DOI:
10.1039/D0BM00656D
(Paper)
Biomater. Sci., 2020,
8, 4206-4215
Synthesis of cyclic graft polymeric prodrugs with heterogeneous grafts of hydrophilic OEG and reducibly conjugated CPT for controlled release†
Received
25th April 2020
, Accepted 2nd June 2020
First published on 3rd June 2020
Abstract
Fabrication of cyclic graft (cg) copolymer-based polymeric prodrugs by conjugation of drug molecules to cg copolymers via a dynamic covalent bond capable of responding to biorelevant signals integrates simultaneously the merits of cg copolymers and polymeric prodrugs for enhanced stability of nanocarriers and precise modulation of drug release kinetics. To completely eliminate the compromised drug conjugation efficiency due to the steric hindrance of hydrophilic grafts, it will be useful to develop cg polymeric prodrugs with heterogeneous grafts composed of hydrophilic polymers and drug species, respectively. For this purpose, we reported in this study the synthesis of cyclic graft polymeric prodrugs with heterogeneous grafts of hydrophilic oligo (ethylene glycol) (OEG) and reducibly conjugated camptothecin (CPT), cg-poly(oligo(ethylene glycol) monomethyl ether methacrylate)-b-poly((2-hydroxyethyl methacrylate)-disulfide link-camptothecin) (cg-P(OEGMA)-b-P(HEMA-SS-CPT), cg-prodrugs), via an integrated strategy of a previously reported diblock copolymer-based template and post-polymerization intermolecular click conjugation of a reducible CPT prodrug. The micelles self-assembled from cg-prodrugs on one hand had sufficient salt stability due to the branched cg structure, and on the other hand showed a reduction-triggered cleavage of the disulfide link for a promoted CPT release. Most importantly, we uncovered two interesting phenomena of the cg-based polymeric prodrugs as delivery vehicles: (i) the dimensions of both self-assemblies formed by the cg and bottlegraft (bg) polymers depend substantially on the molecular size of the cg and bg polymers likely due to the steric hindrance of the grafted structures of the cg and bg molecules and relatively low aggregation number of the self-assembled structures, and (ii) cg-prodrug-based micelles exhibited greater in vitro cytotoxicity against cancer cells despite the lower drug loading content (DLC) than the bg-based analogues, which results primarily from the faster reduction-triggered degradation and drug release as well as the greater cellular uptake efficiency of the former micelle prodrugs. Taken together, the developed cg-prodrugs provide great potential for chemotherapy, and the aforementioned interesting results will definitely inspire more upcoming studies on the future design and development of novel cg polymers for biomedical applications.
Introduction
Cyclic graft (cg) copolymers with radiating grafts densely attached onto a cyclic core have attracted great research interest in recent years due to their unique topology-generated self-assembly behaviors1–5 and better performance for potential biomedical applications relative to the bottlegraft (bg) analogues.6–11 The elegant combination of various controlled living radical polymerization (CRP) techniques and intra- and inter-molecular click coupling has enabled the recent rapid development of cg copolymers12–16 with tailor-made structures and compositions based on various polymer species including cg copolymers with homogeneous12,13,15,17–20 and heterogeneous5,7 grafts. Fabrication of cg copolymer-based polymeric prodrugs by conjugation of drug molecules to the cg copolymers via a dynamic covalent bond capable of responding to biorelevant signals integrates simultaneously the merits of cg copolymers and polymeric prodrugs for enhanced stability of nanocarriers and precise modulation of drug release kinetics. One notable example is Pun's17cg polymeric prodrugs with homogeneous poly(oligo(ethylene glycol) monomethyl ether methacrylate) (P(OEGMA)) grafts for efficient in vitro anticancer drug delivery, but this construct suffered from compromised drug conjugation efficiency and low drug loading capacity of 2–5% due to the steric hindrance of P(OEGMA) grafts. To completely eliminate the negative effect of hydrophilic grafts on drug conjugation, it will be useful to develop cg polymeric prodrugs with heterogeneous grafts composed of hydrophilic polymers and drug species, respectively. For this purpose, we reported in this study the synthesis of cyclic graft polymeric prodrugs with heterogeneous grafts of hydrophilic oligo (ethylene glycol) (OEG) and reducibly conjugated camptothecin (CPT), (cg-P(OEGMA)-b-P(HEMA-SS-CPT), cg-prodrugs), via an integrated strategy of a previously reported diblock copolymer-based template7 and post-polymerization intermolecular click conjugation of a reducible CPT prodrug.21 The self-assembly behaviors and in vitro anticancer efficiency of the resulting cg-prodrugs were further compared with those of the bg-analogues.
Results and discussion
Synthesis and characterization of cg and bg-prodrugs
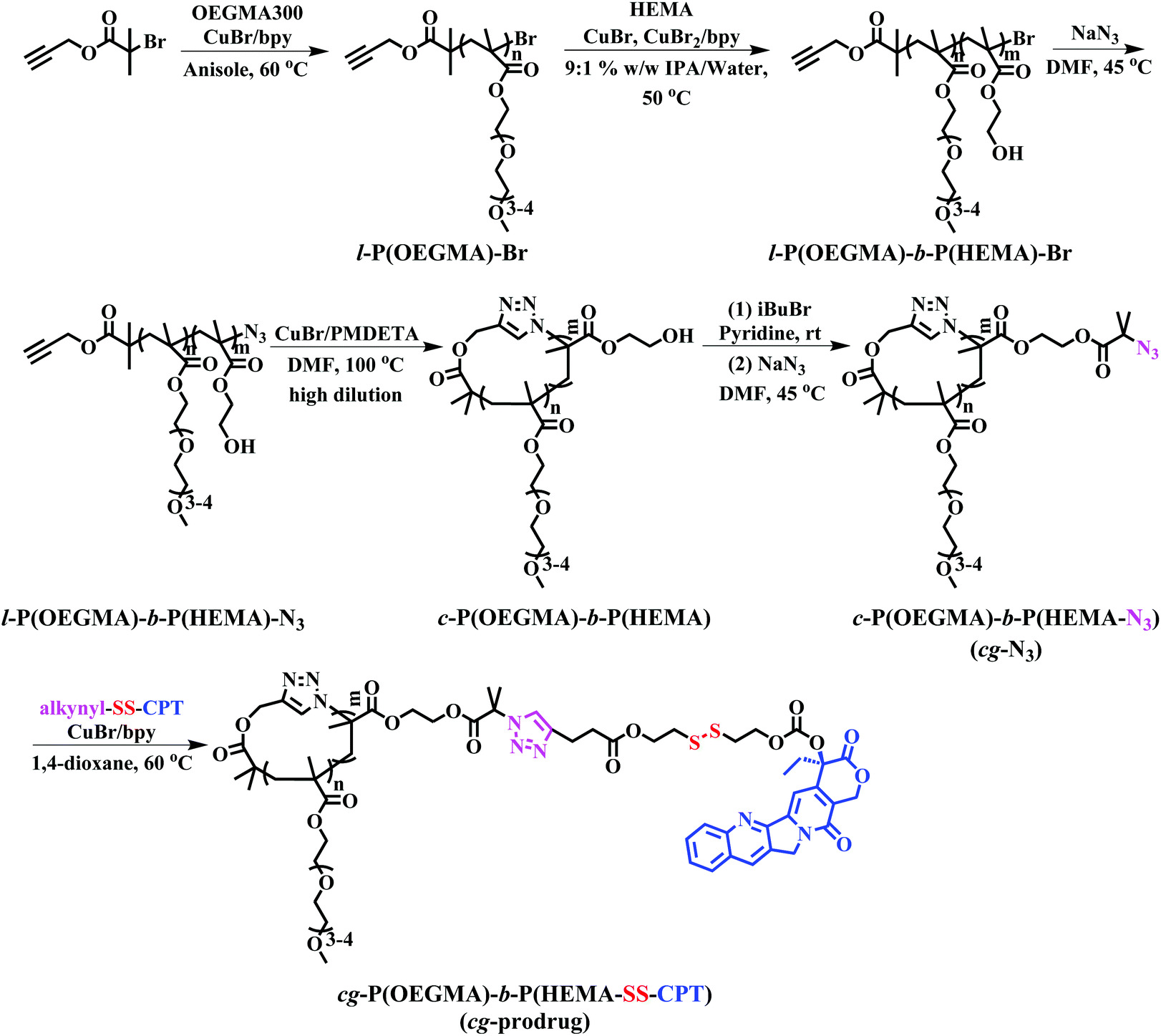
The preparation of cg-prodrug, cg-P(OEGMA)-b-P(HEMA-SS-CPT), consisted mainly of four steps (Scheme 1): (i) synthesis of a linear precursor, l-P(OEGMA)-b-P(HEMA)-N3, by successive ATRPs of OEGMA and HEMA and subsequent azidation of the bromine terminus of the synthesized l-P(OEGMA)-b-P(HEMA)-Br, (ii) intrachain click cyclization of the linear precursor to afford a diblock copolymer-based cyclic template, c-P(OEGMA)-b-P(HEMA), (iii) conversion of the pendant hydroxyl groups of HEMA units in c-P(OEGMA)-b-P(HEMA) to highly reactive azide functions by an esterification reaction with 2-bromoisobutyryl bromide (iBuBr), followed by an azidation with sodium azide, and (iv) a reducible CPT prodrug with a disulfide-linked alkynyl terminus denoted as alkynyl-SS-CPT was conjugated to the cg copolymer with the azide side groups synthesized in the third step via an intermolecular click coupling. The bg analogue, bg-P(OEGMA)-b-P(HEMA-SS-CPT), was synthesized following identical procedures to the aforementioned synthesis of cg-prodrug except using ethyl 2-azidoisobutyrate (EAB) end-capped l-P(OEGMA)-b-P(HEMA)-Br for the drug conjugation in the last step (Scheme S2†).
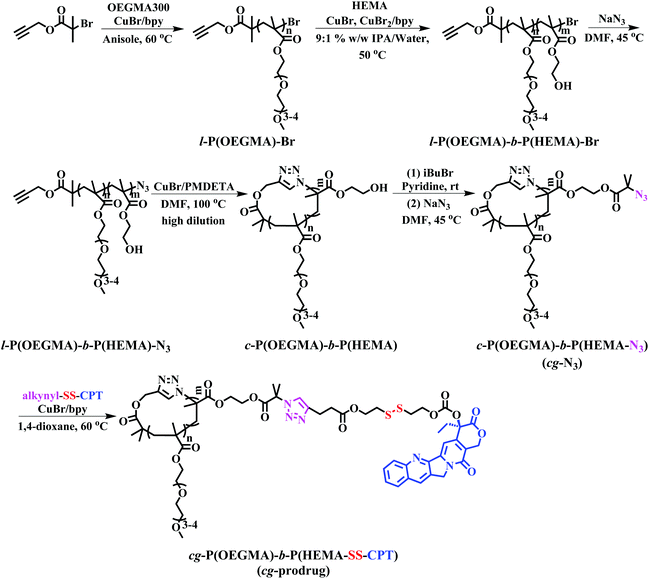 |
| | Scheme 1 Synthesis of cg-prodrug. | |
A precise synthesis of α-alkynyl, ω-azide linear precursors with fully functionalized termini is crucial for the successful synthesis of cyclic polymers with high purity via an intrachain click cyclization.22,23 To ensure the maintenance of the bromine terminus of the linear diblock copolymer prepared by consecutive ATRPs for the subsequent azidation, a low monomer conversion was used for both ATRP processes because of the previously reported successful ATRP production of polymers without any loss of highly reactive chain ends at low monomer conversions.24 Following this synthesis guideline, l-P(OEGMA)-b-P(HEMA)-Br was thus synthesized at relatively low monomer conversions for both polymerization steps. Adoption of optimized reaction conditions for each polymerization step is supported by the kinetic studies (Fig. S2 and S3†).
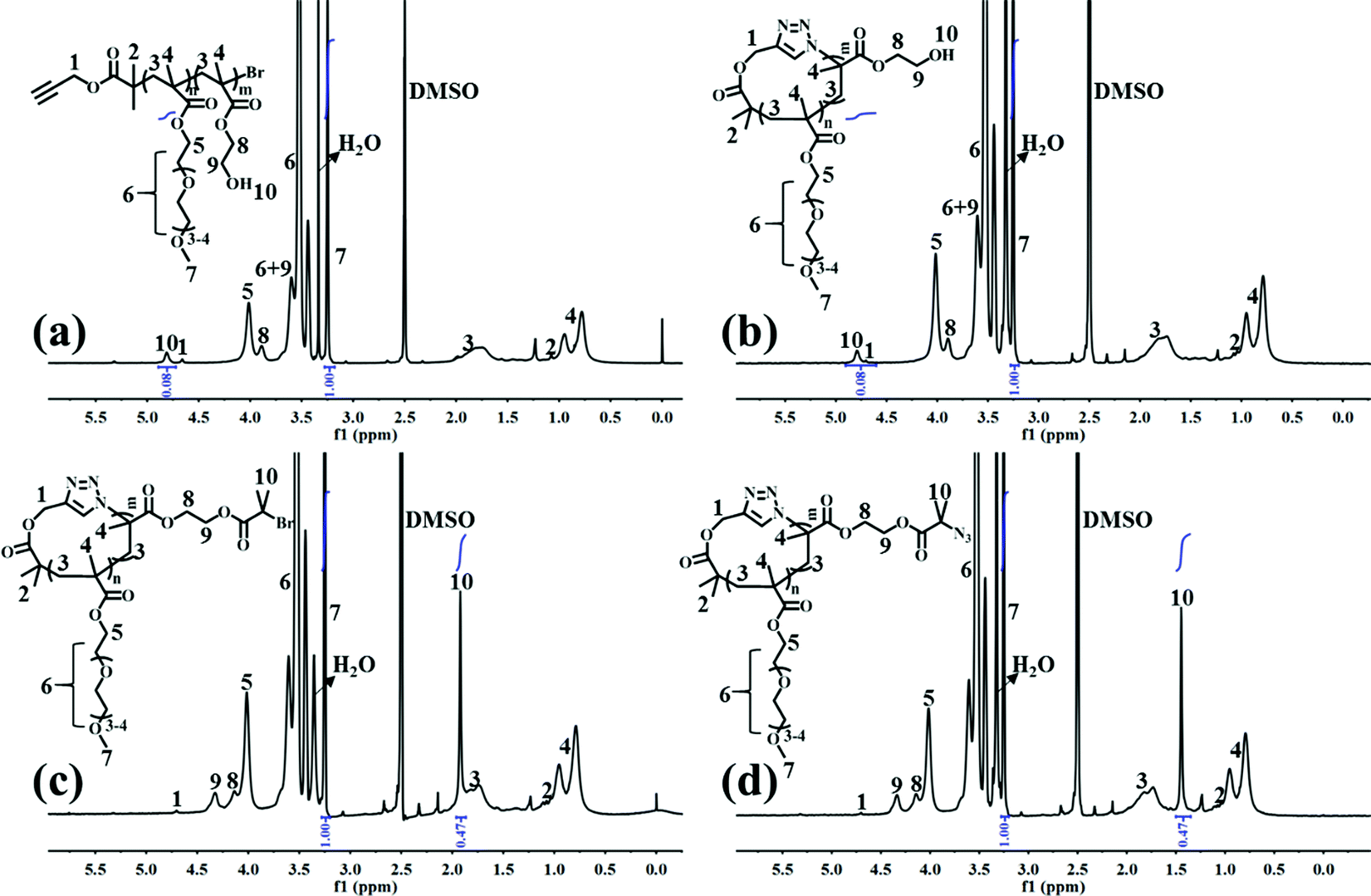
l-P(OEGMA)-Br was first synthesized by ATRP of OEGMA using an alkynyl-terminated initiator.25 All of the l-P(OEGMA)-Br synthesized at various polymerization times show a clear shift of the SEC elution peaks toward a greater molecular weight (MW) (shorter retention times) with unimodal and narrow-distributed MWs (Fig. S2a†). The degree of polymerization (DP) of l-P(OEGMA)-Br was calculated from the monomer conversion, which was determined by the 1H NMR analysis of unpurified reaction mixture. Taking 1.5 h of ATRP as an example (Fig. S1†), the monomer conversion was calculated to be 38.0% based on the following eqn (1), where I1 and I2 are the integrated intensity of the methylene protons in the polymerized OEGMA units and unpolymerized OEGMA monomer, respectively. The DP of l-P(OEGMA)-Br was thus 38 based on a target DP of 100. Next, l-P(OEGMA)-b-P(HEMA)-Br was prepared by ATRP of HEMA using l-P(OEGMA)38-Br as a macroinitiator. Polymerization was conducted following the previously reported procedures with some modifications.26 Note that a small amount of Cu(II) with a one quarter molar ratio to that of Cu(I) was added during polymerization to exert better modulation on the polymerization of HEMA and to minimize any chain termination reactions for the preservation of the terminal bromine group during the polymerization process considering the truly fast ATRP kinetics of HEMA. All of the l-P(OEGMA)-b-P(HEMA)-Br synthesized at various polymerization times show a clear shift of the SEC elution peaks toward a greater MW (shorter retention times) without any tailing at both high- and low-MW sides relative to the l-P(OEGMA)38-Br macroinitiator (Fig. S3a†), supporting the high initiation efficiency of P(OEGMA)-based macroinitiator for a well-controlled ATRP of HEMA. The living characteristics were reflected by the pseudo-first-order kinetics, an almost linear growth of MW with increased monomer conversion and narrow molecular weight distribution (Đ) recorded during the evaluated polymerization process (Fig. S3† and Table 1). The DP of the P(HEMA) block was calculated based on the following eqn (2), where I7 is the integrated intensity of the terminal methyl protons in the side chain of OEGMA units, and I10 is the integrated intensity of the hydroxyl protons of HEMA units (Fig. 1a). Because the P(HEMA) block was used for final CPT conjugation to serve as the hydrophobic moiety of the amphiphilic cg-prodrug, a diblock copolymer with the greatest hydrophilic weight fraction of the synthesized four polymer constructs, i.e., l-P(OEGMA)38-b-P(HEMA)9-Br, was chosen for further reactions to guarantee sufficient colloidal stability for the self-assembled polymeric micelles. Therefore the linear precursor, l-P(OEGMA)38-b-P(HEMA)9-N3, was prepared by azidation of the terminal bromine group with a 20-fold molar excess of sodium azide.27,28
| | | Conv. = I1/(I1 + I2) × 100%, | (1) |
| | | DP of P(HEMA) = 3I10 / I7 × 38. | (2) |
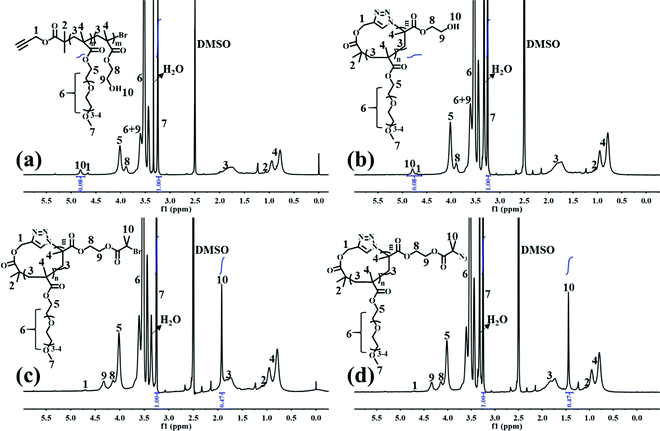 |
