
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePre-melting and the adsorption of formic acid at the air–ice interface at 253 K as seen by NEXAFS and XPS
Astrid
Waldner
a,
Luca
Artiglia
 a,
Xiangrui
Kong
a,
Fabrizio
Orlando
a,
Thomas
Huthwelker
b,
Markus
Ammann
a and
Thorsten
Bartels-Rausch
*a
a,
Xiangrui
Kong
a,
Fabrizio
Orlando
a,
Thomas
Huthwelker
b,
Markus
Ammann
a and
Thorsten
Bartels-Rausch
*a
aLaboratory of Environmental Chemistry, Paul Scherrer Institut, CH-5232 Villigen PSI, Switzerland. E-mail: thorsten.bartels-rausch@psi.ch
bSwiss Light Source (SLS), Paul Scherrer Institut, CH-5232 Villigen PSI, Switzerland
First published on 4th September 2018
Abstract
Interactions between trace gases and ice are important in environmental chemistry and for Earth's climate. In particular, the adsorption of trace gases to ice surfaces at temperatures approaching the melting point has raised interest in the past, because of the prevailing pre-melting. Here, we present Near Edge X-ray Absorption Fine Structure (NEXAFS) spectroscopy data at ambient partial pressure of water to better define the onset temperature of pre-melting at the interfacial region of ice. Further, this study directly compares the interaction between an organic acid common in the atmosphere, formic acid, and that of an aliphatic carbon with ice at 253 K. It makes use of X-ray Photoelectron Spectroscopy (XPS) with its inherent narrow probing depth covering both the surface and near-surface bulk region when detecting electrons. We use the tender X-ray range for excitation to locate the organic species within the interfacial region with an extended probing depth compared to published XPS work. Electron kinetic energy dependent C1s photoemission data indicate that, at low coverage of a few 1014 molecules cm−2, the presence of formic acid is restricted to the upper ice layers of the interfacial region. Increasing the dosage, formic acid penetrates 6–7 nm into the air–ice interface. The presence of the more hydrophobic aliphatic carbon is restricted to the upper ice monolayers. This direct comparison of an organic acid with an aliphatic compound confirms the emerging picture where solutes enter the interfacial region of ice at a depth related to their specific tendency to form solvation shells.
Introduction
Atmospheric and terrestrial ice, including cirrus clouds, sea-ice and snow, are omnipresent in the environment and host a number or chemical reactions. They thus have the capability to heavily influence the atmospheric composition and impact Earth's (bio)geochemical cycles.3–5 Given that the solubility of reactants within the ice crystal structure (bulk ice) is generally negligible, research focused on the air–ice and ice–ice interfaces as compartments where multiphase reactions take place.7 Already in 1840, Faraday suggested the importance of the interfacial region of ice to explain macroscopic processes and postulated that the surface is different from the bulk.8 This interfacial region, which is called a disordered interface, pre-melting, or quasi-liquid layer (QLL), is characterized by changes of the average coordination of the water molecules and thus of the structure of the hydrogen bonding network from that in the crystalline bulk ice. The pre-melting intensifies with increasing temperature close to the melting point of ice and with the presence of solutes.7,9–11 Electron yield NEXAFS has become an established tool to look at the hydrogen-bonding network of water and ice at the interface.9,11 In their pioneering work, Bluhm et al. have quantified the thickness of the quasi-liquid layer on neat ice based on gradual changes of NEXAFS data upon increasing the temperature from 233 K to 271 K.9 The results were interpreted as the quasi-liquid properties reaching deeper into the interfacial region at an onset temperature of between 248 K and 258 K. Here, we present additional data at 253 K to establish the onset of significant liquid character at the interfacial region more precisely.Short chain organics are present in the air above ice, and only limited studies of their interactions with ice are available.12 Formic acid (HCOOH), one of the strongest short chain organic acids abundant in the atmosphere, occurs in mixing ratios of around ∼2 parts per billion in boundary layer air.13 The primary sources of formic acid are photochemical oxidation of volatile organic compounds and biogenic sources such as direct forest emissions.12,14–16 Main formic acid sinks include photochemical oxidation17 as well as dry and wet deposition.12,16,18,19 With an atmospheric lifetime in the atmosphere of 2–4 days,20 formic acid represents a substantial sink for in-cloud OH21 and has significant impact on key chemical budgets such as that of nitrogen oxides. This makes the loss processes of formic acid important for atmospheric chemistry.12 Theoretical and experimental work at temperatures below ∼230 K showed that the partitioning of formic acid to ice is significantly smaller than that of inorganic acids such as HCl, but still larger compared to other volatile organics.22–25 Thus, in presence of ice, partitioning of formic acid has the ability to influence the OH budget and the oxidation capacity of the atmosphere.
We have recently reported on the interaction of formic acid with ice at the molecular level using Near Ambient Pressure X-ray Photoelectron Spectroscopy (NAP-XPS), which probes the chemical composition with high surface sensitivity under equilibrium thermodynamic conditions.26 Depth profiles at 251 K have revealed that the presence of formic acid is restricted to the surface of the interfacial region at 3 × 1014 molecules cm−2.26 Increasing the dosage, formic acid enters 3–6 nm into the interfacial region of ice at 251 K. The presence of formic acid deeper in the interfacial region went along with an increase in the liquid character of the interfacial hydrogen-bonding network.26 We have suggested that the driving force for solutes to enter into the interfacial region and to modify the hydrogen-bonding structure is their demand for water molecules to form energetically favourable solvation shells.26,27 This picture of very localized changes to the interfacial structure nicely explains the increase in disorder in presence of solutes, as shown in the pioneering work by McNeill et al.,10 while suggesting structural differences in the pre-melting of neat ice and impurity induced solvation shells. Interestingly, recent large scale molecular simulations also linked the increase of liquid-like character at the ice interface in presence of solutes with the formation of solvation shells.28 In particular, the increase in liquid-like character was found to be less in presence of an oxygenated organic solute than in presence of ions and this difference was linked to the specific energetic attraction for water molecules. Therefore, we compare the depth profiles of formic acid with those of a more hydrophobic carbon species in this work to investigate the link between the solute's ability to modify the hydrogen-bonding network and its solubility.26,27,29,30 The hydrophobic species is adventitious carbon, a ubiquitous carbon species in many photoemission experiments.31,32 A well-known source are radical reactions of carbonaceous precursors that are initiated by secondary electrons emitted from the ice sample upon X-ray irradiation in the gas phase close to the sample spot.33
Last but not least, the results by Bartels-Rausch et al. have been derived at the SIM beamline (Swiss Light Source, Paul Scherrer Institute) with an upper limit of excitation energy of 1600 eV, corresponding to an electron escape depth† of ∼5 nm.26 This poses the question, whether the observed limit of formic acid to the upper 3–6 nm is given by the constraints to the probing depth of those experiments. Therefore, we probe the presence of formic acid in the interfacial region using tender X-rays (2000–7000 eV) with an upper electron escape depth in the ice of 12 nm in this work.
