
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigner artificial membrane binding proteins to direct stem cells to the myocardium†
Wenjin
Xiao‡
a,
Thomas I. P.
Green‡
ab,
Xiaowen
Liang‡
c,
Rosalia Cuahtecontzi
Delint
ab,
Guillaume
Perry
 d,
Michael S.
Roberts
ce,
Kristian
Le Vay
ab,
Catherine R.
Back
fg,
Raimomdo
Ascione
h,
Haolu
Wang
c,
Paul R.
Race
fg and
Adam W.
Perriman
*ag
d,
Michael S.
Roberts
ce,
Kristian
Le Vay
ab,
Catherine R.
Back
fg,
Raimomdo
Ascione
h,
Haolu
Wang
c,
Paul R.
Race
fg and
Adam W.
Perriman
*ag
aSchool of Cellular and Molecular Medicine, University of Bristol, BS8 1TD, UK. E-mail: chawp@bristol.ac.uk
bBristol Centre for Functional Nanomaterials, University of Bristol, BS8 1FD, UK
cThe University of Queensland Diamantina Institute, The University of Queensland, Translational Research Institute, Woolloongabba, QLD 4102, Australia
dSorbonne Université, Laboratoire d'Electronique et d'Electromagnétisme, L2E, F-75005, Paris, France
eSchool of Pharmacy and Medical Science, University of South Australia, Adelaide, SA 5001, Australia
fSchool of Biochemistry, University of Bristol, BS8 1TD, UK
gBrisSynBio Synthetic Biology Research Centre, University of Bristol, BS8 1TQ, UK
hTranslational Biomedical Research Centre and Bristol Heart Institute, University of Bristol, Bristol, UK
First published on 3rd July 2019
Abstract
We present a new cell membrane modification methodology where the inherent heart tissue homing properties of the infectious bacteria Streptococcus gordonii are transferred to human stem cells. This is achieved via the rational design of a chimeric protein–polymer surfactant cell membrane binding construct, comprising the cardiac fibronectin (Fn) binding domain of the bacterial adhesin protein CshA fused to a supercharged protein. Significantly, the protein–polymer surfactant hybrid spontaneously inserts into the plasma membrane of stem cells without cytotoxicity, instilling the cells with a high affinity for immobilized fibronectin. Moreover, we show that this cell membrane reengineering approach significantly improves retention and homing of stem cells delivered either intracardially or intravenously to the myocardium in a mouse model.
Introduction
There has been significant interest in the development of cell-based therapies for the treatment of disease, due to the major and increasing burden on health outcome and life expectancy. However, challenges remain in relation to cellular delivery mode, efficacy, off-target effects and therapeutic mechanism of action. For example, studies using stem cells in vivo to regenerate myocardium post ischemia have postulated many potential mechanisms linked to cell type, including functional integration with cardiomyocytes, paracrine effects from secreted factors, tissue matrix remodelling via metalloproteases, as well as recruitment of monocytes and macrophages to repair micro-vessels.1–3 For the majority of these mechanisms, it is clear that the therapeutic cells need to be recruited/retained at the site of injury in significant numbers and for a sufficient duration to be able to have an effect. Accordingly, increasing cell homing and retention is likely to be advantageous, especially as myocardial retention of cells delivered via intravenous (IV), intra-arterial (IA), or intramyocardial (IM) injections are associated with only 0% (IV), 2% (IA) and 10–15% (IM) retention up to 3 days post-injection, followed by rapid signal loss over 24 hours.4–6Such low levels of viable cell retention in the myocardium can be rationalized by a number of factors, including a lack of adhesion of the cells to the site of injury, poor targeting by selected delivery mode, high turbulent hemodynamic flow, aggressive or hypoxic environments, and presence of inflammatory cytokines.4,5,7 To combat these effects, several cell immobilization approaches for in vivo site-directed tissue repair have been developed. Cells have been transplanted in soft biocompatible matrices for enhanced retention using gelatin,8 alginate9 and decellularized extracellular matrix (ECM) hydrogels.10 Synthetic matrices comprising cell-supporting scaffolds produced from polyglycerol-sebacate (PGS)11 and poly(octamethylene maleate (anhydride) citrate) (POMaC)12 have also been utilized for implantation. Although these approaches provide the added benefit of high cell numbers, they generally require more invasive surgical procedures when compared with direct cell injection, while the carrier biomaterials may feature unmatched biomechanical properties when compared with the myocardium.13
Cell membrane re-engineering is emerging as a powerful new approach for in vivo tissue repair, where exogenous molecules are directly introduced to the plasma membrane to drive active cell homing to the site of injury. These approaches commonly involve direct covalent modifications of the membrane binding homing motifs, and include antibodies,14,15 selectin-binding peptides16 and polymers.17 Recent studies on increasing homing of therapeutic cells18,19 and vesicles20,21 for heart repair have achieved significant improvement in outcomes with reduced fibrosis and scar size, and increased cellular proliferation and angiogenesis. However, these approaches have drawbacks, including steric hindrance of the homing paratope, non-organ specificity and off-target stimulation of irrelevant sites.22 Accordingly, the conceptual advance described herein is centred on utilising the inherent homing properties of infectious bacteria by immobilizing the cardiac fibronectin (Fn) binding domain of the bacterial adhesin protein CshA on the plasma membrane of therapeutic cells. The adherence of bacteria to biotic or abiotic surfaces is essential for host colonization, persistence, and pathogenicity, and the process is facilitated by bacterial adhesins, which recognize and bind specific partner molecules presented on the surfaces of host cells and other microorganisms.23 Fibronectin, a large and essential multidomain glycoprotein, functioning as a key link between cells and their extracellular matrices, has been recognized as the target for a large number of bacterial adhesins.24 Significantly, the CshA-Fn interaction directs S. gordonii to damaged fibrotic regions of the cardiac endothelium, e.g., infarcted or scarred tissue, damaged or prosthetic heart valves, promoting the onset of infective endocarditis,25–27 and undamaged cardiac Fn. Although Fn is expressed in healthy cardiac tissue, expression levels are increased enormously after myocardial infarction.27,28 Here, the mechanism underpinning the high levels of selectivity exhibited by CshA for cardiac Fn involves an atypical “catch–clamp” process.29 After entering the blood stream, the cell wall-bound adhesin rapidly samples the localised ECM environment using an extended intrinsically disordered low affinity binding site (catch), which, upon binding, activates a second high affinity (clamp) to anchor the bacterium to the immobilized Fn (Fig. 1a).
