects of dusts and aggregation of polymer assemblies. The DLS data were similar to SAXS results, further confirming the confidence of SAXS fitting (Table S1†).
To determine whether the distance between the charged headgroups and the polymer backbone affected self-assembly of the cationic alternating copolymers, a series of poly(As-alt-B1)10 assemblies were characterized via SAXS (Fig. 3). Unlike the hydrocarbon spacings between charged units that collapse in the hydrophobic domain, the side chains bearing charged headgroups are amphiphilic and extend in water due to repulsion between charges. The upturn shape of the SAXS curve in the low-q region (0.01–0.02 Å−1) for the poly(A2-alt-B1)10 assemblies suggests that the copolymers formed aggregates in solution,43 and therefore the data for the assemblies of this polymer were not fitted to the model. The assemblies of the other copolymers in this series showed major and minor structures similar to those observed for the poly(A1-alt-Bs)10 assemblies and therefore were also fitted using a core–shell ellipsoid model with a hard sphere structure factor (Fig. 3). There was no significant relationship between assembly size and the distance between the charged headgroups and the polymer backbone for the 10-mers (Table 2, entries 1, 4, and 5); this result can be explained by the fact that the (As-alt-B1)10 assemblies were relatively small.
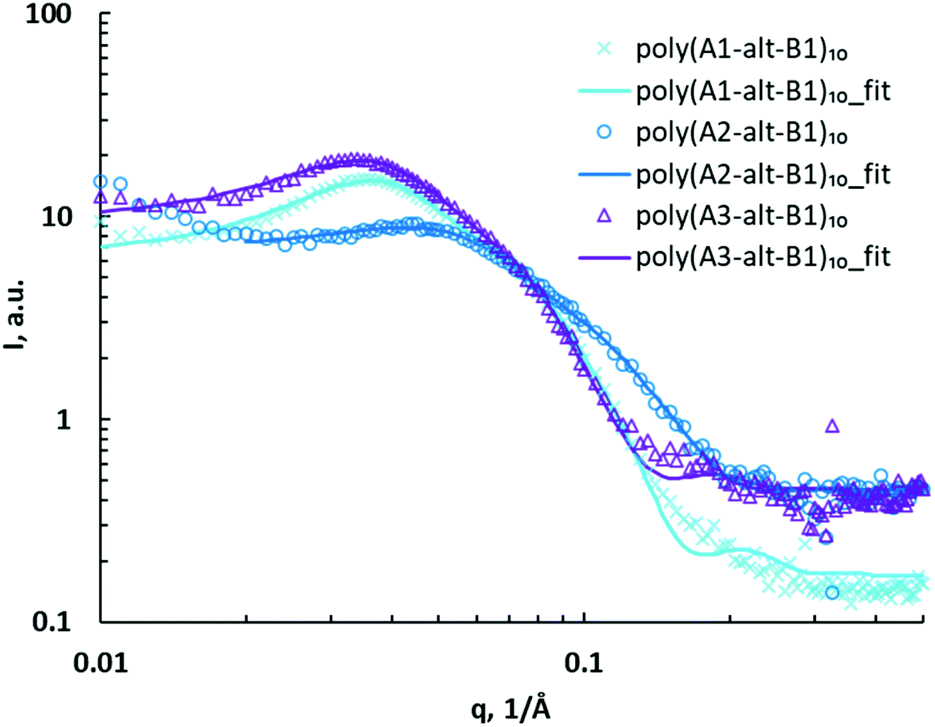 | ||
| Fig. 3 SAXS data for poly(As-alt-B1)10 assemblies (symbols) and fits to a core–shell ellipsoid model with a hard sphere structure factor (curves). | ||
Poly(As-alt-B1)50 assemblies were studied via SAXS, and the data were compared with those for the assemblies of the corresponding 10-mers to determine how DP affected self-assembly. The SAXS data for the 50-mer assemblies showed major and minor peaks in a q region similar to those observed for the poly(As-alt-B1)10 assemblies, indicating that increasing the polymer chain length had little effect on assembly size. The 50-mer assemblies showed significant structural differences due to side-chain variation (Fig. 4). As the distance between the charged headgroups and the hydrophobic backbone increased, both the major peak and the minor peak shifted to lower q, suggesting that the structures enlarged. The core–shell ellipsoid model with a hard sphere structure factor was used to fit the SAXS data for the 50-mer assemblies (Table 2, entries 6–8). The upturn shape at low q (0.01–0.02 Å−1) may have been caused by the formation of aggregation of assemblies that did not fit well with the core–shell ellipsoid model. SAXS fitting further confirmed that the ellipsoidal assemblies formed by the 50-mers were similar in size to or smaller than the corresponding 10-mer assemblies, suggesting that the 50-mer assemblies were more compact. The average sizes of the 50-mer assemblies were also measured by means of DLS, but the diameters obtained in this way were much larger than those determined by SAXS analysis, a discrepancy that may have been due to aggregation of polymer assemblies (Table S1†).
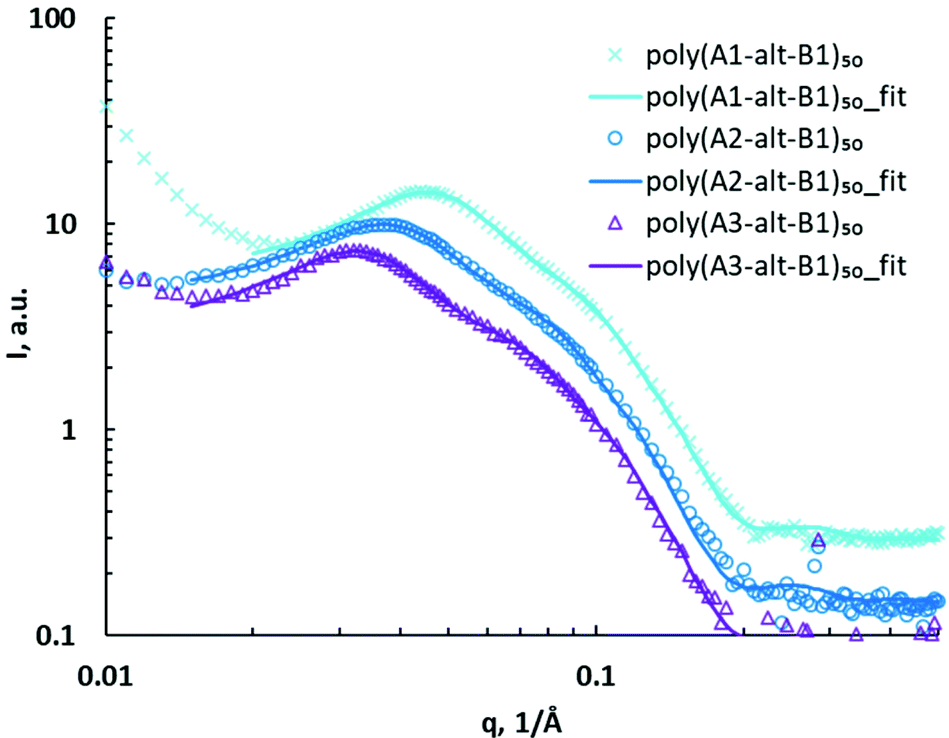 | ||
| Fig. 4 SAXS data for (As-alt-B1)50 assemblies (symbols) and corresponding fits using core–shell ellipsoid model with hard sphere structure factor (curves). | ||
Solutions of 10-mer and 50-mer cationic alternating copolymers in deionized water were dried on grids and analyzed by transmission electron microscopy (TEM) (Fig. S30 and S31†); representative images are shown in Fig. 5. The images of the 10-mers showed hollow ellipsoids (vesicles) with diameters ranging from 15 to 30 nm, which agrees well with the SAXS data (Table 2, entries 1–5). In contrast, the images of the 50-mers showed solid ellipsoids (micelles) with diameters that were consistent with those determined by SAXS analysis (Table 1, entries 6–8). Although the structures of 10-mers appear to be vesicles in TEM, we noted that our SAXS fits could be interpreted as a micelle where the core is not very dense and is highly swollen with solvent. In either case, the 10-mers form an assembly with a high fraction of solvent in the core, and a structure that is less dense than the 50-mers. Taken together, the TEM and SAXS analyses reveal that in deionized water, the 10-mers formed loosely-packed vesicles/micelles, whereas the 50-mers self-assembled into densely-packed micelles with a hydrophobic core and a hydrophilic shell (Fig. 6).32 The TEM images showing solid ellipsoid morphology further support the conclusion, based on the SAXS data, that the assemblies of 50-mers were more compact because the hydrophobic