Experimental
The experiments were performed at the Swiss Light Source (SLS) of the Paul Scherrer Institute using the Near Ambient Pressure Photoemission endstation (NAPP) at the beamlines PHOENIX and SIM.34 The endstation's key components are a differentially pumped electron energy analyser (Scienta R4000 HiPP-2), a sample holder that is temperature controlled by a flow of cooled helium gas and that is embedded in a flow-through cell to maintain a sample environment with well controlled gas pressures.34 The measurements were performed at partial pressures of water of up to 1 mbar to maintain the ice samples in equilibrium with their vapour pressure at the temperatures of the experiment.Dosing of gases
The ice samples were prepared in situ by depositing water from the gas phase onto the gold-coated sample holder. To dose the water from a temperature-stabilized source a capillary dosing approach was used. Approximately 15 ml liquid water (Fluka TraceSelect Ultra; Water ACS reagent, for ultratrace analysis) was filled into a reservoir. A second dosing line with a reservoir holding ∼2.5 ml liquid formic acid (LC-MS ultra by Sigma-Aldrich) was used to independently dose formic acid to the sample. The temperature of the reservoirs and the capillaries’ dimensions determined the vapour flux into the flow-through cell and thus the experimental pressure. We used stainless steel capillaries of about 1 m length and inner diameters from 0.8 to 1.0 mm to dose water vapour, and a 1 m long fused silica capillary (postnova analytics) with an inner diameter of 0.1 or 0.2 mm for formic acid. The Knudsen numbers are far below 0.01 for all capillaries at the high-pressure side, indicating viscous flow. Thus, cross contamination by diffusion into the reservoirs is negligible. Before dosing, both the formic acid and water were degassed by at least 4 freeze–pump–thaw cycles. The purity of formic acid vapour dosed to the experiment was confirmed by proton transfer reaction mass spectrometry (PTRMS) and ion chromatography (IC). Neither PTRMS nor IC showed the presence of any other volatile organic carbon species than formic acid (detection limit: lower ppb range).Preparation of ice samples
Ice samples were kept at 253 K (251 K for X-ray absorption data) or 233 K. The temperature of the ice sample was derived based on its vapour pressure measured with a residual gas analyser mass spectrometer (RGA 100, Stanford Research) located at the differentially pumped section of the electron analyser. The RGA was calibrated by means of a capacitance manometer (MKS Baratron 626A) with an accuracy of 0.25% of the reading and a measurement range from 5 × 10−3 to 10 mbar prior to the experiments. During the experiments, when water and formic acid were dosed simultaneously, the RGA data was used because of its high selectivity as compared to the manometer. To grow ice at 251 K and at 253 K, water was dosed to the flow-through cell at 1 mbar. After ascertaining a stable pressure, the sample holder was cooled until ice nucleation was detected either by a pressure decrease or by visual observation. Onset of ice nucleation was observed at ∼250 K and the ice grew slowly while keeping the oversaturation for about half an hour. For experiments at 233 K, ice growth was triggered at 200 K and 0.1 mbar partial pressure of water. These ice samples were generally opaque due to their more polycrystalline nature. To minimize perturbations due to radiative heating, heating by X-ray beam energy deposition, and the impact of the electron-sampling aperture on the pressure field near the ice surface,35 we kept a slight oversaturation in the flow-through cell.XPS and NEXAFS
Once the ice was equilibrated, the neat sample was characterized by measuring photoemission (PE) and NEXAFS spectra. Then, formic acid was dosed to the flow-through cell at a constant partial pressure for each experiment. The flow of formic acid into the cell was set such that a clear change in the C1s PE spectra acquired at the ice sample surface from the background measurement just prior to dosing was observed. For all PE measurements, dwell time and pass energy were set to 100 ms and 100 eV. The incident photon flux was set to ∼1011 photon s−1 (at 2740 eV). Due to the photoemission events, the surface of the ice sample was charging leading to negative shifts of the measured photoelectron kinetic energy in the 0–15 eV range. Therefore, all C1s PE spectra were referenced to the O1s peak with a binding energy of 533.2 eV. The calibration of the PE intensity ratios, IC1s/IO1s, was performed by measuring gas-phase CO2. A stable pressure of CO2 (0.8 mbar) was admitted to the flow-through cell and the C 1s and O 1s gas-phase spectra were recorded in the absence of a sample in front of the analyser sampling orifice and at the same photon energies used for the ice experiments. The O1s and C1s spectra were fitted in IgorPro's XPST‡ package by symmetric Gaussian–Lorentzian functions with equal peak width for all surface components of the PE spectra. Binding energy constraints of 285 ± 1, 286.5 ± 0.1, 288 ± 0.05, and 289 ± 0.3 eV were used to fit the individual features in the C1s spectra. The partial pressure of formic acid in the flow-through cell was derived based on the capacitance manometer readings of total pressure and the partial pressure of water as determined by the RGA (see above).In addition, we measured partial Auger electron-yield NEXAFS spectra at the O K-edge using a kinetic energy window at the background of the Auger lines (450–470 eV). For these NEXAFS measurements, a pass energy of 20 eV and dwell times of 100 ms were used. The beam polarization vector was rotated 54.7° from the analyser detection axis, which corresponds to the so-called magic angle, to ensure a signal intensity independent of the molecular orientation. In the O K-edge region of most interest, 529 to 542 eV, we used a photon energy step size of 0.2 eV instead of 1 eV. The NEXAFS spectra were normalized to background measurements (I0) to account for variations in beamline flux with photon energy as derived from in situ flux measurements at the refocusing mirror of the beamline, to drifts in the pre-edge region using a linear fit, and to their integrated area between 534 and 545 eV photon energy to facilitate comparison.
Results and discussion
Onset temperature of the QLL
Fig. 1 shows O K-edge NEXAFS spectra at 233 K and 251 K probing changes to the structure of the hydrogen-bonding network at the interfacial region of neat ice with increasing temperature. Generally, the spectra show the typical two-peak feature of ice (main- and post-edge in Fig. 1) in good agreement with spectra reported elsewhere.26,27 Clearly, the spectra at 233 K and 251 K are very similar and distinct from that of liquid water (grey dotted line in Fig. 1).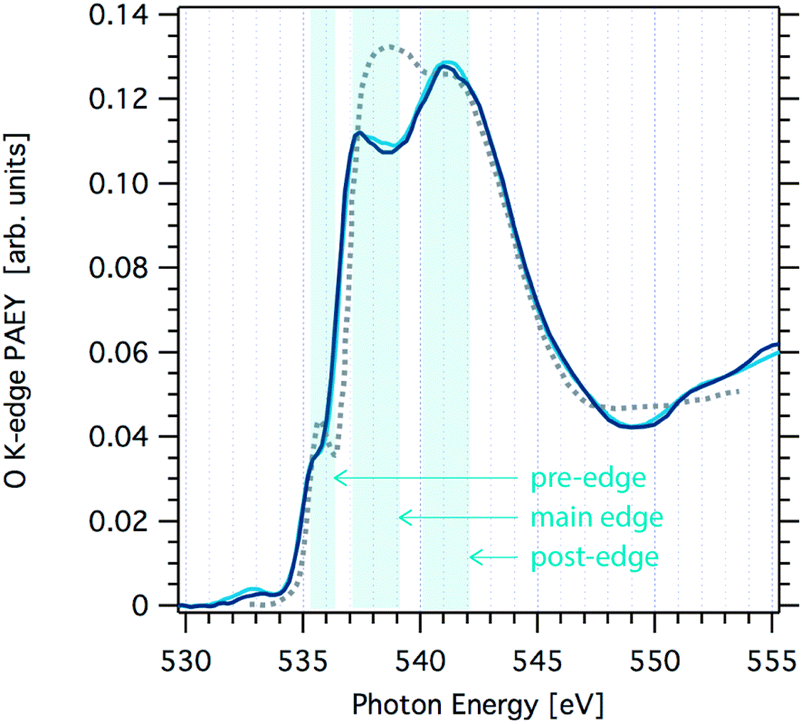 | ||
| Fig. 1 Oxygen K-edge NEXAFS spectra of ice acquired at the SIM beamline at SLS at a temperature of 251 K (light blue line) and 233 K (dark blue line). Also shown is the oxygen K-edge NEXAFS of water at 269 K (grey dotted line) that was not acquired in this work.1 | ||
The origin of the changes in the NEXAFS spectra between that of ice and that of water can be linked to the coordination at the molecular level as follows.9,11 In tetrahedral ice both OH groups of the water molecules are coordinated with neighbouring water molecules, whereas in liquid water this coordination is weaker. The post-edge results from strongly H-bonded OH, thus the post-edge to main edge peak ratio is well pronounced in ice. The O K-edge NEXAFS spectrum of liquid water exhibits a shift towards the main edge with a maximum around 538 eV. A second difference in the NEXAFS spectra of ice and of water is evident in the pre-edge region. The intensity of this pre-edge peak increases with increasing disorder of the water molecules as we go from solid ice to liquid water. It corresponds to a transition to empty states similar to the 4a1 lowest unoccupied molecular orbital of gas-phase water molecules. Because of the dipole selection rule, the intensity of this transition depends on the degree of s- or p-character of the molecular energy state. In ice, oxygen is tetrahedrally coordinated thus symmetry arguments indicate predominant s-symmetry. In liquid, this symmetry is broken, thus the p-character is more probable and the peak intensity increases. Features at 532.5 eV in the spectra, can be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or NO bonds indicating slight contamination of the ice.
O or NO bonds indicating slight contamination of the ice.
At a closer look, a small shift in the slope of the main edge region in the O K-edge NEXAFS of ice is observed towards that seen in liquid water when increasing the temperature from 233 K to 251 K. Reproducibility of NEXAFS spectra acquired one after the other during the same beamtime at constant temperature was excellent, i.e. the spectra were indistinguishable over the whole excitation energy range. Comparing spectra from samples prepared at different days revealed larger fluctuations in particular in the main edge to post-edge intensity ratio. We attribute this to differences in the stability of the ice sample during the NEXAFS acquisition of 30 min duration: minute changes in the ice thickness directly relate to changes in the distance between the electron analyser's sample orifice and the sample surface. This working distance is a key factor of absolute sensitivity in electron spectroscopy. Nevertheless, the slope of the main edge turned out to be a robust feature to differentiate the NEXAFS at 233 K and 251 K. This minor alteration in the NEXAFS indicates that there are only very little changes in the hydrogen-bonding network associated with a temperature increase to 251 K. Based on analogue Auger yield NEXAFS measurements, Bluhm et al. concluded that quasi-liquid is absent at the air–ice interface at temperatures below 248 K.9 Our data presented here expand this data set to higher temperatures and give experimental evidence that the interfacial liquid character does not significantly increase up to 251 K. Clearly, the onset of surface disorder, defined at the temperature where the liquid-like character is found beyond the outermost crystal layer and as seen by NEXAFS, occurs at temperatures above 251 K.
In summary, the interfacial region of ice is at the dawn of the disorder being significantly present within the interfacial region at 251 K. In the following, we will analyse how trace gases adsorb at the interfacial region of ice just prior to the onset of the quasi-liquid layer.
Adsorption of formic acid on ice
Fig. 2 shows changes to the C 1s PE spectrum acquired at the ice interfacial region when increasing the partial pressure of formic acid. Introducing formic acid to the gas phase of the flow-through cell leads to marked increase of carbon at the interfacial region of the ice sample. The PE spectra reveal that the increase is most pronounced in the spectral regions of 285 eV and of 289 eV binding energy and scales with partial pressure of formic acid (Fig. 2). We attribute the peak at 289.3 eV to formic acid.26,36 The broader feature at 285 eV to 287.5 eV reflects C–H (285 eV) and C–OH (288.2 eV) functionalities and can be attributed to adventitious carbon. We found clear experimental evidence that formic acid is a precursor of adventitious carbon at the partial pressure used in this study. Additionally, the intensity of adventitious carbon increased with time during a beamtime also prior to the exposure of the flow-through cell to formic acid and we account this to other carbonaceous traces potentially desorbing from the walls of flow-through cell. Once formic acid had been introduced to the flow-through cell, we observed a memory effect of formic acid such that the spectroscopic feature at 289.3 eV was always present at the ice interfacial region. We account this to desorption of formic acid from the walls of the set-up and subsequent adsorption at the ice surface.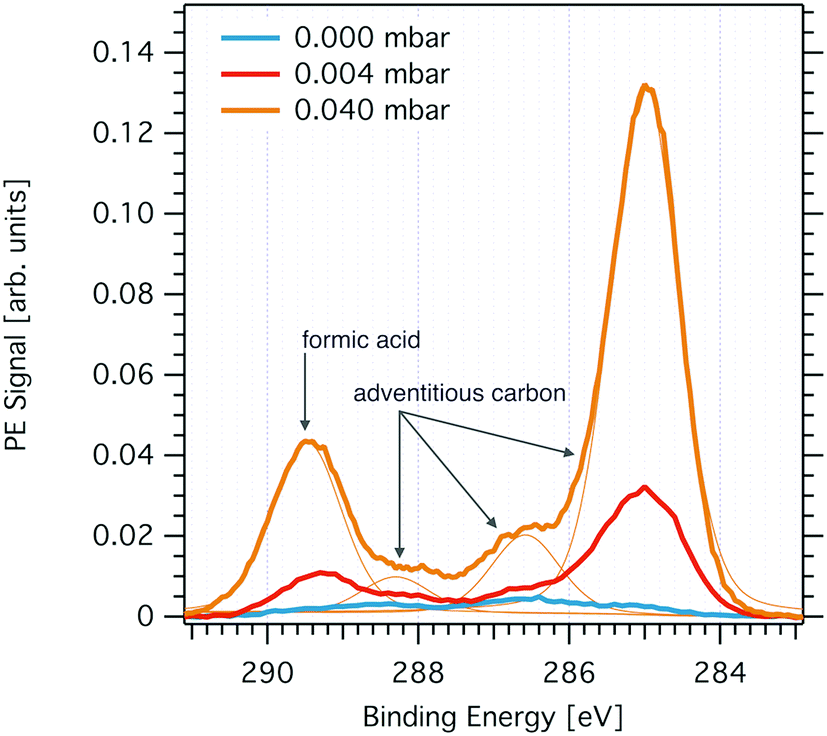 | ||
| Fig. 2 C1s PE spectrum probing the interfacial region of ice at 253 K prior to dosing gas-phase formic acid (light blue solid line), at 0.004 mbar (orange solid line) formic acid partial pressure, and at 0.04 mbar (yellow line). Also shown are the Gaussian–Lorentzian peaks used to fit the PE spectra (thin yellow lines). The feature at 289.3 eV is attributed to formic acid and the features at 285 eV, 288.2 eV, and at 286.6 eV to adventitious carbon. The PE peak at 288.2 eV may also originate from dissociated formic acid (see text for details). Data were acquired at the PHOENIX beamline with 2200 eV X-ray excitation energy. | ||
The origin of the spectroscopic feature at 288 eV in the C1s PE spectrum is less certain. We have found earlier, that the neutral and the dissociated form of acids can be differentiated at the ice interfacial region upon adsorption by means of XPS.27,29 Density functional theory calculations and XPS data taken at liquid water samples have revealed the electron binding energy of deprotonated formic acid to be shifted by −1.3 eV as compared to the binding energy of the neutral acid.36 However, as we cannot exclude that adventitious carbon also contains carbonyl functionalities with typical binding energies of 288 eV, we refrain from analysing this feature further. In summary, the C1s PE spectrum at the interfacial region of ice reveals the presence of adventitious carbon with a dominant aliphatic component and of formic acid. Both spectroscopic features respond to increasing partial pressures of formic acid with an intensity increase reflecting a rise in surface concentration. In the following, we will discuss the distribution of these chemical species with depth at the air–ice interface and their surface concentration as directly seen by XPS. We’d like to note that the precision of the partial pressures given in this work to reflect the gas-phase composition at the sample spot might be rather low. First, capacitance manometers do not work reliable with sticky gases as formic acid. Secondly, formic acid may form dimers at high concentration as used in this work. Third, loss of formic acid at the walls of the flow-through cell during gas-phase transport likely results in a substantially lower partial pressure of formic acid at the ice sample compared to that measured upstream of the sample. Nevertheless, based on these upper partial pressure limits, we conclude that the work was done in the ice stability domain in absence of melting. Freezing point depression data and Henry's law coefficients indicate that melting does not occur below 0.09 mbar and 253 K.26,37 Please note, that traces of formic acid have been observed in the RGA, but were not used for quantification because formic acid fragments strongly in RGA and we have thus not quantified this signal further.
Depth profiles of formic acid and adventitious carbon
To obtain information about the distribution of formic acid in the ice interfacial region, we analysed depth profiles of the calibrated IC1s(FA)/IO1s intensity ratios at increasing photoelectron kinetic energy (Fig. 3 and 4). Given that the electron escape depth increases with the kinetic energy, Fig. 3A and 4 show integrated C1s to O1s PE intensity ratios from the sample surface to increasing probing depths. The first thing to notice in Fig. 3A is that at low dosing of formic acid with an apparent partial pressure of 0.004 mbar smaller IC1s(FA)/IO1s are found than at higher dosing, 0.04 mbar, reflecting the increase in surface coverage with partial pressure of the trace gas. Secondly, the IC1s(FA)/IO1s decreases by about 50% with increasing photoelectron kinetic energy over the range of 3000 eV probed in the experiment at low surface coverage of formic acid. At higher dosage, the IC1s(FA)/IO1s are nearly invariant with increasing electron kinetic energy. Fig. 3B shows how the electron escape depth increases from 6 nm at 2000 eV kinetic energy to 12 nm at 5000 eV in our experiments. The electron escape depth (ED) is a direct measure of the sampling depth and relates to the inelastic mean free path (IMFP) by| ED = IMFP × cos(θ), | (1) |
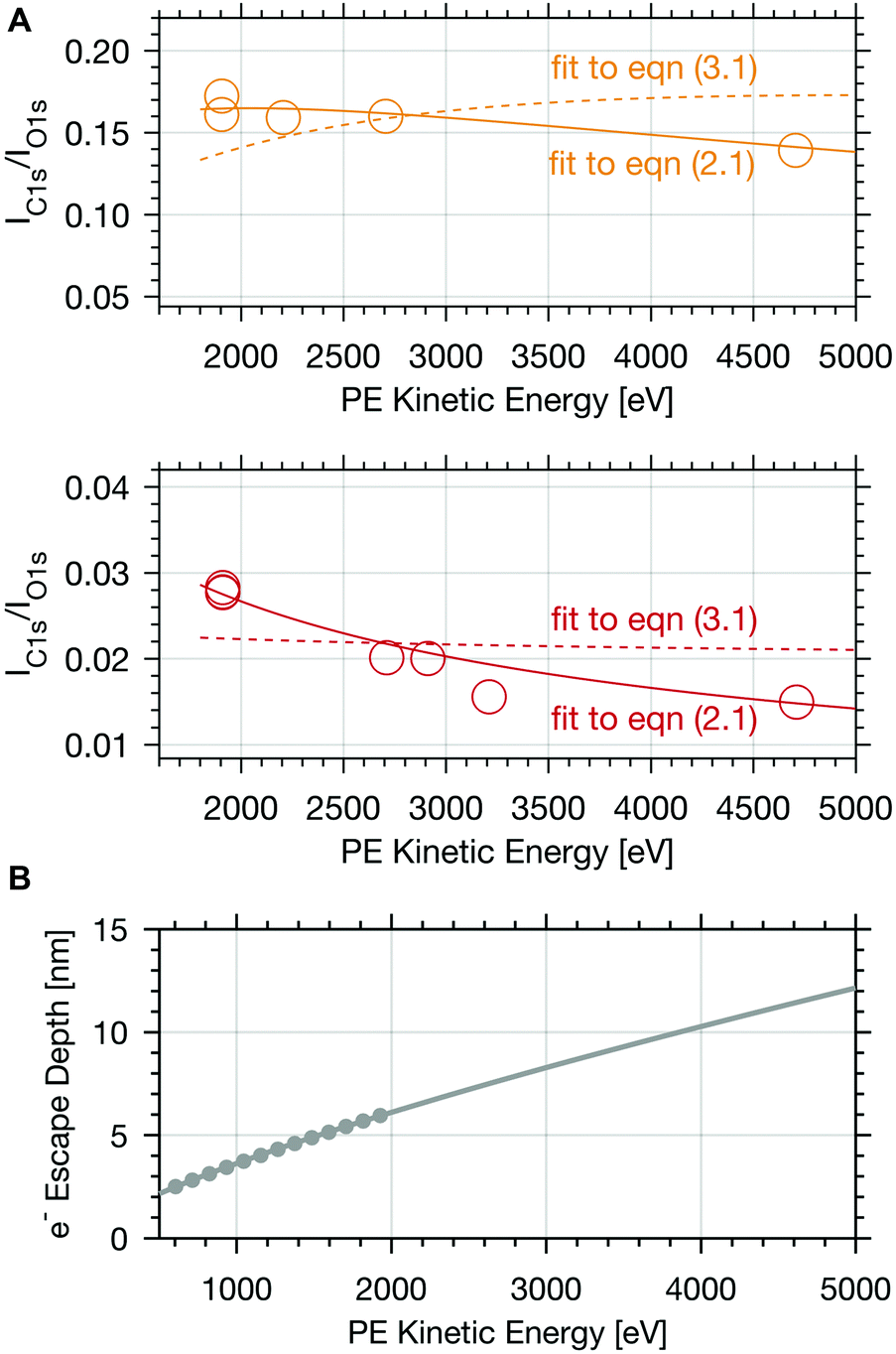 | ||
| Fig. 3 (A) Calibrated PE ratio of the integrated C 1s carboxylic peak area and O 1s peak area as a function of electron kinetic energy at 253 K. The upper panel shows results for experiments with 0.04 mbar formic acid partial pressure (yellow circles) and the lower one gives the data at 0.004 mbar formic acid (orange circles). The coloured lines represent fits to eqn (2.1) (solid line) and of eqn (3.1) (dashed line). (B) Calculated mean electron escape depth (grey dots) in ice versus electron kinetic energy.2 The grey line shows the fit and extrapolation based on the quantitative prediction.6 | ||
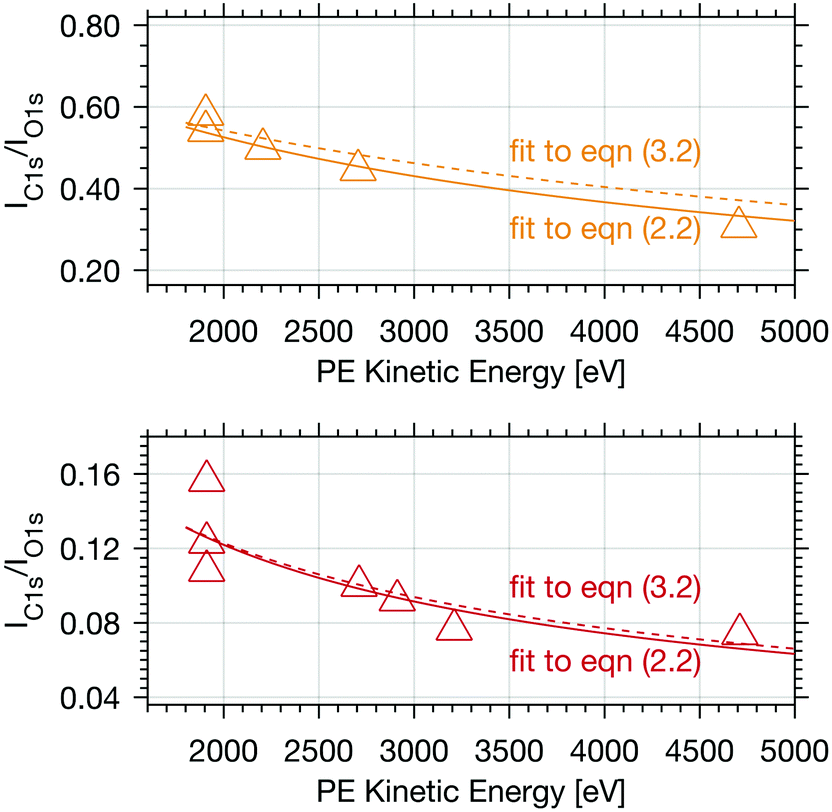 | ||
| Fig. 4 Calibrated PE intensity ratio of the integrated C 1s adventitious carbon peak area and O 1s peak area as a function of electron kinetic energy. The upper panel shows results (yellow triangles) for experiments with 0.04 mbar formic acid and the lower with 0.004 mbar formic acid partial pressure (orange triangles), both at 253 K. The coloured lines represent fits to eqn (2.2) (solid line) and to eqn (3.2) (dashed line). | ||
A species with homogeneous concentration along the entire sampling depth at the interfacial region would show a steady increase of the calibrated IC1s(FA) and IO1s with increasing photon energy, and consequently a constant IC1s(FA)/IO1s in Fig. 3A and 4. The other extreme, a species that is purely surface adsorbed would result in a constant IC1s(FA) regardless of the electron kinetic energy, an increase of IO1s as the number of water molecules grows with probing volume, and thus a strongly decreasing IC1s(FA)/IO1s. Therefore, the steep profile of the integrated signal for 0.004 mbar formic acid in Fig. 3A indicates that the formic acid is predominantly located at the surface and its availability decreases strongly with depth, whereas the steady profile of Fig. 3A (upper panel) indicates significant presence of formic acid also deeper in the ice. This is fully consistent with earlier findings26 and indicates a substantial increase in the penetration depth into the air–ice interface of the formic acid with increasing partial pressure.
Adventitious carbon shows a distinctly different behaviour compared to formic acid with persistent steep profiles at both surface coverages. The IC1s/IO1s of adventitious carbon (Adv.C) decreases to 75% over the first 1000 eV in the experiments with 0.004 mbar formic acid and to 82% in the experiments at 0.04 mbar formic acid. The decline of the depth profile at low dosage agrees perfectly well with the observations of formic acid at the interface where a decline to 76% was observed. These results are in-line with adventitious carbon not entering the interface at considerable depth, but rather covering the ice sample surface.
In summary, this qualitative analysis of the data gives clear experimental evidence of formic acid entering the interfacial region deeper at higher surface coverage. Further, the more hydrophobic adventitious carbon is found more at the surface compared to formic acid at high coverage. For a more in-depth analysis of the concentration of formic acid and adventitious carbon in the interfacial region, a 3-layer model was used to interpret the profiles quantitatively. The model is similar to that presented by Křepelová et al., but to account for the adventitious carbon an additional surface layer was introduced.27 In brief, a homogeneous layer of thickness d(1) ranging from the air–ice interface at 0 nm to a depth of z1 nm contains adventitious carbon with an atomic concentration of nAdv.C(1) and nO(1) oxygen atoms per unit cell. This layer sits on top of a homogeneous 2nd layer with a thickness of d(2) (depth z1 to z2) containing nFA(2) formic acid and nO(2) oxygen atoms per unit cell. The third layer below z2 is infinitely thick consisting of neat ice with an atomic concentration of nO(3) oxygen per unit cell. Now, the measured IC1s(FA)/IO and IC1s(Adv.C)/IO1s can be described by eqn (2.1) and (2.2), respectively. Iterative fit results, minimizing the residuals of IC1s(FA)/IO1s, IC1s(Adv.C)/IO1s, and of IC1s(FA)/IC1s(Adv.C) simultaneously applying the 3-layer model presented above are displayed in Fig. 3A and 4 (solid lines). Tables 1 and 2 also give the constrains to the fits that were used taken that the model system (eqn (2.1 and 2.2)) holding 4 unknown fitting parameters is underdetermined.
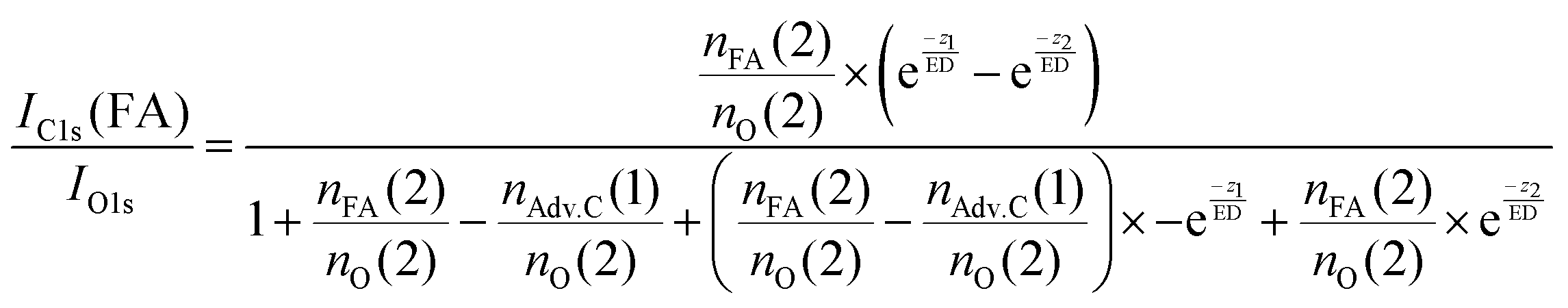 | (2.1) |
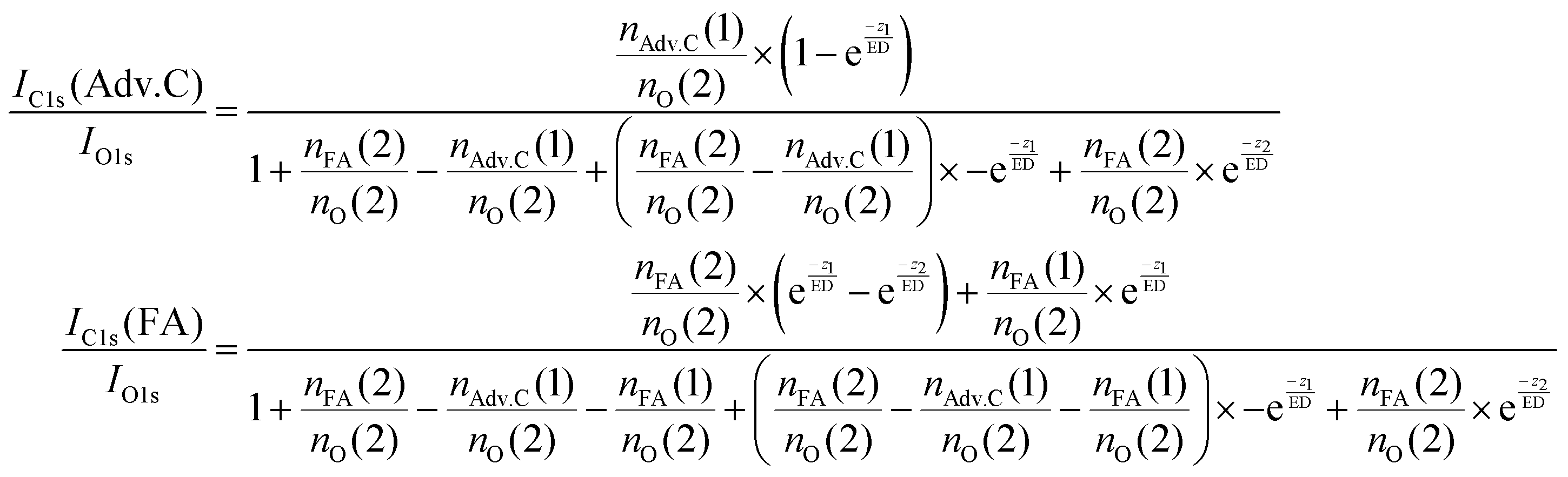 | (2.2) |
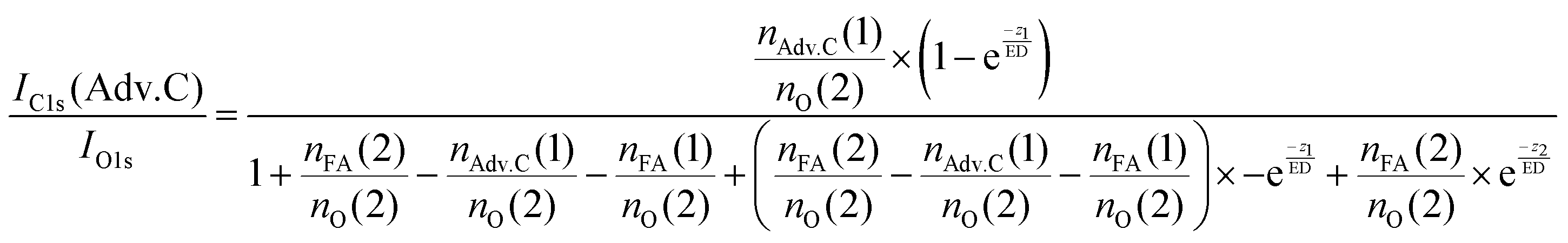 | (3.1) |
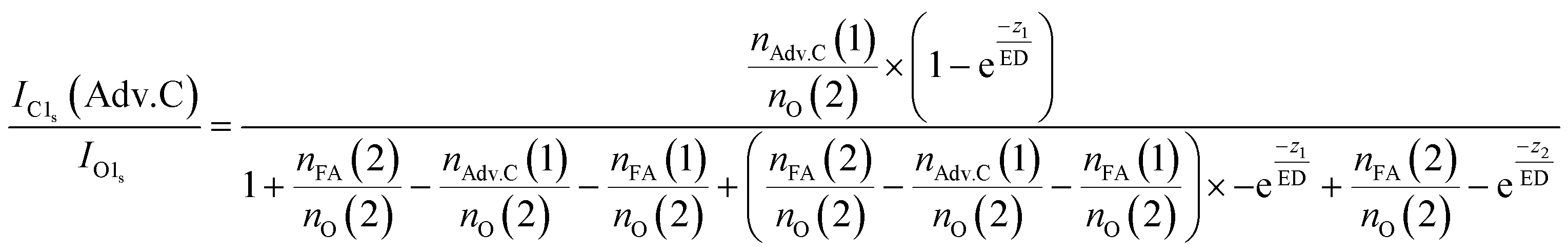 | (3.2) |
| Model | Constrains | d(1) [nm] | d(2) [nm] | [FA]surf [molec. cm−2] | X FA(2) [%] | FA(1)/FA(2) [%] | r 2 |
|---|---|---|---|---|---|---|---|
| Eqn (2.1 and 2.2) | d(1) > 0.3 nm; d(2) > 0.3 nm; X(FA) < 100% | 0.3 | 0.3 | 1.2 × 10 +14 | 16 | — | 0.9 |
| Eqn (2.1 and 2.2) | d(1) > 0.3 nm; d(2) > 0.3 nm; 25% < X(FA) < 100% | 0.4 | 0.3 | 1.7 × 10+14 | 25 | — | 0.9 |
| Eqn (2.1 and 2.2) | 0.3 < d(1) < 1 nm; 1 < d(2) < 4 nm; X(FA) < 100% | 0.3 | 2.3 | 2.0 × 10+15 | 52 | — | 0 |
| Eqn (2.1 and 2.2) | 1 < d(1) < 4 nm; 1 < d(2) < 4 nm; X(FA) < 100% | 1.0 | 0.4 | 4.4 × 10+14 | 100 | — | 0.9 |
| Eqn (3.1 and 3.2) | d(1) > 0.3 nm; d(2) > 0.3 nm; X(FA) < 100% | 0.3 | 4.6 | 9.3 × 10+13 | 0.6 | 15 | 0.6 |
| Eqn (3.1 and 3.2) | 0.3 < d(1) < 0.5 nm; 0.3 < d(2) < 0.5 nm; X(FA) < 100% | 0.3 | 0.5 | 2.1 × 10 +13 | 1.4 | 22 | 0.2 |
| Eqn (3.1 and 3.2) | 1 < d(1) < 4 nm; 0.3 < d(2) < 4 nm; X(FA) < 100% | 1.0 | 4.0 | 1.6 × 10+14 | 1.4 | 34 | 0.3 |
| Eqn (3.1 and 3.2) | 1 < d(1) < 4 nm; 0.3 < d(2) < 0.5 nm; X(FA) < 100% | 1.0 | 0.5 | 2.4 × 10+13 | 1.6 | 56 | 0 |
| Eqn (3.1 and 3.2) | 0.3 < d(1) < 4 nm; 0.3 < d(2) < 4 nm; 11 < X(FA) < 100% | 4.0 | 1.2 | 3.6 × 10+14 | 11 | 1 | 0 |
| Model | Constrains | d(1) [nm] | d(2) [nm] | [FA]surf [molec. cm−2] | X FA(2) [%] | FA(1)/FA(2) [%] | r 2 |
|---|---|---|---|---|---|---|---|
| Eqn (2.1 and 2.2) | d(1) > 0.3 nm; d(2) > 0.3 nm; X(FA) < 100% | 4.9 | 4.9 | 5.9 × 10+15 | 100 | — | 0.8 |
| Eqn (2.1 and 2.2) | 0.3 < d(1) < 2 nm; d(2) > 0.3 nm; X(FA) < 100% | 2.0 | 6.6 | 1.9 × 10 +15 | 11 | — | 0.8 |
| Eqn (2.1 and 2.2) | d(1) > 0.3 nm; d(2) > 0.3 nm; X(FA) < 25% | 3.2 | 5.8 | 3.2 × 10+15 | 25 | — | 0.8 |
| Eqn (3.1 and 3.2) | 0.3 < d(1) < 2 nm; d(2) > 0.3 nm; X(FA) < 100% | 0.3 | 4.2 | 1.6 × 10 +14 | 1 | 12 | 0.8 |
| Eqn (3.1 and 3.2) | 0.3 < d(1) < 2 nm; d(2) > 0.3 nm; 11 < X(FA) < 100% | 2.0 | 2.1 | 8.8 × 10+14 | 18 | 6 | 0 |
| Eqn (3.1 and 3.2) | 0.3 < d(1) < 5 nm; d(2) > 0.3 nm; 11 < X(FA) < 100% | 4.7 | 10.0 | 4.2 × 10+15 | 18 | 18 | 0 |
Thickness of the QLL
For experiments at low formic acid partial pressure, we find that the data are best represented by using a layer thickness of 0.3 nm for both layers (fit to eqn (2.1 and 2.2)). The model also returns the atomic ratio of formic acid to oxygen. Given that formic acid holds two oxygen atoms, this ratio can be converted to the mole fraction of formic acid in the quasi liquid layer of 16% which can be expressed as formal surface coverage of 1 × 1014 molecules cm−2. Constraining the mole fraction to 25%, which corresponds to upper limit of reported mole fraction of an aqueous solution in thermodynamic equilibrium with ice at 253 K,38,39 similar thicknesses of layer d(1) and of d(2) are derived (Table 1). Taken that generally the solubility in crystalline ice is orders of magnitude lower than the detection limit of X-ray photoemission spectroscopy, we interpret the finding as a thin layer of adventitious carbon covering, potentially partially, the ice surface and a thin quasi-liquid at the upper interfacial region into which the formic acid, but not the adventitious carbon dissolves. This result for formic acid is in good agreement to our previous X-ray photoemission work performed with lower electron kinetic energies and consequently limited to probe a thinner fraction of the interfacial region.26 Constraining the fit to a thicker quasi-liquid layer (d(2) > 1 nm) does not result in a fit solution. Constraining the fit to a thicker adventitious carbon layer (d(1) > 1 nm) gives as good fit result, however, the mole fraction of formic acid in the quasi-liquid is unreasonable high as it reaches that of a pure formic acid layer. At high partial pressure, the formic acid penetrates deeper into the ice again in perfect qualitative agreement to our earlier work.26 Here, we get layers with a thickness of about 2–3 nm for layer d(1) and ∼6–7 nm for layer d(2) (Table 2). Obviously, the presence of formic acid is not restricted to the upper few monolayers of the ice but the quasi-liquid spans over a depth of ∼7 nm. Formic acid is thus found at depths of the interfacial region that significantly exceed the depth of the quasi-liquid layer on neat ice at 253 K. The fits to eqn (2) also give the mole fraction of formic acid in the quasi-liquid layer, from which a surface coverage can be derived. The surface coverages of 2–3 × 1015 molecules cm−2 at high formic acid dosage (0.04 mbar) agrees well with the estimate derived based on single measurements as shown in Fig. 2. In this case, calibrating the data to the PE signals of gas phase CO2 acquired at the same kinetic energy, a value of 2–6 × 1015 molecules cm−2 is obtained. This agreement between the fits to eqn (2.1) and (2.2) and the direct observables gives significant confidence toward the model results. Agreement at low dosing is arguably less good: 1 × 1014 molecules cm−2 are derived by fits to eqn (2.1) and (2.2) and 1–2 × 1015 molecules cm−2 are derived directly from the data as presented in Fig. 3A. We account this to the fact that the lowest sampling depth spans over the upper 6 nm at the PHOENIX beamline, which is not surface sensitive enough. Support comes from our earlier more surface sensitive photoemission data26 where a perfect agreement of both surface coverages estimates of 3 × 1014 molecules cm−2 at 0.01 mbar was derived, in good agreement to the results to the fits to eqn (2.1) and (2.2) in this work.This finding supports the conclusion that, at such high concentrations, formic acid induces disorder to the hydrogen-bonding network at the interfacial region resulting in greater depth at which formic acid is found compared to the data at lower formic acid surface coverage. That the depth of formic acid is limited to about 7 nm is an important finding unambiguously showing that the surface disorder is limited to a nanometre scale region even in presence of large amounts of solute. While being in agreement to our previous analysis, this is the first photoemission study to probe the upper 12 nm of the air–ice interface. Our previous work, showing a depth of 3–6 nm at similar formic acid partial pressure reached the beam-lines probing depth limits of 5 nm. These data at high surface coverage of formic acid indicate that also the adventitious carbon might enter the quasi-liquid to some extend as the first layer of the model (d(1)) with a thickness of 2 nm extents further than the upper monolayer of the ice. Therefore, we tested whether a modified model where formic acid is also present in the layer d(1) might represent the data as well. Model eqn (3.1) and (3.2) holds both formic acid and adventitious carbon in their first layer. The interfacial region of ice is thus described by three layers where the upper one holds formic acid and adventitious carbon, the second one formic acid, and the lowest one no solutes. Introducing formic acid into layer 1 results in poorer fits both to the data at low formic acid partial pressure (Table 1 and Fig. 3A) and at high formic acid concentration (Table 2 and Fig. 3A). For example, when constraining the thickness of layer d(1) and of d(2) to less than 0.5 nm, corresponding to the fitting results of eqn (2.1) and (2.2), the fit does not capture the trend in the data (Fig. 3A). The performance of the fit is improved when allowing a thicker layer with d(2) = 4.6 nm, but overall fits to eqn (2.1) and (2.2) were found to reproduce the data better. The impact of the presence of formic acid in layer d(1) on the fitted profiles of formic acid is significantly more evident than that on the fitted profiles of adventitious carbon (Fig. 4). Despite the uncertainty in these underdetermined systems, these model runs support the conclusion that the upper layer holds no formic acid.