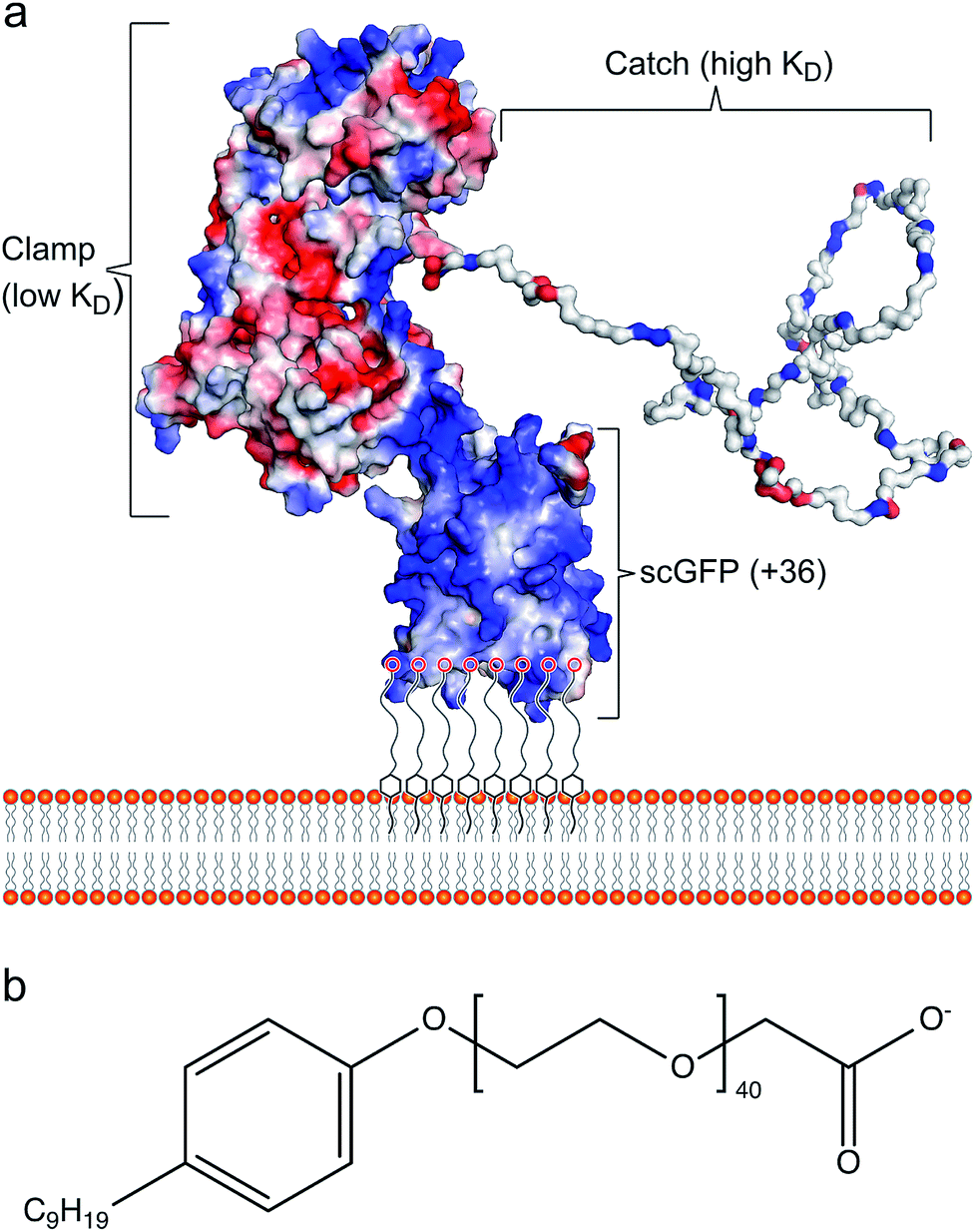 | ||
| Fig. 1 The artificial plasma membrane binding construct. (a) Schematic showing the interaction of the polymer surfactant conjugated supercharged fusion [CshA_scGFP][S] construct with the bilayer of the cytoplasmic membrane. The structure shows the surface charge potential, highlighting the high positive charge (blue) on the supercharged GFP domain and the negative charge (red) on the CshA globular domain, as well as the “catch” and “clamp” domains of CshA. (b) The structure of the anionic polymer surfactant glycolic acid ethoxylate 4-nonylphenyl ether (S), which electrostatically complexes with the supercharged GFP moiety. | ||
The ability to readily display the CshA binding motif on the membrane of therapeutic cells, which has been evolutionarily optimized to recognize and bind Fn in the cardiac endothelium with high affinity, would provide a new direction for cell-based myocardial therapy. Accordingly, a designer CshA chimera construct was converted into an artificial membrane binding protein using a protein surface reengineering methodology recently developed in our laboratories.30 Here, the CshA Fn-binding domain was fused to a supercharged green fluorescent protein (scGFP), which was then electrostatically conjugated to polymer surfactant molecules to produce a cytoplasmic membrane active polymer surfactant corona. Significantly, this new artificial membrane binding construct was not cytotoxic, spontaneously inserted into the plasma membrane of human mesenchymal stem cells (hMSCs), did not elicit a hematologic response in mice, and directed hMSCs, delivered either intracardially or intravenously, to the mouse myocardium.
Results and discussion
Synthesis and characterisation of the protein–polymer surfactant construct
The gene comprising the non-repetitive domains (NR1, NR2, and NR3) of CshA (Fig. S1A†) fused to a +36 scGFP was synthesized (Table S1†), and the chimera CshA_scGFP (Fig. 1a) was expressed in E. coli and purified using immobilized metal affinity chromatography (IMAC) and size exclusion chromatography (SEC) (Fig. S1B†). Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry gave a molecular weight of 112.6 kDa (Fig. S1C†), which confirmed the presence of the intact CshA_scGFP construct. Electrostatically driven conjugation between the cationic residue sidechains and the anionic polymer surfactant glycolic acid ethoxylate 4-nonylphenyl ether ([S], Fig. 1b) was performed over a range of protein cationic sites![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :polymer surfactant ratios (PCS:PS; 1:1.4, 1:2.1 and 1:2.8), followed by dialysis. UV-vis spectra from the resulting dialysed constructs (Fig. S2A†) showed strong absorbances arising from the scGFP fluorophore (487 nm) and the aromatic groups of both the protein and the nonylphenyl moiety of the surfactant (270–280 nm). The protein and polymer surfactant molar extinction coefficients were used to evaluate the final PCS:PSs, which were 1:0.6, 1:1.1 and 1:1.6, indicating the removal of unbound surfactant molecules during dialysis. Sedimentation velocity analytical ultracentrifugation (SV-AUC) experiments were performed on the three formulations at 487 nm and 280 nm to monitor the chimera and the polymer surfactant independently. Significantly, electrostatic surfactant conjugation of CshA_scGFP to yield [CshA_scGFP][S] increased the sedimentation coefficient (Abs 487 nm) of the primary feature from 4.1 to 4.8 S at a PCS:PS of 1:0.6 (Fig. 2a and Table S2†). Moreover, increasing the PCS:PS to 1:1.1 or 1:1.6 did not increase the sedimentation coefficient of this feature, which indicated that the surfactant binding sites on the CshA_scGFP fusion were saturated. Indeed, the emergence of a feature at ca. 1.6 S at a PCS:PS of 1:1.1 (Abs 280 nm), which increased in magnitude at 1:1.6, signified the presence of free surfactant (Fig. 2b and Table S2†). Accordingly, a PCS:PS of 1:0.6 was used for all subsequent experiments.
:polymer surfactant ratios (PCS:PS; 1:1.4, 1:2.1 and 1:2.8), followed by dialysis. UV-vis spectra from the resulting dialysed constructs (Fig. S2A†) showed strong absorbances arising from the scGFP fluorophore (487 nm) and the aromatic groups of both the protein and the nonylphenyl moiety of the surfactant (270–280 nm). The protein and polymer surfactant molar extinction coefficients were used to evaluate the final PCS:PSs, which were 1:0.6, 1:1.1 and 1:1.6, indicating the removal of unbound surfactant molecules during dialysis. Sedimentation velocity analytical ultracentrifugation (SV-AUC) experiments were performed on the three formulations at 487 nm and 280 nm to monitor the chimera and the polymer surfactant independently. Significantly, electrostatic surfactant conjugation of CshA_scGFP to yield [CshA_scGFP][S] increased the sedimentation coefficient (Abs 487 nm) of the primary feature from 4.1 to 4.8 S at a PCS:PS of 1:0.6 (Fig. 2a and Table S2†). Moreover, increasing the PCS:PS to 1:1.1 or 1:1.6 did not increase the sedimentation coefficient of this feature, which indicated that the surfactant binding sites on the CshA_scGFP fusion were saturated. Indeed, the emergence of a feature at ca. 1.6 S at a PCS:PS of 1:1.1 (Abs 280 nm), which increased in magnitude at 1:1.6, signified the presence of free surfactant (Fig. 2b and Table S2†). Accordingly, a PCS:PS of 1:0.6 was used for all subsequent experiments.
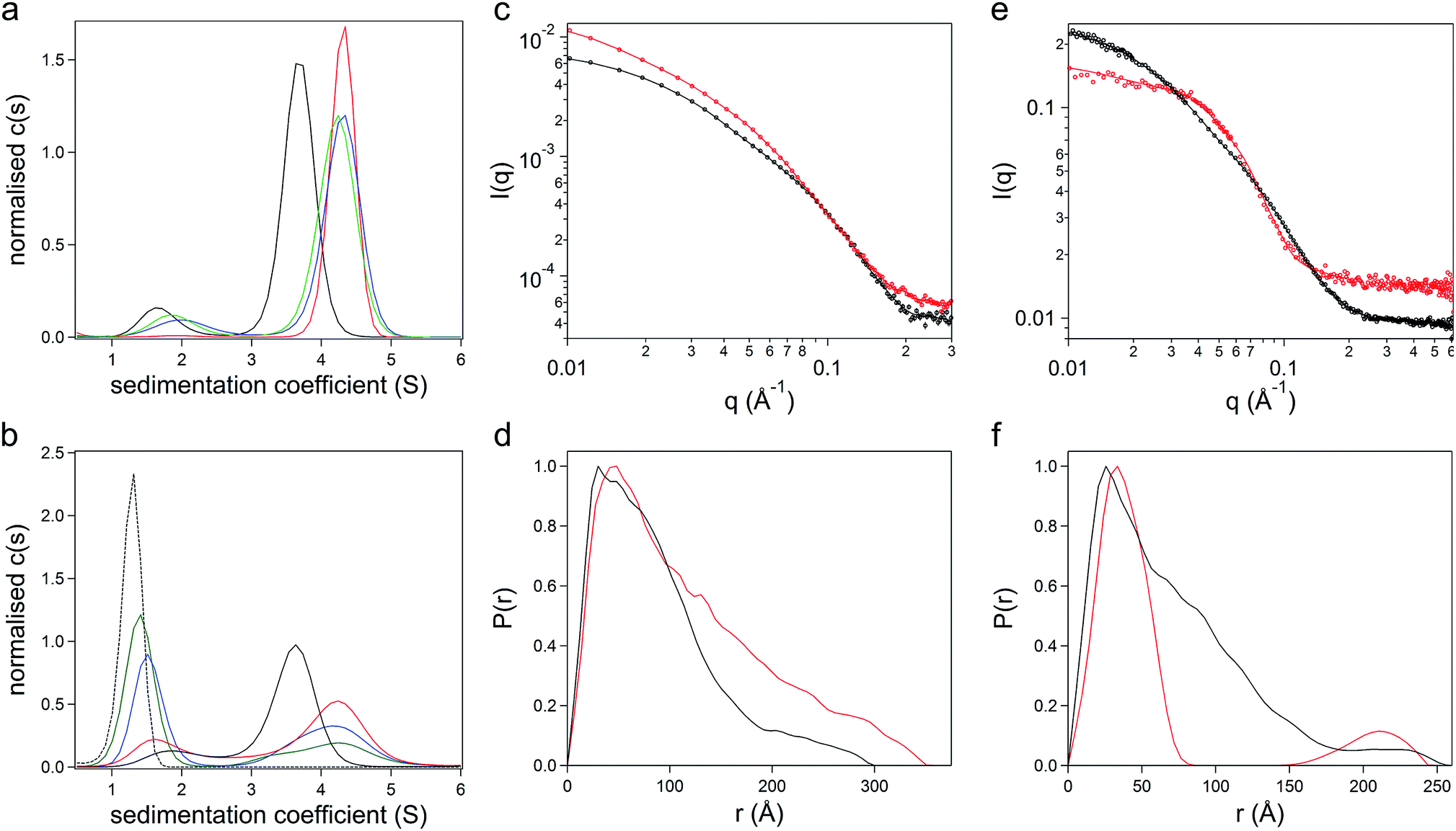 | ||
| Fig. 2 The structure and conformation of the artificial membrane binding construct. Sedimentation velocity analytical ultracentrifugation (SV-AUC) sedimentation coefficient {c(s)} distributions at (a) 487 nm and (b) 280 nm. The distributions show the overlay of the surfactant [S] (black dashed line), CshA_scGFP (black line) and [CshA_scGFP][S] at protein cationic sites:polymer surfactant ratios of 0.6:1 (red line), 1.1:1 (blue line) and 1.6:1 (green line). (c) Synchrotron radiation small angle X-ray scattering (SR-SAXS) data from CshA_scGFP (black symbols) and [CshA_scGFP][S] (red symbols) at 25 °C. The corresponding P(r) distribution fits (solid lines) were obtained using Bayesapp. (d) Pair-distance distribution functions (P(r)) derived from SR-SAXS of CshA_scGFP (black trace) and [CshA_scGFP][S] (red trace). (e) Small angle neutron scattering (SANS) data (symbols) with P(r) distribution fits (solid lines) obtained using Bayesapp. SANS was used to probe the structure of CshA_scGFP in 100% D2O (black) and [CshA_scGFP][S] in 40% D2O (red) at 25 °C. (f) Pair-distance distribution functions (P(r)) derived from SANS data fitting of CshA_scGFP in 100% D2O (black trace) and [CshA_scGFP][S] in 40% D2O (red trace). | ||
The formation of the sub-stoichiometric [CshA_scGFP][S] complex (PCS:PS of 1:0.6) can be rationalized by the anisotropic distribution of cationic surface charge on the CshA_scGFP construct (Fig. 1a). Here, the scGFP membrane binding region displays a high density of cationic surface charge (+36 at pH 7.5), which would favour cooperative surfactant binding through hydrophobic tail burial beneath the PEG-rich regions on the protein surface.31 Conversely, the CshA motif has a theoretical net charge of −30 at pH 7.5. This was reflected in the low zeta potential of the CshA_scGFP construct, which was +0.9 mV at pH 7.5, and decreased to −15.2 mV after the positive surface charges were neutralized via electrostatic surfactant conjugation. The formation of a compact polymer surfactant corona was also supported by dynamic light scattering (DLS) experiments, which showed a 2 nm increase in the hydrodynamic diameter (Fig. S2B†), increasing from approximately 10 nm for CshA_scGFP to 12 nm for [CshA_scGFP][S].
To study the constructs in higher resolution, synchrotron radiation small angle X-ray scattering (SR-SAXS) and neutron scattering (SANS) experiments were performed. For the CshA_scGFP fusion construct, fitting the SAXS scattering pattern (Fig. 2c) yielded a radius of gyration of 7.4 ± 0.4 nm with an axial ratio of 10.6 ± 0.4 (Table S3†). Moreover, the resulting pair-distance distribution function (P(r)) rapidly decayed to a distance of approximately 18 nm before extending out to a Dmax of 29 nm (Fig. 2d). This “long tail” in the P(r) function and high axial ratio can be reconciled by the dynamic intrinsically disordered NR1 “catch” domain in CshA,29 and the corresponding Porod exponent of 2.1 ± 0.1 (Table S3†) confirmed the high level of flexibility. SAXS experiments performed on [CshA_scGFP][S] (Fig. 2c) showed that polymer surfactant conjugation resulted in a 2.4 nm increase in the radius of gyration (cf. CshA_scGFP), which was accompanied by a reduction in the axial ratio (from 10.6 to 8.2 ± 0.1, Table S3†) and an increase in the flexibility of the construct (Porod exponent of 1.8 ± 0.1; Table S3†). SANS data fitting (Fig. 2e) also yielded parameters that were consistent with a highly elongated structure for CshA_scGFP (Table S3†), with a radius of gyration of 6.1 nm, and a P(r) function with Dmax of ca. 25 nm (Fig. 2f). Experiments performed on [CshA_scGFP][S] at the protein contrast match point (40:60 D2O:H2O) gave a bimodal P(r) function (Fig. 2f), which indicated that although the majority of surfactant chains were associated with the scGFP moiety, some electrostatic assembly had occurred near the CshA domain.
The effect of surfactant electrostatic conjugation on protein structure was also investigated using synchrotron radiation circular dichroism (SRCD) and fluorescence spectroscopies. Deconvolution of the Far-UV SRCD spectrum from the CshA_scGFP fusion (Fig. S2C†) gave a secondary structure distribution that was representative of the constituent protein components, with high levels of disordered structure from NR1 of CshA29 (48.6%), as well as beta structure from scGFP32 (21.2%) and alpha helical content from NR2 and NR3 of CshA29 (16.1%) (Fig. S2D†). Significantly, surfactant conjugation to yield [CshA_scGFP][S] produced a SRCD spectrum that could be superimposed on that of the fusion protein (Fig. S2C†), signifying that no major changes in the secondary structure distribution had occurred (Fig. S2E†). The persistence of protein structure after polymer surfactant conjugation was also supported by fluorescence spectroscopy measurements, which showed that both CshA_scGFP and [CshA_scGFP][S] retained the characteristic fluorescent signature of scGFP,33 with an excitation peak at 487 nm and a corresponding emission peak at 510 nm (Fig. S2F†).
The interaction of the artificial membrane binding construct with hMSCs
The potential cytotoxicity of the CshA_scGFP fusion and the [CshA_scGFP][S] conjugate was evaluated by performing cell viability experiments using bone marrow-derived hMSCs. Here, hMSCs could be incubated in either construct up to protein concentrations as high as 12 μM, without significant cell death (Fig. 3a). Accordingly, an incubation concentration of 4 μM was used for all subsequent experiments, and hMSCs incubated with either CshA_scGFP or [CshA_scGFP][S] retained their ability to undergo self-renewal over a two week period (Fig. 3b). Significantly, live cell widefield microscopy experiments showed rapid labelling of the plasma membrane of the hMSCs after incubation with either CshA_scGFP or [CshA_scGFP][S] (Fig. 3c and d). scGFP_CshA was also capable of labelling hMSCs in a mechanism mediated by electrostatic attraction to anionic sulphated proteoglycans on the plasma membrane, which is consistent with studies of supercharged proteins.30,32,33 Moreover, supernatant depletion assays performed on the constructs after incubation with the hMSCs showed approximately 2 billion and 0.7 billion constructs per cell for CshA_scGFP and [CshA_scGFP][S], respectively (Fig. S4A†). Fluorescence activated flow cytometry measurements showed complete labelling of the hMSCs populations by both constructs, with no significant difference in the total fluorescence intensity between the samples (Fig. S4B†). As the flow cytometry assays could not discriminate between constructs on the plasma membrane or in the cytosol, time course live-cell widefield fluorescence microscopy experiments were conducted, which showed that the [CshA_scGFP][S] persisted at the plasma membrane for at least 12 hours (Fig. 3f). Conversely, the CshA_scGFP was rapidly endocytosed over the same period (Fig. 3e), which is consistent with a recent report by Krishnan et al.34 that described a strong cationic surface charge dependency for tissue and cell uptake of supercharged green fluorescent proteins. The cardiogenic differentiation potency of hMSCs after the [CshA_scGFP][S] modification was also investigated. After exposed to 5-azacytidine, the [CshA_scGFP][S] modified hMSCs acquired phenotype of cardiomyocytes as shown by the expression of cardiac biomarkers including cardiac transcription factor Nkx2.5, alpha-cardiac actin and cardiac troponin T (Fig. S5†).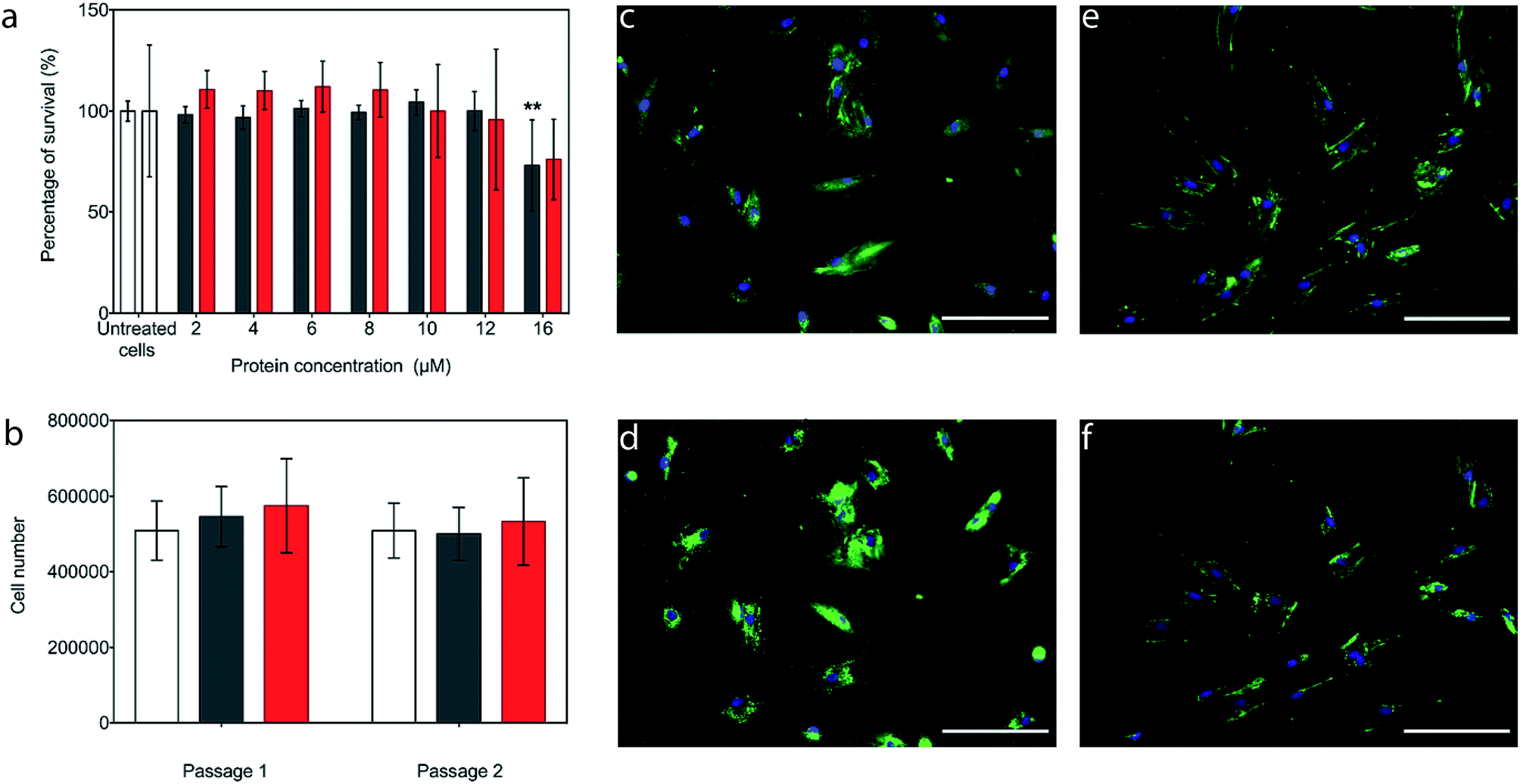 | ||
| Fig. 3 The interaction of the constructs with the hMSC plasma membrane. (a) (3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay showing the MSC viability (via the total metabolic activity) as a function of CshA_scGFP (black) or [CshA_scGFP][S] (red) incubation concentration. The bars represent the mean average and the error bars represent the standard deviation calculated using hMSCs from 3 different patients (n = 3). The groups were tested using a one-way ANOVA followed by a two-tailed equal variance Student's t-test. A p-value of less than 0.05 and 0.01 considered significant (*) and highly significant (**), respectively, when compared to the untreated cells. (b) hMSC proliferation after incubation for 15 minutes with either with 4 μM CshA_scGFP (black) or 4 μM [CshA_scGFP][S] (red), and the untreated cell controls (white). The treated cells retained their proliferation capability for 14 days in culture, indicating CshA_scGFP and [CshA_scGFP][S] had no long-term effects on cell self-renewal. The statistical approach was the same as for (a). Live cell fluorescence widefield microscopy images showing the cell membrane affinity of (c) CshA_scGFP (green) imaged immediately after 15 minutes incubation, and (d) after a further 12 hours, which shows the result of significant endocytosis. Analogous experiments performed after incubation with [CshA_scGFP][S] (green) show persistence fluorescence at the plasma membrane when (e) imaged immediately after 15 minutes incubation, and (f) after a further 12 hours. Cell nuclei were stained with a Hoechst dye (blue). Scale bar = 100 μm. | ||
Modified hMSC fibronectin adhesion assays
Following the demonstration of persistent and efficient hMSC membrane labelling, static adhesion assays were performed to assess the ability of the modified cells to bind to immobilized human fibronectin (Fn). Here, hMSCs were seeded onto Fn-coated substrates and bovine serum albumin (BSA) blocked substrates were used as controls. Widefield microscopy performed on samples showed hMSCs firmly adhered to the Fn-coated dishes after 6 hours of incubation, with the typical flat fibroblastic morphology of hMSCs (Fig. S6†). Significantly, quantitative biochemical assays revealed that the cells labelled with either construct exhibited an increase in adhesion, with [CshA_scGFP][S] producing a two-fold increase in hMSC adherence compared to untreated hMSCs (Fig. 4a). To explore the process adhesion under dynamic conditions, a biomimetic microfluidic adhesion assay was performed to model interactions between circulating modified hMSCs and immobilized Fn. Here, the microfluidic channel was coated with Fn and controls blocked using BSA. By flowing hMSCs at physiologically-relevant shear stresses (1–5 dyne cm−2 (ref. 35)), a general trend was observed, whereby fewer cells adhered to Fn at higher shear stresses (Movies S1, S2 and S3†). Significantly, CshA_scGFP and [CshA_scGFP][S] both conveyed significantly higher adherence of hMSCs to Fn at shear stresses of 1 and 1.5 dyne cm−2 when compared to untreated hMSCs (Fig. 4b and c). This confirmed that the Fn binding propensity of CshA could be effectively transferred from Streptococcus gordonii to human stem cells in vitro.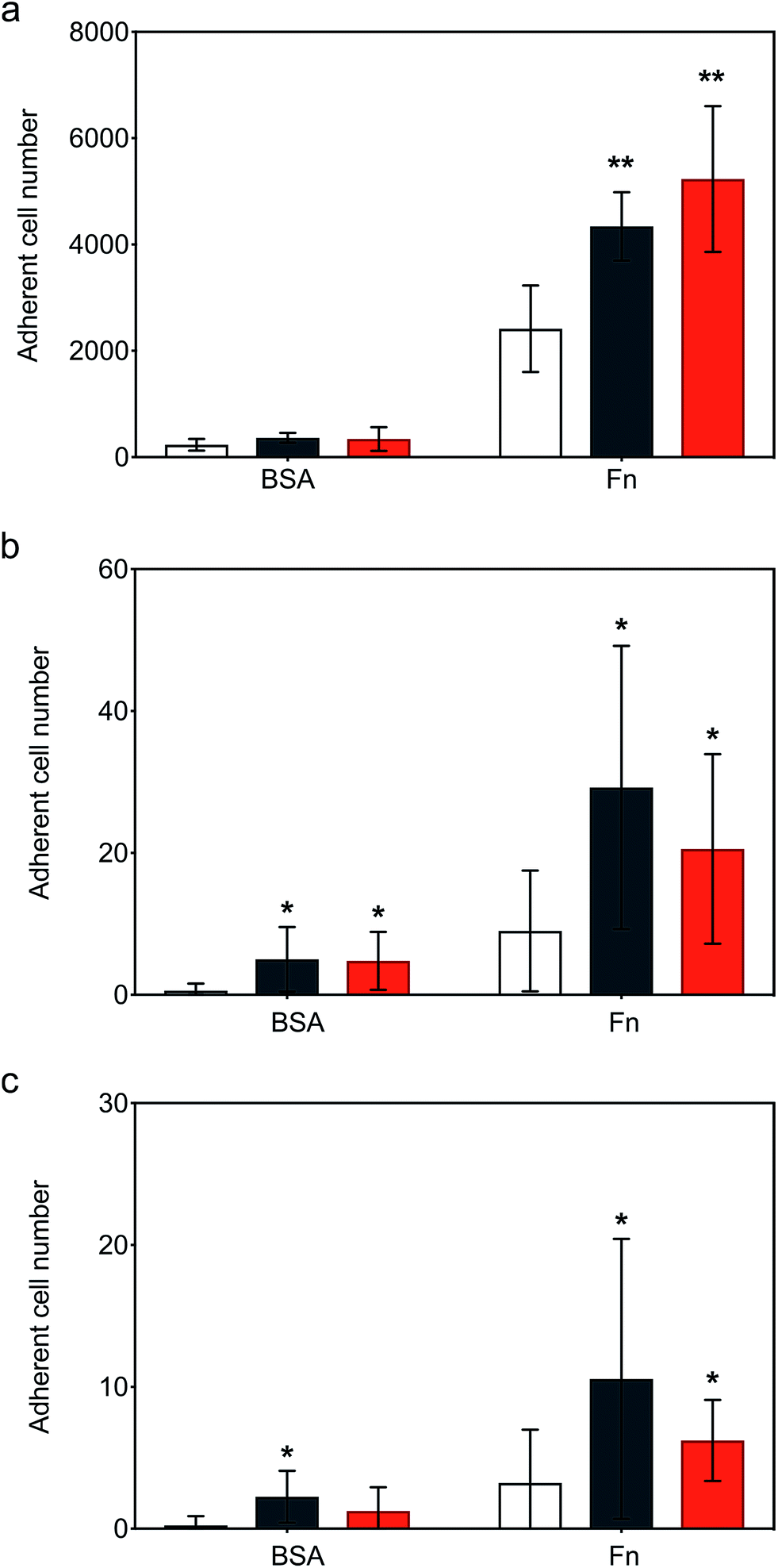 | ||
| Fig. 4 Static and dynamic cell adhesion on immobilised fibronectin (Fn). (a) Static adhesion assay showing the hMSC adherence to immobilised Fn after cells were incubated for 15 minutes with 4 μM of either CshA_scGFP (black) or [CshA_scGFP][S] (red), respectively, and compared with untreated cell controls (white). Dynamic fluidic adhesion assay showing the hMSCs adherence to Fn at shear stress of (b) 1 and (c) 1.5 dyne cm−2, respectively. The bars represent the mean and the error bars represent the standard deviation calculated using hMSCs from 3 different patients (n = 3). Comparison of differences was tested using one-way ANOVA followed by a two-tailed, equal variance Student's t-test with a p-value of less than 0.05 and 0.01 considered significant (*) and highly significant (**), respectively, compared to the untreated cells on either BSA or Fn coating. | ||
hMSCs homing in vivo
In light of the positive Fn adhesion results, an in vivo mouse model was used to assay the propensity of the [CshA_scGFP][S] construct to promote stem cell homing to the myocardium. It was hypothesised that as the fibronectin sequence is remarkably well conserved among higher animals,24 that sufficient targets for the CshA would still be present. Prior to the in vivo administration of the modified hMSCs, the hemocompatibility and systemic toxicity of the [CshA_scGFP][S] construct were evaluated in mice. Significantly, no granulocyte activation, thrombocyte activation, or hemolysis was observed in whole blood samples at 2 hours and 24 hours after the intravenous injection of [CshA_scGFP][S] (Table S4†). Accordingly, [CshA_scGFP][S] modified hMSCs were intravenously transplanted into mice and the systemic toxic effects were evaluated by examining the pathological changes of the major organs after 4 weeks. No notable differences were observed in the heart, lung, kidney or liver among these groups (Fig. S7†), suggesting that [CshA_scGFP][S] modified hMSCs had no acute toxic effects on normal tissues in vivo. The CshA sequence is derived from a commensal organism, which may help reduce immunogenicity. Moreover, as the membrane functionalization is transient (endocytosed by the transplanted cell), this may also help to reduce immunogenicity. For future work, and eventual clinical translation of this system, it will be essential to perform reactivity assays on T- and B-cell proliferation from peripheral human blood after exposure to the chimera-polymer surfactant conjugates as well as complementary enzyme-linked immunosorbent assays (ELISA).The distribution of hMSCs and [CshA_scGFP][S] modified hMSCs in mouse organs after intracardiac injection (Movie S4†) was explored at the cellular level using confocal microscopy, where cells were labelled with red CMPTX cell tracker before injection. Representative images of untreated hMSCs and [CshA_scGFP][S] modified hMSCs in frozen heart sections at 2 and 24 hours after the intracardiac injection showed that the [CshA_scGFP][S] modified hMSCs retained their fluorescence for 24 hours in vivo and could be readily distinguished against the tissue background (Fig. 5a–f). Quantitative PCR assays for human-specific Alu sequences36 were performed to evaluate the number of [CshA_scGFP][S] modified hMSCs and control hMSCs at 2 hours and 24 hours after either intracardiac or intravenous injection. Significantly, an in vivo cytokinetic model37 was applied to the resulting data to elucidate the in vivo spatiotemporal disposition of the administered hMSCs, which showed that the tissue:plasma distribution coefficient of hMSCs in the heart had increased by 100% after modification with [CshA_scGFP][S] for both intracardiac and intravenous injection (Fig. 6a and b, Table S5†). Moreover, hMSC accumulation in the lung did not differ between untreated hMSCs and [CshA_scGFP][S] modified cells (Fig. 6c and d), which indicated that the process involved cell homing and not simply retention.
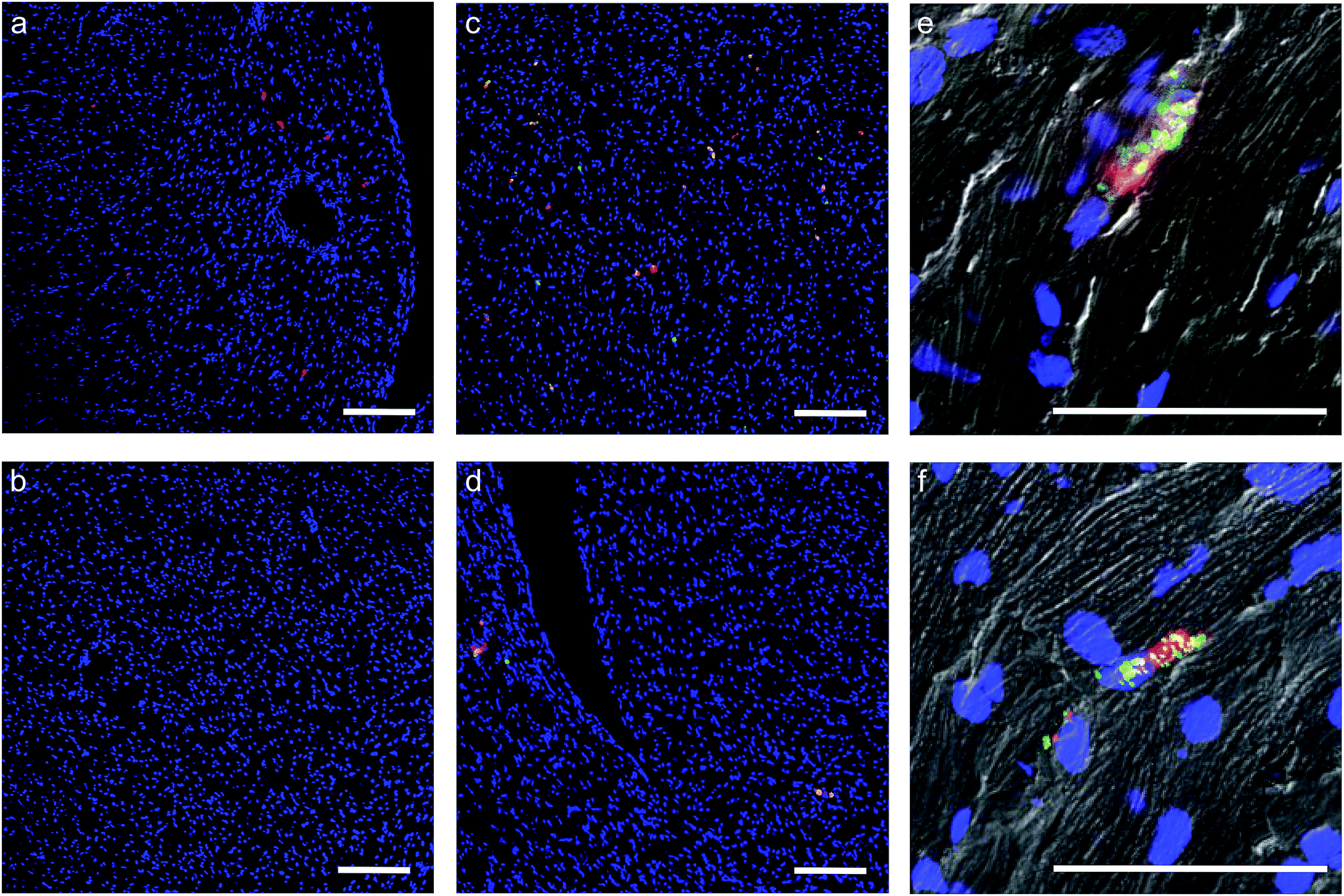 | ||
| Fig. 5 The effect of the construct on hMSCs homing and retention in a murine heart. Representative laser scanning confocal microscopy images from DAPI (blue) stained 10 μm sections of murine heart after intracardiac injection of 2000000 untreated hMSCs or [CshA_scGFP][S]-modified hMSCs. All hMSCs were stained with CMPTX cell tracker (red) before injection. (a) Control unmodified hMSCs after 2 hours, and (b) after 24 hours. (c) [CshA_scGFP][S]-modified hMSCs after 2 hours, and (d) after 24 hours. (e) [CshA_scGFP][S]-modified hMSCs after 24 hours. High magnification composite (brightfield/fluorescence) images show the presence of the [CshA_scGFP][S] construct (green) on the cells (red) after (e) 2 hours and (f) after 24 hours. Scale bar = 200 μm. | ||
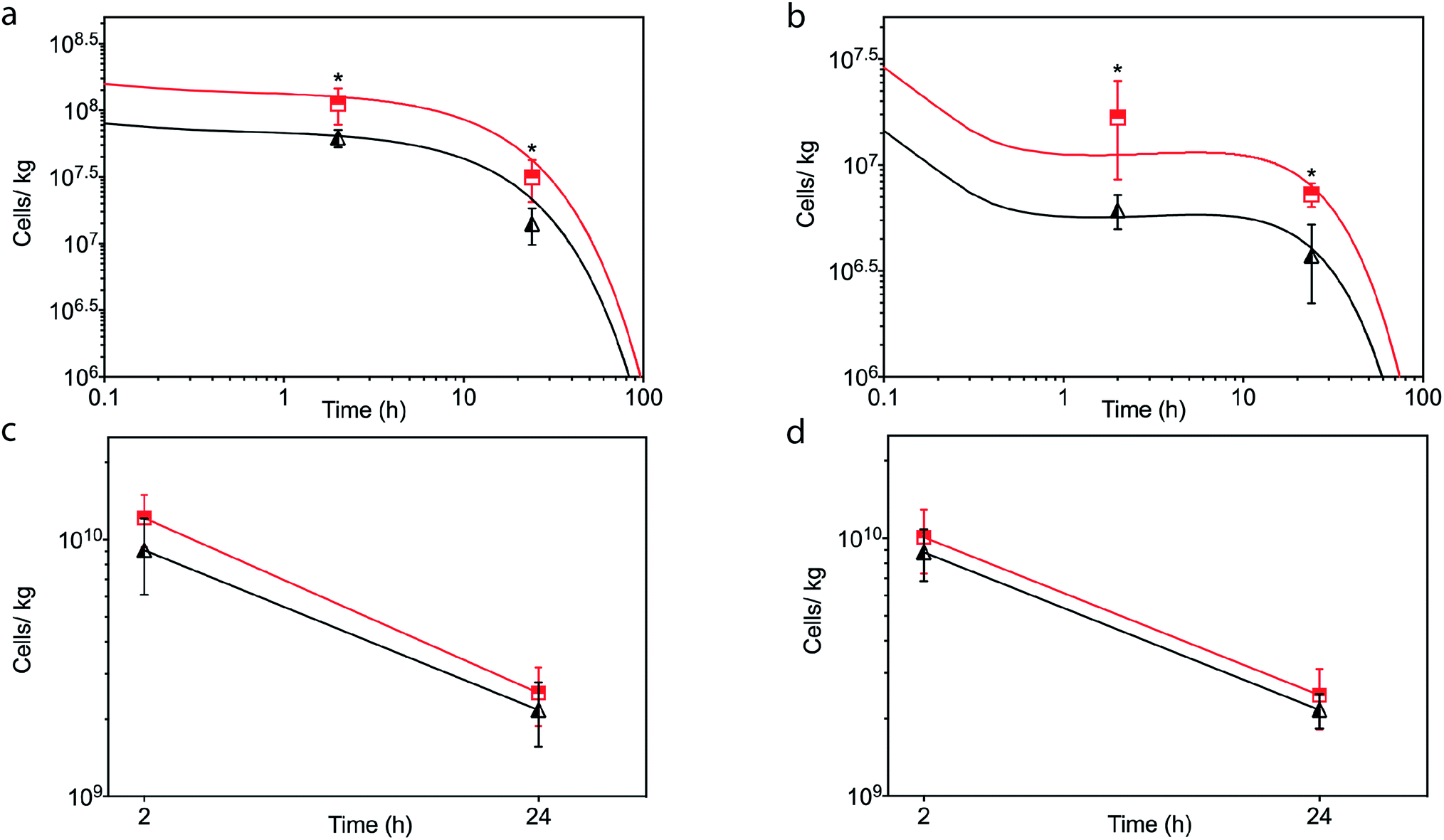 | ||
| Fig. 6 In vivo kinetics of untreated hMSCs (black) and [CshA_scGFP][S] modified hMSCs (red) in the heart after (a) intracardiac or (b) intravenous injection of 2 × 106 cells. The solid line represents the concentration–time profile of the hMSCs simulated by the physiologically based kinetic model while the closed triangles or squares represent measured biodistribution data. Concentration of untreated hMSCs (black) and [CshA_scGFP][S] modified hMSCs (red) in the lung at 2 hour and 24 hour after (c) intracardiac and (d) intravenous injection. Concentration of cells is expressed as number of cells per kilogram of tissue. The triangles and squares represent the mean average and the error bars represent the standard deviation calculated using cells from 3 mice (n = 3), and comparison of differences was tested using a two-tailed, unequal variance Student's t-test with a p-value of less than 0.05 and 0.01 considered significant (*) and highly significant (**), respectively. | ||
Conclusions
In this work, we rationally designed, built and characterised a protein chimera-polymer surfactant construct with cardiac tissue homing properties, which spontaneously inserted into the plasma membrane of hMSCs. Where this system differs considerably from non-homing approach is two-fold. Firstly, we hijacked the innate ability of Streptococcus gordonii to home to cardiac tissue by displaying an artificial membrane binding analogue of the bacterial adhesin CshA on the surface of hMSCs. Bacterial adhesins exhibit exquisite selectivity for target molecules and recognize molecular motifs in a lock-and-key fashion, in line with enzymes and immunoglobulins.23 Accordingly, our approach could be readily extended to include other adhesins that exhibit specific protein–protein interactions to drive homing of therapeutic cells to different organs, e.g., brain, kidney, liver, and related acute or chronic disease. Secondly, we demonstrated that a supercharged protein motif can be used to drive the electrostatic assembly of a cell membrane active polymer surfactant corona. This hydrophobic nonylphenyl chain membrane insertion mechanism presents a number of key advantages. The process is facile, non-cytotoxic and does not require genetic modifications of the cells. Moreover, as our cell membrane engineering display system is completely independent of the cell type, it could be readily applied to other therapeutic cells, including endothelial progenitor cells, bone marrow-derived mononuclear cells, and pluripotent stem cells.38In summary, we demonstrate a completely bacterial adhesin-based homing technology with several key advantages, such as ease of synthesis and handling of both the anchor region and homing molecule, large construct display number (billions) and persistence (>12 hours) on the cell surface, no loss of the biological function of protein after membrane insertion, and high specificity for in vitro/in vivo affinity to Fn. We show that the construct is not cytotoxic, does not elicit a hematologic response in mice, and directs engineered hMSCs delivered either intracardially or intravenously to home selectively to the myocardium, without a concomitant increase in the number of cells in the lungs.
Conflicts of interest
A. W. P. is the Founder, a Director and a shareholder of CytoSeek, a company engaged in the development of cell membrane reengineering. W. X., T. I. P. G. and R. C. D. are shareholders of CytoSeek. Work in the Perriman laboratories at the University of Bristol is supported in part by CytoSeek. The remaining authors declare no competing interests.Acknowledgements
We would like to acknowledge the Wolfson Bioimaging Centre (BBSRC Alert 13 capital grant BB/L014181/1) and the Flow Cytometry Facility at the University of Bristol. We also would like to acknowledge the Diamond Light Source (UK) for access to Far-UV SRCD at beamline B23 and SAXS at beamline B21, as well as the ILL (France) for SANS at beamline D22. We would like to thank Dr D. Frankel for assisting with the microfluidics, Dr B. Carter for assisting with Fig. 1 and SANS, Dr W. Zhang for assisting with the protein purification, G. Day for assisting with the BCA assay, M. Doran for providing the hMSCs, S. Roy for assisting with the imaging, and B. Tse for assisting with the in vivo experiments. We thank EPSRC Doctoral Training Centre Grant EP/G036780/1 for funding Dr T. I. P. G. and the EPSRC (Early Career Fellowship EP/K026720/1), the UKRI (Early Career Fellowship EP/K026720/1 and Future Leaders Fellowship MR/S016430/1) for support for Dr A. W. P. This work was also supported by grants from the Australian National Health and Medical Research Council (APP1126091, to M. S. R. and APP1141121, to H. W.). The datasets corresponding to this Manuscript and Supporting Information are available at DOI: 10.5523/bris.21w0xiagciryg2b1gcdaxn5z2q.References
- M. Angelos and D. Kaufman, Curr. Opin. Organ Transplant., 2015, 20, 663–670 CAS.
- S. A. Fisher, C. Doree, A. Mathur and E. Martin-Rendon, Circ. Res., 2015, 116, 1361–1377 CrossRef CAS PubMed.
- G. Condorelli, U. Borello, L. De Angelis, M. Latronico, D. Sirabella, M. Coletta, R. Galli, G. Balconi, A. Follenzi, G. Frati, M. G. Cusella De Angelis, L. Gioglio, S. Amuchastegui, L. Adorini, L. Naldini, A. Vescovi, E. Dejana and G. Cossu, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10733–10738 CrossRef CAS PubMed.
- D. Hou, E. A. S. Youssef, T. J. Brinton, P. Zhang, P. Rogers, E. T. Price, A. C. Yeung, B. H. Johnstone, P. G. Yock and K. L. March, Circulation, 2005, 112, 150–157 Search PubMed.
- Y. L. Tang, Y. Tang, Y. C. Zhang, K. Qian, L. Shen and M. I. Phillips, J. Am. Coll. Cardiol., 2005, 46, 1339–1350 CrossRef CAS PubMed.
- S. H. Li, T. Y. Y. Lai, Z. Sun, M. Han, E. Moriyama, B. Wilson, S. Fazel, R. D. Weisel, T. Yau, J. C. Wu and R. K. Li, J. Thorac. Cardiovasc. Surg., 2009, 137, 1225–1233 CrossRef PubMed.
- J. M. Karp and G. S. Leng Teo, Cell Stem Cell, 2009, 4, 206–216 CrossRef CAS PubMed.
- D. Feyen, R. Gaetani, J. Deddens, D. van Keulen, C. van Opbergen, M. Poldervaart, J. Alblas, S. Chamuleau, L. van Laake, P. Doevendans and J. Sluijter, Adv. Healthcare Mater., 2016, 5, 1071–1079 CrossRef CAS PubMed.
- T. Dvir, A. Kedem, E. Ruvinov, O. Levy, I. Freeman, N. Landa, R. Holbova, M. S. Feinberg, S. Dror, Y. Etzion, J. Leor and S. Cohen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14990–14995 CrossRef CAS PubMed.
- J. M. Singelyn, P. Sundaramurthy, T. D. Johnson, P. J. Schup-Magoffin, D. P. Hu, D. M. Faulk, J. Wang, K. M. Mayle, K. Bartels, M. Salvatore, A. M. Kinsey, A. N. Demaria, N. Dib and K. L. Christman, J. Am. Coll. Cardiol., 2012, 59, 751–763 CrossRef CAS PubMed.
- R. Ravichandran, J. R. Venugopal, S. Sundarrajan, S. Mukherjee, R. Sridhar and S. Ramakrishna, Nanotechnology, 2012, 23, 385102 CrossRef PubMed.
- M. Montgomery, S. Ahadian, L. Davenport Huyer, M. Lo Rito, R. A. Civitarese, R. D. Vanderlaan, J. Wu, L. A. Reis, A. Momen, S. Akbari, A. Pahnke, R. K. Li, C. A. Caldarone and M. Radisic, Nat. Mater., 2017, 16, 1038–1046 CrossRef CAS PubMed.
- F. Wang and J. Guan Jianjun, Adv. Drug Delivery Rev., 2010, 62, 784–797 CrossRef CAS PubMed.
- I. K. Ko, T. J. Kean and J. E. Dennis, Biomaterials, 2009, 30, 3702–3710 CrossRef CAS PubMed.
- K. Cheng, D. Shen, M. T. Hensley, R. Middleton, B. Sun, W. Liu, G. De Couto and E. Marbán, Nat. Commun., 2014, 5, 4880 CrossRef CAS PubMed.
- C. Y. Lo, B. R. Weil, B. A. Palka, A. Momeni, J. M. Canty and S. Neelamegham, Biomaterials, 2016, 74, 19–30 CrossRef CAS PubMed.
- Y. Teramura, Y. Kaneda, T. Totani and H. Iwata, Biomaterials, 2008, 29, 1345–1355 CrossRef CAS PubMed.
- Y. He, Y. Guo, Y. Xia, Y. Guo, R. Wang, F. Zhang, L. Guo, Y. Liu, T. Yin, C. Gao, E. Gao, C. Li, S. Wang, L. Zhang, W. Yan and L. Tao, Am. J. Physiol.: Heart Circ. Physiol., 2018, 316, H233–H244 CrossRef PubMed.
- Z. Li, D. Shen, S. Hu, T. Su, K. Huang, F. Liu, L. Hou and K. Cheng, ACS Nano, 2018, 12, 12193–12200 CrossRef CAS PubMed.
- T. J. Antes, R. C. Middleton, K. M. Luther, T. Ijichi, K. A. Peck, W. J. Liu, J. Valle, A. K. Echavez and E. Marbán, J. Nanobiotechnol., 2018, 16, 1–15 CrossRef PubMed.
- A. Vandergriff, K. Huang, D. Shen, S. Hu, M. T. Hensley, T. G. Caranasos, L. Qian and K. Cheng, Theranostics, 2018, 8, 1869–1878 CrossRef CAS PubMed.
- T. J. Kean, P. Lin, A. I. Caplan and J. E. Dennis, Stem Cells Int., 2013, 2013, 732742 Search PubMed.
- P. Klemm and M. A. Schembri, Int. J. Med. Microbiol., 2000, 290, 27–35 CrossRef CAS PubMed.
- B. Henderson, S. Nair, J. Pallas and M. A. Williams, FEMS Microbiol. Rev., 2011, 35, 147–200 CrossRef CAS PubMed.
- R. McNab, A. R. Holmes, J. M. Clarke, G. W. Tannock and H. F. Jenkinson, Infect. Immun., 1996, 64, 4204–4210 CAS.
- N. S. Jakubovics, J. L. Brittan, L. C. Dutton and H. F. Jenkinson, Microbiology, 2009, 155, 3572–3580 CrossRef CAS PubMed.
- M. H. Konstandin, H. Toko, G. M. Gastelum, P. Quijada, A. De La Torre, M. Quintana, B. Collins, S. Din, D. Avitabile, M. Völkers, N. Gude, R. Fässler and M. A. Sussman, Circ. Res., 2013, 113, 115–125 CrossRef CAS PubMed.
- A. Van Dijk, H. W. M. Niessen, W. Ursem, J. W. R. Twisk, F. C. Visser and F. J. VanMilligen, Cell Tissue Res., 2008, 332, 289–298 CrossRef CAS PubMed.
- C. R. Back, M. N. Sztukowska, M. Till, R. J. Lamont, H. F. Jenkinson, A. H. Nobbs and P. R. Race, J. Biol. Chem., 2017, 292, 1538–1549 CrossRef CAS PubMed.
- J. P. K. Armstrong, R. Shakur, J. P. Horne, S. C. Dickinson, C. T. Armstrong, K. Lau, J. Kadiwala, R. Lowe, A. Seddon, S. Mann, J. L. R. Anderson, A. W. Perriman and A. P. Hollander, Nat. Commun., 2015, 6, 7405 CrossRef CAS PubMed.
- A. P. S. Brogan, R. B. Sessions, A. W. Perriman and S. Mann, J. Am. Chem. Soc., 2014, 136, 16824–16831 CrossRef CAS PubMed.
- M. S. Lawrence, K. J. Phillips and D. R. Liu, J. Am. Chem. Soc., 2007, 129, 10110–10112 CrossRef CAS PubMed.
- D. B. Thompson, J. J. Cronican and D. R. Liu, in Methods in Enzymology, Elsevier Inc., 1st edn, 2012, vol. 503, pp. 293–319 Search PubMed.
- Y. Krishnan, H. A. Rees, C. P. Rossitto, S.-E. Kim, H.-H. K. Hung, E. H. Frank, B. D. Olsen, D. R. Liu, P. T. Hammond and A. J. Grodzinsky, Biomaterials, 2018, 183, 218–233 CrossRef CAS PubMed.
- A. M. Malek, S. L. Alper and S. Izumo, JAMA, J. Am. Med. Assoc., 1999, 282, 2035–2042 CrossRef CAS PubMed.
- R. H. Lee, A. A. Pulin, M. J. Seo, D. J. Kota, J. Ylostalo, B. L. Larson, L. Semprun-Prieto, P. Delafontaine and D. J. Prockop, Cell Stem Cell, 2009, 5, 54–63 CrossRef CAS PubMed.
- H. Wang, X. Liang, Z. P. Xu, D. H. G. Crawford, X. Liu and M. S. Roberts, Sci. Rep., 2016, 1–12 Search PubMed.
- D. A. Lerman, A. Nasri, U. Kiddy Levente and P. Bruno, Eur Cardiol., 2016, 11, 43–48 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02650a |
| ‡ These authors contributed equally to the work and are joint first authors. |
| This journal is © The Royal Society of Chemistry 2019 |