| | Fig. 1
1H NMR spectra of (a) l-P(OEGMA)38-b-P(HEMA)9-Br, (b) c-P(OEGMA)38-b-P(HEMA)9, (c) c-P(OEGMA)38-b-P(HEMA-Br)9, and (d) c-P(OEGMA)38-b-P(HEMA-N3)9 in DMSO-d6. | |
Table 1 Summary of l-P(OEGMA)-b-P(HEMA)-Br prepared by ATRP of HEMA using l-P(OEGMA)38-Br as a macroinitiator
| Run |
Time (min) |
DPa |
Conv. (%) |
Real structure |
M
w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (kDa) b (kDa) |
Đ
|
|
Determined by 1H NMR analysis.
Determined by SEC-MALLS.
|
| 1 |
30 |
9 |
11.3 |
l-P(OEGMA)38-b-P(HEMA)9-Br |
19.1 |
1.1 |
| 2 |
45 |
15 |
18.8 |
l-P(OEGMA)38-b-P(HEMA)15-Br |
19.8 |
1.1 |
| 3 |
60 |
20 |
25.0 |
l-P(OEGMA)38-b-P(HEMA)20-Br |
20.4 |
1.1 |
| 4 |
75 |
24 |
30.0 |
l-P(OEGMA)38-b-P(HEMA)24-Br |
21.2 |
1.1 |
c-P(OEGMA)38-b-P(HEMA)9 was later prepared by intrachain Cu(I)-catalyzed azide–alkynyl cycloaddition (CuAAc) of the linear precursor following reported procedures.29 The cyclic diblock copolymer shows the same polymer composition as the linear precursor, as evidenced by the identical ratio of the integrated intensity of the OEGMA units to that of the HEMA units in the 1H NMR spectra (Fig. 1b). Successful cyclization was confirmed by a clear shift of the SEC elution trace of the synthesized cyclic copolymer toward a longer retention time (lower MW) as well as a discernible absence of the characteristic absorption band at ∼2100 cm−1 ascribed to the terminal azide group after cyclization compared to those of the linear precursor (Fig. S4†).
Multivalent pendant azide reactive sites for various potential functionalizations were introduced to the resulting cyclic diblock copolymer by esterification and subsequent azidation reactions of the hydroxyl groups of P(HEMA) with excessive iBuBr and sodium azide, respectively, to ensure the full transition, which is supported by the complete disappearance of the characteristic signal assigned to the hydroxyl group of HEMA units and the expected ratio of the integrated intensity of peak 10 ascribed to the methyl groups near bromine to that of peak 7 assigned to the pendant terminal methyl protons of the OEGMA units (Fig. 1c and d). c-P(OEGMA)38-b-P(HEMA-Br)9 and c-P(OEGMA)38-b-P(HEMA-N3)9 (cg-N3) share almost identical MW values, both of which are greater than that of c-P(OEGMA)38-b-P(HEMA)9 (Fig. S5a†).
Note that the use of an alkynyl-functionalized initiator in the consecutive two-step ATRP synthesis of l-P(OEGMA)-b-P(HEMA)-Br led to a partial occurrence of certain side reactions, including the addition of the generated radicals to the alkynyl terminus and Glaser coupling between the alkynyl termini, which produced a negligible portion of the high-molecular weight moiety in l-P(OEGMA)-b-P(HEMA)-Br, especially at the relatively low monomer conversions. However, further esterification of HEMA units with iBuBr contributed to a significant increase of the molecular weight of this part, resulting in the appearance of a small shoulder at high molar masses for both cyclic and linear polymers of P(OEGMA)-b-P(HEMA-Br).
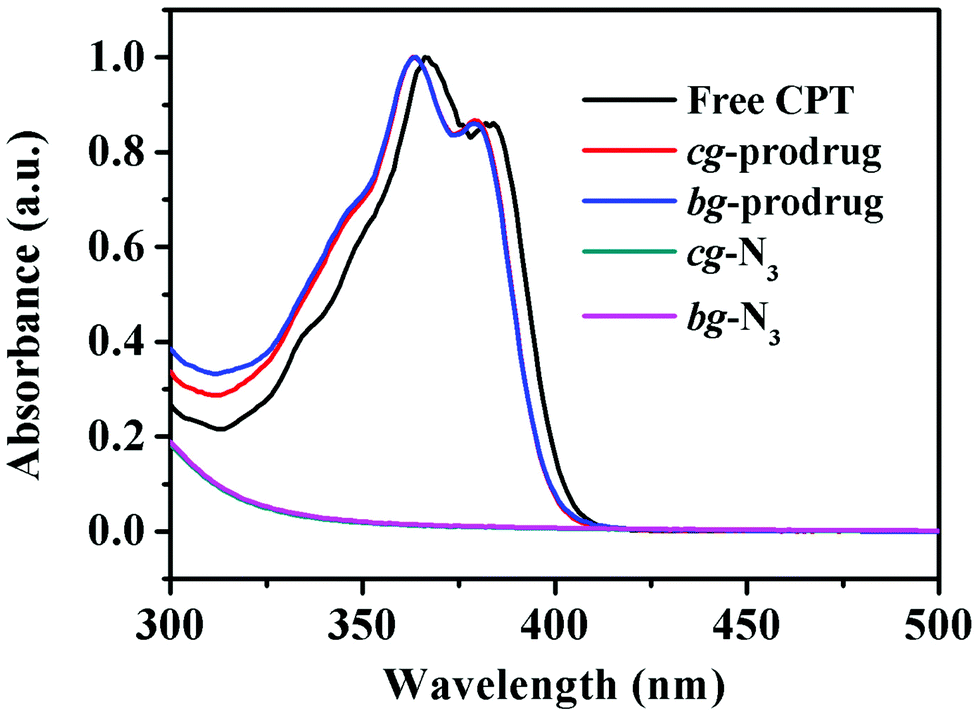
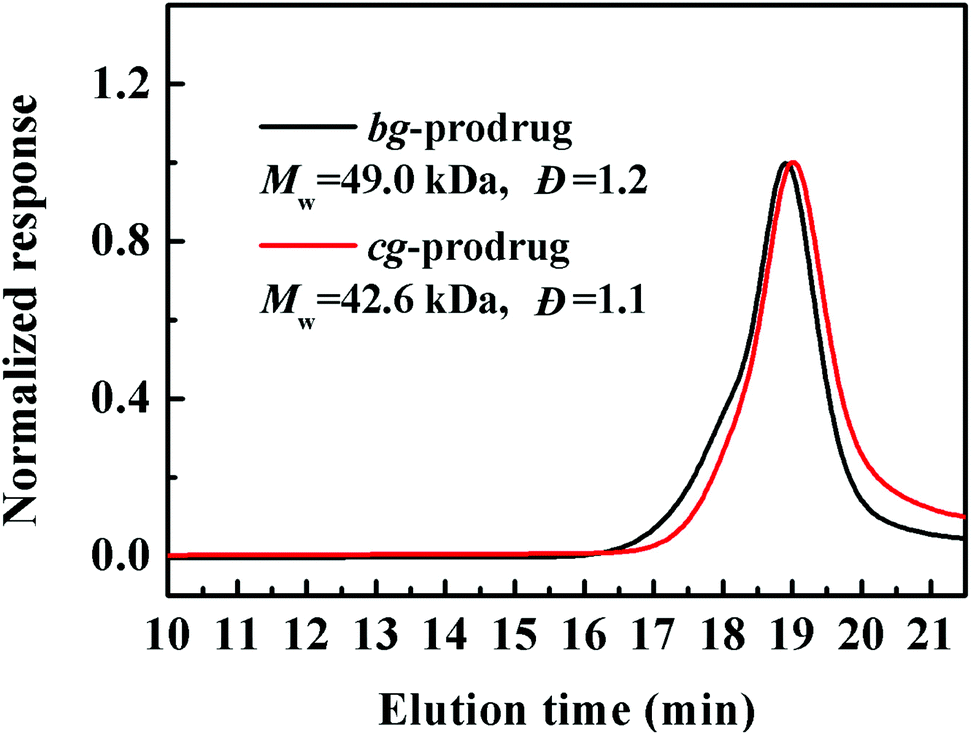
Finally, alkynyl-SS-CPT was conjugated to cg-N3 by intermolecular click coupling to yield the target cg-prodrug. The successful conjugation of CPT molecules is supported by recording a notable characteristic UV absorbance peak centered at 365 nm for cg and bg-prodrugs, which is almost identical to that of the free CPT molecules after the conjugation of CPT to cg-N3 and bg-N3 (Fig. 2), and by the appearance of a new signal at 7.6 ppm (peak 17) attributed to the proton of the click-formed triazole ring and the presence of CPT signals at 7.3–8.4 ppm (assigned to the aromatic protons) in the 1H NMR spectra of the cg-prodrug (Fig. 3). More importantly, both cg and bg-prodrugs show unimodal and narrow-distributed MWs (Fig. 4), confirming the successful preparation of the two prodrug formulations without any side reactions in the drug conjugation process.
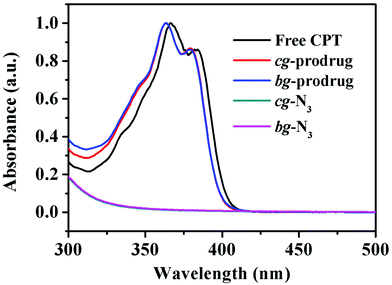 |
| | Fig. 2 UV-vis spectra of free CPT, cg-N3, bg-N3, and cg and bg-prodrugs in DMSO. | |
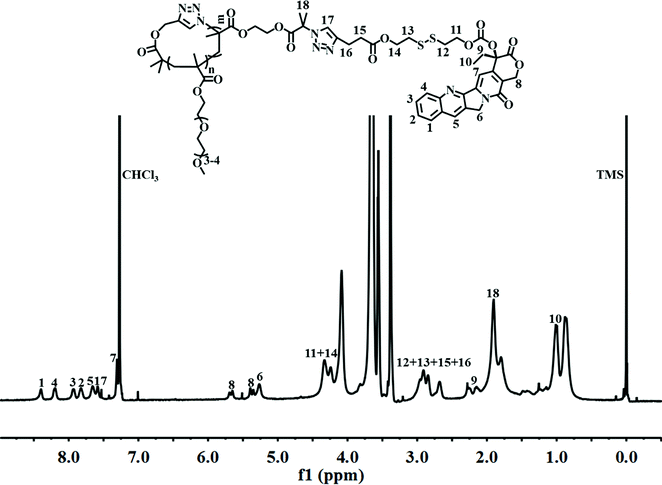 |
| | Fig. 3
1H NMR spectrum of cg-prodrug in CDCl3. | |
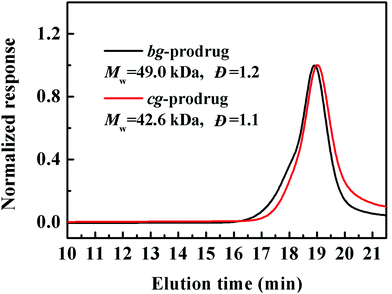 |
| | Fig. 4 SEC elution traces of cg and bg-prodrugs. | |
The actual drug loading content (DLC) of cg and bg-prodrugs was determined to be 12.3% and 13.9%, respectively, by using a UV–vis spectrometer based on the standard calibration working curves (Fig. S11b†) and eqn (3), in which mCPT is the mass of the conjugated CPT that was calculated using the absorption measured by the UV–vis spectrometer, and mcg- or bg-prodrugs is the mass of cg- or bg-prodrugs, respectively. The theoretical DLC of cg and bg-prodrugs was calculated to be 16.3% and 17.4%, respectively, based on eqn (4) assuming the full occupation of the reactive azide functions by CPT molecules without any consideration of the negative effect resulting from the steric hindrance. In eqn (4), 348.35 is the molecular weight of CPT, and nazide is the molar amount of azide functions in cg or bg-N3. Therefore, the conjugation efficiency of CPT was finally determined by eqn (5) and all of the results are listed in Table 2. The slightly greater conjugation efficiency of the bg-prodrug than that of the cg analogue is reasonably attributed to the stronger steric hindrance of cg relative to bg formulations.7
| | | Actual DLC = mCPT/mcg or bg-prodrugs × 100%, | (3) |
| | | Theoretical DLC = 348.35 × nazide/mcg or bg-prodrugs × 100%, | (4) |
| | | Conjugation efficiency of CPT = actual DLC/theoretical DLC × 100%. | (5) |
Table 2 Summary of the calculated parameters of cg and bg-prodrugs
| Sample |
Actual DLC (wt%) |
Theoretical DLC (wt%) |
Conjugation efficiency (%) |
The number of CPT units |
Polymer structure |
|
cg-Prodrug |
12.3 |
16.3 |
75.5 |
6.8 |
cg-P(OEGMA)38-b-P(HEMA9-SS-CPT6.8) |
|
bg-Prodrug |
13.9 |
17.4 |
79.9 |
8.0 |
bg-P(OEGMA)38-b-P(HEMA9-SS-CPT8.0) |
Self-assembly behaviors of cg and bg-prodrugs
The critical micelle concentrations (CMCs) of both formulations were determined using pyrene as a fluorescent probe, and were determined to be 14.5 and 9.1 μg mL−1 for cg and bg-prodrugs, respectively (Fig. S10†). The greater CMC value of the cg-prodrug relative to the bg analogue results likely from the different packing behaviors caused by the cg and bg copolymers with different topologies.30–32 The cyclic polymers are entropically disfavored in the micellar state, because two block junctions must be located in the core–fringe interface in the micelles self-assembled from cyclic copolymers compared to the self-assembly behavior of bg copolymers with only one block junction.7,22
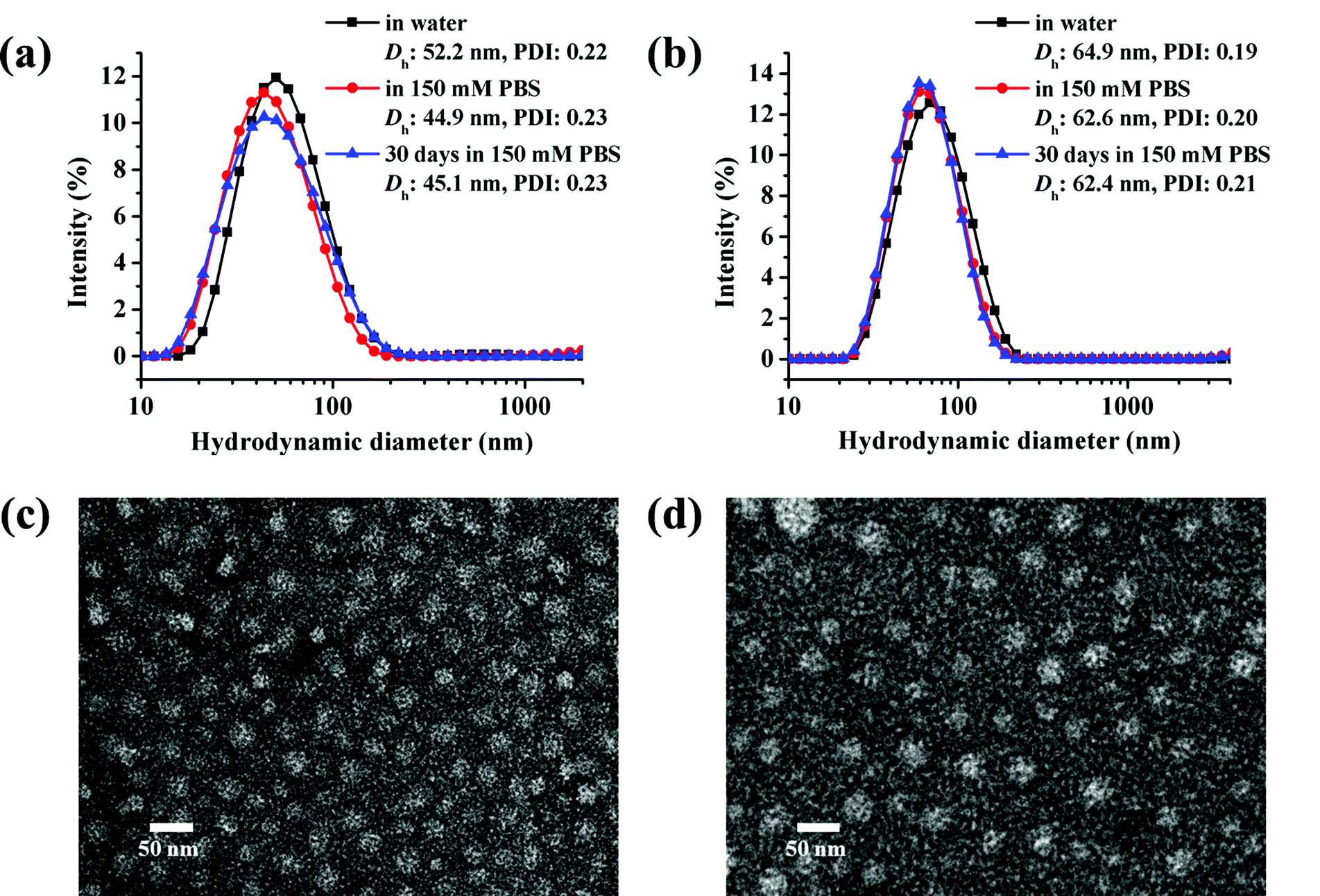