Discussion
The O K-edge NEXAFS spectra at 251 K and at 233 K were dominated by features associated to the structure of the hydrogen bonding network of ice suggesting that pre-melting is little evolved and does not reach deep into the interfacial region up to a temperature of 251 K. An onset temperature of the QLL, defined as the temperature where a liquid-like structure of the hydrogen bonding network is found deep in the interfacial region, above 251 K supports recent sum frequency generation data indicating the QLL at depth beyond the uppermost layer of water molecules at about 257 K.40 The small amount of water molecules experiencing a quasi-liquid environment at 251 K is also in agreement with recent large-scale molecular simulations showing an incomplete coverage of liquid-like water at the ice interfacial region at 260 K.28 Taken together, these studies suggest that the ice interface prior to adsorption of formic acid does not feature pronounced pre-melting. Uncertainty in the precise depth calibration of partial Auger-electron yield NEXAFS might come from the wide kinetic energy range, and from secondary electrons that have had multiple scattering events contributing to the measured intensity.The adsorption of formic acid to ice at temperatures below ∼230 K and at low dosage can be well described by Langmuir isotherms with a saturation coverage of 2–3 × 1014 molecules cm−2.23–25 The XPS data presented here show that formic acid remains at the outermost edge of the interfacial region upon partitioning to roughly the saturation coverage. This XPS result gives thus direct evidence of surface adsorption à la Langmuir. Support comes from molecular dynamics simulations41 and from grand canonical Monte Carlo calculations23 indicating that formic acid forms two exceptionally strong hydrogen bonds with rigid ice surfaces up to a Langmuir saturation coverage.
Increasing the partial pressure of formic acid and thus the apparent surface concentration to 2–3 × 1015 molecules cm−2, we can state that formic acid penetrates deeper into the ice, while the adventitious carbon remains at the upper ice surface. We interpret this as adventitious carbon covering the ice sample surface with a quasi-liquid layer below, which solvates the formic acid. Quantification based on a homogenous quasi-liquid layer gives a thickness of 7 nm at maximum. The adventitious carbon is mainly composed of aliphatic C–H groups (Fig. 2) and thus rather hydrophobic. Hydrophobic compounds have been found not to interact strongly with ice surfaces: laser induced fluorescence work and molecular dynamics calculations revealed that the spectra of polycyclic aromatic hydrocarbons at ice surfaces resemble that of the neat crystal and that these compounds favour self-association rather than forming strong hydrogen bonds with the ice.42,43 That formic acid enters deeper into the interfacial region once the Langmuir saturation coverage is reached is at odds with number density profiles derived with grand canonical Monte Carlo calculations that showed multilayer adsorption on rigid ice.23 Molecular dynamics calculations in presence of pre-melting have revealed a high tendency of neutral formic acid molecules to enter and dissolve in the interfacial region.41 It appears therefore likely that the increased flexibility of the hydrogen bonding network at 253 K facilitates the formation of two strong hydrogen bonds41 as solvation shells are formed within the interfacial region. Formic acid–formic acid bonds and hydrogen bonds with the ice surface beyond the Langmuir monolayer coverage are weak.23 Semantically, we would refer to the interfacial region where the concentration of solutes exceeds that of a solid-solution in crystalline ice as QLL. A similar QLL thickness was observed in presence of HCl. Nota bene, a thickness of the QLL of 8 nm was derived based on XPS data at a surface concentration below a formal monolayer.29 At such low coverage, XPS data reveals that formic acid and acetic acid remain at the upper surface only in this and in earlier work.26,27 Apparently, strong acids attract water molecules from the ice more easily to solvate than weaker acids. This striking ability of strong acids to modify the hydrogen bonding network at the interfacial region of ice is also evident from NEXAFS data in presence of HNO344 and of HCl,29 and from Raman data in presence of HCl and of HNO3.45 The interpretation is that the overall energetics of the system favour strong acids forming solvation shells deeper in the interfacial region where the hydrogen bonding network becomes increasingly ice-like. The overall ice-like structure of the QLL is supported by recent sum frequency generation data comparing spectral features of supercooled water, ice and the QLL40 and of NEXAFS data in presence of solutes revealing a dominating ice-like feature in the spectra of the interfacial region.26,29,44
Conclusions
The results presented here confirm that the tendency to enter the interfacial region of a specific compound is linked to its surface concentration and to its hydrophilicity. The driving force for solutes to enter into the interfacial region is their demand for water molecules to form energetically favourable solvation shell configurations. Thus flexibility in the hydrogen-bonding network is a prerequisite for solutes to enter and dissolve in the interfacial region. Taking full advantage of the extended electron escape depth at the PHOENIX beamline, the thickness of the quasi-liquid layer holding formic acid was confirmed to be 7 nm. The neat ice did show only very minor liquid-like features at 251 K, indicating that the structure of the quasi-liquid layer's hydrogen bonding network is predominately ice-like.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the SLS for provision of beamtime at the PHOENIX and SIM beamlines. We acknowledge the Swiss National Science Foundation (grant #149629). XK thanks the Swedish Research Council (grant 2014-6924). The work is part of the PhD thesis of Astrid Waldner at ETHZ (examiner: Thomas Peter).Notes and references
- J. A. Sellberg, S. Kaya, V. H. Segtnan, C. Chen, T. Tyliszczak, H. Ogasawara, D. Nordlund, L. G. M. Pettersson and A. Nilsson, J. Chem. Phys., 2014, 141, 034507 CrossRef PubMed.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standard ReferenceDatabase 69, http://webbook.nist.gov/, accessed January 2013.
- A. M. Grannas, A. E. Jones, J. Dibb, M. Ammann, C. Anastasio, H. J. Beine, M. Bergin, J. Bottenheim, C. S. Boxe, G. Carver, G. Chen, J. H. Crawford, F. Domine, M. M. Frey, M. I. Guzman, D. E. Heard, D. Helmig, M. R. Hoffmann, R. E. Honrath, L. G. Huey, M. Hutterli, H. W. Jacobi, P. Klan, B. Lefer, J. McConnell, J. Plane, R. Sander, J. Savarino, P. B. Shepson, W. R. Simpson, J. R. Sodeau, R. von Glasow, R. Weller, E. W. Wolff and T. Zhu, Atmos. Chem. Phys., 2007, 7, 4329–4373 CrossRef.
- A. M. Grannas, C. Bogdal, K. J. Hageman, C. Halsall, T. Harner, H. Hung, R. Kallenborn, P. Klán, J. Klánová, R. W. Macdonald, T. Meyer and F. Wania, Atmos. Chem. Phys., 2013, 13, 3271–3305 CrossRef.
- A. Steffen, T. Douglas, M. Amyot, P. Ariya, K. Aspmo, T. Berg, J. Bottenheim, S. Brooks, F. Cobbett, A. Dastoor, A. Dommergue, R. Ebinghaus, C. Ferrari, K. Gardfeldt, M. E. Goodsite, D. Lean, A. J. Poulain, C. Scherz, H. Skov, J. Sommar and C. Temme, Atmos. Chem. Phys., 2008, 8, 1445–1482 CrossRef.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1994, 21, 165–176 CrossRef.
- T. Bartels-Rausch, H. W. Jacobi, T. F. Kahan, J. L. Thomas, E. S. Thomson, J. P. D. Abbatt, M. Ammann, J. R. Blackford, H. Bluhm, C. Boxe, F. Domine, M. M. Frey, I. Gladich, M. I. Guzmán, D. Heger, T. Huthwelker, P. Klán, W. F. Kuhs, M. H. Kuo, S. Maus, S. G. Moussa, V. F. McNeill, J. T. Newberg, J. B. C. Pettersson, M. Roeselová and J. R. Sodeau, Atmos. Chem. Phys., 2014, 14, 1587–1633 CrossRef.
- T. Huthwelker, M. Ammann and T. Peter, Chem. Rev., 2006, 106, 1375–1444 CrossRef PubMed.
- H. Bluhm, D. F. Ogletree, C. S. Fadley, Z. Hussain and M. Salmeron, J. Phys.: Condens. Matter, 2002, 14, L227–L233 CrossRef.
- V. F. McNeill, T. Loerting, F. M. Geiger, B. L. Trout and M. J. Molina, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9422–9427 CrossRef PubMed.
- A. Nilsson, D. Nordlund, I. Waluyo, N. Huang, H. Ogasawara, S. Kaya, U. Bergmann, L. Å. Näslund, H. Öström, P. Wernet, K. J. Andersson, T. Schiros and L. G. M. Pettersson, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 99–129 CrossRef.
- D. B. Millet, M. Baasandorj, D. K. Farmer, J. A. Thornton, K. Baumann, P. Brophy, S. Chaliyakunnel, J. A. de Gouw, M. Graus, L. Hu, A. Koss, B. H. Lee, F. D. Lopez-Hilfiker, J. A. Neuman, F. Paulot, J. Peischl, I. B. Pollack, T. B. Ryerson, C. Warneke, B. J. Williams and J. Xu, Atmos. Chem. Phys., 2015, 15, 6283–6304 CrossRef.
- J. Liu, X. Zhang, E. T. Parker, P. R. Veres, J. M. Roberts, J. A. de Gouw, P. L. Hayes, J. L. Jimenez, J. G. Murphy, R. A. Ellis, L. G. Huey and R. J. Weber, J. Geophys. Res., 2012, 117, D00V21 CrossRef.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, J. H. Kroll, J. H. Seinfeld and P. O. Wennberg, Atmos. Chem. Phys., 2009, 9, 1479–1501 CrossRef.
- P. Neeb, F. Sauer, O. Horie and G. K. Moortgat, Atmos. Environ., 1997, 31, 1417–1423 CrossRef.
- T. Stavrakou, J. F. Müller, J. Peeters, A. Razavi, L. Clarisse, C. Clerbaux, P. F. Coheur, D. Hurtmans, M. De Mazière, C. Vigouroux, N. M. Deutscher, D. W. T. Griffith, N. Jones and C. Paton-Walsh, Nat. Geosci., 2011, 5, 26–30 CrossRef.
- D. J. Jacob and S. C. Wofsy, J. Geophys. Res., 1988, 93, 1477 CrossRef.
- W. L. Chameides and D. D. Davis, Nature, 1983, 304, 427–429 CrossRef.
- M. O. Andreae, R. W. Talbot, T. W. Andreae and R. C. Harriss, J. Geophys. Res., 1988, 93, 1616–1624 CrossRef.
- F. Paulot, D. Wunch, J. D. Crounse, G. C. Toon, D. B. Millet, P. F. DeCarlo, C. Vigouroux, N. M. Deutscher, G. G. Abad, J. Notholt, T. Warneke, J. W. Hannigan, C. Warneke, J. A. de Gouw, E. J. Dunlea, M. De Mazière, D. W. T. Griffith, P. Bernath, J. L. Jimenez and P. O. Wennberg, Atmos. Chem. Phys., 2011, 11, 1989–2013 CrossRef.
- D. J. Jacob, J. Geophys. Res., 1986, 91, 9807 CrossRef.
- N. Pouvesle, M. Kippenberger, G. Schuster and J. N. Crowley, Phys. Chem. Chem. Phys., 2010, 12, 15544–15550 RSC.
- P. Jedlovszky, G. Hantal, K. Neuróhr, S. Picaud, P. N. M. Hoang, P. von Hessberg and J. N. Crowley, J. Phys. Chem. C, 2008, 112, 8976–8987 CrossRef.
- P. von Hessberg, N. Pouvesle, A. K. Winkler, G. Schuster and J. N. Crowley, Phys. Chem. Chem. Phys., 2008, 10, 2345–2355 RSC.
- A. Symington, R. A. Cox and M. A. Fernandez, Z. Phys. Chem., 2010, 224, 1219–1245 CrossRef.
- T. Bartels-Rausch, F. Orlando, X. Kong, L. Artiglia and M. Ammann, ACS Earth Space Chem., 2017, 1, 572–579 CrossRef.
- A. Křepelová, T. Bartels-Rausch, M. A. Brown, H. Bluhm and M. Ammann, J. Phys. Chem. A, 2013, 117, 401–409 CrossRef PubMed.
- A. Hudait, M. T. Allen and V. Molinero, J. Am. Chem. Soc., 2017, 139, 10095–10103 CrossRef PubMed.
- X. Kong, A. Waldner, F. Orlando, L. Artiglia, T. Huthwelker, M. Ammann and T. Bartels-Rausch, J. Phys. Chem. Lett., 2017, 8, 4757–4762 CrossRef PubMed.
- D. E. Starr, D. Pan, J. T. Newberg, M. Ammann, E. G. Wang, A. Michaelides and H. Bluhm, Phys. Chem. Chem. Phys., 2011, 13, 19988–19996 RSC.
- T. L. Barr and S. Seal, J. Vac. Sci. Technol., A, 1995, 13, 1239–1246 CrossRef.
- M. Krzywiecki, L. A. Grządziel, P. Powroźnik, M. Kwoka, J. Rechmann and A. Erbe, Phys. Chem. Chem. Phys., 2018, 20, 16092–16101 RSC.
- F. F. Tao and L. Nguyen, Phys. Chem. Chem. Phys., 2018, 20, 9812–9823 RSC.
- F. Orlando, A. Waldner, T. Bartels-Rausch, M. Birrer, S. Kato, M.-T. Lee, C. Proff, T. Huthwelker, A. Kleibert, J. van Bokhoven and M. Ammann, Top. Catal., 2016, 59, 591–604 CrossRef.
- H. Bluhm, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 71–84 CrossRef.
- M. A. Brown, F. Vila, M. Sterrer, S. Thürmer, B. Winter, M. Ammann, J. J. Rehr and J. A. van Bokhoven, J. Phys. Chem. Lett., 2012, 3, 1754–1759 CrossRef PubMed.
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef.
- D. R. Lide, CRC Handbook of Chemistry and Physics, http://hbcponline.com/, March 2018.
- J. Timmermans, Physico-Chemical Constants of Binary Systems in Concentrated Solutions, Volume 4 – Systems with Inorganic + Organic or Inorganic Compounds (Excepting Metallic Derivatives), http://app.knovel.com/hotlink/toc/id:kpPCCBSC21/physico-chemical-constants, March 2018.
- M. A. Sánchez, T. Kling, T. Ishiyama, M.-J. van Zadel, P. J. Bisson, M. Mezger, M. N. Jochum, J. D. Cyran, W. J. Smit, H. J. Bakker, M. J. Shultz, A. Morita, D. Donadio, Y. Nagata, M. Bonn and E. H. G. Backus, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 227–232 CrossRef PubMed.
- M. Compoint, C. Toubin, S. Picaud, P. N. M. Hoang and C. Girardet, Chem. Phys. Lett., 2002, 365, 1–7 CrossRef.
- D. Ardura, T. F. Kahan and D. J. Donaldson, J. Phys. Chem. A, 2009, 113, 7353–7359 CrossRef PubMed.
- T. F. Kahan, S. N. Wren and D. J. Donaldson, Acc. Chem. Res., 2014, 47, 1587–1594 CrossRef PubMed.
- A. Křepelová, J. Newberg, T. Huthwelker, H. Bluhm and M. Ammann, Phys. Chem. Chem. Phys., 2010, 12, 8870–8880 RSC.
- T. F. Kahan, J. P. Reid and D. J. Donaldson, J. Phys. Chem. A, 2007, 111, 11006–11012 CrossRef PubMed.
Footnotes |
| † The electron escape depth (ED) is a direct measure of the sampling depth in XPS. |
| ‡ XPST is an add-on to IgorPro to analyse X-ray Photoelectron Spectroscopy (XPS) data. Written by Dr Martin Schmid, Philipps University Marburg. http://www.igorexchange.com/project/XPStools |
| This journal is © the Owner Societies 2018 |