
Power of protein/tRNA functional assembly against aberrant aggregation†
Charles
Bou-Nader
a,
Ludovic
Pecqueur
a,
David
Cornu
b,
Murielle
Lombard
a,
Manuela
Dezi
c,
Magali
Nicaise
d,
Christophe
Velours
d,
Marc
Fontecave
a and
Djemel
Hamdane
 *a
*a
aLaboratoire de Chimie des Processus Biologiques, CNRS-UMR 8229, Collège De France, Université Pierre et marie Curie, 11 place Marcelin Berthelot, 75231 Paris Cedex 05, France. E-mail: djemel.hamdane@college-de-france.fr; Tel: +331-44271645
bInstitut de Biologie Intégrative de la Cellule, CNRS, CEA, Université Paris-Saclay, Gif-sur-Yvette, France
cInstitut de Minéralogie, Physique des Matériaux et Cosmo chimie, UMR 7590, CNRS, Université Pierre et Marie Curie, 4, place Jussieu, 75005, Paris, France
dMacromolecular Interaction Platform of I2BC, UMR 9198, 1 avenue de la terrasse, 91191 Gif Sur Yvette, France
First published on 26th September 2017
Abstract
Understanding the mechanisms of protein oligomerization and aggregation is a major concern for biotechnology and medical purposes. However, significant challenges remain in determining the mechanism of formation of these superstructures and the environmental factors that can precisely modulate them. Notably the role that a functional ligand plays in the process of protein aggregation is largely unexplored. We herein address these issues with an original flavin-dependent RNA methyltransferase (TrmFO) used as a protein model since this protein employs a complex set of cofactors and ligands for catalysis. Here, we show that TrmFO carries an unstable protein structure that can partially mis-unfold leading to either formation of irregular and nonfunctional soluble oligomers endowed with hyper-thermal stability or large amorphous aggregates in the presence of salts. Mutagenesis confirmed that this peculiarity is an intrinsic property of a polypeptide and it is independent of the flavin coenzyme. Structural characterization and kinetic studies identified several regions of the protein that enjoy conformational changes and more particularly pinpointed the N-terminal subdomain as being a key element in the mechanisms of oligomerization and aggregation. Only stabilization of this region via tRNA suppresses these aberrant protein states. Although protein chaperones emerged as major actors against aggregation, our study emphasizes that other powerful mechanisms exist such as the stabilizing effect of functional assemblies that provide an additional layer of protection against the instability of the proteome.
Introduction
Proteins are inherently flexible entities, which is key for their proper function in cells.1 The ability to fold, to stay under a stable native state or to alternate between active and inactive conformations is all dictated by the intrinsic properties of the polypeptide sequence and by the physical and chemical properties of the milieu. All of this is described by the so called protein's free energy landscape that schematically represents the accessible folding states of a specific protein.2,3 Some of these states can be highly reactive and functionally vital while others can be deleterious such as folding intermediates that act as kinetic traps or misfolded conformations.4–8 The latter are prone to oligomerization leading to the formation of cytotoxic species.9,10 Depending on the conformation of these precursors and the physico-chemical properties of the environment, proteins can undergo multiple ways of self-interaction leading to a large polymorphism of oligomers and aggregates.11–13 This is particularly the case for large proteins that can experience during the course of their conformational changes a higher probability to populate aberrant states that are precursors of oligomers.14–16 Therefore, it is important to (i) detect metastable intermediate states and (ii) to characterize them for a better understanding of the competing balance between the physiological function and pathological disorders.TrmFO is a large tRNA-modifying enzyme that has recently been discovered in Gram+ bacteria and in certain mycoplasma.17,18 This enzyme catalyzes the methylation of uridine 54 to ribothymidine (m5U54), one of the most conserved modified bases in tRNAs. While in all other organisms m5U54 is catalyzed by a classical S-adenosylmethionine-based methyltransferase, TrmFO employs an extremely challenging and unprecedented methylation mechanism that involves a set of several cofactors, including the flavin adenine dinucleotide (FAD), methylene tetrahydrofolate and NADPH.19–21 Moreover, the crystallographic structure shows that this enzyme adopts a complex topology with the presence of a domain inserted within the FAD binding domain splitting this latter into two subdomains22 (Fig. 1A). Such a structural arrangement has been proposed to be selected during evolution to promote cooperative folding between domains and thus to avoid areas of the landscape that lead to kinetic trapping and misfolding.23,24 However, we recently showed that the tertiary structure of TrmFO is loosely packed and thus quite unstable.25 Here we show that TrmFO from Bacillus subtilis is an unstable protein that can rapidly lose some of its secondary structure and then self-assemble into stable oligomers or large amorphous aggregates depending on the ionic strength of the milieu. These non-functional oligomers have the particularity of presenting various morphologies with the presence of amyloid-like assemblies and also irregular and compact spheroidal oligomers. Computational prediction as well as structural characterization by mild-proteolysis coupled with ESI-MS analysis allowed us to identify the N-terminal protein region as being a key structural element in determining the shape of the oligomers and in the oligomerization selectivity with respect to the aggregation pathway. Remarkably, stabilization of this region by tRNA reshapes drastically the conformational space of the enzyme towards a more secure energy landscape demonstrating the relevance of functional assemblies against misunfolding events.
 | ||
| Fig. 1 Thermal stability of the holo- and apoprotein forms of Bacillus subtilis TrmFO measured using circular dichroism spectroscopy. (A) Structural model of TrmFO from Bacillus subtilis. The FAD binding domain is subdivided into two subdomains (Nt-subdomain in pink and Ct-subdomain in purple) by an inserted domain (in dark green). The FAD cofactor is represented in yellow sticks. The two catalytic cysteines, C53 and C226 as well as the tyrosine Y346 stacked against FAD isoalloxazine ring are shown in ball sticks. (B) Chemical structure of the different FAD species observed in a fresh protein preparation of wild type TrmFO. The species 1 is oxidized FAD while 2 is a N5 alkylated two-electron reduced FAD reduced covalently bound to the polypeptide of TrmFO via the conserved C53 residue. Form 3 is the one electron oxidized product of 2. The mutant C53A is a holoprotein form of TrmFO containing exclusively the species 1 while Y346A and Y346F are apoprotein forms of TrmFO. (C) Thermal denaturation curves of wild-type and mutant TrmFO recorded by circular dichroism at 222 nm. The blue curves are those followed at low ionic strength in 10 mM sodium phosphate pH 8 while curves in red are obtained at high ionic strengths in a 50 mM HEPES buffer, pH 7.5, 100 mM ammonium acetate. (D) Histograms representing T1/2 values in °C of wild type TrmFO and mutants in the presence of 10 mM sodium phosphate, pH 8 (black) or in the presence of 50 mM HEPES, pH 7.5, 100 mM ammonium acetate (gray) which is the activity buffer of the enzyme. | ||
Materials and methods
Experimental procedures
Preparation of Δ1–59 Y346A TrmFO Y346A was obtained by a mild proteolysis treatment of the full length Y346A protein. Briefly, this latter mutant was reconstituted to a holoenzyme form by adding 2 eq. of FAD. Then, the resulting holoenzyme was subjected to mild trypsinolysis for 2 hours at room temperature with a ratio of 1/200 trypsin in 50 mM sodium phosphate pH 8 and 200 mM NaCl. The reaction was quenched by adding 2 mM PMSF and the truncated protein was further purified by gel filtration on a S200 increase 10-300 GL (GE). The purity of the shortened protein was further checked by SDS-PAGE showing a purity >95%.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 15 minutes at 4 °C prior to measurements. 50 μL of samples were loaded in the cuvette and covered by 20 μL of PCR oil to prevent evaporation. Denaturation ramp was conducted from 20 to 60 °C by a step of 1 °C with an equilibration time of two minutes. Each temperature point corresponds to a 100 s effective measurement time. For the TrmFO-tRNA complex, 4 μM TrmFO was mixed with 20 μM tRNA in 10 mM sodium phosphate pH 8 and let on ice for 15 minutes before centrifugation at 20000g for 15 minutes at 4 °C.
:1) and electrophoresis was conducted for 1 h at 4 °C and 100 V. RNAs were colored with a 0.1% toluidine solution.
000g for 15 minutes at 4 °C prior to measurements. 50 μL of samples were loaded in the cuvette and covered by 20 μL of PCR oil to prevent evaporation. Denaturation ramp was conducted from 20 to 60 °C by a step of 1 °C with an equilibration time of two minutes. Each temperature point corresponds to a 100 s effective measurement time. For the TrmFO-tRNA complex, 4 μM TrmFO was mixed with 20 μM tRNA in 10 mM sodium phosphate pH 8 and let on ice for 15 minutes before centrifugation at 20000g for 15 minutes at 4 °C.
:1) and electrophoresis was conducted for 1 h at 4 °C and 100 V. RNAs were colored with a 0.1% toluidine solution.
Bands corresponding to the TrmFO protein were excised and subjected to enzymatic digestion. Briefly, protein bands were excised and extensively washed with CH3CN and 25 mM NH4HCO3. The excised bands were treated with 100 μL 10 mM DTT at 57 °C for 30 minutes. The DTT was removed and 100 μL of 55 mM iodoacetamide was added for cysteine carbamidomethylation. The reaction was allowed to proceed at room temperature for 30 minutes. The supernatant was removed, the washing procedure was repeated and the gel slices were dried. We added 20 μL of 10 ng μL−1 trypsin or Glu-C (Promega) diluted in 25 mM NH4HCO3, and the mixture was incubated overnight at room temperature. Peptides were extracted in 60% acetonitrile and 0.1% (v/v) formic acid and 0.5 μL were mixed with an equal volume of α-cyano-4-hydroxycinnamic acid (10 mg mL−1, 50% CH3CN; Sigma-Aldrich). Crystals were obtained using the dried droplet method and Trypsin- and GluC-generated peptide mixtures were analyzed using a MALDI-TOF-TOF 5800 (ABSciex). Survey scans were performed using delayed extraction (390 ns) in reflector mode for a total of 15000 shots. Peak lists were processed and analyzed with ProteinPilot software (ABSciex), using the MASCOT algorithm. The search parameters were as follows: Y346A TrmFOBS sequence, digest reagent Glu-C (cleavage at the C-terminal of D/E) or trypsin and semi-trypsin, cysteine carbamidomethylation and oxidation (methionine and tryptophan) were considered respectively a complete and variable modification and peptide mass tolerance was set at 10 ppm.
The fluorescence of free or oligomer bound ANS was measured at λex = 370 nm between 380–700 nm using a slit width of excitation and emission at 5/5 mm, respectively. The final concentrations of ANS and soluble oligomers of TrmFO in the assays were 100 and 2 μM, respectively, in 10 mM potassium phosphate pH 8. Oligomers of TrmFO were obtained as described just above. Oligomers and ANS were incubated for 10 minutes before acquisition of a spectrum.
The test with the Congo red was followed by absorbance spectroscopy on a carry 50. The final concentration of the Congo red and the oligomers of Y346A TrmFO were 5 and 20 μM, respectively.
Results
Structural instability is a peculiarity of both holo- and apoTrmFO proteins
We have previously shown that TrmFO is a unique flavoenzyme since it is isolated with different FAD species including the oxidized form and a two- and one-electron reduced alkylated flavins that are covalently bound to the polypeptide chain via the conserved cysteine 5320,21 (Fig. 1B). In order to determine the contribution of FAD to the stability of TrmFO, the thermal stability of the wild type protein was analyzed along with that of the C53A mutant, which has the particularity of carrying only non-covalently bound oxidized FAD,20 and two other mutants, Y346A and Y346F, which are apoprotein forms of TrmFO.26 The stability of TrmFO was evaluated by measuring the circular dichroism signal at 222 nm, assigned to the alpha helix, as a function of temperature and under low ionic strength conditions (10 mM sodium phosphate pH 8). As shown in Fig. 1C, the temperature increase resulted in a cooperative loss of the dichroic signal characterized by a low value of the half-transition temperature (T1/2). The latter value ranges from 36 ± 2 °C for the least stable Y346F protein to 38.5 ± 2.5 °C for the most stable one, which is the C53A mutant (Fig. 1D). Surprisingly, the signal variation observed with the holo- or apoproteins was too low to account for a full denaturation process suggesting that it was very likely that TrmFO had unfolded only partially. Moreover, the thermally-induced transition was irreversible since the starting signal was never recovered.To further investigate this phenomenon, the thermal stability of the wild type protein and the three mutants was studied using differential scanning calorimetry (DSC). As shown in Fig. 2, the T1/2 values obtained by DSC were dependent on the temperature ramping rate ν (Fig. 2), which is known to be a typical peculiarity of the thermal denaturation reaction controlled by a kinetic phenomenon.27,28 Accordingly, apparent activation energy (Ea) can be determined from the Arrhenius like-plot ln(ν/T1/22) versus 1/T1/2. The calculated Ea values are 339 ± 15, 360 ± 20, 352 ± 10 and 300 ± 15 kJ mol−1 for the wild type, C53A, Y346A and Y346F proteins, respectively. Interestingly, the wild type TrmFO is less stable than the C53A mutant. In addition, the apoprotein Y346A is more stable than the wild type and the apoprotein Y346F by +13 kJ mol−1 and +52 kJ mol−1, respectively. The apoprotein Y346A and the holoprotein C53A have comparable stability. These results indicate that (i) the covalent linkage between C53 and the alkylated FAD destabilizes the protein, (ii) the FAD cofactor is not important for the stability of TrmFO (illustrated by similar apparent Ea between C53A and Y346A) and (iii) introduction of a bulky and hydrophobic residue at position 346 such that phenylalanine in the FAD binding pocket can destabilize the protein. This statement is also supported by the fact that the Y346W variant is not soluble at all (data not shown).