The micelles based on cg and bg-prodrugs with a final polymer concentration of 0.2 mg mL−1 that is well above the CMC values for both formulations were next fabricated via a nanoprecipitation method by adding the polymer solution prepared in DMSO dropwise into water followed by extensive dialysis against deionized water to remove any organic solvent. The mean size and size distribution (also termed as polydispersity index, PDI) of the two micelle prodrugs in an aqueous phase were determined by dynamic light scattering (DLS), which were found to be 52.2 nm and 0.22, and 64.9 nm and 0.19, respectively (Fig. 5a and b). The smaller size of micelles based on cg-prodrugs relative to micelles of bg-prodrugs is most likely attributed to the smaller dimension of the cg-prodrug than that of the bg construct that is supported by the SEC-MALLS-determined root-mean-square z-average radius of gyration, 〈Rg〉 value of cg (11.2 nm) and bg (15.1 nm)-prodrugs. Therefore, the results show that the dimension of both self-assemblies formed by the cg and bg polymers depends substantially on the molecular size of cg and bg polymers likely due to the steric hindrance of the grafted structures of cg and bg molecules and the relatively low aggregation number of the self-assembled structures. The salt stability of both micelle prodrugs was next evaluated in PBS (pH 7.4, 150 mM). The apparent stability of both micelle constructs is supported by the almost identical size distribution upon a shift of the incubation media from water to PBS as well as an unaltered dimension during the assessed 30 days (Fig. 5a and b).
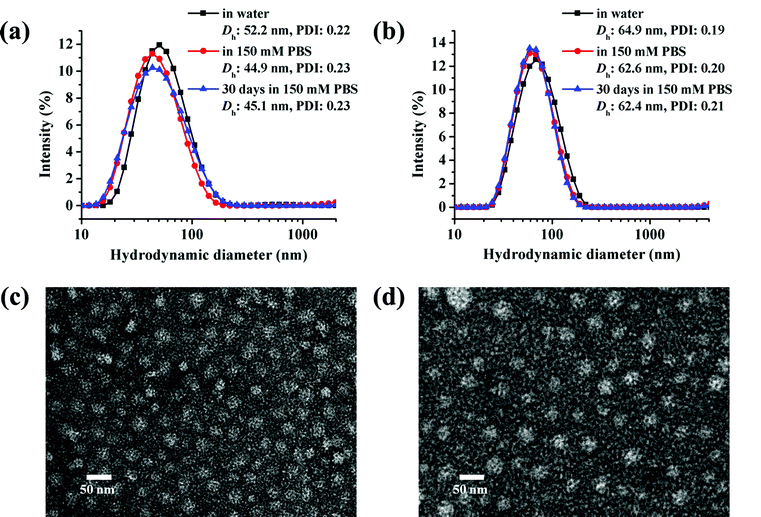 |
| | Fig. 5 Size distributions and TEM images of micelles self-assembled by cg (a and c) and bg-prodrugs (b and d) at a fixed polymer concentration of 0.2 mg mL−1. | |
Transmission electron microscopy (TEM) observation was used to provide a morphological insight into the self-assembled nanoparticles formed by cg and bg-prodrugs. Both polymers can form well-dispersed micelles with a regular spherical shape. The mean diameter was estimated to be approximately 20 and 25 nm for micelles of cg and bg-prodrugs, respectively, based on the TEM observations (Fig. 5c and d). The smaller size visualized by TEM than that determined by DLS is attributed to the dehydrated status of TEM samples.33,34
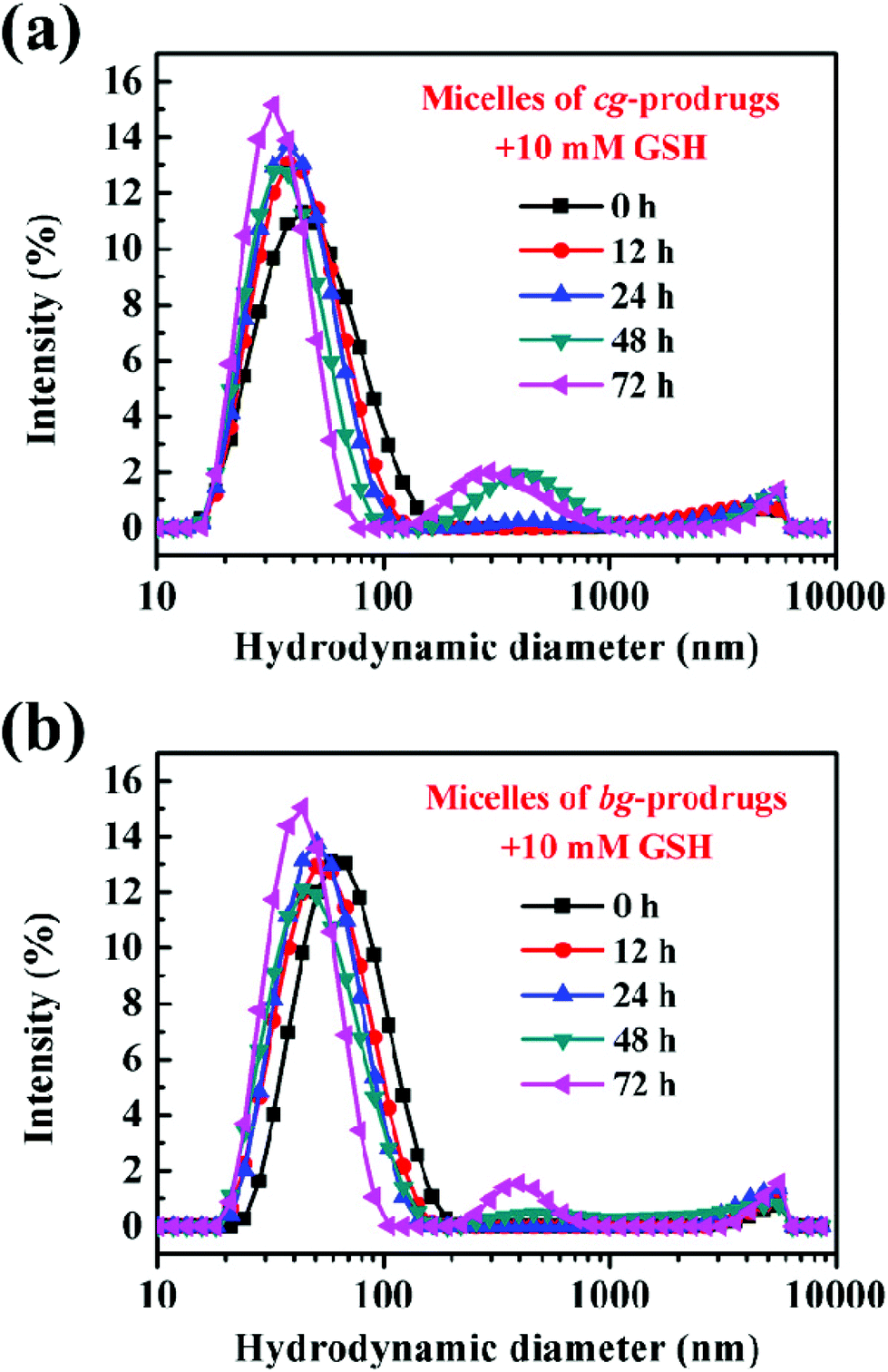
To demonstrate the reduction-responsive degradation of micelles formed by cg and bg-prodrugs, the size change of both micelles incubated with 10 mM GSH was monitored by DLS at different incubation intervals (Fig. 6). The unimodal size population centered at approximately 50 nm with a narrow distribution clearly evolved to the bimodal populations centered at around 40 and 400 nm with broad distributions for both micelles, which confirms the reduction-triggered cleavage of the disulfide link for CPT release from the parent micelle prodrugs35–37 and the reassembly of the prodrug residues with greater hydrophilic weight fractions. Interestingly, the notable degradation of micelles based on cg-prodrugs commenced sometime after 12 h of incubation, which is faster than that of the bg-analogues with a starting time between 24 and 48 h. The lower DLC of cg-prodrugs seems to account substantially for the reduction-triggered promoted re-assembly process considering a speedier cleavage of low amounts of disulfide links by the consumption of the same amount of GSH.