segments clustered and coiled in the core. The difference between the morphologies of the 10-mer and 50-mer assemblies can be explained in terms of the critical packing parameter (p), which is an important property of self-assembled aggregates. This parameter, which can be used as a theoretical framework for determining the type of aggregates formed by amphiphilic molecules, is given by the following equation:
| p = v/al |
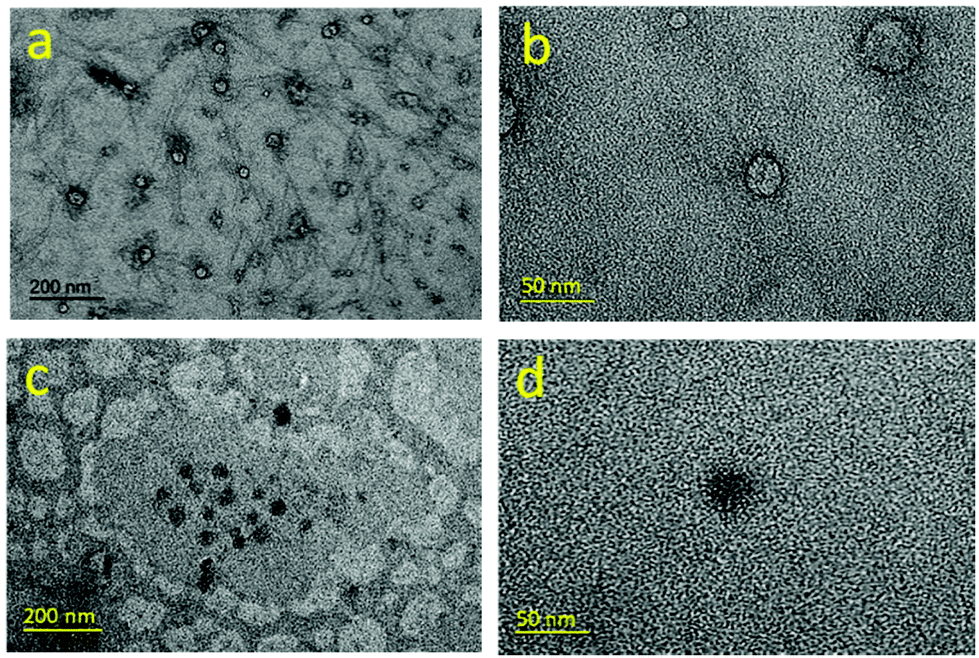 | ||
| Fig. 5 TEM images for poly(A2-alt-B1)10 (a, b) and poly(A2-alt-B1)50 (c, d). 1 wt% samples in deionized water were dried on copper grids for TEM imaging. | ||
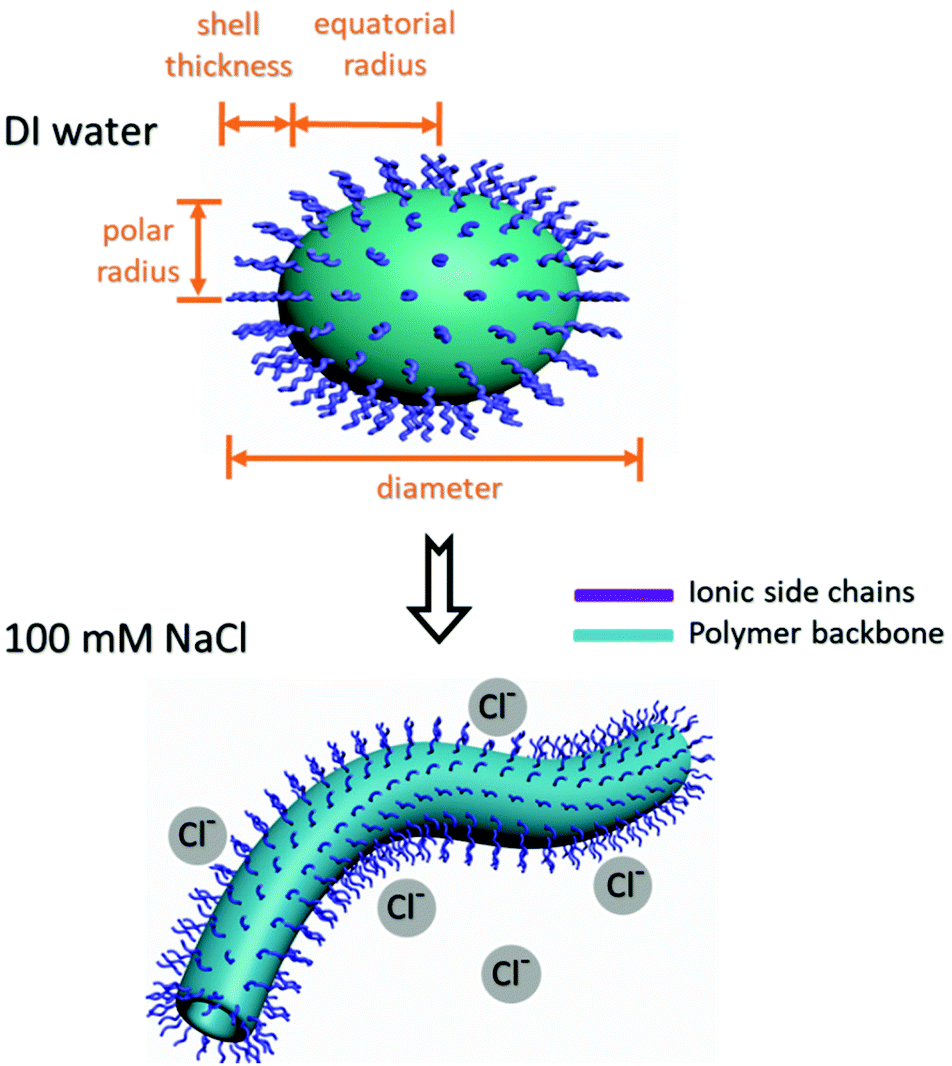 | ||
| Fig. 6 Schematic illustration of ellipsoidal vesicles/micelles self-assembled from 10-mers and 50-mers in deionized (DI) water (upper panel where the core and the shell consisted of hydrophobic and hydrophilic segments, respectively. Both the 10-mer and the 50-mers self-assembled into cylinders in 100 mM aqueous NaCl (lower panel); both assemblies transformed into cylinders in 100 mM NaCl solution. | ||
Self-assembly of copolymers in 100 mM NaCl was also characterized by means of SAXS, and under these conditions, the SAXS curves showed no major peaks (Fig. S32†). The data for all polymers (in Fig. 1) were well fitted by a cylinder model44 (as shown by the goodness-of-fit data for the cylinder lengths and radii listed in Table S2†). These copolymers formed ellipsoidal micelles/vesicles in water because they have charged headgroups. In contrast, in 100 mM NaCl solution, the ions could partially screen the inter-headgroup repulsion, and as a result, the polymers formed cylindrical micelles/vesicles instead of ellipsoidal micelles/vesicles (Fig. 6).45 There was no obvious trend in the length or radius of the cylindrical micelles as DP, lengths of side chains and lengths of spacers change, which is consistent with the fact that cylindrical polymer micelles are usually polydisperse because their growth is sensitive to copolymer chain length and to the temperature and ionic strength of the medium.45
The above-described cationic alternating copolymers had Cl− as a counterion. In later experiments, the Cl− in selected copolymers was replaced with SO42− or PO43− to determine whether the charge of the counterion affected self-assembly of the copolymers in deionized water. Complete exchange of the counterions was confirmed by elemental analysis, which revealed that there was no residual Cl− or any other impurities in the aqueous polymer solutions. The resulting copolymers were characterized by means of SAXS, and the data were fitted to a cylinder model (Table S3 and Fig. S33†). The screening effect introduced by counterions increased as the counterion charge was increased from −1 to −3; as a result, the cationic side chains were more shielded from inter-headgroup repulsion, and thus cylinders formed in water instead of ellipsoids. There was no specific trend of morphological changes of assemblies when the counterion charges increased from −2 to −3.
Conclusions
We synthesized a series of cationic amphiphilic alternating copolymers and investigated various factors with the potential to affect their self-assembly, including DP, the distance between charged units along the polymer backbone, the distance between the charged headgroups and the backbone, and solvents and counterions. SAXS and TEM results indicated that the copolymers self-assembled into loosely-packed ellipsoidal vesicles/micelles when the DP was 10 and into densely-packed ellipsoidal micelles when the DP was increased to 50. The assemblies increased in size as the spacing between charged groups was increased; increasing the distance between the charged headgroups and the backbone had little effect on the morphology of the 10-mer assemblies but caused the 50-mer assemblies to increase in size. Switching the solvent from deionized water to 100 mM aqueous NaCl and increasing the charge number of the counterion resulted in the copolymer morphology changing from ellipsoidal to cylindrical, owing to charge-screening effects. This study of factors that affect self-assemblies of ionic alternating copolymers could potentiate deeper understanding of sequence-specific macromolecules with complex architectures and shed light on the development of new therapeutics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by NSF CHE1609494 (NSS), NIH R01GM097971 (NSS) and ACS PRF 55729-ND9 (SRB). The SAXS measurements used beamline 16-D, LIX, of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. LIX is part of the Center for BioMolecular Structure (CBMS), primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010). TEM characterization in this work made use of the ThINC facility of AERTC at Stony Brook University. Shearson Editorial Services (Cornwall, NY, USA) provided English language editing of the text of this paper.References
- A. Rösler, G. W. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- K. Holmberg, in Amphiphilic block copolymers: self-assembly and applications, ed. P. Alexandridis and B. Lindman, Elsevier, Amsterdam, 2000, ch. 13, pp. 305–318 Search PubMed.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343–1355 CrossRef CAS PubMed.
- Y.-C. Tan, K. Hettiarachchi, M. Siu, Y.-R. Pan and A. P. Lee, J. Am. Chem. Soc., 2006, 128, 5656–5658 CrossRef CAS PubMed.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- A. M. de Graff, M. J. Hazoglou and K. A. Dill, Structure, 2016, 24, 329–336 CrossRef CAS PubMed.
- U. Wendler, J. Bohrisch, W. Jaeger, G. Rother and H. Dautzenberg, Macromol. Rapid Commun., 1998, 19, 185–190 CrossRef CAS.
- J.-I. Murata, Y. Ohya and T. Ouchi, Carbohydr. Polym., 1996, 29, 69–74 CrossRef CAS.
- Q. Yang, S. Wang, P. Fan, L. Wang, Y. Di, K. Lin and F.-S. Xiao, Chem. Mater., 2005, 17, 5999–6003 CrossRef CAS.
- M. A. Wolfert, E. H. Schacht, V. Toncheva, K. Ulbrich, O. Nazarova and L. W. Seymour, Hum. Gene Ther., 1996, 7, 2123–2133 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- J. N. Israelachvili, in Intermolecular and Surface Forces, ed. J. N. Israelachvili, Academic Press, San Diego, 3rd edn, 2011, vol. III, ch. 19, pp. 503–534 Search PubMed.
- M. Muthukumar, C. Ober and E. Thomas, Science, 1997, 277, 1225–1232 CrossRef CAS.
- D. G. Bucknall and H. L. Anderson, Science, 2003, 302, 1904–1905 CrossRef CAS PubMed.
- M. G. McKee, J. M. Layman, M. P. Cashion and T. E. Long, Science, 2006, 311, 353–355 CrossRef CAS PubMed.
- D. W. Löwik and J. C. van Hest, Chem. Soc. Rev., 2004, 33, 234–245 RSC.
- O. V. Borisov, E. B. Zhulina, F. A. Leermakers and A. H. Müller, in Self organized nanostructures of amphiphilic block copolymers I, ed. A. H. E. Müller and O. V. Borisov, Springer-Verlag Berlin Heidelberg, 2011, vol. 241, pp. 57–129 Search PubMed.
- S. Förster, N. Hermsdorf, C. Böttcher and P. Lindner, Macromolecules, 2002, 35, 4096–4105 CrossRef.
- A. Sundararaman, T. Stephan and R. B. Grubbs, J. Am. Chem. Soc., 2008, 130, 12264–12265 CrossRef CAS PubMed.
- D. J. Pochan, Z. Chen, H. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94–97 CrossRef CAS PubMed.
- B. L. Sanchez-Gaytan, S. Li, A. C. Kamps, R. J. Hickey, N. Clarke, M. Fryd, B. B. Wayland and S.-J. Park, J. Phys. Chem. C, 2011, 115, 7836–7842 CrossRef CAS.
- J.-H. Ryu, R. Roy, J. Ventura and S. Thayumanavan, Langmuir, 2010, 26, 7086–7092 CrossRef CAS PubMed.
- P. Guenoun, H. T. Davis, M. Tirrell and J. W. Mays, Macromolecules, 1996, 29, 3965–3969 CrossRef CAS.
- K. Khougaz, I. Astafieva and A. Eisenberg, Macromolecules, 1995, 28, 7135–7147 CrossRef CAS.
- J. E. Laaser, Y. Jiang, D. Sprouse, T. M. Reineke and T. P. Lodge, Macromolecules, 2015, 48, 2677–2685 CrossRef CAS.
- A. S. Lee, V. Bütün, M. Vamvakaki, S. P. Armes, J. A. Pople and A. P. Gast, Macromolecules, 2002, 35, 8540–8551 CrossRef CAS.
- G. L. Sternhagen, S. Gupta, Y. Zhang, V. John, G. J. Schneider and D. Zhang, J. Am. Chem. Soc., 2018, 140, 4100–4109 CrossRef CAS PubMed.
- J. Teng and E. R. Zubarev, J. Am. Chem. Soc., 2003, 125, 11840–11841 CrossRef CAS PubMed.
- A. P. Schenning, C. Elissen-Roman, J.-W. Weener, M. W. Baars, S. J. van der Gaast and E. Meijer, J. Am. Chem. Soc., 1998, 120, 8199–8208 CrossRef CAS.
- T. Wei, C. Chen, J. Liu, C. Liu, P. Posocco, X. Liu, Q. Cheng, S. Huo, Z. Liang and M. Fermeglia, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2978–2983 CrossRef CAS PubMed.
- V. Percec, D. A. Wilson, P. Leowanawat, C. J. Wilson, A. D. Hughes, M. S. Kaucher, D. A. Hammer, D. H. Levine, A. J. Kim and F. S. Bates, Science, 2010, 328, 1009–1014 CrossRef CAS PubMed.
- E. N. Savariar, S. V. Aathimanikandan and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 16224–16230 CrossRef CAS PubMed.
- C. Li, C. Chen, S. Li, T. Rasheed, P. Huang, T. Huang, Y. Zhang, W. Huang and Y. Zhou, Polym. Chem., 2017, 8, 4688–4695 RSC.
- Y. Morishima, T. Kobayashi, S. Nozakura and S. E. Webber, Macromolecules, 1987, 20, 807–813 CrossRef CAS.
- J. Chen, C. Yu, Z. Shi, S. Yu, Z. Lu, W. Jiang, M. Zhang, W. He, Y. Zhou and D. Yan, Angew. Chem., 2015, 127, 3692–3696 CrossRef.
- V. V. Vasilevskaya, A. A. Klochkov, A. A. Lazutin, P. G. Khalatur and A. R. Khokhlov, Macromolecules, 2004, 37, 5444–5460 CrossRef CAS.
- W. Wang, J. Ding, C. Xiao, Z. Tang, D. Li, J. Chen, X. Zhuang and X. Chen, Biomacromolecules, 2011, 12, 2466–2474 CrossRef CAS PubMed.
- M. Ueda, A. Hashidzume and T. Sato, Macromolecules, 2011, 44, 2970–2977 CrossRef CAS.
- L. Chen, L. Li and N. S. Sampson, J. Org. Chem., 2018, 83, 2892–2897 CrossRef CAS PubMed.
- J. Zhang, G. Li and N. S. Sampson, ACS Macro Lett., 2018, 7, 1068–1072 CrossRef CAS PubMed.
- L. Tan, K. A. Parker and N. S. Sampson, Macromolecules, 2014, 47, 6572–6579 CrossRef CAS PubMed.
- G. Li and N. S. Sampson, Macromolecules, 2018, 51, 3932–3940 CrossRef CAS PubMed.
- L. Mourey, J. D. Pédelacq, C. Fabre, H. Causse, P. Rougé and J. P. Samama, Proteins: Struct., Funct., Bioinf., 1997, 29, 433–442 CrossRef CAS.
- J. S. Pedersen, Adv. Colloid Interface Sci., 1997, 70, 171–210 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and surface forces, Academic Press, Oxford, 3rd edn, 2015 Search PubMed.
- N. P. Truong, J. F. Quinn, M. V. Dussert, N. B. Sousa, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2015, 4, 381–386 CrossRef CAS.
- N. P. Truong, M. R. Whittaker, A. Anastasaki, D. M. Haddleton, J. F. Quinn and T. P. Davis, Polym. Chem., 2016, 7, 430–440 RSC.
- S. Y. Khor, N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2017, 6, 1013–1019 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, characterization results including NMR, GPC, SAXS, TEM, DLS. See DOI: 10.1039/d0py00782j |
| This journal is © The Royal Society of Chemistry 2020 |