 | ||
| Fig. 2 Arrhenius-like plots obtained by differential scanning calorimetry for the two-state reaction of thermal-induced denaturation of wild type TrmFO and its mutants. | ||
Taken together, TrmFO has a relatively fragile structure that can partially unfold at relatively mild temperatures and most importantly, the non-covalently bound flavin coenzyme has no major contribution to such a structural change. However, our results are consistent with the fact that the covalent bond between the alkylated flavin and the polypeptide decreases the stability of the wild-type protein probably by affecting the conformational dynamics of the active site loop and by promoting greater exposure of the FAD binding pocket.29 Consequently, this structural instability could play an important physiological role in allowing productive conformational changes to take place during catalysis, with nonetheless a risk of exposure of reactive zones within the protein scaffold.4
Partially unfolded TrmFO with a non-native structure induces an oligomerization process
We further investigated the structural instability of TrmFO by recording the far-UV CD spectrum of the protein as a function of temperature in 10 mM sodium phosphate pH 8 (Fig. 3A). To avoid spectral perturbation caused by the flavin cofactor, we decided to focus our characterization on the apoprotein using the Y346A variant as it is obviously more stable than both the wild type and Y346F proteins (Fig. 1C and 2). Interestingly, and as anticipated, the increase of temperature converted TrmFO to a thermally stable and partially unfolded species that still contained a substantial amount of secondary structure (Fig. 3A). We then examined whether the partially unfolded TrmFO gave soluble oligomers which possibly protected it against further denaturation. To this end, the structural unfolding kinetics triggered by the addition of the folded protein to 10 mM sodium phosphate pH 8 at 50 °C was measured by CD at 222 nm and was then compared to the light scattering kinetics recorded under similar conditions. As shown in Fig. 3B, while the variation of the CD signal was too rapid to be detected by our experimental setting (dead time of the mixing within a few seconds), in contrast, the light scattering kinetics of TrmFO appeared much slower and was characterized by a biphasic shape with a fast phase accounting for ∼19%, k1 = 1.26 min−1 and a slow phase with k2 = 0.046 min−1 (Table 1). Obviously, our results demonstrate that the partial loss of the TrmFO structure preceded the oligomerization process suggesting that the formation of at least one or more partially unfolded intermediates with a non-native structure self-assembled to give stable oligomers. | ||
| Fig. 3 Partial denaturation of the secondary structure of TrmFO in 10 mM sodium phosphate, pH 8. (A) The Far-UV spectrum of Y346A TrmFO recorded between 190–250 nm at different temperatures ranging from 20 °C (bold blue) to 80 °C (light bold blue). (B) Normalized kinetics of temperature-induced partial loss of the secondary structure of Y346A TrmFO recorded by CD at 222 nm and at 50 °C (in Blue). Temperature-induced light scattering kinetics of Y346A TrmFO followed by absorbance at 260 nm and at 50 °C. | ||
| k 1, min−1 | k 2, min−1 | |
|---|---|---|
| TrmFO full length | ||
| 10 mM NaP8 | 1.26 ± 0.2 | 0.05 ± 0.006 |
| 10 mM NaP8 + 0.2 M NaCl | 1.4 ± 0.15 (↑) | 0.1 ± 0.01 (↓) |
| 10 mM NaP8 + tRNA | — | — |
| 10 mM NaP8 + tRNA-RNAseA | 0.8 ± 0.09 | 0.02 ± 0.002 |
| Δ1–59 TrmFO | ||
| 10 mM NaP8 | 0.7 ± 0.06 (↑) | 0.01 ± 0.001 (↓) |
| 10 mM NaP8 + 0.2 M NaCl | 0.6 ± 0.08 | 0.005 ± 0.0001 |
| 10 mM NaP8 + tRNA | 0.84 ± 0.03 (↑) | 0.21 ± 0.015 (↓) |
| 10 mM NaP8 + tRNA-RNAseA | 0.22 ± 0.07 (↑) | 0.026 ± 0.0005 (↓) |
Characterization of TrmFO soluble oligomers
To confirm and characterize the oligomerization process of TrmFO, the thermal-induced partial unfolding reaction of wild type and Y346A TrmFO was followed by dynamic light scattering (DLS). As shown in Fig. 4A, the normalized scattered intensity I/I0 increased with temperature, which was indicative of an oligomerization process for both the holo- and apoproteins. The sigmoidal shape of the signal reached a stable plateau at T > 38 °C with a mid-temperature transition comparable to that determined by circular dichroism. Thus, partial unfolding of both holo- and apo-TrmFO led to oligomer formation. Quantitative information on the hydrodynamic molecular sizes of the oligomers was obtained from the size distribution diagram analysis. At 20 °C, the folded wild type and Y346A proteins adopt a globular shape with a Rh value of 3.08 ± 0.1 nm similar to that calculated from the crystal structure of Thermus thermophilus TrmFO (PDB: 3G5Q, Rh = 3.0 nm) (Fig. 4B).22 As the thermal denaturation of the protein proceeded, the initial folded ensemble shifted towards larger species characterized by a stable Rh value of ∼46 nm for the wild type and 23 nm for Y346A at a protein concentration of 0.2 mg mL−1 (Fig. 4B). These oligomers were thermally stable and did not disassemble at high temperatures confirming that these species acted as a kinetic trap during the partial unfolding process. | ||
| Fig. 4 Characterization of TrmFO oligomerization by dynamic light scattering (DLS). (A) Normalized scattered intensity I/I0 measured by DLS for wild type and Y346A TrmFO (0.2 mg mL−1) in black solid circles and open circles, respectively. (B) Intensity size distribution derived from the NNLS analysis of the sample at 20, 36 and 48 °C for the wild type (bottom box) and Y346A (upper box) TrmFO. | ||
The oligomerization of TrmFO was further analyzed by size exclusion column (SEC) and SEC-MALLS (Fig. 5 and Fig. S1, ESI†). The folded wild type TrmFO and mutants eluted at ∼14.8 mL (Ve) on the SEC. Analysis of MALLS gave Mw ∼ 47.2 kDa and Rh ∼ 2.7 nm consistent with the presence of monomers in solution (Fig. S1, ESI†). Following heat treatment, the wild type, C53A, Y346A and Y346F proteins were all converted to oligomers that were eluted at Ve ∼ 8.1 mL (Fig. 5). In the case of the Y346A mutant, we confirmed by MALLS the superstructure of the oligomers exhibiting an Rh ∼ 22 nm (Fig. S1, ESI†) consistent with that determined by DLS. The SEC profiles recorded at 450 nm, a wavelength specific to FAD, demonstrated that the oligomers of wild type and C53A holoproteins did not contain the flavin coenzyme, which has most likely been released from TrmFO during the heat treatment.
 | ||
| Fig. 5 Size exclusion profile of folded and heated TrmFO. Analysis was carried on a Superdex 200 increase 10/300 GL in 10 mM sodium phosphate, pH 8, 100 mM NaCl (red curve) and after heating up to 70 °C in 10 mM sodium phosphate pH 8 (black curve). The units are reported in a.u. × 10−3. The respective molecular weights and hydrodynamic radius (Rh) of TrmFO determined by SEC-MALLS are shown in Fig. S1 (ESI†). | ||
The catalytic cysteine 226 was proposed to form a covalent complex with the tRNA substrate during catalysis.20 In order to eliminate the possibility that formation of a disulfide bond between the reactive cysteines may be at the origin of the oligomerization process, we tested that whether the C226A single mutant and the C53A/C226A double mutant could form oligomers when they were heated in 10 mM sodium phosphate pH 8. As shown in Fig. 5, the cysteine mutants also formed soluble oligomers. Hence, TrmFO's ability to self-assemble into these aberrant superstructures is neither a property of the apoprotein nor caused by any mutation tested but it is indeed an intrinsic feature of the polypeptide.
Finally, we investigated whether these oligomers retained the capability of binding tRNA. As illustrated by the gel shift assay (Fig. S2, ESI†), the oligomers did not bind tRNA suggesting that TrmFO is inactive under such an oligomeric state.
From now on, we will focus mainly on the study of the Y346A protein, which we will hereafter call TrmFO.
Morphology of TrmFO oligomers
Negative staining electron microscopy was performed to further characterize the shape of the oligomers. The soluble oligomers obtained by heat treatment of TrmFO at 0.1 mg mL−1 appeared as small amyloid-like aggregates with a beaded morphology (Fig. 6A). In contrast, at 1 mg mL−1, the soluble oligomers were instead irregular particles with a diameter of ∼50 nm (Fig. 6B). To verify their amylogenic nature, we tested whether the soluble oligomers of TrmFO could alter the fluorescence signal of thioflavin and the absorbance spectrum of Congo red, both known to be specific probes for the detection of amyloid aggregates. Interestingly, thioflavin appeared to bind to TrmFO oligomers since the maximum fluorescence intensity increases by 18-fold as compared to the fluorescence intensity of the probe in the presence of folded TrmFO (Fig. 6D) suggesting that these oligomers may potentially carry a cross-β structure. Moreover, the same oligomers are able to bind the Congo red as evidenced by the red shift of its absorbance spectrum in their presence (Fig. 6E). The physico-chemical nature of the oligomers was further characterized by the 8-anilinonaphthalene-1-sulfonic acid (ANS) fluorescence, a classical probe for hydrophobicity screening. As shown in Fig. 6F, a large increase in fluorescence associated with an important blue-shift was observed when the probe was incubated with TrmFO's oligomers. The maximum passed from 525 nm for the free probe to 475 nm in the oligomeric TrmFO. Thus, the surface of the oligomers carries hydrophobic pockets that are accessible to the solvent.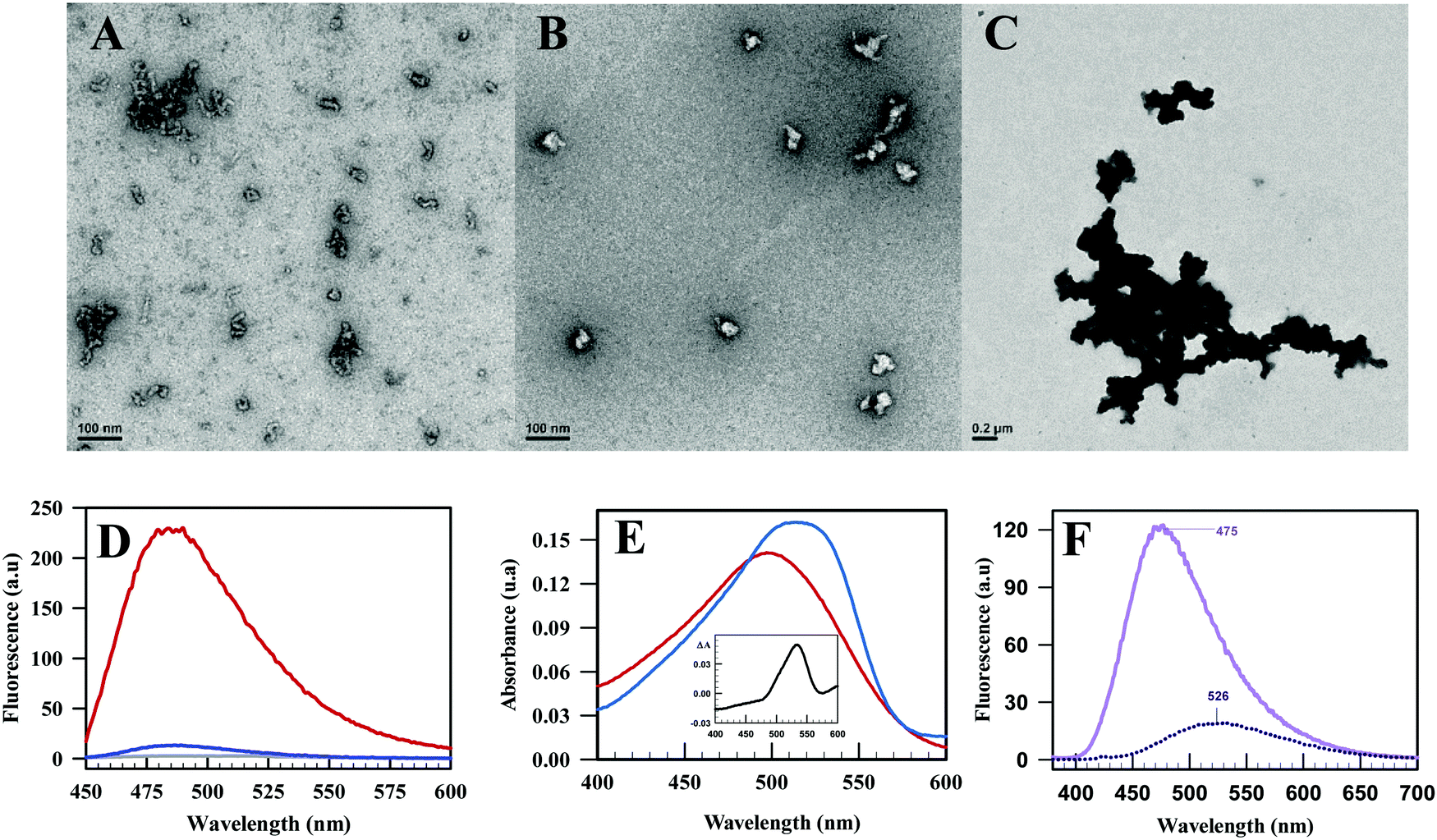 | ||
| Fig. 6 Negative staining electron microscopy of temperature-induced Y346A TrmFO oligomers in 10 mM sodium phosphate pH 8 with a protein concentration of 0.1 mg mL−1 (A), 1 mg mL−1 (B) and in the presence of 200 mM NaCl (C). (D) Fluorescence emission spectra of thioflavin alone (purple), in the presence of folded Y346A TrmFO (blue) and in the presence of soluble oligomers of Y346A TrmFO (red). (E) Absorbance spectra of Congo red alone (red) and in the presence of Y346 TrmFO oligomers (blue). The inset is the delta spectrum between the oligomers bound and free Congo red. (F) Emission fluorescence spectra of free ANS (blue dotted line) and bound to the soluble oligomers of Y346A TrmFO (light purple). | ||
Role of ionic strength in TrmFO aggregation
The apparent instability of TrmFO was seemingly incompatible with its capacity to efficiently methylate tRNAs at 37 °C.20,30 However, the activities were previously assayed with a buffer containing much higher ionic strength, which might have further stabilized the enzyme. Thus, we first examined the contribution of the activity buffer (50 mM HEPES, pH 7.5, 100 mM ammonium acetate) to the thermal stability of TrmFO wild type, C53A, Y346A and Y346F. The resulting thermal profiles of these proteins also showed a single cooperative transition during the course of thermal denaturation (Fig. 1C). Nonetheless, three major distinctive features characterized them. Firstly, this buffer drastically stabilized the folded state of both TrmFO and its mutants as demonstrated by the large increase of the T1/2 values (>12 °C for the four proteins, Fig. 1D). Secondly, a turbidity of the heated samples associated with the formation of aggregates was observed at the end of the denaturation reaction. Thirdly, the variation of the ellipticity signal between the native and the denatured states was much larger in the presence of the activity buffer (>2-fold increase) when compared to that in 10 mM sodium phosphate. This thermal aggregation is attributed to the ionic strength since the same behavior was observed when the experiments were repeated in 10 mM sodium phosphate pH 8 supplemented with various NaCl concentrations (Fig. S3A, ESI†) or in the presence of other salts such as Na2SO4 or KCl. Therefore, the thermal aggregation is not salt specific but it is rather promoted by the ionic strength. The far-UV CD spectrum of TrmFO was also recorded as a function of temperature in 0.2 M NaCl (Fig. S3B, ESI†). Interestingly, although we cannot exclude that some of the signal loss can be caused by light diffusion during the formation of heavy aggregates, it is nevertheless clear that NaCl enabled TrmFO to be gradually converted to an unfolded state different from that observed at low ionic strength by the temperature.To estimate the size of such aggregates, thermal denaturation of TrmFO was carried out in the presence of 0.2 M NaCl and followed by DLS. As expected, the reaction led to the formation of heavy aggregates with Rh in the order of micrometer (Fig. S4, ESI†). The heavy aggregates obtained by thermal-induced denaturation in the presence of 0.2 M NaCl were also analyzed by EM (Fig. 6C). Close inspection of the images revealed that the unfolded protein formed amorphous aggregates with various diameters that collapsed into large agglomerates of several microns.
We also verified whether the soluble oligomers obtained at low ionic strength could aggregate in the presence of NaCl. Interestingly, when the latter were heated in 0.2 M NaCl, no heavy precipitation was observed. Moreover, analysis of the resulting solution on gel filtration and DLS showed that the oligomers were converted into larger species of about 100 nm but remained much smaller than the heavy aggregates of TrmFO observed in the presence of salts. It can therefore be concluded that the salt-induced aggregates do not form from the soluble oligomers of partially unfolded TrmFO.
Structural characterization of the soluble oligomers of TrmFO
Mapping the structure of the partially unfolded TrmFO within soluble oligomers is crucial to understand the mechanism of protein oligomerization and aggregation. Mild proteolysis can be a powerful technique to evaluate the conformational flexibility of the polypeptide and thus to identify unfolded regions of the protein. Eventually, identification of the cleavage site by mass spectrometry might allow an atomic structural resolution of these regions. Hence, oligomer mild trypsinolysis kinetics were monitored and compared to those of the folded TrmFO control (Fig. S5, ESI†). SDS-PAGE analysis of the solution revealed that TrmFO was quite resistant and underwent, among the forty four potential proteolytic sites, only one at R58, which is a residue located on the active site flexible loop close to the FAD binding site. In contrast, the partially unfolded protein was rapidly hydrolyzed. As the proteolysis proceeded, the protein was converted to several lower mass fragments (at least 8 fragments). Fragments 2 and 3 were those that appeared immediately suggesting a high degree of exposure and/or flexibility. In contrast, fragment 8 was generated slowly but remained quite stable over time. All bands were submitted to trypsin proteolysis and analyzed by MALDI peptide mass fingerprinting (Fig. S6, ESI†). The MS results identified K30, K111, R181, K237 and K409 as being new cleavage sites (see the ESI†) and these residues are reported on the structural model of TrmFO (Fig. 7A). Thus these sites were initially buried in the folded structure and became solvent accessible in the partially unfolded state of TrmFO within the oligomers.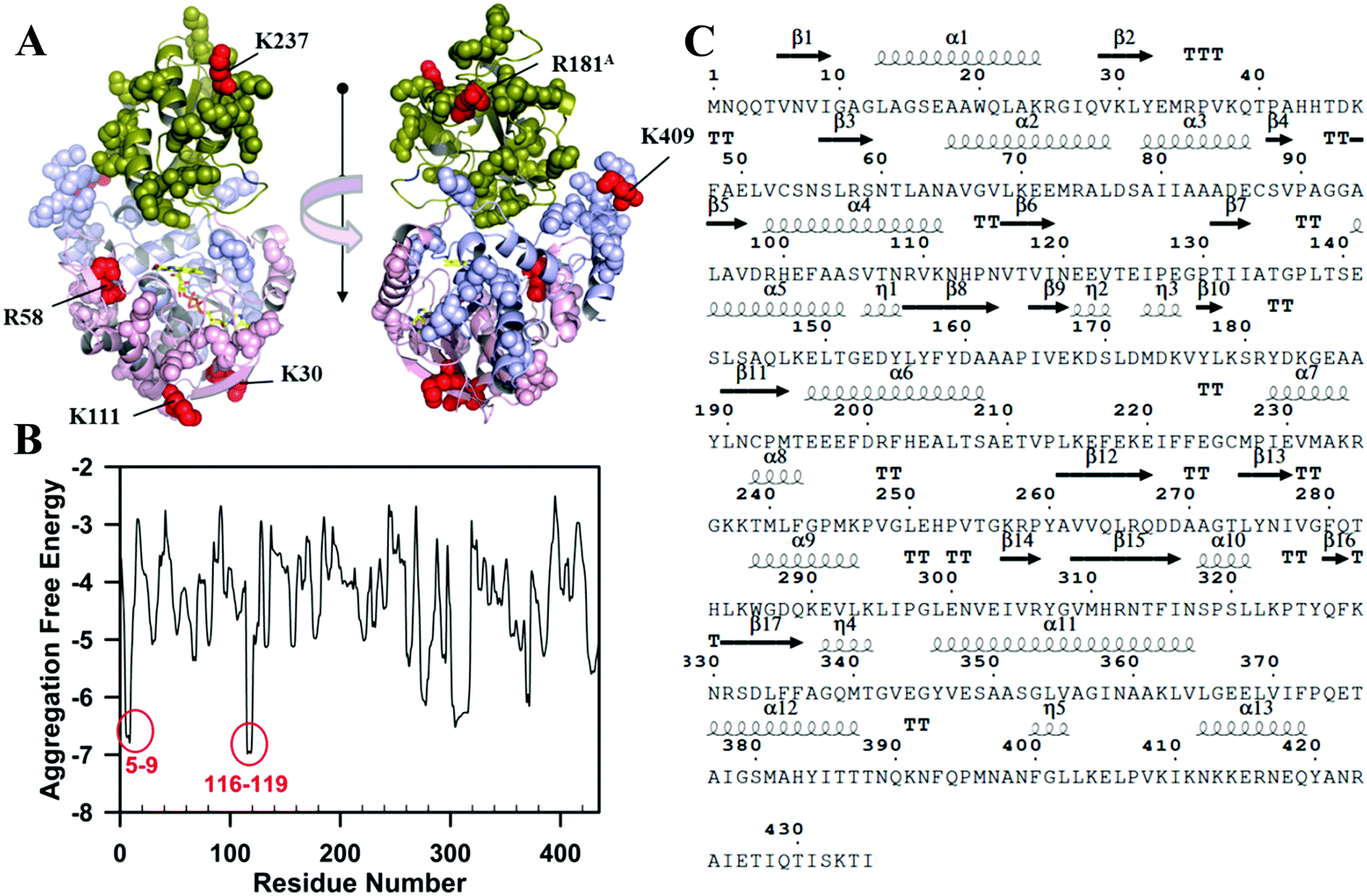 | ||
| Fig. 7 Identification of the regions of TrmFO that have undergone local unfolding during thermal denaturation. (A) Location of the residues accessible to the trypsin (shown as red spheres) are shown on the structural model of Bacillus subtilis TrmFO. (B) Predicted aggregation free energy profile of Y346A TrmFO showing the regions that are prone to aggregation. One energy unit is equivalent to 2 kBT at room temperature that is 1.192 kcal mol−1. For highly confident region detection, the top energy = 22 and energy cut-off <−2.8 were used to produce 90% specificity and 30% sensitivity. (C) Amino-acids primary sequence of TrmFO from Bacillus subtilis. | ||
Role of the N-terminal subdomain in oligomerization and aggregation reactions
To further analyze the aggregation-prone properties of TrmFO, prediction of protein regions carrying a high propensity to aggregate were made using the PASTA algorithm.31 As shown in Fig. 7B, the aggregation free energy profile incriminated several regions but two of them comprising residues 5–9 and 116–119 drew particular attention because (i) they belong to the largest parallel beta sheet of the protein (Fig. 7C), and these latter are known as driver structures in aggregation events, (ii) the N-terminal region of TrmFO was specifically identified as dynamic and solvent accessible in the partially unfolded state trapped under an oligomeric species (Fig. 7A). To evaluate experimentally the respective contribution of these regions in the oligomerization and aggregation reactions, a truncated version of TrmFO deleted from the first 59 residues (Δ1–59 TrmFO) was generated by mild trypsinolysis of the full length protein and then purified on a gel filtration. In this protein form, the 5–9 residues are expected to be removed while 116–119 remain. In the inset of Fig. 8A, the SDS-PAGE of the resulting Δ1–59 TrmFO showed an acceptable level of purity for further characterization. The SEC elution profile revealed Ve ∼ 15.07 mL slightly larger than that observed with the full length enzyme and thus consistent with a shorten version. The circular dichroism of this freshly prepared protein was that of a folded state indicating that the truncation did not perturb the rest of the protein (Fig. 8B). However, we observed that when Δ1–59 TrmFO was left 48 hours at 4 °C, its secondary structure was converted into a random coil while that of full length TrmFO remained unchanged. Thus this result demonstrates that the truncated protein is kinetically unstable and unfolds with time. Therefore, all the following experiments were performed on a freshly purified batch of protein. The intrinsic aggregation propensity of Δ1–59 TrmFO protein was assessed by measuring the light scattering kinetics at 50 °C in the absence or in the presence of 0.2 M NaCl (Fig. 8C and D). While the signal increase was biphasic in the full length form with a fast phase at 1.26 min−1 and a slow phase at 0.046 min−1, in the case of the truncated TrmFO, the increase of the light scattering was monophasic and occurred at ∼0.7 min−1 in the absence of salt (Fig. 8A and Table 1). However, the latter is distinguished by the fact that after reaching a maximum at 10 min, its signal decreased with k = 0.012 min−1. This signal drop suggested the formation of heavier oligomers. Consistently, electron microscopy showed that heated Δ1–59 TrmFO generated larger oligomers (∼100 nm diameters) characterized by a remarkable new regular and spheroidal shape (Fig. 8E). Furthermore, Δ1–59 TrmFO seemed to aggregate more slowly in the presence of NaCl (Fig. 8D and Table 1), according to a biphasic kinetics (A1 = 27%, k1 = 0.6 min−1 and A2 = 73%, k2 = 0.005 min−1) with no evidence for formation of heavy aggregate particles as those observed with the full length protein (Fig. 8F). Altogether, these results are consistent with the fact that the 1–59 residues (i) contain key elements for the heavy aggregation pathway and (ii) are responsible for the inhomogeneity of the oligomer morphologies. | ||
| Fig. 8 Characterization of the oligomerization process of Δ1–59 TrmFO. (A) Gel filtration profile of Δ1–59 TrmFO generated from mild trypsinolysis of Y346A TrmFO. The gel filtration was done on a Superdex 200 increase 10/300 GL in 10 mM sodium phosphate, pH 8, 100 mM NaCl. The inset is a SDS-PAGE showing the bands corresponding to full length Y346A TrmFO and to Δ1–59 TrmFO. (B) Far-UV CD spectra of full length Y346A TrmFO (red), Δ1–59 TrmFO (blue) and Δ1–59 TrmFO incubated at 4 °C for 48 hours (dark pink) (C) Kinetic of oligomerization of full length Y346A TrmFO (red) and Δ1–59 TrmFO (blue) in the absence of salt at 50 °C followed at 260 nm with a protein concentration of 0.2 mg mL−1. (D) Kinetic of oligomerization of full length Y346A TrmFO (red) and Δ1–59 TrmFO (blue) in the presence of 200 mM NaCl at 50 °C followed at 800 nm with a protein concentration of 0.2 mg mL−1. (E) and (F) are the negative staining electron microscopy of temperature-induced Δ1–59 TrmFO oligomers in 10 mM sodium phosphate pH 8 without NaCl and supplemented with 200 mM NaCl, respectively. | ||
Role of functional assembly in oligomerization and aggregation of TrmFO
In general ligand binding that stabilizes the native state can modify the free energy landscape of proteins and as a consequence inhibit aggregation events.32 First, NADPH and CH2THF ligands were tested but they both did not inhibit enzyme oligomerization (Fig. S7, ESI†). Then, we examined the effect of tRNA. Remarkably, when the TrmFO/tRNA complex was heated, SEC analysis showed the presence of exclusively the folded TrmFO and tRNA and confirmed the absence of oligomers (Fig. S8, ESI†). In addition, the absence of light scattering with the complex offered even more compelling evidence of the protective role of tRNA against oligomerization (Fig. 9A). This effect required a fully structured tRNA since oligomerization of TrmFO was not stopped by tRNA fragments obtained upon digestion by RNAse A. Moreover, this treatment allowed the recovery of an aggregation kinetics similar to that carried out in the absence of tRNA (Fig. 9A and Table 1).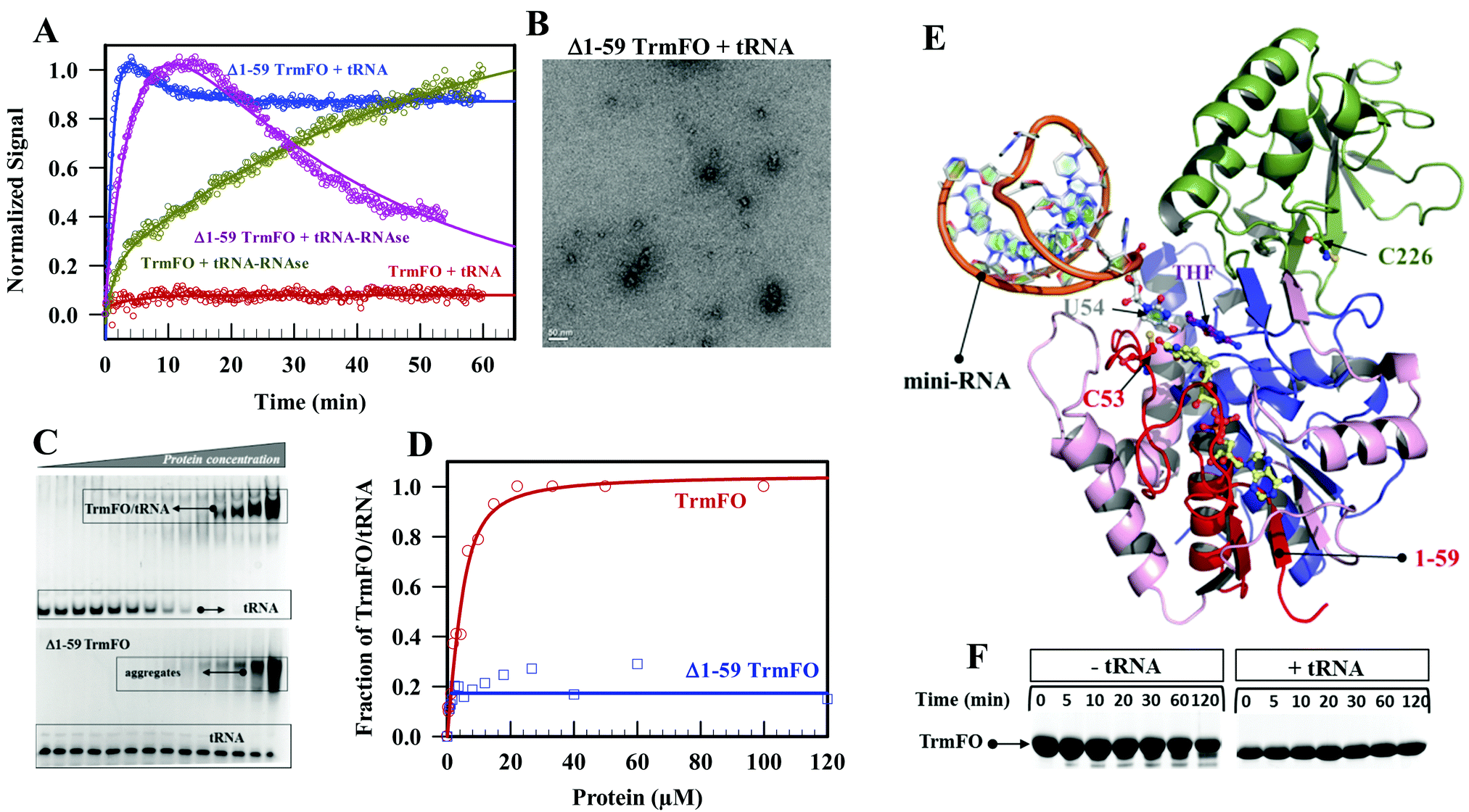 | ||
| Fig. 9 Protecting role of tRNA against TrmFO oligomerization. (A) Kinetics of TrmFO oligomerization in the absence and the presence of 1 eq. of bulk Bacillus subtilis tRNA or in the presence of pretreated tRNA with RNAse A. (B) Negative staining electron microscopy of Δ1–59 TrmFO heated with tRNA. (C) Electrophoretic mobility shift assay of tRNA in the presence of increasing concentration of Y346A TrmFO (upper box) or Δ1–59 TrmFO (lower box). (D) tRNA binding isotherm of Y346A TrmFO (red) and Δ1–59 TrmFO (blue). (E) Structural model of Bacillus subtilis TrmFO holoenzyme in complex with a miniRNA performed as described in ref. 22. The Nt and Ct FAD binding subdomain is shown in pink and purple, respectively, whereas the inserted domain is shown in green. The 1–59 first residues within the Nt-subdomain are red colored. The FAD is in yellow sticks. The model shows U54 in gray flipped-out from the T-loop of the miniRNA. (F) Effect of tRNA on the mild-trypsinolysis kinetics of Y346A TrmFO. | ||
We also tested whether the tRNA protected Δ1–59 TrmFO. While tRNA did not avoid oligomerization of TrmFO, it nonetheless reduced the size of the protein oligomers as demonstrated by the lack of the important sedimentation phase observed without tRNA (Fig. 9A and Table 1). This was fully supported by electron microscopy pictures showing that large spheroidal oligomers of Δ1–59 TrmFO were absent (Fig. 9B). This inhibition also required a fully structured tRNA since when tRNA was predigested by RNAse A, Δ1–59 TrmFO behaved as in the absence of tRNA. We then asked if such impediment resulted from a specific mechanism of action. Therefore, the interaction between the truncated protein and tRNA was measured. As shown by the gel shift assay and the resulting isotherms (Fig. 9C and D), while TrmFO binds tRNA, Δ1–59 TrmFO did not. Likewise, tRNA acted via a specific functional assembly in the case of full length TrmFO while for Δ1–59 TrmFO, a non-specific mechanism was implicated.
Discussion
Herein, we showed that TrmFO from B. subtilis carries a risky conformational energy landscape that can be converted into a safer formulation upon tRNA binding.Under low ionic strength, the folded state of the protein seemed to be easily perturbed at physiological temperatures (Fig. 1). Under these conditions, the folded protein was irreversibly converted to at least one partially unfolded intermediate that retained a substantial amount of native secondary structure of TrmFO (Fig. 1B, 2 and 3A). Regardless of whether this is an ON- or OFF-pathway intermediate, it is a reactive species that self-assembled into nonfunctional oligomers (Fig. 3B). This observed instability is neither a property of the apoprotein nor caused by the mutations tested since the wild type holoproteins formed oligomers when heated (Fig. 1 and 5).
Instead, it is an intrinsic feature of the polypeptide.
The biphasic nature of the oligomerization kinetics demonstrates that following the formation of this aggregation prone intermediate, a fast polymerization event occurred prior to slow assemblage into larger oligomers (Fig. 3B). The latter were stable and were characterized by a hydrodynamic radius varying between ∼20–30 nm depending on the protein concentration (Fig. 4). Interestingly, at 0.1 mg mL−1 of TrmFO, their morphology resembled that of curled proto-fibrils (Fig. 6A). In contrast, at 1 mg mL−1, the shape evolved and became more irregular and compact (Fig. 6B). This may indicate the existence of multiple competing pathways of assembly. The effect of the protein concentration on the heterogeneous morphology of aggregates was also observed with β2-microglobulin. It was shown that worm-like fibrils of this protein, which form rapidly during assembly, are kinetically trapped species, formed via a non-nucleated pathway that is explicitly distinct from that leading to the formation of the relatively rigid long-straight fibrils classically associated with amyloid.33 The amyloidic nature of TrmFO aberrant assemblies was supported by the large increase of thioflavin fluorescence produced in their presence as well as the binding of the Congo red (Fig. 6D and E). In addition, these oligomers present a low degree of hydrophobic packing that allows the binding of ANS (Fig. 6F) as for the case of the toxic oligomers of HypF-N, a small domain of E. coli HypF known for its ability to be easily destabilized in vitro and to form spherical oligomers, protofibrils and amyloid-like fibrils.10
Additional evidence for such a risky conformational energy landscape was given by the thermal denaturation reaction in the presence of salts (Fig. 1C). It is known that marginal changes in the solution conditions can severely impact the aggregation pathway. Accordingly, we showed that salts promoted another thermal-induced unfolding pathway of TrmFO leading to large aggregates (Fig. S5, S6 and Fig. 6C, ESI†). The same observation was recently made with the aggregation profile of human carbonic anhydrase II in the presence of salt.34 Indeed, the soluble oligomers obtained at low ionic strength cannot be converted into these large amorphous aggregates when heated in the presence of salts. Although the exact mechanism of NaCl-induced unfolding and the subsequent formation of large aggregate is enigmatic at this stage, it is nonetheless known that salt ions enhance intermolecular interactions by weakening repulsive electrostatic interactions via (i) a Debye–Hückel screening effect, (ii) by direct ion binding, (iii) by perturbation of the protein hydration shell or by a combination of several of these effects.
Our results suggest that the mechanisms of oligomerization and aggregation are somehow different. This is supported by the fact that the truncation of the first 59 residues of the protein: (i) abolished the ability of TrmFO to generate NaCl-induced large aggregates (Fig. 8D and F), (ii) promoted the formation of new homogeneous oligomers characterized by a larger diameter and with a regular spheroidal shape (Fig. 8C and E). In that respect, the 5–9 beta-strand predicted as an aggregation-prone structure may be important to the aggregation pathway. On the other hand, removal of this structure had probably favored unmasking of oligomerization-promoting regions such as the 116–119 residues being part of the long N-terminal beta strand of the FAD binding domain and that could participate in non-native beta–beta strand intermolecular interactions. Indeed, the fact that these oligomers induce a large increase of thioflavin fluorescence is indicative of the presence of a cross beta-structure characteristic of amyloid-type aggregates. Such a mechanism of oligomerization is not uncommon and has been postulated for several proteins. For instance, in the case of lysozyme, loosening of the C-helix favors exposure of the beta-strand of the beta-domain of the protein allowing the formation of spherical aggregates.35 The same mechanism has also been observed for several pro-aggregating globular proteins involved in several human pathologies.35 Structure determination by NMR of a low-populated on-pathway folding intermediate showed that the mutation-induced disorder in the carboxyl terminus exposes the aggregation-prone amino-terminal beta strand which by non-native interactions can derail folding and initiate fibrillation.36
Another important result of our study is the report of an unprecedented role played by the tRNA partner against oligomerization/aggregation of TrmFO. Likewise, the specific tRNA binding promoted a new stable conformation of TrmFO. Although, there is no structure of the TrmFO/tRNA complex to judge the nature of such a stabilizing conformational change, extensive mutagenesis and molecular modelling studies have delineated the RNA binding interface on the protein.22,37 We reproduced the identical TrmFO–miniRNA complex model obtained from the manual docking of a tRNA T-arm onto the TrmFO–THF structure of thermus thermophilus (Fig. 9E). In this docking model, the T-arm analog could be accommodated by the positively-charged, concave surface of TrmFO without serious steric hindrances. Interestingly, the N-terminal subdomain that we showed here as being implicated in the oligomerization/aggregation reactions of TrmFO constitutes a part of its tRNA binding site. Accordingly, trypsin-induced mild proteolysis showed that tRNA protects TrmFO from the single cleavage at position R59 (Fig. 9F). It is therefore conceivable that within the functional complex, the thermal fluctuation of this region is drastically reduced thereby minimizing eventually from misunfolding events at physiological temperatures. Partial deletion of this subdomain disturbed the tRNA binding and abolished the specific RNA protecting activity (Fig. 9C and D). However, by preventing the oligomerization of this truncated form to proceed toward the formation of larger oligomers (Fig. 9A and B), tRNA was somehow acting as an unspecific protecting agent wherein its protective role has been decoupled from the function. Moreover, this role required a structured RNA as demonstrated by the aggregation kinetics performed by tRNA digested with RNAse. Our results are in full agreement with the major chaperone activity of RNAs acting against aggregation of proteins that are not related to RNA metabolism.38,39 For instance, RNA was found 300-fold more effective at preventing luciferase or citrate synthase aggregation than the well-established GroEL chaperone system. Recently, structured RNAs have been shown as potential suppressors of p53 aggregation, a protein that does not bind RNA and is involved in many human cancers.40 Although the precise mechanism for such unspecific protection is unclear, the fact that RNA species can bind to unfolded proteins, as for instance in the ribosome, and assist them during the folding reaction suggest that hydrophobic interactions between the nucleic bases and the polypeptide play an important role in this unspecific chaperone activity.41,42 Several RNA species carry various modified bases that modulate the nucleotide hydrophobicity. Thus, it would be an outstanding issue to uncover the potential role of these modifications in their chaperone activity. Since RNA modifications are tightly regulated by the cellular metabolism,43 a potential RNA chaperone activity controlled by these modifications could guarantee to the cell an ingenious alternative way to regulate the instability of the proteome to particular stress. This intertwined notion of specific cellular fitness is again supported by the recent study uncovering that in contrast tRNA-modifying enzymes can also behave as RNA chaperones.44
Altogether, we herein demonstrated that large proteins carry a risky conformational energy landscape that can be modulated by the ionic strength and the physiological partners. Biophysical characterization together with computational prediction allowed us to propose the potential conformational events that control the oligomerization polymorphism and the aggregation pathways. In addition, our study revealed that the own tRNA substrate diverts conformational instability of the protein toward a safer energy landscape, which may afford an effective and economic cellular protection.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Centre National de la Recherche Scientifique, the university Pierre et Marie Curie as well as the French State Program ‘Investissements d’Avenir’ (ANR-15-CE11-0004-01 MIGRATE).Notes and references
- D. D. Boehr, R. Nussinov and P. E. Wright, Nat. Chem. Biol., 2009, 5, 789–796 CrossRef CAS PubMed
.
- H. Frauenfelder, S. G. Sligar and P. G. Wolynes, Science, 1991, 254, 1598–1603 CAS
- K. A. Dill and H. S. Chan, Nat. Struct. Biol., 1997, 4, 10–19 CrossRef CAS PubMed
- A. Gershenson, L. M. Gierasch, A. Pastore and S. E. Radford, Nat. Chem. Biol., 2014, 10, 884–891 CrossRef CAS PubMed
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed
- P. L. Clark, Trends Biochem. Sci., 2004, 29, 527–534 CrossRef CAS PubMed
- R. Wetzel, Cell, 1996, 86, 699–702 CrossRef CAS PubMed
- T. R. Jahn and S. E. Radford, Arch. Biochem. Biophys., 2008, 469, 100–117 CrossRef CAS PubMed
- V. N. Uversky and A. L. Fink, Biochim. Biophys. Acta, 2004, 1698, 131–153 CrossRef CAS PubMed
- S. Campioni, B. Mannini, M. Zampagni, A. Pensalfini, C. Parrini, E. Evangelisti, A. Relini, M. Stefani, C. M. Dobson, C. Cecchi and F. Chiti, Nat. Chem. Biol., 2010, 6, 140–147 CrossRef CAS PubMed
- M. Tanaka and Y. Komi, Nat. Chem. Biol., 2015, 11, 373–377 CrossRef CAS PubMed
- S. Saha and S. Deep, J. Phys. Chem. B, 2014, 118, 9155–9166 CrossRef CAS PubMed
- S. Saha and S. Deep, Phys. Chem. Chem. Phys., 2016, 18, 18934–18948 RSC
- H. W. He, J. Zhang, H. M. Zhou and Y. B. Yan, Biophys. J., 2005, 89, 2650–2658 CrossRef CAS PubMed
- Y. B. Yan, J. Zhang, H. W. He and H. M. Zhou, Biophys. J., 2006, 90, 2525–2533 CrossRef CAS PubMed
- J. H. Han, S. Batey, A. A. Nickson, S. A. Teichmann and J. Clarke, Nat. Rev. Mol. Cell Biol., 2007, 8, 319–330 CrossRef CAS PubMed
- D. Hamdane, H. Grosjean and M. Fontecave, J. Mol. Biol., 2016, 428, 4867–4881 CrossRef CAS PubMed
- M. Lombard and D. Hamdane, Arch. Biochem. Biophys., 2017 DOI:10.1016/j.abb.2017.06.011
- C. Bou-Nader, C. David, V. Guerineau, M. Fontecave and D. Hamdane, Angew. Chem., Int. Ed., 2017, 56, 12523–12527 CrossRef CAS PubMed
- D. Hamdane, M. Argentini, D. Cornu, B. Golinelli-Pimpaneau and M. Fontecave, J. Am. Chem. Soc., 2012, 134, 19739–19745 CrossRef CAS PubMed
- D. Hamdane, E. Bruch, S. Un, M. Field and M. Fontecave, Biochemistry, 2013, 52, 8949–8956 CrossRef CAS PubMed
- H. Nishimasu, R. Ishitani, K. Yamashita, C. Iwashita, A. Hirata, H. Hori and O. Nureki, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8180–8185 CrossRef CAS PubMed
- E. A. Shank, C. Cecconi, J. W. Dill, S. Marqusee and C. Bustamante, Nature, 2010, 465, 637–640 CrossRef CAS PubMed
- V. V. Giri Rao and S. Gosavi, PLoS Comput. Biol., 2014, 10, e1003938 Search PubMed
- D. Hamdane, C. Velours, D. Cornu, M. Nicaise, M. Lombard and M. Fontecave, Phys. Chem. Chem. Phys., 2016, 18, 20410–20421 RSC
- D. Hamdane, C. Bou-Nader, D. Cornu, G. Hui-Bon-Hoa and M. Fontecave, Biochemistry, 2015, 54, 4354–4364 CrossRef CAS PubMed
- M. Costas, D. Rodriguez-Larrea, L. De Maria, T. V. Borchert, A. Gomez-Puyou and J. M. Sanchez-Ruiz, J. Mol. Biol., 2009, 385, 924–937 CrossRef CAS PubMed
- M. Goyal, T. K. Chaudhuri and K. Kuwajima, PLoS One, 2014, 9, e115877 Search PubMed
- D. Hamdane, M. Argentini, D. Cornu, H. Myllykallio, S. Skouloubris, G. Hui-Bon-Hoa and B. Golinelli-Pimpaneau, J. Biol. Chem., 2011, 286, 36268–36280 CrossRef CAS PubMed
- D. Hamdane, V. Guerineau, S. Un and B. Golinelli-Pimpaneau, Biochemistry, 2011, 50, 5208–5219 CrossRef CAS PubMed
- I. Walsh, F. Seno, S. C. E. Tosatto and A. Trovato, Nucleic Acids Res., 2014, 42, W301–W307 CrossRef CAS PubMed
- S. M. Johnson, R. L. Wiseman, Y. Sekijima, N. S. Green, S. L. Adamski-Werner and J. W. Kelly, Acc. Chem. Res., 2005, 38, 911–921 CrossRef CAS PubMed
- W. S. Gosal, I. J. Morten, E. W. Hewitt, D. A. Smith, N. H. Thomson and S. E. Radford, J. Mol. Biol., 2005, 351, 850–864 CrossRef CAS PubMed
- P. Gupta and S. Deep, RSC Adv., 2015, 5, 95717–95726 RSC
- F. Chiti and C. M. Dobson, Nat. Chem. Biol., 2009, 5, 15–22 CrossRef CAS PubMed
- P. Neudecker, P. Robustelli, A. Cavalli, P. Walsh, P. Lundstrom, A. Zarrine-Afsar, S. Sharpe, M. Vendruscolo and L. E. Kay, Science, 2012, 336, 362–366 CrossRef CAS PubMed
- R. Yamagami, K. Yamashita, H. Nishimasu, C. Tomikawa, A. Ochi, C. Iwashita, A. Hirata, R. Ishitani, O. Nureki and H. Hori, J. Biol. Chem., 2012, 287, 42480–42494 CrossRef CAS PubMed
- S. Horowitz and J. C. A. Bardwell, RNA Biol., 2016, 1–4 Search PubMed
- B. E. Docter, S. Horowitz, M. J. Gray, U. Jakob and J. C. A. Bardwell, Nucleic Acids Res., 2016, 44, 4835–4845 CrossRef CAS PubMed
- P. S. Kovachev, D. Banerjee, L. P. Rangel, J. Eriksson, M. M. Pedrote, M. Martins-Dinis, K. Edwards, Y. Cordeiro, J. L. Silva and S. Sanyal, J. Biol. Chem., 2017, 292, 9345–9357 CrossRef CAS PubMed
- C. M. Kaiser, D. H. Goldman, J. D. Chodera, I. Tinoco, Jr. and C. Bustamante, Science, 2011, 334, 1723–1727 CrossRef CAS PubMed
- S. I. Choi, K. Ryu and B. L. Seong, RNA Biol., 2009, 6, 21–24 CrossRef CAS PubMed
- M. Helm and J. D. Alfonzo, Chem. Biol., 2014, 21, 174–185 CrossRef CAS PubMed
- L. C. Keffer-Wilkes, G. R. Veerareddygari and U. Kothe, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14306–14311 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: SEC-MALLS, thermal denaturation in the presence of NaCl followed by CD and DLS, mild proteolysis coupled to LC-MS analysis, gel filtrations furnished. See DOI: 10.1039/c7cp05599d |
| This journal is © the Owner Societies 2017 |