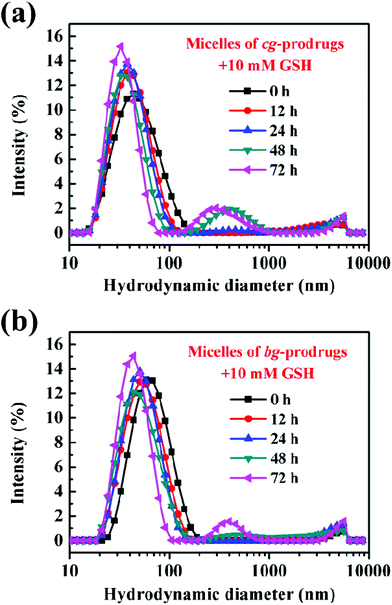 |
| | Fig. 6 Reduction-triggered size changes of micelles based on cg (a) and bg-prodrugs (b) at various incubation periods with 10 mM GSH. | |
In vitro drug release study
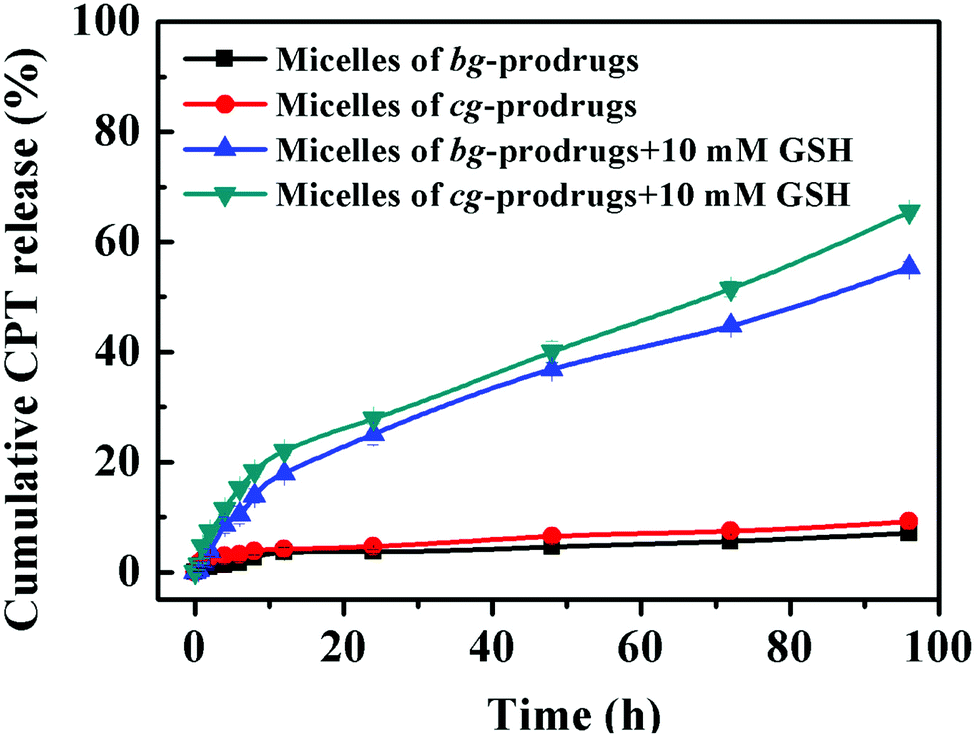
To further validate reduction-sensitive drug release behaviors, an in vitro drug release study of micelles of cg and bg-prodrugs was investigated in PBS (pH 7.4, 150 mM) at 37 °C in the absence and presence of 10 mM GSH (Fig. 7). Both micelle prodrugs had very good protection of the conjugated drug with less than 10% CPT release in 96 h without incubation of GSH because of the enhanced stability of the conjugated drug molecules for a minimized premature drug release and off-target associated side effects.38 The slow hydrolysis of the carbonate bond in PBS likely accounts for the slight cumulative drug release in the absence of GSH.39 However, incubation with 10 mM GSH significantly promoted CPT release to 65% and 55% for micelles of cg and bg-prodrugs in the same period, respectively, due to the reduction-triggered cleavage of CPT from the parent polymer. The faster drug release behaviors of cg-prodrug-based micelles than that of the micelles based on bg analogues agrees well with the faster degradation behaviors of the former micelle construct (Fig. 6).
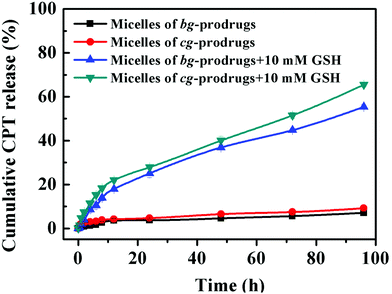 |
| | Fig. 7
In vitro drug release profiles of micelles of cg and bg-prodrugs under different conditions at 37 °C. | |
In vitro cellular uptake and cytotoxicity studies
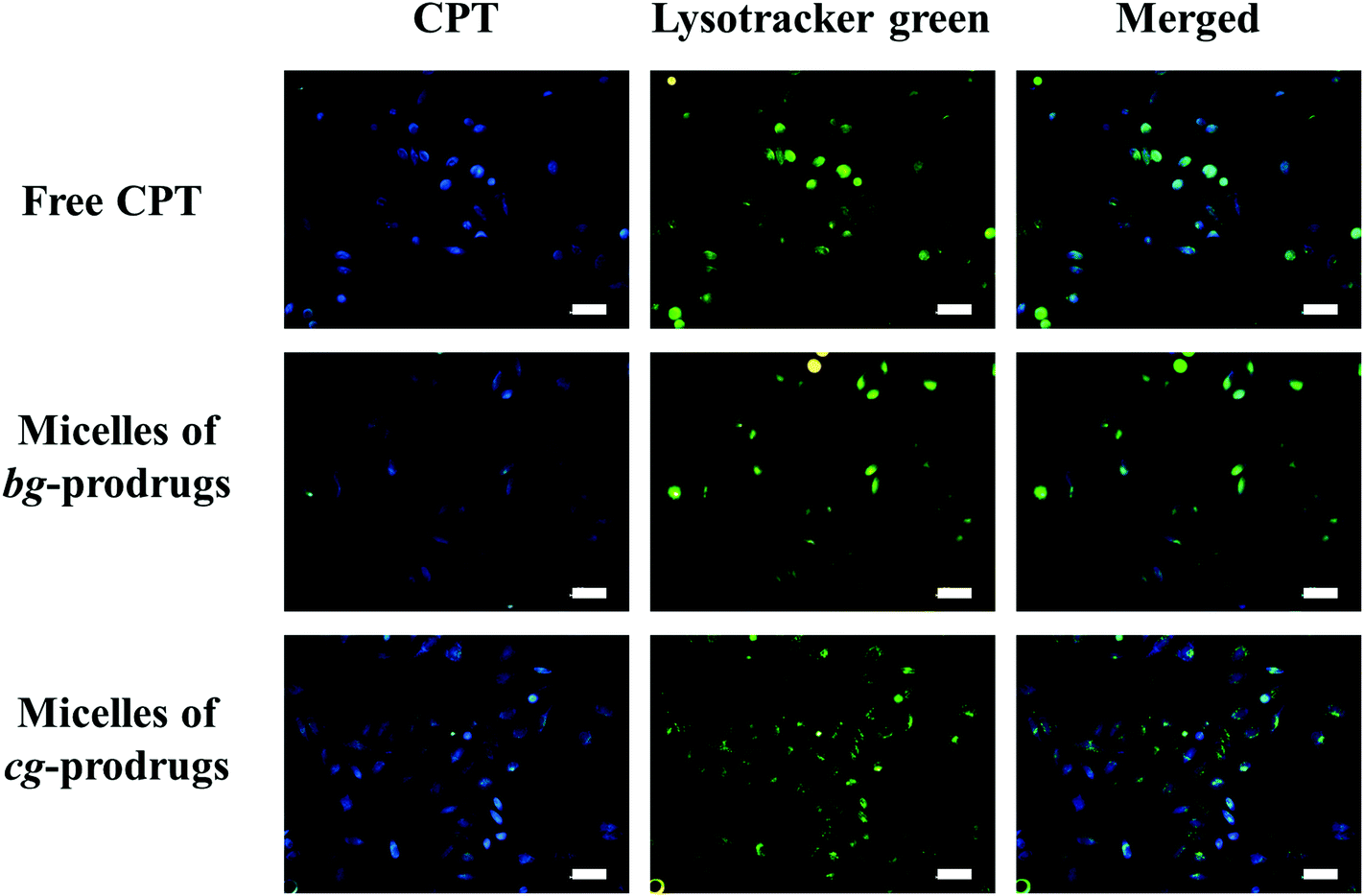
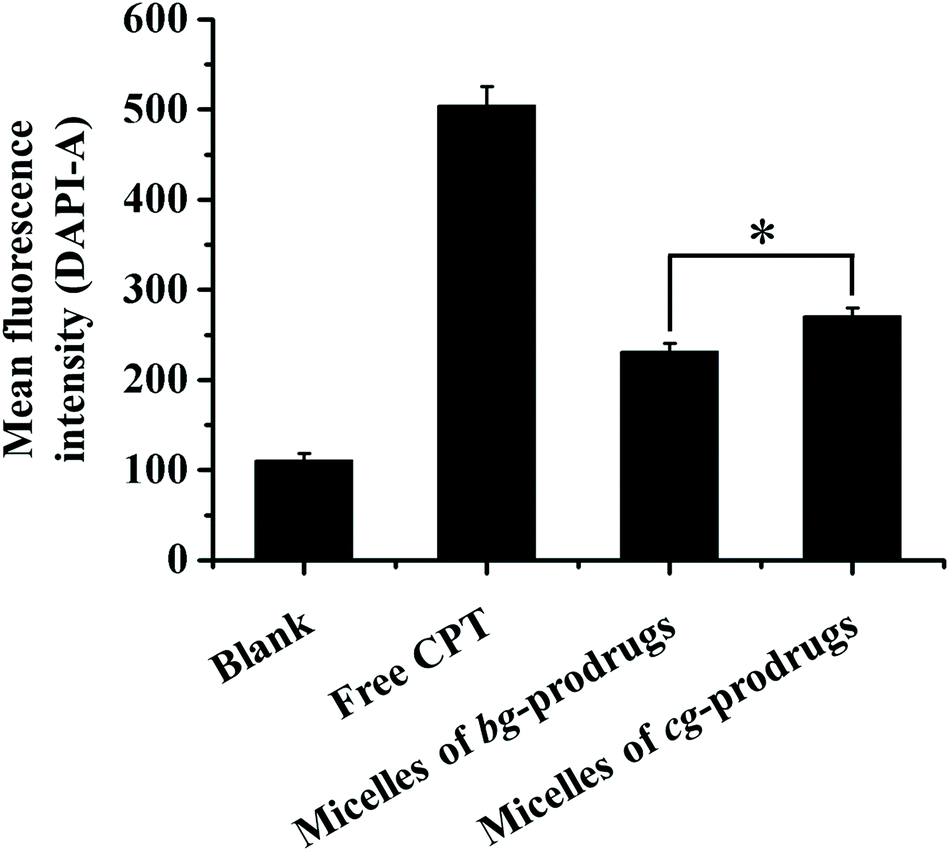
To investigate the intracellular trafficking of cg and bg-prodrug-based micelles, the cellular uptake properties of the two micelle prodrugs with an equivalent CPT amount of 25 μg mL−1 were visualized by fluorescence microscopy in HeLa cells. Note that the cells were stained with LysoTracker Green to distinguish from the blue fluorescence of CPT. The clear visualization of the overlapped blue and green colors in the merged images after 4 h of incubation (Fig. 8) confirms efficient endocytosis of both micelle formulations and the subsequent substantial localization of either micelle prodrugs or the released CPT molecules in the cytoplasm. The average fluorescence density per cell area of free CPT, micelles of bg-prodrugs, and micelles of cg-prodrugs was further determined to be 0.125, 0.088, and 0.095 per pixel, respectively, by semi-quantitative analysis of confocal images using ImageJ software. The cells treated with micelles of cg and bg-prodrugs presented weaker fluorescence intensity than those treated with free CPT due to their different internalization mechanisms (endocytosis vs. direct membrane permeation). The stronger fluorescence intensity of cg-prodrug-based micelles suggests the greater cellular uptake efficiency of this formulation than that of the bg-based analogues. The cellular uptake efficiency was further quantified by flow cytometry (FCM) analysis (Fig. 9). The greater cellular uptake efficiency of the micelles of cg-prodrugs than that of the bg-prodrug-based analogues with statistical significance agrees well with the fluorescence observation, and is possibly attributed to the smaller size of the micelles of cg-prodrugs for faster and greater cellular internalization.40
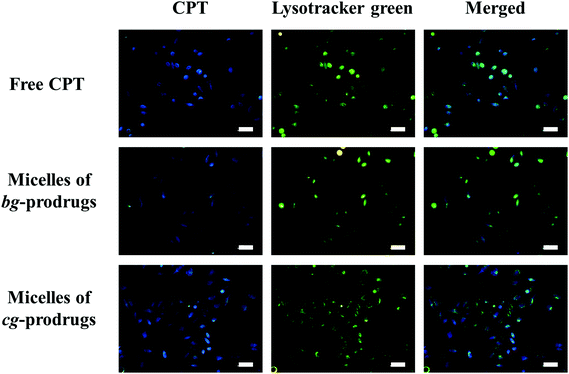 |
| | Fig. 8 Fluorescence images of HeLa cells incubated with free CPT and micelles of bg and cg-prodrugs for 4 h. The scale bars correspond to 50 μm in all the images. | |
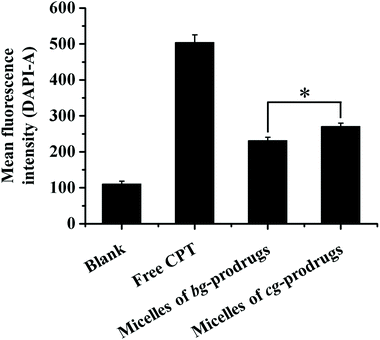 |
| | Fig. 9 Quantitative measurements of the mean fluorescence intensity after incubation with free CPT and micelles of bg and cg-prodrugs in HeLa cells via flow cytometry analysis (4 h incubation, CPT concentration = 25 μg mL−1, and 10000 cells were counted). The data were expressed as mean ± SD, n = 3, Student's t-test, *p < 0.01. | |
Finally, in vitro cytotoxicity of cg-N3, bg-N3, and micelles of cg and bg-prodrugs was evaluated by a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) cell viability assay in HeLa cells (Fig. S12† and Fig. 10). Both polymer precursors, cg-N3 and bg-N3, were non-toxic to cells with a high viability above 90% at all the examined polymer concentrations. The half-maximal inhibitory concentration (IC50) of free CPT, micelles of cg-prodrugs, and micelles of bg-prodrugs is determined to be 1.72 (1.31, 2.25), 3.96 (3.40, 4.61), and 5.35 (4.60, 6.22) μg mL−1, respectively. Although both micelle prodrugs exhibited less in vitro cytotoxic activity than the free CPT likely due to their different internalization mechanisms mentioned above and the release kinetics of free drug from the micelle prodrugs, the micelle constructs with enhanced stability can prolong the blood circulation and enhance the passive accumulation of CPT molecules at the tumor site via an enhanced permeability and retention (EPR) effect as well as reduce the systematic side effects of free CPT. More importantly, cg-prodrug-based micelles exhibited greater cytotoxicity (lower IC50 value) despite the lower DLC than the bg-based analogues, which results primarily from the faster reduction-triggered degradation and drug release as well as the greater cellular uptake efficiency of the former micelle prodrugs.
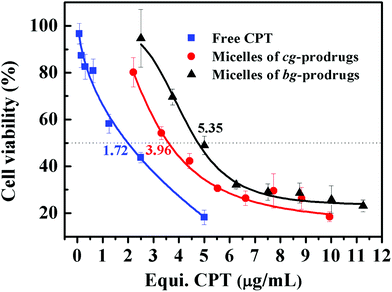 |
| | Fig. 10
In vitro viability of HeLa cells treated with free CPT and micelles of cg and bg-prodrugs at various concentrations for 72 h of incubation. | |
Conclusions
In summary, we reported in this study the synthesis of cg-prodrugs with heterogeneous grafts of hydrophilic OEG and reducibly conjugated camptothecin (CPT) via an integrated strategy of a previously reported diblock copolymer-based template and post-polymerization intermolecular click conjugation of a reducible CPT prodrug to completely eliminate the compromised drug conjugation efficiency due to the steric hindrance of hydrophilic grafts. The micelles self-assembled from the cg-prodrugs had sufficient salt stability and reduction-triggered cleavage of the disulfide link for a promoted CPT release, simultaneously. Most importantly, we revealed that the dimension of both self-assemblies formed by the cg and bg polymers depends substantially on the molecular size of the cg and bg polymers and the cg-prodrug-based micelles exhibited greater in vitro cytotoxicity against cancer cells despite the lower DLC than the bg-based analogues. Therefore, the developed cg-prodrugs provide great potential for chemotherapy, and the aforementioned interesting results will definitely inspire more upcoming studies on the future design and development of novel cg polymers for biomedical applications.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (51473072 and 21504035), the Thousand Young Talent Program, and the Open Research Fund of State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, and the Chinese Academy of Sciences.
References
- L. Gao, Z. Ji, Y. Zhao, Y. Cai, X. Li and Y. Tu, ACS Macro Lett., 2019, 8, 1564–1569 CrossRef CAS.
- B. J. Ree, Y. Satoh, K. Sik Jin, T. Isono, W. Jong Kim, T. Kakuchi, T. Satoh and M. Ree, NPG Asia Mater., 2017, 9, e453–e453 CrossRef CAS.
- M. Schappacher and A. Deffieux, Science, 2008, 319, 1512 CrossRef CAS PubMed.
- J. Yang, R. Wang and D. Xie, Macromolecules, 2019, 52, 7042–7051 CrossRef CAS.
- F. Zhou, Z. Zhang, G. Jiang, J. Lu, X. Chen, Y. Li, N. Zhou and X. Zhu, Polym. Chem., 2016, 7, 2785–2789 RSC.
- Y. Cheng, H. Wei, J.-K. Y. Tan, D. J. Peeler, D. O. Maris, D. L. Sellers, P. J. Horner and S. H. Pun, Small, 2016, 12, 2750–2758 CrossRef CAS PubMed.
- Z. Liu, Y. Huang, X. Zhang, X. Tu, M. Wang, L. Ma, B. Wang, J. He, P. Ni and H. Wei, Macromolecules, 2018, 51, 7672–7679 CrossRef CAS.
- X. Y. Tu, C. Meng, Y. F. Wang, L. W. Ma, B. Y. Wang, J. L. He, P. H. Ni, X. L. Ji, M. Z. Liu and H. Wei, Macromol. Rapid Commun., 2018, 39, 1700744 CrossRef PubMed.
- B. Chen, K. Jerger, J. M. J. Fréchet and F. C. Szoka, J. Controlled Release, 2009, 140, 203–209 CrossRef CAS PubMed.
- N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox, J. M. J. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef CAS PubMed.
- M. C. Arno, R. J. Williams, P. Bexis, A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Biomaterials, 2018, 180, 184–192 CrossRef CAS PubMed.
- X. Y. Tu, C. Meng, X. L. Zhang, M. G. Jin, X. S. Zhang, X. Z. Zhao, Y. F. Wang, L. W. Ma, B. Y. Wang, M. Z. Liu and H. Wei, Macromol. Biosci., 2018, 18, 1800022 CrossRef PubMed.
- S. Zhang, L. Yin, W. Zhang, Z. Zhang and X. Zhu, Polym. Chem., 2016, 7, 2112–2120 RSC.
- S. Zhang, Y. Tezuka, Z. Zhang, N. Li, W. Zhang and X. Zhu, Polym. Chem., 2018, 9, 677–686 RSC.
- B. Mu, Q. Li, X. Li, S. Pan, Y. Zhou, J. Fang and D. Chen, Polym. Chem., 2016, 7, 6034–6038 RSC.
- R. J. Williams, A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Macromolecules, 2016, 49, 2802–2813 CrossRef CAS PubMed.
- C. E. Wang, H. Wei, N. Tan, A. J. Boydston and S. H. Pun, Biomacromolecules, 2016, 17, 69–75 CrossRef CAS PubMed.
- H. Wei, C. E. Wang, N. Tan, A. J. Boydston and S. H. Pun, ACS Macro Lett., 2015, 4, 938–941 CrossRef CAS PubMed.
- X. Fan, G. Wang and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1361–1367 CrossRef CAS.
- H. Zhang, W. Wu, X. Zhao and Y. Zhao, Macromolecules, 2017, 50, 3411–3423 CrossRef CAS.
- L. Zheng, Y. Wang, X. Zhang, L. Ma, B. Wang, X. Ji and H. Wei, Bioconjugate Chem., 2018, 29, 190–202 CrossRef CAS PubMed.
- Z. Ge, Y. Zhou, J. Xu, H. Liu, D. Chen and S. Liu, J. Am. Chem. Soc., 2009, 131, 1628–1629 CrossRef CAS PubMed.
- L. Gao, J. Oh, T. Chang, D. Chen, X. Li, X. Yang, Y. Tu, X. Zhu and C. Y. Li, Polymer, 2016, 101, 379–387 CrossRef CAS.
- J.-F. Lutz and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 897–910 CrossRef CAS.
- D. Han, X. Tong, Y. Zhao, T. Galstian and Y. Zhao, Macromolecules, 2010, 43, 3664–3671 CrossRef CAS.
- P. Yang and S. P. Armes, Macromol. Rapid Commun., 2014, 35, 242–248 CrossRef CAS PubMed.
- H. Wei, D. S. H. Chu, J. Zhao, J. A. Pahang and S. H. Pun, ACS Macro Lett., 2013, 2, 1047–1050 CrossRef CAS PubMed.
- X. Wan, T. Liu and S. Liu, Biomacromolecules, 2011, 12, 1146–1154 CrossRef CAS PubMed.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS PubMed.
- H. Iatrou, N. Hadjichristidis, G. Meier, H. Frielinghaus and M. Monkenbusch, Macromolecules, 2002, 35, 5426–5437 CrossRef CAS.
- E. Minatti, P. Viville, R. Borsali, M. Schappacher, A. Deffieux and R. Lazzaroni, Macromolecules, 2003, 36, 4125–4133 CrossRef CAS.
- G. E. Yu, Z. Yang, D. Attwood, C. Price and C. Booth, Macromolecules, 1996, 29, 8479–8486 CrossRef CAS.
- X. Zhang, M. Zhang, M. Wang, H. Peng, Q. Hua, L. Ma, B. Wang and H. Wei, Bioconjugate Chem., 2018, 29, 2239–2247 CrossRef CAS PubMed.
- X. Zhang, X. Zhang, L. Sun, F. Liu, M. Wang, J. Peng, Y. Wang, L. Ma, B. Wang and H. Wei, Chem. Commun., 2018, 54, 13495–13498 RSC.
- X. Hu, J. Hu, J. Tian, Z. Ge, G. Zhang, K. Luo and S. Liu, J. Am. Chem. Soc., 2013, 135, 17617–17629 CrossRef CAS PubMed.
- L. Dai, R. Cai, M. Li, Z. Luo, Y. Yu, W. Chen, X. Shen, Y. Pei, X. Zhao and K. Cai, Chem. Mater., 2017, 29, 6976–6992 CrossRef CAS.
- M. Hou, Y. E. Gao, X. Shi, S. Bai, X. Ma, B. Li, B. Xiao, P. Xue, Y. Kang and Z. Xu, Acta Biomater., 2018, 77, 228–239 CrossRef CAS PubMed.
- Y. Shen, E. Jin, B. Zhang, C. J. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan, E. Van Kirk and W. J. Murdoch, J. Am. Chem. Soc., 2010, 132, 4259–4265 CrossRef CAS PubMed.
- S. J. Moon, S. V. Govindan, T. M. Cardillo, C. A. D'Souza, H. J. Hansen and D. M. Goldenberg, J. Med. Chem., 2008, 51, 6916–6926 CrossRef CAS PubMed.
- T. Chang, M. S. Lord, B. Bergmann, A. Macmillan and M. H. Stenzel, J. Mater. Chem. B, 2014, 2, 2883–2891 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and schemes of synthesis of alkynyl-SS-CPT and bg-prodrug; ATRP kinetic studies for the synthesis of l-P(OEGMA)-Br and l-P(OEGMA)-b-P(HEMA)-Br; the additional SEC-MALLS data; 1H NMR spectra; CMC determination of cg and bg-prodrug-based micelles; absorption spectra of CPT in DMSO at different concentrations and the calibration working curve of CPT. See DOI: 10.1039/d0bm00656d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab