 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBand-selective dynamics in charge-transfer excited iron carbene complexes
Pavel
Chábera
 a,
Lisa A.
Fredin
b,
Kasper S.
Kjær
a,
Nils W.
Rosemann
a,
Linnea
Lindh
a,
Om
Prakash
c,
Yizhu
Liu
c,
Kenneth
Wärnmark
c,
Jens
Uhlig
a,
Villy
Sundström
a,
Arkady
Yartsev
a and
Petter
Persson
*b
a,
Lisa A.
Fredin
b,
Kasper S.
Kjær
a,
Nils W.
Rosemann
a,
Linnea
Lindh
a,
Om
Prakash
c,
Yizhu
Liu
c,
Kenneth
Wärnmark
c,
Jens
Uhlig
a,
Villy
Sundström
a,
Arkady
Yartsev
a and
Petter
Persson
*b
aDivision of Chemical Physics, Department of Chemistry, Lund University, Box 124, SE-22100 Lund, Sweden
bDivision of Theoretical Chemistry, Department of Chemistry, Lund University, Box 124, SE-22100 Lund, Sweden. E-mail: Petter.Persson@teokem.lu.se
cCenter for Analysis and Synthesis (CAS), Department of Chemistry, Lund University, Box 124, SE-22100 Lund, Sweden
First published on 18th February 2019
Abstract
Ultrafast dynamics of photoinduced charge transfer processes in light-harvesting systems based on Earth-abundant transition metal complexes are of current interest for the development of molecular devices for solar energy conversion applications. A combination of ultrafast spectroscopy and first principles quantum chemical calculations of a recently synthesized iron carbene complex is used to elucidate the ultrafast excited state evolution processes in these systems with particular emphasis on investigating the underlying reasons why these complexes show promise in terms of significantly extended lifetimes of charge transfer excited states. Together, our results challenge the traditional excited state landscape for iron-based light harvesting transition metal complexes through radically different ground and excited state properties in alternative oxidation states. This includes intriguing indications of rich band-selective excited state dynamics on ultrafast timescales that are interpreted in terms of excitation energy dependence for excitations into a manifold of charge-transfer states. Some implications of the observed excited state properties and photoinduced dynamics for the utilization of iron carbene complexes for solar energy conversion applications are finally discussed.
Introduction
Characterization of ultrafast excited-state dynamics is crucial for understanding and rationally improving the performance of a wide range of emerging solar energy conversion technologies, including molecular systems for both photovoltaic and solar fuels applications.1 Many molecular-based approaches utilize the favourable photoelectrochemical properties of transition metal complexes (TMCs) that have been developed both for light-harvesting and photocatalytic functions.2 TMCs can, in particular, utilize charge-transfer (CT) excited states as a first step towards generating high-energy CT states. The intrinsic capabilities of many transition metal complexes to sustain a range of oxidation states also make them suitable as central components for electrochemical and photoelectrochemical oxidation and reduction reactions as a key part of many photocatalytic reaction schemes.3Octahedral second and third row transition metal complexes sporting nd6 (with n = 4 or 5) electronic configurations on the metal have maintained a central role for many light-harvesting and molecular photochemistry applications for several decades, owing in large part to the large ligand field splitting (LFS) in which the low-spin nd6 electronic ground state configuration is stabilized. This has yielded many complexes with long-lived high-energy metal-to-ligand charge transfer (MLCT) excited states that have been widely used for photophysical and photochemical applications.2 This particularly includes the famous family of RuII polypyridyl complexes such as Ru(bpy)3 derivatives used extensively as photosensitizers, e.g. in molecular photovoltaics.4,5 Other prominent examples of photoactive d6 complexes are based on the third-row species ReI, OsII, and PtIII which are also interesting for light-emitting applications.6
It has, however, increasingly been recognized that it is desirable for environmentally friendly and sustainable large-scale solar energy applications to replace rare and expensive second and third row transition metal complexes with more Earth-abundant alternatives.7–11 This has prompted significant interest in developing first row transition metal complexes with good photophysical and photochemical properties to ideally rival and replace those from the more well-established second and third row transition metals. This has led to the emergence in recent years of several interesting photoactive first row transition metal complexes of, for example, copper, chromium, and iron.12–16 As the lighter and abundant congener of ruthenium and osmium, iron looks prima facie to be an obvious candidate to develop photofunctional first row transition metal complexes. Progress in this direction has, however, until very recently been largely hampered by poor excited state properties, including a preponderance for substantial and ultrafast excited state energy losses,17,18 with high-energy metal-to-ligand charge-transfer excited states in iron polypyridyl complexes typically decaying on a few picoseconds or faster timescale.19,20 This has remained rather discouraging compared to the abundance of ruthenium polypyridyl complexes maintaining high excited state energies for hundreds of nanoseconds or longer.2
The photophysics and photochemistry of many first–row transition metal complexes, including traditional octahedral FeII polypyridyl complexes, suffer severely from an intrinsically weaker ligand field splitting for 3d complexes compared to their 4d and 5d counter-parts.21 Even the favourable 3d6 configuration in octahedral FeII complexes is typically not sufficient to avoid serious problems with ultrafast deactivation of high energy excited singlet and triplet 1/3MLCT states to low-energy high-spin (quintet) metal centred (5MC) scavenger states.22 Complexes with low-energy high-spin MC states are of some considerable interest in themselves as spin cross-over (SCO) complexes, and they are investigated in the particular context of photoinduced formation as light-induced excited state spin transition (LIESST) systems.23 This has prompted several investigations to elucidate the ultrafast dynamics and excited state cascade of photoexcited iron complexes.17 Recently, this has also been complemented by ultrafast X-ray spectroscopic investigations capable of probing local structure and spin dynamics involving the iron centre.24–26 Experimental progress together with increasingly advanced quantum chemical calculations and dynamics simulations have yielded a wealth of valuable insight into the ultrafast early deactivation dynamics in such complexes.27–31
In the last five years, a new ligand motif featuring strongly sigma-donating N-heterocyclic carbenes (NHCs) has been realized synthetically and demonstrated through a combination of time-resolved optical characterization and calculations to achieve dramatically improved excited state properties of iron complexes.32,33 This has provided a promising path for development also of iron-based photosensitizers for a variety of photoinduced electron transfer applications. Initial synthesis and characterization of octahedral FeII complexes with up to four carbene ligands yielded excited charge-transfer state lifetimes of up to a few tens of picoseconds;34–36 significantly longer than the typical ultrafast decay of Fe-polypyridyl LIESST complexes on a few ps or faster timescale, though still quite short compared to e.g. many champion RuII photosensitizers. This excited state lifetime improvement to a few tens of ps excited state lifetimes was sufficient for a recent proof-of-principle demonstration of efficient photoinduced interfacial electron injection from an anchored iron carbene photosensitizer to a semiconducting TiO2.37
Another step towards more broadly useful iron-based photosensitizers was recently achieved through a synthetic design that involved full octahedral coordination using a hexa-carbene motif in a Fe(btz)3 complex with the chemical structure shown in Fig. 1. Interestingly, this complex was found to be sufficiently stable in both oxidation states II and III to allow both ground state absorption (Fig. 1b) as well as excited state dynamics characterization. FeII and FeIII correspond to 3d6 and 3d5 electronic configurations or the iron centre, respectively.38,39 The FeIII(btz)3 complex was first reported in 2017 as a quite rare case of a 3d5 complex exhibiting interesting photophysics, with a record ∼100 ps charge-transfer excited state lifetime involving a 2LMCT state as illustrated in Fig. 2.38 Spin-allowed decay to the doublet ground state (2GS) also showed the first persistent room temperature photoluminescence (PL) in the visible part of the electromagnetic spectrum from such an iron complex, albeit with a rather low PL quantum yield (ΦPL ∼ 10−4).38 The corresponding FeII species FeII(btz)3 was subsequently also shown to have a long room-temperature excited state lifetime of several hundred picoseconds (τ ∼500 ps), following a more conventional MLCT excitation scheme for a 3d6 complex (Fig. 2).39 A new hexa-carbene FeIII complex was furthermore recently shown to feature enhanced photoluminescence from an excited CT state with a nanosecond lifetime, as well as being capable of reductive and oxidative bimolecular electron transfer reactions from the photoexcited 2LMCT state.40 The extension of the excited state lifetimes of the MLCT and LMCT charge transfer states for iron complexes into the hundreds of picosecond range and longer thus open up a range of photophysical and photochemical applications for iron carbene complexes.
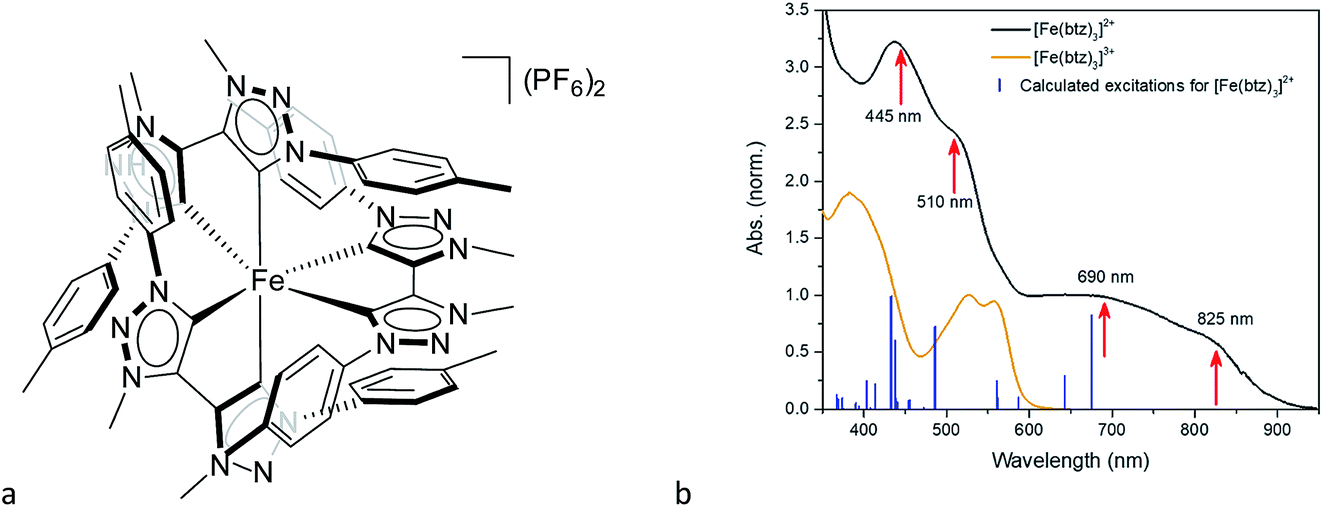 | ||
| Fig. 1 (a) Chemical structure of the FeII(btz)3 complex, and (b) steady state spectra of FeII(btz)3 and FeIII(btz)3 in acetonitrile. The red arrows indicate the excitation wavelengths for transient absorption experiments. | ||
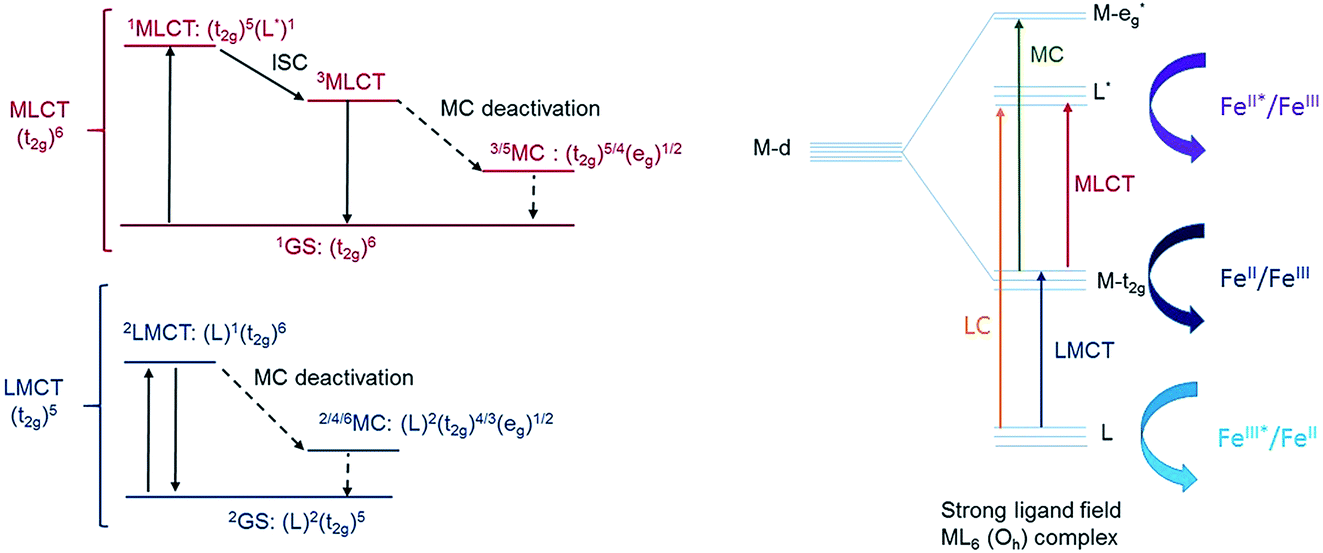 | ||
| Fig. 2 Simplified Jablonski-type diagram for low-spin FeII 3d5 and FeIII 3d6 complexes (left), together with a schematic illustration of key electronic levels and photoexcitation processes involved in the photophysics (right). Curved arrows indicate opportunities to drive subsequent bimolecular or interfacial electron transfer processes (far right). | ||
It is, in this context, quite remarkable that the same strategy of increasing the σ-donation of the ligand surrounding the iron centre was effective for both FeII and FeIII as shown in the case of Fe(btz)3. It is therefore interesting to develop a more comprehensive comparison of the excited state dynamics of these two types of excited state cascades. In order to achieve this, we here report new results, in particular pertaining to dynamics of FeII(btz)3 following excitations at different excitation energies, together with some computational results detailing electronic structure properties in these photosensitizers. Together with recent time-resolved and computational results that we have reported for these complexes, this allows us to present a more comprehensive discussion focused on the ultrafast excited state dynamics of this novel type of promising photosensitizer.
Methods
Experimental
The experimental setup for recording broadband transient absorption spectra was based on a Ti:Sapphire amplified laser system (Spitfire XP Pro, Spectra Physics) operating at a 1 kHz repetition rate, producing ∼80 fs pulses centered at 796 nm. The pump beam was tuned by an optical parametric amplifier (TOPAS C, Light Conversion) to excite the sample at various wavelengths (445 nm, 510 nm, 690 nm, and 825 nm) with typical fluency ∼3 × 1015 photons per pulse per cm2 and maximum fluency not exceeding 1 × 1016 photons per pulse per cm2 which is well below absorption saturation density ∼5 × 1017 photons per cm2. A power dependency check was also performed by varying the excitation intensity by a factor of 25 (100 to 2500 μW) with no deviation of the observed ΔA signal from linear behaviour. A super-continuum white-light was used as probe, generated by focusing the NIR signal from a TOPAS C into a 5 mm sapphire plate. The desired timing between excitation and probe pulses was achieved using a computer-controlled delay line (Aerotech, 10 ns). The pump and the probe beams were overlapped on the sample with their relative polarization set to the magic-angle (54.7°) by a Berek polarization compensator placed in the pump beam path. The sample was placed in a quartz 1 mm path length cuvette with an automated sample mover to avoid sample photodamage, which was also checked by measuring the absorption spectra of the sample before and after each experiment. The probe and reference beams were collimated on the entrance aperture of a prism-based, double-beam spectrograph, and detected by a double diode-array detection system (Pascher Instruments). Correction of the data as well as fitting of the transient absorption datasets was carried out using both an in-house written Python routine and the analysis software DAFit (Pascher Instruments) and the individual kinetics were fitted using the Origin software.Computational
First principles quantum chemical calculations have been performed at the Density Functional Theory (DFT) and time-dependent DFT (TD-DFT) levels of theory for ground- and excited-state properties, respectively. The presented computational results were obtained using the B3LYP* modification41 of the standard B3LYP hybrid density functional42 and employing standard 6-311G basis sets with polarization functions,43 as well as a polarizable continuum solvation model for acetonitrile.44 All calculations were performed using the Gaussian 09 program package.45Results
In this results section, we first consider the linear absorption spectra of both FeII(btz)3 and FeIII(btz)3 as a basis for the subsequent discussions. Next, new time-resolved optical spectroscopy results for the excited state dynamics of Fe(btz)3 are presented. In particular, we present new transient absorption (TA) results for the FeII(btz)3 complex in acetonitrile following excitations at different energies. Finally, absorption spectra are complemented by a presentation of selected results from quantum chemical calculations of both FeII and FeII oxidation states of this complex. Together, this forms a basis for a comparative analysis of the excited state dynamics of this complex in the complementary FeII (3d6) and FeIII (3d5) oxidation states presented in the subsequent discussion below.Linear absorption
The linear absorption spectra of FeII(btz)3 and FeIII(btz)3, shown in Fig. 1b, clearly demonstrate that, in contrast to many other TMCs, this new class of Fe-carbene TMCs exhibit two distinct CT absorption bands on the red side of the strong ligand-based absorption below ∼350 nm. There are, furthermore, indications of several sub-band features of vibrational or electronic nature that appear as shoulders or double peaks in the CT regions of the absorption spectra. Additionally, the blue CT band in FeII(btz)3 has at least twice as strong maximum absorption strength compared to the red band for both oxidation states. The measured CT absorption bands agree qualitatively with the calculated singlet–singlet TD-DFT spectrum for FeII(btz)3 presented as sticks for comparison.Transient absorption
Transient absorption data has been recorded for the FeII(btz)3 complex as shown in Fig. 3. In particular, this includes transient data following excitations at 445 nm, 510 nm, 690 nm, and 825 nm, complementing the published data for excitation at 800 nm at key points in the Fe(II) absorption spectrum (Fig. 1).39 The excitations into different parts of the visible absorption spectrum probe the dynamics following excitations into CT states of different electronic nature and with different initial excess energies compared to the absorption threshold.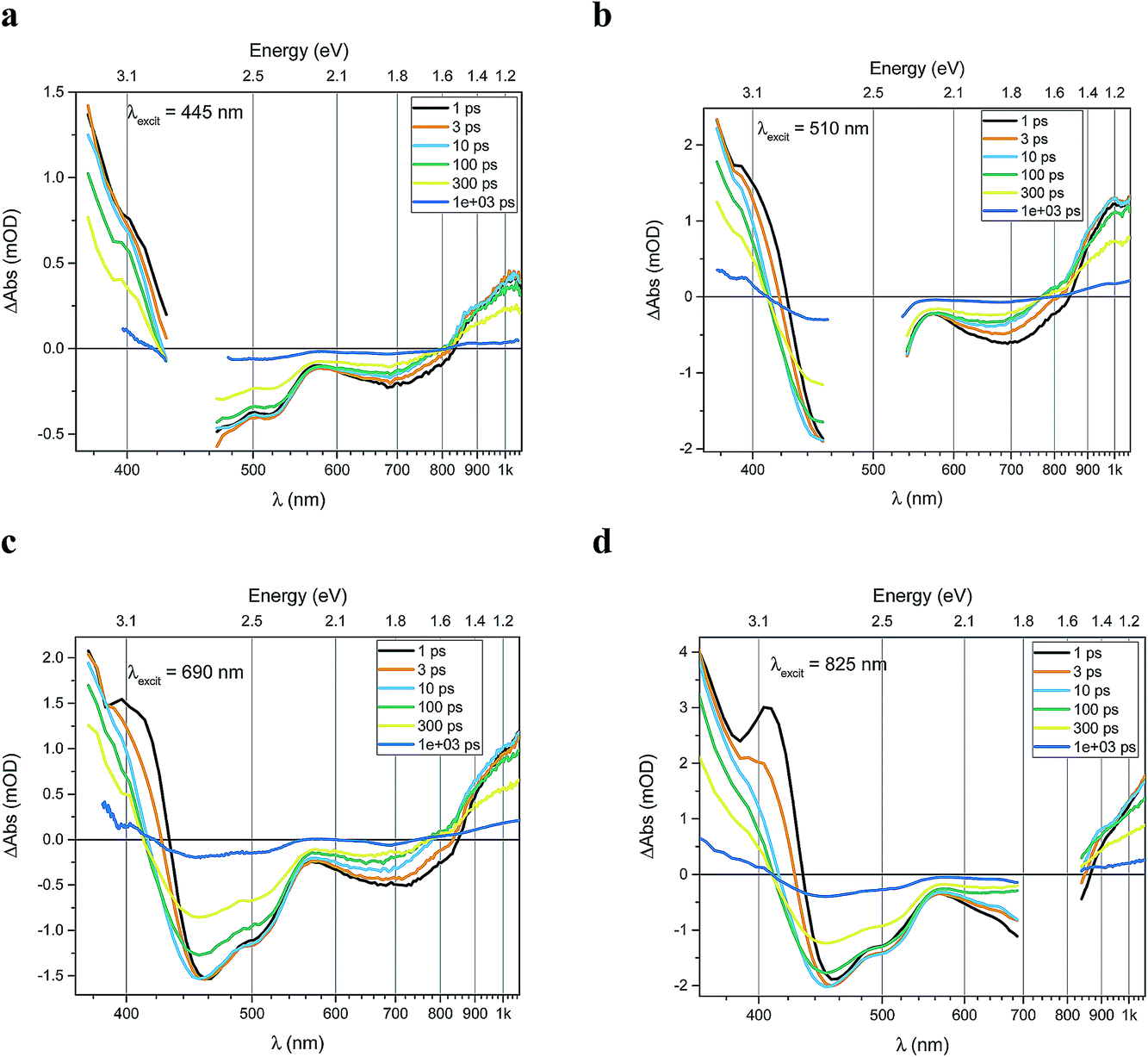 | ||
| Fig. 3 Transient absorption spectra of FeII(btz)3 as measured, after correction for GVD (chirp) and background subtraction. Spectra were recorded following excitations at (a) 445 nm, (b) 510 nm, (c) 690 nm and (d) 825 nm, with excitation densities of 2.4 × 1015, 3.7 × 1015, 3.3 × 1015 and 1.1 × 1016 photons per pulse per cm2, respectively. Excitation scatter region is omitted for clarity. For all panels the x-axis (wavelength) is in reciprocal values (linear steps in energy). | ||
Several main features are recognizable throughout as relatively persistent features that survive the full dynamics, also for the different excitation energies. TA dynamics are brought out further in the analysis shown in Fig. 4, which compares the spectral shape of the TA at 1 and 20 ps for all excitation energies after accounting for the excitation densities.
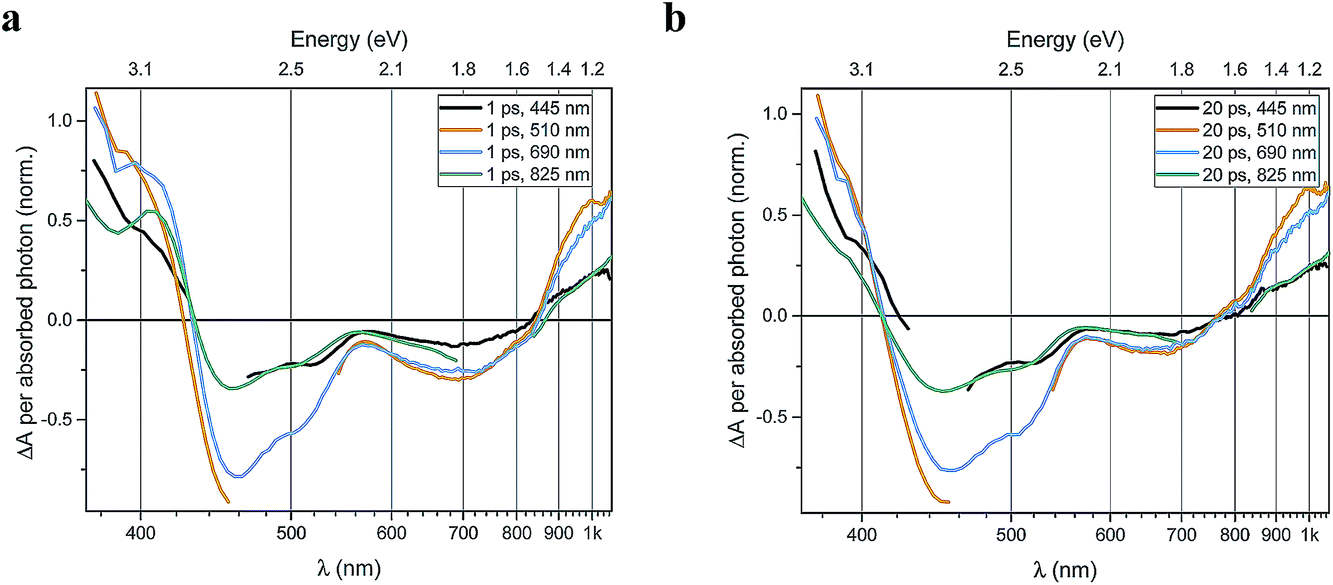 | ||
| Fig. 4 Transient absorption spectra of FeII(btz)3 at (a) 1 ps, and (b) 20 ps normalized by the number of absorbed photons per cm2 at each respective excitation wavelength. The x-axis (wavelength) is in reciprocal values (linear steps in energy). | ||
The main spectral features for all the four excitation energies are similar to what was recently outlined for 800 nm excitation in our paper introducing this complex.39 Briefly, this covers broad ground state bleach (GSB) features similar to an inverted absorption spectrum in the ca 450–800 nm region of the spectrum, together with strong excited state absorption (ESA) in the blue part of the spectrum giving a positive signal above ca 425 nm as well as some ESA in the red part of the spectrum that turns positive beyond the GSB region for wavelengths longer than ca 800 nm. The blue ESA band is apparently relatively strong, as the positive TA signal is observed in the spectral range where one would expect a pronounced bleach signal based on the linear absorption spectrum. Clearly, the overlapping ESA band must have a sharp rise towards the blue to overshoot the expected bleach shoulder, and instead give rise to a strong net positive TA signal.
A closer look at the spectral features in Fig. 3 and 4 reveals some noticeable similarities and differences in terms of the detailed shape and evolution of these TA spectra. The 510 nm and 690 nm mid-CT band spectra are, in particular, very similar throughout. The TA results for all the excitations furthermore show a qualitatively similar GSB to the TA for the recently reported energy excitation at 800 nm. There is a noticeable reduction in the intensity of the ESA feature at ∼408 nm at early timescales for the higher energy excitations compared to the corresponding features for the lowest excitation energy at 825 nm. There is, however, little further spectral evolution after the first 20 ps, as evident for excitations at all four excitation energies. Thus, this behaviour is very similar for the higher energy excitations compared to what has already been established for excitation at 800 nm.39 It is interesting to note that although similar, the main features of the differential spectra appear to have excitation energy dependent amplitudes.
As a final step in the analysis of the excitation energy dependent dynamics, we have investigated different aspects of the multi-exponential kinetics for different combinations of pump and probe wavelengths in further detail as shown in Fig. 5, together with an associated summary of selected key kinetics components listed in Table 1. The single kinetic analysis is focused on identifying specific decay component in the TA data. As some of the decay components only contribute significantly at some probe wavelengths it is not always possible to fit all the time regions in one kinetic with current S/N ratio in order to receive satisfactory fits. Thus only selected unambiguous components are listed in Table 1. It can be generally noted that the dynamics in several places show signs of an ultrafast component on a sub-ps timescale that is potentially interesting in terms of ISC etc, but in all transient spectra and kinetics discussed in this study, we do not discuss further the very early dynamics that occur during the first few 100s of fs due to rather strong contribution of the coherent and solvent related experimental artefacts. For excitation energies into both main charge-transfer bands, the major part of the decay originates from a long-term component with τ ∼540 ps, as e.g. clearly evident for the 1000 nm kinetics for all excitation energies, and overall in good agreement with what we recently reported for excitation at 800 nm. There is also clear evidence for a ∼3 ps component at selected spectral regions in the more comprehensive TA obtained here, as seen e.g. for the 420 nm probes with different excitation energies presented in panels Fig. 5c and d; again in broad agreement with the results in our initial study based on 800 nm excitation. We also identify an additional new kinetic component with τ ∼50 ps as part of the early dynamics. This component is most clearly pronounced in the displayed TA data for the 408 nm kinetics following 825 nm excitation while appearing gradually less pronounced for higher energy excitations. Overall, the kinetics measured at different excitations thus look qualitatively similar, but the contribution of the different decay components is quantitatively distinct. For example, all kinetics look very similar as probed at 1000 nm, while there are clear differences in the kinetics probed at 408 nm for the different excitation energies. Some implications of the rich dynamics observed here is further considered in the discussion below.
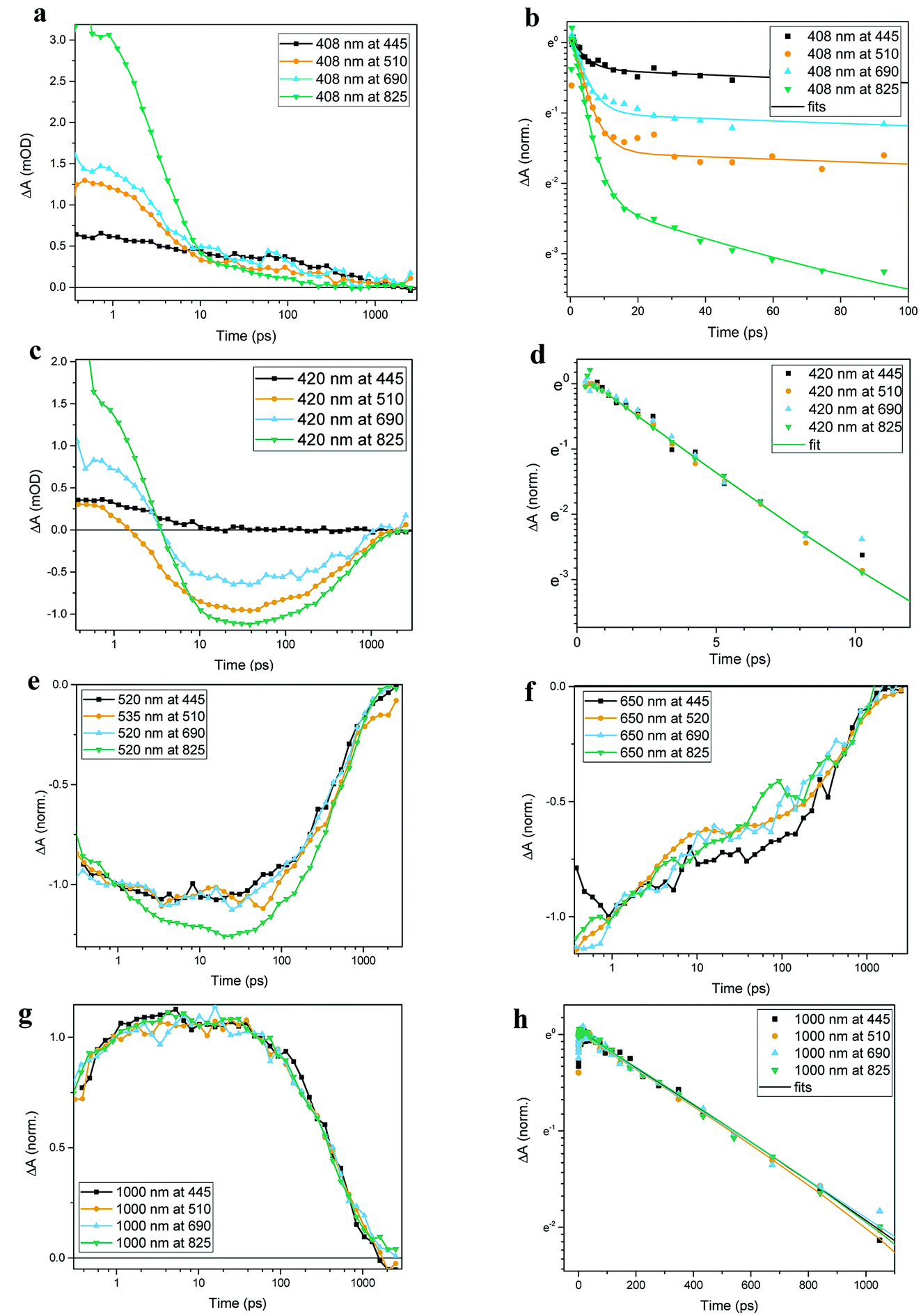 | ||
| Fig. 5 Comparison of FeII(btz)3 kinetics following excitations at different wavelengths. Results are shown for kinetics recorded at (a and b) 408 nm, (c and d) 420 nm, (e) 520 nm, (f) 650 nm, and (g and h) 1000 nm following excitations at 445 nm, 510 nm, 690 nm, and 825 nm. Solid lines represent multi-exponential fits. Note that graphs in panels (a), (c), (e), (f) and (g) are plotted in ΔA(lin)/time (log) scale while graphs in panels (b), (d), and (h) are shown in ΔA(ln)/time (lin) scale. All kinetics except for panels (a) and (c) have been normalised for visual clarity. | ||
| λexcitation (nm) | λ probe (nm) | τ 1 (ps) | τ 2 (ps) | τ 3 (ps) |
|---|---|---|---|---|
| 445, 510, 690, 825 | 1000 | 540 ± 35 | ||
| 445, 510, 690, 825 | 420 | 3.2 ± 0.10 | ||
| 445, 510, 690 | 408 | 3.8 ± 0.81 | 540 ± 35 | |
| 825 | 408 | 3.5 ± 0.36 | 54.0 ± 5.67 | 540 ± 35 |
Electronic structure
Quantum chemical calculations performed at the DFT and TD-DFT level of theory provide information about the electronic states involved in the excited state cascade. Some key results were presented for both FeIII(btz)3 and FeII(btz)3 already as part of the initial characterization of the respective species and their excited state properties.38,39 Selected computational results, including some new information, are presented here, to put results from both species on common ground to facilitate a common discussion about the similarities and differences in fundamental electronic structure properties pertinent for understanding the excited state dynamics.First, it can be noted that the change in oxidation state from FeII to FeIII has a profound influence both on the ground state electronic structure and the excited state manifold. The FeII carbene complexes are characterized as a regular low-spin 3d6 species, i.e. with an electronic configuration of (t2g)6 that is similar e.g. to many well-known RuII polypyridyl complexes.2 This features a singlet ground state and 1/3MLCT excited states. For traditional FeII complexes with weaker LFS, initial excitation into MLCT excited states have been extensively discussed to decay to low-energy metal centred triplet and quintet states (3MC and 5MC) on ultrafast timescales.17,21,22,27,46 This is typically followed by slower ground state recovery from meta-stable MC states on the timescale of hundreds of picosecond or slower.47 Dramatically different excited state dynamics has instead been observed for FeII carbene complexes, including assignment of extended excited state lifetimes of 3MLCT states of ∼10 ps or longer, and followed by fast ground state recovery on similar timescales. The changes in excited state dynamics are largely attributed to a strong destabilization of the metal centred states in the iron carbene complexes due to the strong sigma-donating nature of the carbenes.14 This altered photophysics also allowed a recent demonstration of efficient electron injection into a TiO2 substrate.37 The electronic structure for FeII(btz)3 features a characteristic 3MLCT-like spin density for the photofunctional charge-transfer state (Fig. 6a).
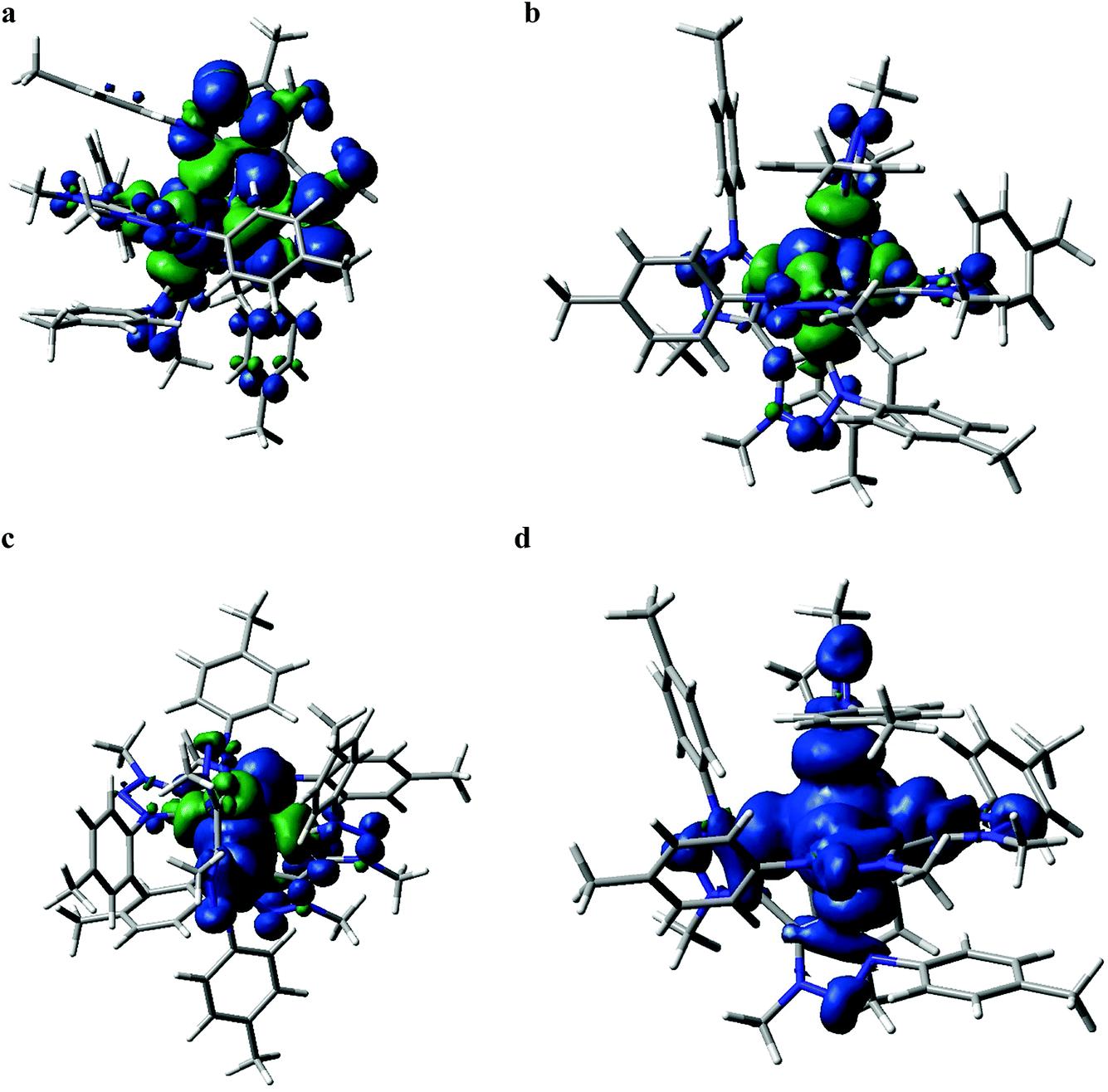 | ||
| Fig. 6 Spin density plots (isovalue 0.0004) of selected FeII(btz)3 and FeIII(btz)3 states from B3LYP*/6-311G(d)|MeCN calculations. Specific oxidation and electronic state assignments: (a) FeII:3MLCT, (b) FeIII:2GS, (c) FeIII:4MC, (d) FeIII:6MC. | ||
The electronic structure properties of FeIII(btz)3 can be understood in simple terms as originating from a one-electron oxidation of the FeII(btz)3 complex in which one electron is removed from the iron center so that it takes on what could recently been characterized as a low-spin 3d5 electronic configuration, i.e. with a (t2g)5 population on iron in quasi-octahedral symmetry notation (Fig. 2). From this follows that the ground state is a doublet (Fig. 6b), and that the excited state manifold will consist of doublet, quartet, and hextet excited states (limiting the number of unpaired electrons to five for simplicity). With a hole in the (t2g) levels, the lowest FeIII(btz)3 excitation was characterized as a doublet ligand-to-metal charge transfer state.38 Metal centered quartet and hextet states could also be identified computationally, and involve population of eg-levels on the iron center. Such intermediate- and high-spin MC states are naturally stabilized by elongated metal–ligand bonds, similar to the MC states for d6 complexes.1 Calculated spin density plots of such 4MC and 6MC states for FeIII(btz)3 are shown in Fig. 6c and d panels, respectively.
It is thus clear that the FeII and FeIII forms of iron carbene complexes like Fe(btz)3 differ fundamentally in their basic electronic properties, excitations, and excited state manifolds. In particular, the altered electronic structure effectively yields a reversal of the fundamental direction of the lowest charge-transfer excitations, and while there are metal centered scavenger states that facilitate excited state deactivation for both oxidation states, there are fundamental differences in the spin properties of the states and state crossings involved in the excited state cascade. It is therefore interesting to compare the excited state dynamics for the same complex in the two different oxidation states.
One potentially promising aspect of the photophysics of the FeIII(btz)3 complex for photosensitizer applications is that the excited state lifetime of the 2LMCT state was characterized experimentally to be largely unaffected by the excitation energy over a significant part of the visible absorption band (probed between ca 400 and 550 nm).39 This facilitates for the complex to perform well as a photosensitizer for a broad range of solar illumination. Here, the dynamics of the FeII(btz)3 complex is also further explored experimentally via TA to enable a comparative discussion of the photophysics of the two oxidation states. It is in this context relevant to consider the excited state manifold that can be accessed at different excitation energies also for this FeII complex. Fig. 7 provides a graphical illustration of singlet and triplet excited states in the accessible energy region obtained from singlet–singlet and singlet–triplet TD-DFT calculations, respectively.
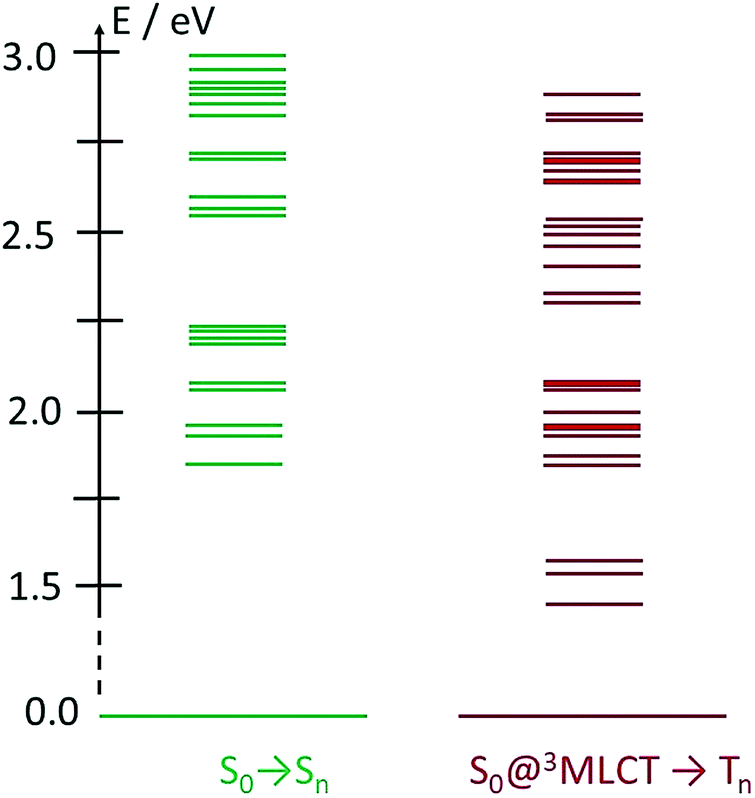 | ||
| Fig. 7 TD-DFT excited state calculations for FeII(btz)3. Shown excitations include singlet excitations at S0 geometry (left, green), and triplet excitations from the S0 state at the 3MLCT geometry (right, red). Energies and assignments from Chábera et al.39 | ||
Discussion
Conventionally, the main electronic states considered for excited state evolution in d6 TMCs include the singlet ground state (1GS), 1MLCT, 3MLCT, 3MC and, for complexes with weak LFS, 5MC, and in many cases modelling excited state dynamics as an interplay of these states has provided a satisfactory picture of the overall spectral changes associated with the excited state evolution including depopulation of the initial charge transfer state (loss of ESA) and ground state recovery (loss of GSB).Our results for Fe(btz)3 provide evidence that a more sophisticated description may generally be needed for a comprehensive understanding of the photoinduced dynamics of iron carbenes. A strong indication of the rather special features of Fe(btz)3 is apparent already from analysis of the absorption spectra. In contrast to MLCT absorption of many typical TMCs, we record two well-resolved CT transitions centered at 450 and 700 nm, assigned to population of two different CT bands in FeII(btz)3. For FeII, one can furthermore identify two high-energy shoulders at ∼520 and ∼825 nm for the two 1MLCT absorption bands, respectively. We can attribute this rich CT absorption appearance as an effect of the strong σ-donating effect of the carbene ligands which causes an up-shift in the energy of the highest occupied metal-based t2g levels of the ground state (correlated also to a shift towards more negative ground state reduction potentials).1 This shifts the CT transition energies towards red to the extent that more of the CT manifold appears clearly below the more intense intra-ligand transitions. This observation is in very good agreement with the predictions of our TD-DFT calculations which, as outlined above, indicate that there is a broad range of charge-transfer excitations that cover the visible spectral region in the experimental linear absorption spectrum.
Having clearly distinct CT bands that are well separated in energy and furthermore have very different transition strength, provides an opportunity to investigate to what extent the dynamics following excitation to different CT states are similar or different. In the following discussion, we will first consider separately FeII and FeIII cases before our final conclusions relating to the emergence of a more comprehensive view of the photoinduced dynamics of the iron carbenes.
As described in the results section, we observe differences in the excited state dynamics in FeII(btz)3 after excitation into the two 1MLCT bands. In particular, we observe some clear changes of the TA spectral shapes for different excitations, as exemplified by the more pronounced ∼408 band in the ESA from excitations at low excitation energy at early timescales. Following the established picture of early FeII excited state evolution, we assume very efficient depopulation of the 1MLCT due to fast ISC on a sub-picosecond timescale (not resolved in this study).46 Our observations suggest that excitation into different parts of the 1MLCT bands leads to different population dynamics and branching ratio between the 3MLCT and 3MC excited states. Initially hot conversion of 3MLCT into 3MC probably does not occur for delay times longer than ∼1 ps. In agreement with our previous assignments for excitation at ∼800 nm, we tentatively associate some changes to the blue part of the ESA at early times (see Fig. 3–5) mainly with intramolecular vibration relaxation (IVR).39 Hot population of the energetically accessible 3MC is likely to be limited to the early times prior to such IVR. It is tempting to conclude that population of the 3MC occurs from 1MLCT or “hot” 3MLCT whereas the relaxed 3MLCT has a lower probability of converting into 3MC. This interpretation of the very fast excited state processes is visualized schematically in Fig. 8. This suggests that the efficiency of the 3MC population is very sensitive to the exact crossings of the 1MLCT and 3MLCT with the 3MC state in terms of the activation energy and structural rearrangement associated with such a transition.
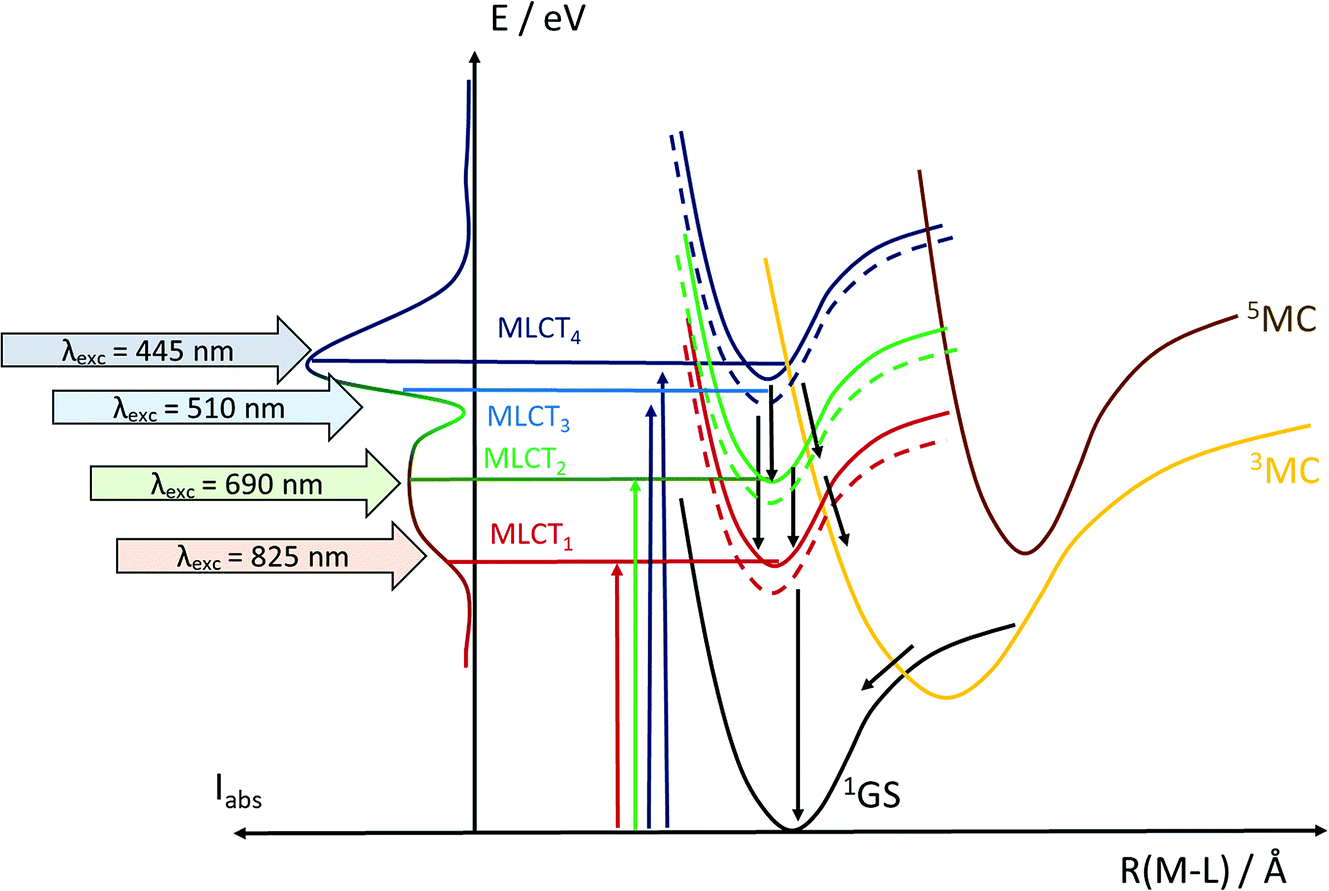 | ||
| Fig. 8 Excited state manifold in the charge-transfer excitation energy region for FeII(btz)3, indicating the different excitation energies/wavelengths employed experimentally to probe the excited state dynamics (red, green, light blue and blue arrows), as well as selected potential decay paths (black arrows). Singlet and triplet forms of representative MLCT states in the charge-transfer manifold energy region are distinguished as solid and dashed lines, respectively. | ||
Finally, irrespective of the rate and amplitude of this few tens of ps component, the overall relaxation of the excited complexes to the ground state occurs with the same rate within our signal-to-noise (S/N). These observations agree with dynamics on a manifold of 3MLCT excited states, including energy relaxation within this manifold that occurs on timescales up to a few tens of ps. Based on the combined information from ground state absorption, first principles calculations, and time-resolved excited state dynamics, we infer that a consistent and complete interpretation of the results require a manifold of 1MLCT and 3MLCT excited states to be considered.
Taken together, the simultaneous excitation and probing of different parts of the CT manifold presented in this investigation provides a first glimpse of rich, and in some aspects band-selective, dynamics in these iron carbene complexes with remarkably long-lived (>100 ps) excited states. From the current understanding of the excited state manifold of these unconventional iron complexes the full dynamics potentially involves a combination of several excited state processes including ultrafast electronic processes such as IVR, IC, ISC within the manifold of excited states, together with molecular and solvent structural dynamics. The current evidence from the TA dynamics suggests that it will be important to carry out further work to fully elucidate key aspects of the ultrafast mechanisms responsible for the dynamics observations, including unambiguous assignments of the ultrafast dynamics and detailed interplay between initial singlet–triplet ISC as well as the deactivation cascade from CT to MC states for a full comparison with the ultrafast LIESST dynamics in conventional Fe complexes typically characterized by ultrafast ISC and CT → MC conversion. A range of further experimental and computational investigative steps appear promising to facilitate a better understanding, including complementary optical characterization e.g. including low-temperature characterization, time-resolved optical measurements with better time-resolution in the ultrafast domain, and studies of dynamics in different solvent environments, as well as complimentary experimental techniques in different energy regimes including e.g. time-resolved X-ray dynamics, as well as more sophisticated theoretical treatments such as high-level quantum dynamics simulations.
It is finally interesting to also compare these new results for FeII to those recently reported for the FeIII form of the Fe(btz)3 complex. Similar to FeII, two clearly separated transitions to CT states in the ground state absorption spectrum can be identified: a high energy band centered at ∼400 and a low energy band with two peaks at 525 and 560 nm. No significant excitation wavelength dependency in the TA spectra and kinetics was, however, noticed. It is important to note here that the excited state energy evolution does not exhibit fast and efficient ISC from the 2LMCT state. On the contrary, after optical excitation from the doublet ground state (2GS) to the 2LMCT state, the primary energy relaxation occurs within the same excited state. This is evident as the excited state is emissive and exhibits a rather small Stokes shift. After reaching the bottom of the 2LMCT manifold, the excited state population undergoes either radiative or non-radiative transition to the ground state in competition with activated ISC transition to 4MC scavenger states. The relative efficiencies of the above listed transitions determine the excited state lifetime (identical to the ground state recovery time), as well as the yield of the photoluminescence.
Conclusions
Efforts to develop iron carbene photosensitizer complexes have recently yielded very significant improvements in excited state lifetimes for both conventional 3d6 FeII carbene complexes, and more unconventional low-spin 3d5 FeIII complexes. These results were obtained in rapid succession for structurally similar FeII and FeIII complexes and appear to rely on the same ligand design strategy to maximize the strong σ-donation by hexa-carbene environments around the metal in the two different formal oxidation states of the iron center. Our results on the excited state properties and dynamics of the Fe(btz)3 molecule with iron in both oxidation state II and III provide an excellent opportunity to compare promising photophysics and photochemistry of iron carbene photosensitizers in the two different electronic configurations 3d5 and 3d6. Until recently, only a relatively limited number of d5 2LMCT complexes, e.g. Re(II) and Tc(II), had received significant attention as potentially interesting for driving photocatalytic processes.48–50 Our combined results for the Fe(btz)3 complex suggest that the photophysics and photochemistry of these complexes provide interesting information regarding ultrafast dynamics following excitations into a manifold of charge-transfer excited states that may be relevant more broadly.One key finding for the time-resolved TA dynamics of FeII(btz)3 following higher energy excitations (from 825 nm to 445 nm excitation wavelengths) is that there remains a long-lived ground state recovery with a time constant of ca 500 ps independent of excitation wavelength accounting for a major part of the decay dynamics. This photophysical consistency in terms of a stable long-term excited state feature following excitation into the charge-transfer manifold of excited states/bands is similar to what was recently shown in our investigation of the FeIII(btz)3 form (in the range of 550–400 nm excitation wavelengths) where there was a ca 100 ps long component dominating the deactivation and ground state recovery regardless of excitation energy. This common feature of the two oxidation states is potentially quite important for the photofunctionality of the iron carbenes e.g. for light-harvesting in solar energy conversion applications profiting from exposure to the full solar spectrum, and not just limited to absorption in a small spectral region.
The detailed investigation of the ultrafast dynamics of the FeII(btz)3 complex through multi-exponential fitting of selected TA kinetics, however, also reveals some interesting additional, smaller but clear, contributions to the dynamics on timescales of a few tens of ps that was not previously discussed. The quantum chemical calculations provide general support for the presence of a manifold of low excited-state energy metal-to-ligand charge transfer states, e.g. involving electronic rearrangements either among the (t2g)5 electrons or low-energy reduced 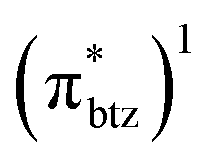 ligand orbitals in the MLCT configuration, at energies that become accessible for the higher energy excitations. It is therefore in itself not surprising that the complex excited state landscape leads to some additional fast dynamics, e.g. involving internal conversion or intersystem crossing. With the present TA and computational results, a detailed interpretation of the short-lived intermediate dynamics was not attempted. Instead, we note that given the current interest in hot dynamics in transition metal complexes in general, coupled with the current high interest in developing Earth-abundant photosensitizers based on iron, our new findings point to the importance of further investigations to characterize the ultrafast dynamics in this type of iron complexes with rich dynamics and extended excited state lifetimes in greater detail.
ligand orbitals in the MLCT configuration, at energies that become accessible for the higher energy excitations. It is therefore in itself not surprising that the complex excited state landscape leads to some additional fast dynamics, e.g. involving internal conversion or intersystem crossing. With the present TA and computational results, a detailed interpretation of the short-lived intermediate dynamics was not attempted. Instead, we note that given the current interest in hot dynamics in transition metal complexes in general, coupled with the current high interest in developing Earth-abundant photosensitizers based on iron, our new findings point to the importance of further investigations to characterize the ultrafast dynamics in this type of iron complexes with rich dynamics and extended excited state lifetimes in greater detail.
A comparison of the ultrafast charge-transfer dynamics and excited state cascade of FeII and FeIII species finally provides a rare opportunity to compare largely complementary excited state dynamics including issues, e.g. about how internal de-excitation differ in charge-transfer excited manifolds that differ in the fundamental charge-transfer direction (MLCT versus LMCT in FeII and FeIII species, respectively). The comparative analysis also points to fundamental differences in energy relaxation paths and state-crossings for species with fundamentally different sets of spin states (even versus odd electronic state multiplicities) that has significant impact on light-harvesting and light-emitting photofunctionality. Further studies of ultrafast dynamics of iron complexes with long-lived charge transfer excited states thus look important for providing further in-depth understanding relevant to the continued development of photofunctional iron complexes. More generally, it remains interesting to conduct investigations of the ultrafast photoinduced dynamics also for other transition metal complexes with unconventional excited state properties and novel ultrafast dynamics.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The Swedish Research Council (VR), the Swedish Energy Agency (Energimyndigheten), the Swedish Foundation for Strategic Research (SSF), and the Knut and Alice Wallenberg (KAW) Foundation are all acknowledged for financial support. The Swedish supercomputing centers LUNARC and NSC are gratefully acknowledged for providing computing facilities through the SNIC allocation system.References
- C. S. Ponseca, P. Chábera, J. Uhlig, P. Persson and V. Sundström, Ultrafast Electron Dynamics in Solar Energy Conversion, Chem. Rev., 2017, 117(16), 10940–11024 CrossRef CAS PubMed.
- V. Balzani, G. Bergamini, S. Campagna and F. Puntoriero, Photochemistry and Photophysics of Coordination Compounds: Overview and General Concepts, Top. Curr. Chem., 2007, 280, 1–36 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Powering the Planet: Chemical Challenges in Solar Energy Utilization, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(43), 15729–15735 CrossRef CAS PubMed.
- G. J. Meyer, Molecular Approaches to Solar Energy Conversion with Coordination Compounds Anchored to Semiconductor Surfaces, Inorg. Chem., 2005, 44(20), 6852–6864 CrossRef CAS PubMed.
- A. Hagfeldt and M. Graetzel, Light-Induced Redox Reactions in Nanocrystalline Systems, Chem. Rev., 1995, 95(1), 49–68 CrossRef CAS.
- V. W.-W. Yam and K. M.-C. Wong, Luminescent Metal Complexes of D6, D8 and D10 Transition Metal Centres, Chem. Commun., 2011, 47(42), 11579 RSC.
- L. A. Büldt and O. S. Wenger, Luminescent Complexes Made from Chelating Isocyanide Ligands and Earth-Abundant Metals, Dalton Trans., 2017, 46, 15175–15177 RSC.
- B. Bozic-Weber, E. C. Constable and C. E. Housecroft, Light Harvesting with Earth Abundant D-Block Metals: Development of Sensitizers in Dye-Sensitized Solar Cells (DSCs), Coord. Chem. Rev., 2013, 257(21–22), 3089–3106 CrossRef CAS.
- O. S. Wenger, Photoactive Complexes with Earth-Abundant Metals, J. Am. Chem. Soc., 2018, 140(42), 13522–13533 CrossRef CAS PubMed.
- P. Sutra and A. Igau, Emerging Earth-Abundant (Fe, Co, Ni, Cu) Molecular Complexes for Solar Fuel Catalysis, Curr. Opin. Green Sustain. Chem., 2018, 10, 60–67 CrossRef.
- B. M. Hunter, H. B. Gray and A. M. Müller, Earth-Abundant Heterogeneous Water Oxidation Catalysts, Chem. Rev., 2016, 116(22), 14120–14136 CrossRef CAS PubMed.
- T. Bessho, E. C. Constable, M. Graetzel, R. A. Hernandez, C. E. Housecroft, W. Kylberg, M. K. Nazeeruddin, M. Neuburger and S. Schaffner, An Element of Surprise-Efficient Copper-Functionalized Dye-Sensitized Solar Cells, Chem. Commun., 2008, 32, 3717–3719 RSC.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, [Cr(Ddpd)2]3+: A Molecular, Water-Soluble, Highly NIR-Emissive Ruby Analogue, Angew. Chem., Int. Ed., 2015, 54(39), 11572–11576 CrossRef CAS PubMed.
- Y. Liu, P. Persson, V. Sundström and K. Wärnmark, Fe N-Heterocyclic Carbene Complexes as Promising Photosensitizers, Acc. Chem. Res., 2016, 49, 1477–1485 CrossRef CAS PubMed.
- M. Abrahamsson, M. J. Lundqvist, H. Wolpher, O. Johansson, L. Eriksson, J. Bergquist, T. Rasmussen, H. C. Becker, L. Hammarström and P. O. Norrby, et al., Steric Influence on the Excited-State Lifetimes of Ruthenium Complexes with Bipyridyl-Alkanylene-Pyridyl Ligands, Inorg. Chem., 2008, 47(9), 3540–3548 CrossRef CAS PubMed.
- M. Abrahamsson, Solar Energy Conversion Using Iron Polypyridyl Type Photosensitizers – a Viable Route for the Future?, in Photochemistry, ed. A. Albini and E. Fasani, RSC, Cambridge, 2017, vol. 44, pp. 285–295, ISBN: 978-1-78262-543-8, 10.1039/9781782626954.
- J. E. Monat and J. K. McCusker, Femtosecond Excited-State Dynamics of an Iron(II) Polypyridyl Solar Cell Sensitizer Model, J. Am. Chem. Soc., 2000, 122(17), 4092–4097 CrossRef CAS.
- S. M. Fatur, S. G. Shepard, R. F. Higgins, M. P. Shores and N. H. Damrauer, A Synthetically Tunable System To Control MLCT Excited-State Lifetimes and Spin States in Iron(II) Polypyridines, J. Am. Chem. Soc., 2017, 139, 4493–4505 CrossRef CAS PubMed.
- W. Zhang, R. Alonso-Mori, U. Bergmann, C. Bressler, M. Chollet, A. Galler, W. Gawelda, R. G. Hadt, R. W. Hartsock and T. Kroll, et al., Tracking Excited-State Charge and Spin Dynamics in Iron Coordination Complexes, Nature, 2014, 509(7500), 345–348 CrossRef CAS PubMed.
- A. Cannizzo, C. J. Milne, C. Consani, W. Gawelda, C. Bressler, F. Van Mourik and M. Chergui, Light-Induced Spin Crossover in Fe(II)-Based Complexes: The Full Photocycle Unraveled by Ultrafast Optical and X-Ray Spectroscopies, Coord. Chem. Rev., 2009, 254, 2677–2686 CrossRef.
- W. Zhang, R. Alonso-Mori, U. Bergmann, C. Bressler, M. Chollet, A. Galler, W. Gawelda, R. G. Hadt, R. W. Hartsock and T. Kroll, et al., Tracking Excited-State Charge and Spin Dynamics in Iron Coordination Complexes, Nature, 2014, 509(7500), 345–348 CrossRef CAS PubMed.
- J. K. Mccusker and A. Vlcek, Ultrafast Excited-State Processes in Inorganic Systems, Acc. Chem. Res., 2015, 48, 1207–1208 CrossRef CAS PubMed.
- Q. Sun, S. Mosquera-Vasquez, L. M. Daku, L. Guénée, H. A. Goodwin, E. Vauthey and A. Hauser, Experimental Evidence of Ultrafast Quenching of the 3 MLCT Luminescence in Ruthenium(II) Tris-Bipyridyl Complexes via a 3dd State, J. Am. Chem. Soc., 2013, 135, 13660–13663 CrossRef CAS PubMed.
- M. Chergui and E. Collet, Photoinduced Structural Dynamics of Molecular Systems Mapped by Time-Resolved X-ray Methods, Chem. Rev., 2017, 117(16), 11025–11065 CrossRef CAS PubMed.
- T. J. Penfold, M. Pápai, T. Rozgonyi, K. B. Møller and G. Vankó, Probing Spin–vibronic Dynamics Using Femtosecond X-Ray Spectroscopy, Faraday Discuss., 2016, 194(0), 731–746 RSC.
- D. Leshchev, T. C. B. Harlang, L. A. Fredin, D. Khakhulin, Y. Liu, E. Biasin, M. G. Laursen, G. E. Newby, K. Haldrup and M. M. Nielsen, et al., Tracking the Picosecond Deactivation Dynamics of a Photoexcited Iron Carbene Complex by Time-Resolved X-Ray Scattering, Chem. Sci., 2018, 9, 405–414 RSC.
- W. Zhang and K. J. Gaffney, Mechanistic Studies of Photoinduced Spin Crossover and Electron Transfer in Inorganic Complexes, Acc. Chem. Res., 2015, 48, 1140–1148 CrossRef CAS PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Spin-Vibronic Mechanism for Intersystem Crossing, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- C. Sousa, M. Alías, A. Domingo and C. de Graaf, Deactivation of Excited States in Transition-Metal Complexes: Insight from Computational Chemistry, Chem.–Eur. J., 2019, 25(5), 1152–1164 CrossRef CAS PubMed.
- J. Nance, D. N. Bowman, S. Mukherjee, C. T. Kelley and E. Jakubikova, Insights into the Spin-State Transitions in [Fe(Tpy)2]2+: Importance of the Terpyridine Rocking Motion, Inorg. Chem., 2015, 54, 11259–11268 CrossRef CAS PubMed.
- I. M. Dixon, F. Alary, M. Boggio-Pasqua and J.-L. Heully, The (N4C2)2– Donor Set as Promising Motif for Bis(Tridentate) Iron(II) Photoactive Compounds, Inorg. Chem., 2013, 52(23), 13369–13374 CrossRef CAS PubMed.
- Y. Liu, P. Persson, V. Sundström and K. Wärnmark, Fe N-Heterocyclic Carbene Complexes as Promising Photosensitizers, Acc. Chem. Res., 2016, 49(8), 1477–1485 CrossRef CAS PubMed.
- L. Liu, T. Duchanois, T. Etienne, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, A New Record Excited State 3 MLCT Lifetime for Metalorganic Iron(II) Complexes, Phys. Chem. Chem. Phys., 2016, 18(18), 12550–12556 RSC.
- Y. Liu, T. Harlang, S. E. Canton, P. Chábera, K. Suárez-Alcántara, A. Fleckhaus, D. A. Vithanage, E. Göransson, A. Corani and R. Lomoth, et al., Towards Longer-Lived Metal-to-Ligand Charge Transfer States of Iron(II) Complexes: An N-Heterocyclic Carbene Approach, Chem. Commun., 2013, 49(49), 6412–6414 RSC.
- Y. Liu, K. S. Kjaer, L. A. Fredin, P. Chabera, T. Harlang, S. E. Canton, S. Lidin, J. Zhang, R. Lomoth and K. E. Bergquist, et al., A Heteroleptic Ferrous Complex with Mesoionic Bis(1,2,3-Triazol-5-Ylidene) Ligands: Taming the MLCT Excited State of Iron(II), Chem.–Eur. J., 2015, 21(9), 3628–3639 CrossRef CAS PubMed.
- L. A. Fredin, M. Papai, E. Rozsalyi, G. Vanko, K. Warnmark, V. Sundstrom and P. Persson, Exceptional Excited-State Lifetime of an Iron(II)-N-Heterocyclic Carbene Complex Explained, J. Phys. Chem. Lett., 2014, 5(12), 2066–2071 CrossRef CAS PubMed.
- T. C. B. Harlang, Y. Liu, O. Gordivska, L. A. Fredin, C. S. Ponseca, P. Huang, P. Chábera, K. S. Kjaer, H. Mateos and J. Uhlig, et al., Iron Sensitizer Converts Light to Electrons with 92% Yield, Nat. Chem., 2015, 7(11), 883–889 CrossRef CAS PubMed.
- P. Chabera, Y. Liu, O. Prakash, E. Thyraug, A. El Nahhas, A. Honarfar, S. Essen, L. A. Fredin, T. C. B. Harlang and K. S. Kjaer, et al., A Low-Spin Fe(III) Complex with 100-Ps Ligand-to-Metal Charge Transfer Photoluminescence, Nature, 2017, 543, 695 CrossRef CAS PubMed.
- P. Chábera, K. S. Kjaer, O. Prakash, A. Honarfar, Y. Liu, L. A. Fredin, T. C. B. Harlang, S. Lidin, J. Uhlig and V. Sundström, et al., FeIIHexa N-Heterocyclic Carbene Complex with a 528 Ps Metal-To-Ligand Charge-Transfer Excited-State Lifetime, J. Phys. Chem. Lett., 2018, 9(3), 459–463 CrossRef PubMed.
- K. S. Kjær, N. Kaul, O. Prakash, P. Chábera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K.-E. Bergquist and L. Häggström, et al., Luminescence and Reactivity of a Charge-Transfer Excited Iron Complex with Nanosecond Lifetime, Science, 2019, 363(6424), 249–253 CrossRef PubMed.
- M. Reiher, O. Salomon and B. A. Hess, Reparameterization of Hybrid Functionals Based on Energy Differences of States of Different Multiplicity, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed.
- A. D. Becke, Density functional Thermochemistry III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- T. H. Dunning and P. J. Hay, Gaussian Basis Sets for Molecular Calculations, in Methods of Electronic Structure Theory, Springer US, Boston, MA, 1977, pp. 1–27 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- G. Auböck and M. Chergui, Sub-50-Fs Photoinduced Spin Crossover in [Fe(Bpy)3]2+, Nat. Chem., 2015, 7(8), 629–633 CrossRef PubMed.
- W. Zhang and K. J. Gaffney, Mechanistic Studies of Photoinduced Spin Crossover and Electron Transfer in Inorganic Complexes, Acc. Chem. Res., 2015, 48, 22 CrossRef PubMed.
- A. S. Del Negro, C. J. Seliskar, W. R. Heineman, S. E. Hightower, S. A. Bryan and B. P. Sullivan, Highly Oxidizing Excited States of Re and Tc Complexes, J. Am. Chem. Soc., 2006, 128(51), 16494–16495 CrossRef CAS PubMed.
- S. Paulson, B. P. Sullivan and J. V. Caspar, Luminescent Ligand-to-Metal Charge-Transfer Excited States Based on Pentamethylcyclopentadienyl Complexes of Tantalum, J. Am. Chem. Soc., 1992, 6905–6906 CrossRef CAS.
- Y. F. Lee and J. R. Kirchhoff, Absorption and Luminescence Spectroelectrochemical Characterization of a Highly Luminescent Rhenium(I1) Complex, J. Am. Chem. Soc., 1994, 116(14), 3599–3600 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |