
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen activation and catalytic aerobic oxidation by Mo(IV)/(VI) complexes with functionalized iminophenolate ligands†
Niklas
Zwettler
,
Martina E.
Judmaier
,
Lara
Strohmeier
,
Ferdinand
Belaj
and
Nadia C.
Mösch-Zanetti
*
Institute of Chemistry, Inorganic Chemistry, University of Graz, Schubertstrasse 1, 8010 Graz, Austria. E-mail: nadia.moesch@uni-graz.at
First published on 27th June 2016
Abstract
Synthesis of molybdenum(VI) dioxido complexes 1–3, coordinated by one or two functionalized iminophenolate ligands HL1 or HL2, bearing a donor atom side chain or a phenyl substituent, respectively, allowed for systematic investigation of the oxygen atom transfer (OAT) reactivity of such complexes towards phosphanes. Depending on stoichiometry and employed phosphane (PMe3 or PPh3), different molybdenum(IV) and molybdenum(V) complexes 4–7 were obtained. Whereas molybdenum(IV) complexes 4 and 5, bearing a terminal PMe3 ligand, readily reacted with molecular O2 to form oxido peroxido complexes 8 and 9, phosphane free μ-oxido bridged dinuclear molybdenum(V) complexes 6 and 7 proved to be stable towards oxidation with molecular O2 under ambient conditions. Single-crystal X-ray diffraction analyses revealed different isomeric structures in the solid state for dioxido complexes 1 and 2 in comparison with oxido phosphane complex 5, dinuclear oxido μ-oxido complex 6 and oxido peroxido complexes 8 and 9, pointing towards an isomeric rearrangement during OAT. Compounds 1 and 2 were furthermore tested for their ability to catalyze the aerobic oxidation of PMe3 and PPh3. A significant difference in catalytic activity has been observed in the oxidation of PMe3, where complex 1 bearing donor atom functionalized ligands led to higher conversion and selectivity than complex 2 coordinated by phenyl iminophenolate ligands. In the oxidation of PPh3, complex 2 leads to higher conversion compared to 1. In a control experiment, phenyl-based dinuclear μ-oxido complex 7, derived from complex 2, was found to be catalytically active, which suggests a lower energy barrier for disproportionation into [MoO(L)2] and [MoO2(L)2] in comparison with methoxypropylene based compound 6, a prerequisite for subsequent reactivity toward molecular O2.
Introduction
The chemistry of molybdenum, as an abundant and biologically relevant transition metal, has been well established over the course of the past few decades. Especially in biomimetic chemistry, much effort has been made to elucidate and mimic the structure and function of various molybdoenzymes.1 An important class among these enzymes is represented by molybdenum oxotransferases such as DMSO reductase or xanthine oxidase, which contain a molybdenum(VI) metal center coordinated by an oxido ligand, as well as depending on the enzyme, one or two molybdopterin ligands.2 These oxotransferases catalyze oxygen atom transfer reactions via a molybdenum(IV)/molybdenum(VI) redox cycle.3 Whereas early modelling approaches for the corresponding active sites focused on sulfur rich dithiolene ligands, several other structurally diverse ligand systems have been explored.3,4 The ease of such molybdenum compounds to undergo oxygen atom transfer made them also interesting in industrial applications. Especially for the catalytic oxidation of alkenes to epoxides, several highly efficient systems exist nowadays.5In general, these catalysts use H2O2 or organic peroxides such as tert-butyl hydroperoxide (TBHP) as terminal oxidants. In our group, Mo(VI) dioxido systems were investigated for their applicability in oxygen atom transfer (OAT) reactions as well as epoxidation catalysis.6–8,9 In that course, iminophenolate based complexes incorporating different functionalities were developed. While amides as internal hydrogen bond donors led to unprecedented C–C and C–N coupling behavior upon coordination,10 the introduction of donor atoms (ether or amine) led to Mo(VI) catalysts for highly selective oxidation of a broad scope of alkene substrates with TBHP.7
Nevertheless, one of the major drawbacks of to date reported catalysts is the necessity of a terminal oxidant. To develop more sustainable catalytic processes it is of great interest to allow for the use of molecular oxygen and in further consequence air as a benign alternative to other oxidants. Not only is oxygen abundant, cheap and environmentally harmless, its use also reduces the formation of undesired side products.11 Whereas the activation of molecular oxygen has been described and thoroughly investigated for a variety of transition metals (e.g. iron, manganese),12 the number of molybdenum complexes that activate molecular oxygen is very limited. Only a few examples with full structural characterization have been disclosed,13,14 among them two examples are from our group.15,16 In general there are only a few examples of homogeneously catalyzed aerobic oxidation reactions using a Mo catalyst and these are severely hampered by disadvantages such as high catalyst loadings or the need of a co-catalyst.17
Since the number of molybdenum oxido peroxido compounds originating from oxygen activation is scarce, it is important to gain further insight into the electronic and structural parameters affecting the applicability of Mo(IV)/Mo(VI) systems for oxygen atom transfer as well as oxygen activation. Our group has thus an ongoing research interest in the OAT and oxygen activation reactivity of Mo(IV)/Mo(VI) systems based on different iminophenolates, with special emphasis on the identification and unequivocal structural elucidation of all involved species to be able to predict and modify their reactivity. In further consequence this is beneficial for the development of systems for catalytic aerobic oxidation reactions.
Herein we present two new molybdenum(IV)/(VI) based systems coordinated by functionalized iminophenolate ligands. The chosen substituents are a methoxypropylene as well as a phenyl group. Whereas the donor functionality potentially acts as a “protection group” for vacant coordination sites, the phenyl group was chosen as an electron withdrawing substituent with steric demand, allowing for the comparison with a previously published system with electron donating tert-butyl substituents and thus electronic influences.16 Two new molybdenum(VI) dioxido complexes employing these ligands were synthesized and investigated for their behavior in OAT reactions with the tertiary phosphanes PMe3 and PPh3 and subsequent reactivity towards molecular oxygen. Structural characterization via single-crystal X-ray diffraction analyses allowed assessing the isomeric structures of the participating species. Furthermore, catalytic oxidation of the phosphanes by molecular oxygen revealed significant reactivity differences depending on phosphanes and catalysts employed. Both investigated systems surpassed the PMe3 conversion obtained using a previously published system.16
Results and discussion
Ligand synthesis
Previously published ligands HL1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 and HL219 were prepared using well established synthetic protocols. Briefly, 1.1 equiv. of a primary amine bearing the desired functionality were added to a methanolic solution of 3,5-di-tert-butyl-2-hydroxy-benzaldehyde and stirred overnight at reflux temperature. Whereas HL1 was obtained after evaporation of the solvent as a yellow oil, HL2 was recovered as an orange crystalline solid after filtration (Scheme 1). Their analytic data is in agreement with the literature.18,19
18 and HL219 were prepared using well established synthetic protocols. Briefly, 1.1 equiv. of a primary amine bearing the desired functionality were added to a methanolic solution of 3,5-di-tert-butyl-2-hydroxy-benzaldehyde and stirred overnight at reflux temperature. Whereas HL1 was obtained after evaporation of the solvent as a yellow oil, HL2 was recovered as an orange crystalline solid after filtration (Scheme 1). Their analytic data is in agreement with the literature.18,19
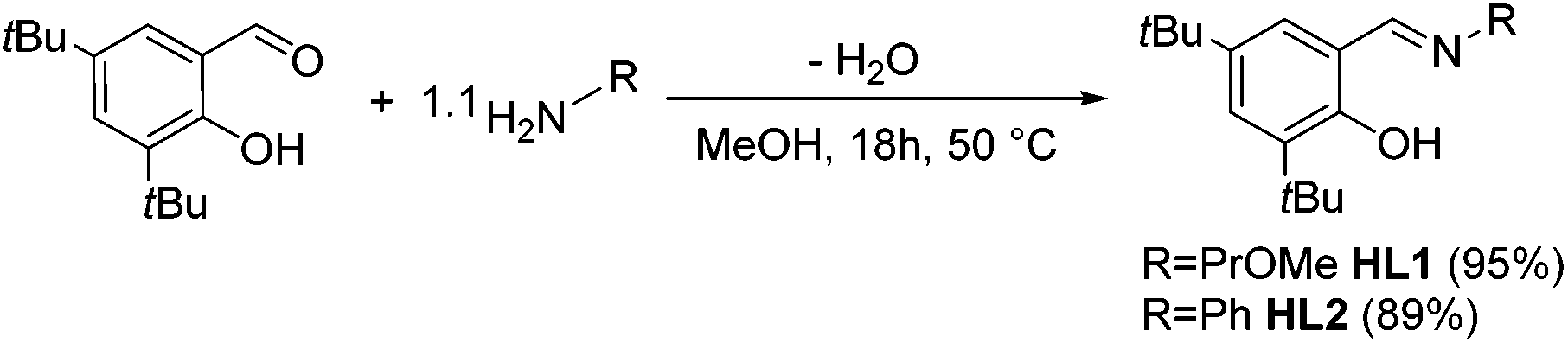 | ||
| Scheme 1 Synthesis of the iminophenolate ligands HL1 and HL2. | ||
Synthesis of molybdenum dioxido complexes
For the synthesis of molybdenum(VI) complexes of the type [MoO2(L)2], synthetic procedures using [MoO2(acac)2],20 [MoO2Cl2] and [MoO2Cl2(DME)]21 (DME = dimethoxyethane) as metal sources were investigated.A reaction of two equiv. of HL1 and HL2, respectively, with the metal precursors [MoO2Cl2(DME)] and [MoO2Cl2] in the presence of excess NEt3 in toluene, led to the targeted [MoO2(L)2] complexes 1 and 2 in good yields (Scheme 2).
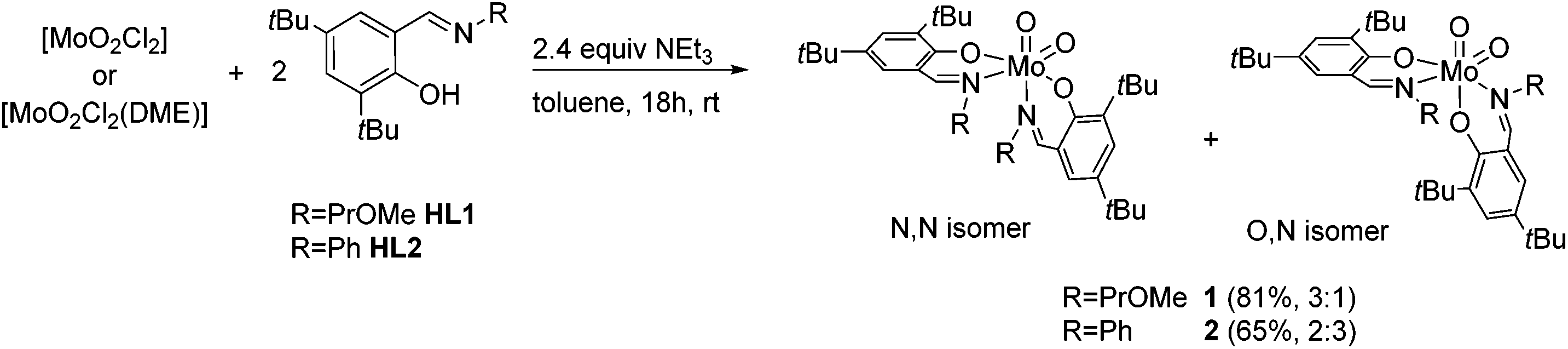 | ||
| Scheme 2 Synthesis of molybdenum(VI) dioxido complexes 1 and 2 and their respective isomeric ratios (N,N:O,N). | ||
Generally, there are three possible isomers for molybdenum(VI) compounds with a cis dioxido arrangement coordinated by two iminophenolate ligands as depicted in Fig. 1. For complexes 1 and 2, 1H NMR data in C6D6 are consistent with the existence of two isomers in solution. One set of ligand resonances corresponds to isomer A as confirmed by single-crystal X-ray diffraction analysis. In principle also the O,O isomer B would feature one set of resonances but this would require a rotation of both ligands upon crystallization which seems highly unlikely. The non-equivalent ligand arrangement of the second isomer C in solution gives rise to two additional sets of ligand resonances. The ratio of the two isomeric forms is strongly dependent on the imine substituent. Whereas in the compound bearing the methoxypropylene-substituted ligands, the N,N form is predominant with a ratio of 3:1, in the complex with the phenyl-substituted ligand, the major isomer corresponds to the O,N form with a ratio of 2:3 (Scheme 2). In compound 1, the signals for the minor isomer are considerably broadened, as previously observed,7 whereas no significant broadening is observed in the 1H NMR spectrum of 2 at room temperature.
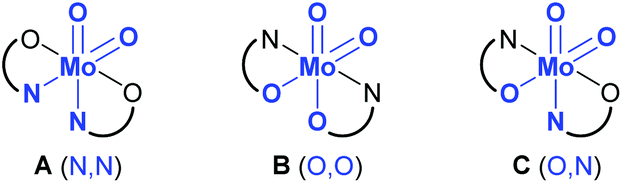 | ||
| Fig. 1 Possible isomers A–C for a cis molybdenum(VI) dioxido complex coordinated by two iminophenolate ligands, and atoms trans to oxido groups in brackets. | ||
Dissolution of single crystals of 2 and immediate recording of 1H NMR spectra led to the same isomeric ratio in several attempts, suggesting a dynamic equilibrium in solution. Subsequent variable temperature 1H NMR spectroscopy of complex 2 in C6D6 (Fig. S6†) showed a significant broadening of the O,N isomeric resonances upon heating accompanied by a slight change in the isomeric ratio (N,N:O,N approx. 2:3 at 9 °C to 5:6 at 50 °C). Similar results have been reported previously for a complex closely related to 1 (R = methoxyethylene).7 A more pronounced effect can be observed in the 1H NMR spectra of single crystals of 2 in different solvents (CD3CN, CD2Cl2, Fig. S7†), where the isomeric N,N:O,N ratios are 1:1.2 (CD2Cl2) and 1.7:1 (CD3CN), respectively, as recently observed for Re(V) iminophenolate complexes,22 further corroborating a dynamic equilibrium in solution.
Upon using [MoO2(acac)2] as a precursor, compound 1 was obtained in significant lower yield and compound 2 did not form. However, the monosubstituted complex [MoO2(acac)(L2)] (3) could be isolated in 34% yield which could not be improved with excess ligand (Scheme 3). NMR spectroscopy clearly features resonances for one iminophenolate and one acetylacetonate ligand.
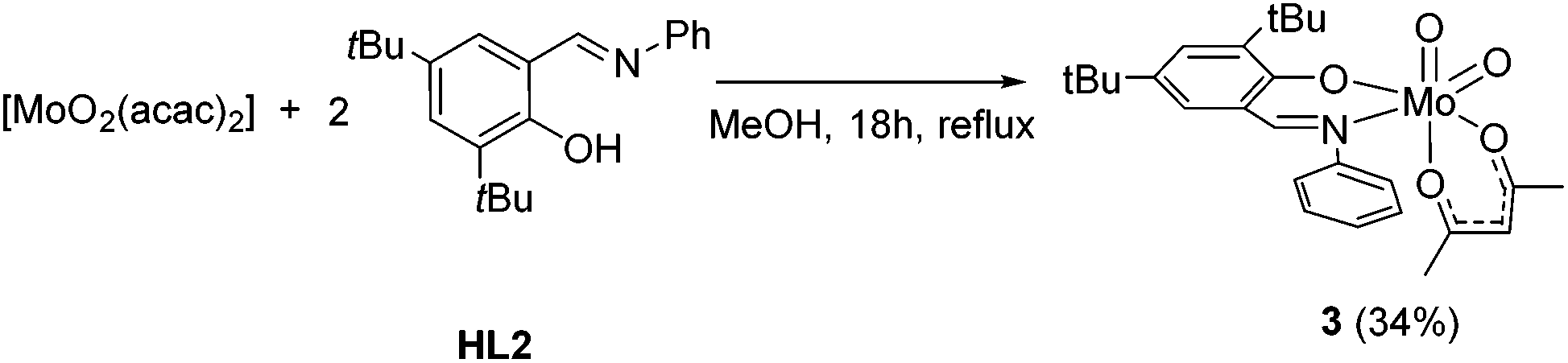 | ||
| Scheme 3 Synthesis of monosubstituted complex 3. | ||
The moisture sensitive complexes 1–3 are well soluble in most organic solvents including aliphatic hydrocarbons. Spectroscopic data (1H, 13C NMR and FT-IR spectroscopy), elemental analyses as well as single-crystal X-ray diffraction analyses confirm their structures.
OAT reactivity of complexes 1–3
The reaction of Mo(VI) dioxido compounds 1 or 2 with excess PMe3 (5 equiv.) led to phosphane coordinated Mo(IV) oxido compounds 4 and 5 in very good yields (Scheme 4). Similar to the 1H NMR resonances for 1 and 2, also for these oxido phosphane compounds two distinct species are observed in solution, reflected by two sets of resonances for two non-equivalent arranged ligands each. For both isomers, the coordinated PMe3 molecule gives rise to a distinct resonance in the decoupled 31P NMR spectra of 4 and 5.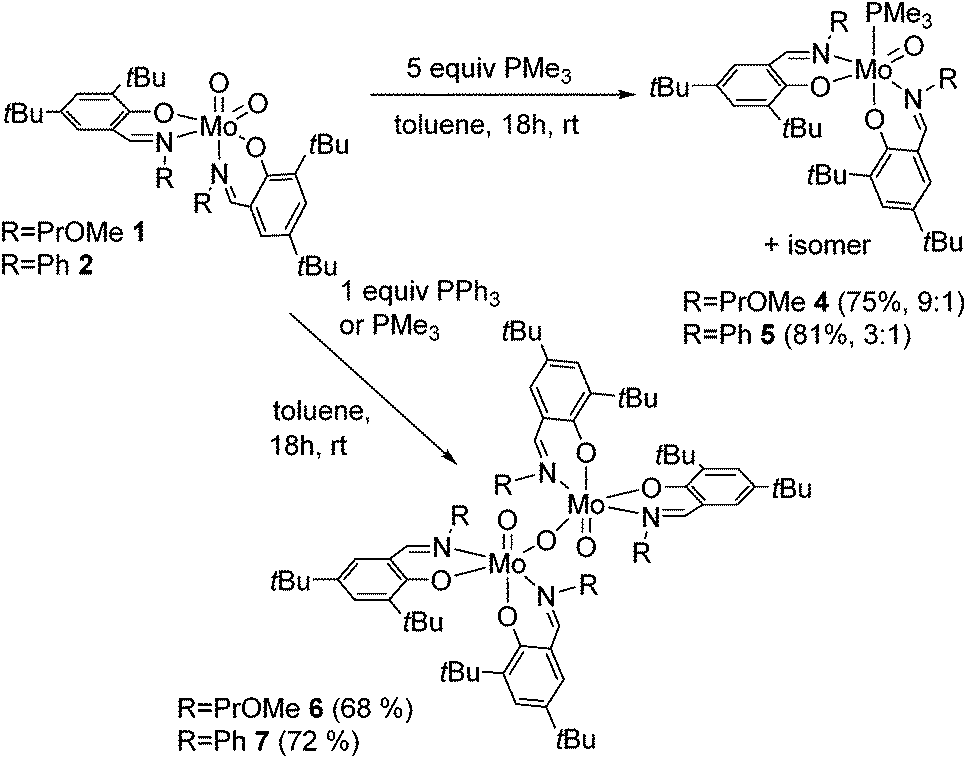 | ||
| Scheme 4 OAT reactions of complexes 1 and 2 with PMe3 and PPh3 with isomeric ratios for complexes 4 and 5. For clarity, only one possible isomer is depicted for all complexes. | ||
The isomeric ratios for the oxido phosphane complexes are found to be 9:1 (4) and 3:1 (5), thus revealing a significant shift in the isomeric ratio for 4 in comparison with 1 (ratio 3:1), whereas the isomeric ratio for 5 is in a similar range than for 2 (ratio 3:2). As all possible isomers exhibit non-equivalent ligand surroundings, routine NMR spectroscopy does not allow for the determination of specific isomers. However, we were able to obtain single-crystals suitable for X-ray diffraction analysis of 5, which revealed it to be the O,O isomeric form (type B, Fig. 1) depicted in Scheme 4.
This is in contrast to the solid state structures of 1 and 2 which displayed the N,N isomeric form (type A, Fig. 1) and points towards an isomeric rearrangement during the transformation of 2 to 5. The rearrangement is likely caused by higher steric demand of the phosphane ligand, which is corroborated by the exclusive O,O isomeric structure of a previously reported system featuring ligands with high steric demand (R = tBu).16 Dissolution of single crystals and immediate recording of a 1H NMR spectrum showed an isomerically pure compound and allowed for the identification of the major isomer in solution as the O,O isomeric form. Interestingly, a 1H NMR spectrum of the same sample after 24 h still showed only resonances corresponding to the O,O isomer, accompanied by traces of oxidized complex 9. This suggests that the two isomers found for 5 are not in a dynamic equilibrium or equilibrate only slowly. Whereas similar unambiguous evidence via the solid state structure cannot be provided for compound 4, we assume that it exhibits similar properties, especially due to the observation of the O,O structural motif in the molecular structure of both corresponding oxido peroxido compounds 8 and 9 (vide infra).
The reactions of dioxido compounds 1 and 2, respectively, with the more bulky PPh3 or with stoichiometric amounts of PMe3 led to species 6 and 7 in good yields (Scheme 4). Proton NMR spectroscopy reveals two sets of ligand resonances for 6 and 7 corresponding to a non-equivalent arrangement of the ligands, furthermore both compounds adopt a single isomer in solution. Neither 6 nor 7 shows any resonances in the 31P NMR spectra and both are thus phosphane free. Single-crystal X-ray diffraction analysis of 6 identified the compound as a μ-oxido bridged dinuclear oxidomolybdenum(V) complex as shown in Scheme 4, complex 7 being likely of a similar structure. The reaction of the monosubstituted complex 3 with PMe3 or PPh3, respectively, led to the formation of several unidentified products.
Complexes 4 and 5 are very well and complexes 6 and 7 are well soluble in organic solvents including aliphatic hydrocarbons. Whereas 4 and 5 are highly sensitive towards air and moisture, 6 and 7 are stable towards air and traces of moisture. In solution, 4 and 5 tend to decompose within hours (4) to days (5). The compounds were characterized via1H, 13C, 31P NMR and FT-IR spectroscopy as well as elemental analyses, confirming their structures. Complexes 5 and 6 have additionally been characterized via single-crystal X-ray diffraction analysis (vide infra).
Activation of molecular dioxygen
Complexes 4 and 5 cleanly reacted with molecular dioxygen, indicated by a quick color change from red-brown to orange-red, to form oxido peroxido compounds 8 and 9 in excellent yields (Scheme 5). Compound 8 is alternatively accessible in a one-pot reaction from 1via the addition of excess PMe3 (1 M solution in toluene) and subsequent stirring overnight under an O2 atmosphere, similar to a previously reported procedure.16 Compounds 6 and 7 were virtually non-reactive toward molecular oxygen under ambient conditions.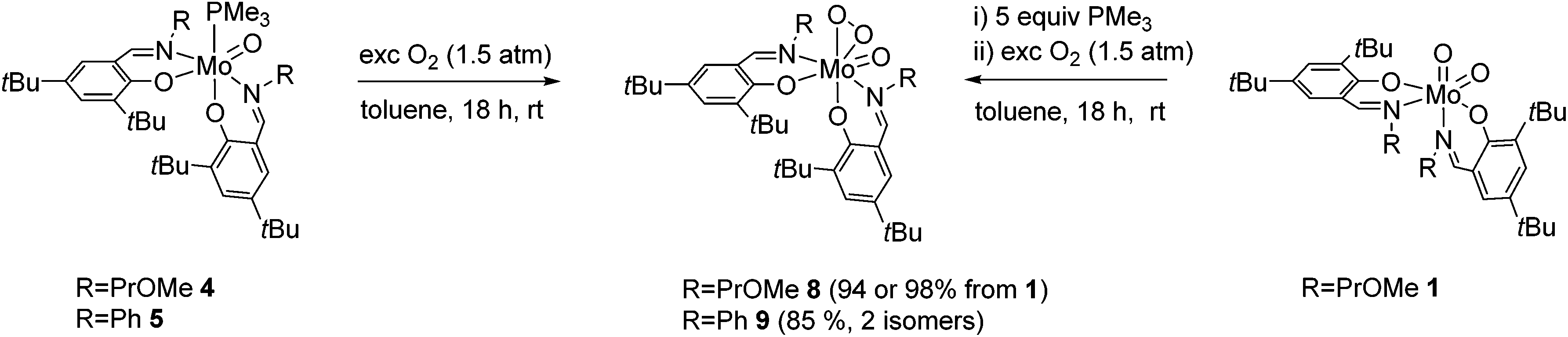 | ||
| Scheme 5 Activation of molecular oxygen to form oxido peroxido compounds 8 and 9. | ||
Exposure of 6 and 7 to molecular oxygen at 80 °C led to the formation of a mixture of dioxido and oxido peroxido complexes 1/8 and 2/9, respectively, accompanied by severe decomposition to free ligands and NMR inactive species. The reaction of 6 or 7 with O2 requires a preceding disproportionation reaction forming Mo(IV)O (and Mo(VI)O2), which is corroborated by the necessity of high temperatures and the resulting product mixture.
Whereas, in contrast to complexes 1, 2, 4 and 5, compound 8 exists as single isomer in solution, as evidenced by 1H and 13C NMR spectroscopy, complex 9 also exists as a mixture of two isomers in solution in an approximate ratio of 4:1.
Similar to the solid state structure of 5, both oxido peroxido compounds were identified as the O,O isomer (type B, Fig. 1) via single-crystal X-ray diffraction analysis. It is feasible to assign the O,O form to the structure of 8 also in solution because only a single isomer is observed. Dissolution of single crystals of 9 and immediate recording of a 1H NMR spectrum showed only resonances for one species, which we thus also assign to the O,O isomer. Interestingly, a 1H NMR spectrum of the same sample after 24 h revealed the initial isomeric ratio of 4:1 pointing towards a (slow) dynamic equilibrium as previously observed in molybdenum(IV) isocyanide complexes.23 Comparing the structure of the oxido peroxido compounds 8 and 9 with previously published cis oxido peroxido complexes originating from O2 activation, it is evident that the O,O isomeric form is the preferred conformation for such compounds in the solid state.6,15,16 Complexes 8 and 9 are well soluble in most organic solvents and soluble in aliphatic hydrocarbons. Both complexes are stable towards air but sensitive towards moisture.
Catalytic oxidation of phosphanes
Complexes 1 and 2 were tested for their ability to catalyze the aerobic oxidation of phosphanes, namely trimethyl phosphane and triphenyl phosphane, according to Scheme 6. The conditions used were 1 mol% catalyst with an excess of dry O2 gas (1.5 atm) in C6D6 at room temperature. Blank experiments under identical conditions but without catalysts led to conversions of PMe3 and PPh3 to <5%. In the aerobic oxidation of PMe3, catalyst 1 was found to be significantly more active compared to 2 (65 vs. 35% conversion, Table 1) and more active than the previously described tert-butyl based system (19%).16 Catalyst 2 proved to be unselective as only 25% of OPMe3 was formed together with 10% of one side product. NMR spectroscopy revealed it to be methyl dimethylphosphinate (OP(OMe)Me2) where an additional oxygen atom was inserted into one P–C bond (Fig. S37 and 38†). This is highly interesting as it demonstrates for the first time the transfer of two oxygen atoms from a molybdenum oxido peroxido species to one substrate molecule. | ||
| Scheme 6 Molybdenum(VI)-catalyzed aerobic oxidation of phosphanes, [Mo] = 1 or 2. | ||
In contrast, PPh3 was oxidized selectively to OPPh3 by 2, but only 23% was converted. Interestingly, catalyst 1 was virtually non-reactive for the oxidation of PPh3 (8%). To our further understanding of this reactivity difference, we subsequently tested the dimeric complexes 6 and 7 in the catalytic oxidation of PPh3. Phenyl-based compound 7 led to a fourfold higher yield of OPPh3 (12% vs. 3%) in comparison with methoxypropylene based complex 6. This is in good agreement with the activity difference of 1 and 2 in the aerobic oxidation of PPh3 and suggests lower activation energy for the disproportionation of 7, a requirement for reactivity with O2, in comparison with 6. The temperature increase in the catalytic oxidation of PPh3 was tested but due to significant autooxidation at 50 °C, as observed in a blank experiment, no reliable results could be obtained. To confirm the participation of the [MoO(O2)L2] species in the catalytic oxidation, we reacted complex 9 with 2 equiv. of PMe3 in a control experiment. Proton NMR measurement after 6 h of reaction time showed a mixture of 9 and the phosphane complex 5 together with OPMe3 and residual PMe3 but no dioxido species 2 (Fig. S45†). These results confirm the oxidation capability of complex 9 and are in good agreement with previous observations.16
Molecular structures
The molecular structures of molybdenum(VI) dioxido complexes 1–3, molybdenum(IV) oxido phosphane complex 5, molybdenum(V) oxido μ-oxido complex 6 as well as molybdenum(VI) oxido peroxido complexes 8 and 9 were determined by single-crystal X-ray diffraction analysis. The molecular views of 1–3 are given in Fig. 2, those of 5 and 6 in Fig. 3, and those of 8 and 9 in Fig. 4. Selected bond lengths and angles for complexes 1–3, 5, 6, 8 and 9 are provided in Table 2, and full crystallographic details such as structure refinement as well as experimental details are provided in the ESI.† | ||
| Fig. 2 Molecular views (50% probability level) of 1 (top), 2 (middle) and 3 (bottom); hydrogen atoms as well as solvent molecules are omitted for clarity. | ||
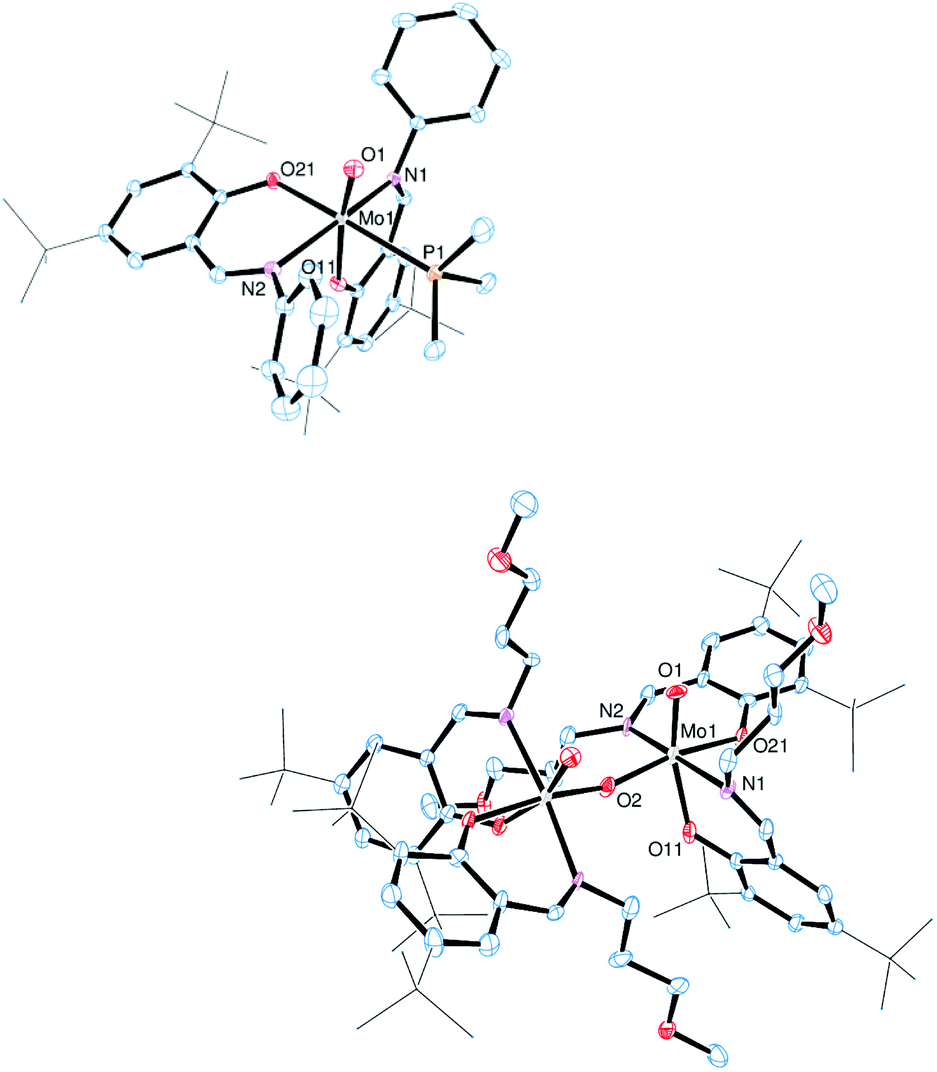 | ||
| Fig. 3 Molecular views (50% probability level) of 5 (top), and 6 (bottom); hydrogen atoms as well as solvent molecules are omitted for clarity, and tert-butyl substituents are depicted as a wireframe. For disordered fragments, only atoms with the higher site occupation factors are depicted. In complex 6, only atoms in the asymmetric unit are labelled. | ||
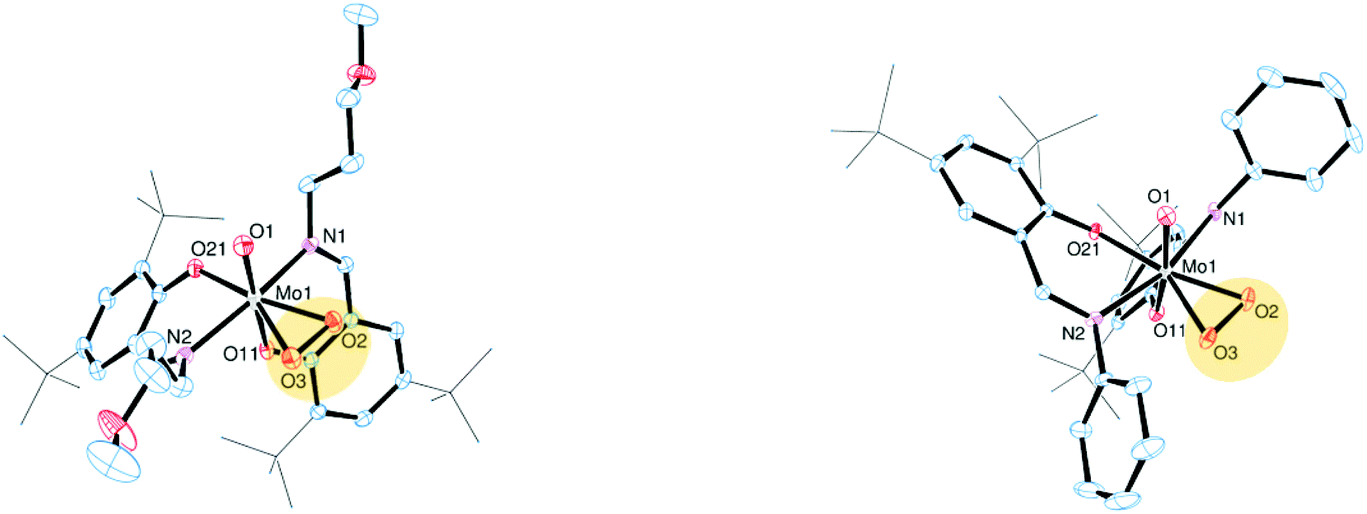 | ||
| Fig. 4 Molecular views (50% probability level) of 8 (left) and 9 (right); hydrogen atoms as well as solvent molecules are omitted for clarity, and tert-butyl substituents are depicted as a wireframe. For disordered fragments, only atoms with the higher site occupation factors are depicted. | ||
| 1 | 2 |
3a |
5 | 6 | 8 | 9 | |
|---|---|---|---|---|---|---|---|
| a Bond length and angle range given due to four distinct molecules in the unit cell. b O2 = μ-oxido. | |||||||
| Mo1–O1 | 1.7099(11) | 1.707(3) | 1.7049(13)–1.7097(13) | 1.7037(13) | 1.694(6) | 1.642(2) | 1.6928(13) |
| Mo1–O2 | 1.7099(11) | 1.706(3) | 1.7028(13)–1.7109(13) | — | 1.8883(18)b | 1.981(2) | 1.9581(13) |
| Mo1–O3 | — | — | — | — | — | 2.012(2) | 1.9301(13) |
| Mo1–O11 | 1.9729(11) | 1.946(3) | 1.9243(12)–1.9304(13) | 2.0660(14) | 2.054(6) | 2.0413(12) | 2.0166(12) |
| Mo1–O21 | 1.9729(11) | 1.946(2) | — | 2.0816(11) | 2.063(5) | 2.0262(12) | 2.0493(12) |
| Mo1–N1 | 2.3319(12) | 2.375(3) | 2.3974(15)–2.4021(15) | 2.1272(13) | 2.182(6) | 2.1810(15) | 2.2137(15) |
| Mo1–N2 | 2.3319(12) | 2.394(3) | — | 2.1769(13) | 2.155(6) | 2.1852(15) | 2.1654(15) |
| O2–O3 | — | — | — | — | — | 1.430(3) | 1.4425(18) |
| O1–Mo1–O2 | 107.82(8) | 105.98(13) | 104.17(6)–104.51(7) | — | 100.5(3) | 99.53(9) | 100.97(6) |
| O1–Mo1–O3 | — | — | — | — | — | 100.03(9) | 104.14(6) |
| O11–Mo1–O21 | 161.05(6) | 152.33(11) | — | 86.95(5) | 79.9(2) | 81.01(5) | 77.09(5) |
| N1–Mo1–N2 | 75.13(6) | 84.47(10) | — | 164.99(5) | 174.3(3) | 160.74(5) | 164.52(5) |
In complexes 1–3, the molybdenum atoms are coordinated in a distorted octahedral fashion by two bidentate ligands and two terminal oxido ligands. In complexes 1 and 2, which are coordinated by two iminophenolate ligands, the ligand arrangement corresponds to the N,N isomer (type A, Fig. 1) whereas in monosubstituted compound 3 one imine nitrogen as well as one acetylacetonate oxygen is trans to the oxido ligands. The molybdenum oxido bond lengths observed in 1–3 are within the expected ranges (Table 2).24
Compound 5, which is obtained via OAT from 2 to PMe3, is coordinated by two bidentate iminophenolate ligands, one terminal phosphane and one terminal oxido ligand in a distorted octahedral fashion. Interestingly, the arrangement of the iminophenolate ligands differs from the parent compound. In 5, the phenolate oxygens are trans to the phosphane and oxido ligands, respectively, which correspond to the O,O isomer (type B, Fig. 1). The Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length is 1.7037(13) Å and the Mo–P bond length is 2.5289(6) Å, thus both are rather long but comparable to previously structurally characterized molybdenum(IV) oxido phosphane complexes (Table 2).8,25
O bond length is 1.7037(13) Å and the Mo–P bond length is 2.5289(6) Å, thus both are rather long but comparable to previously structurally characterized molybdenum(IV) oxido phosphane complexes (Table 2).8,25
Compound 6, obtained via OAT from 1 to PPh3 displays a μ-oxido bridged dimeric structure with two molybdenum(V) oxido metal centers each coordinated by two bidentate ligands, a terminal oxido ligand as well as the μ-oxido ligand in a distorted octahedral fashion. The complex is symmetric around the bridging oxido ligand with a Mo1–O2–Mo1i angle of 157.2(5)° as well as twisted about the Mo1⋯·Mo1′ connecting line (O1–Mo1⋯Mo1′–O1′ −31.1(3)°, N1–Mo1⋯·Mo1′–N2′ −42.8(3)°), rendering the two terminal oxido ligands in a gauche-like conformation in the solid state. The arrangement of the ligands around the metal centers resemble the O,O isomer (Fig. 1) with the phenolate oxygens trans to the oxido and μ-oxido ligands, respectively. The MoO and Mo–(μ-O) bond lengths are within the expected ranges (Table 2).24,26
Compounds 8 and 9 are coordinated by two bidentate iminophenolate ligands, a terminal oxido ligand and a η2 side-on coordinated peroxido ligand in a distorted octahedral fashion.
Both compounds adopt the O,O isomeric form in the solid state, with the phenolate oxygen atoms trans to the oxido and peroxido ligands, respectively. Whereas the MoO bond lengths for 8 and 9 as well as the Mo–O (peroxido) bond lengths in 9 are similar to previously reported Mo(VI) oxido peroxido compounds, the Mo–O (peroxido) distances in 8 are elongated with 2.012(2) and 1.981(2) Å as well as 2.132(5) and 2.056(5) Å for two disordered arrangements, respectively.13,15,16
From comparison of the bond lengths of the phenyl iminophenolate derived complexes 2, 5, 6 and 9, it is evident that the different ligand arrangements in 2 (N,N isomer, type A, Fig. 1) as well as 5, 6 and 9 (O,O type arrangement, Fig. 1) have a pronounced impact on the Mo–N bond lengths which are 2.375(3) and 2.394(3) Å in 2, 2.1272(13) and 2.1769(13) Å in 5, 2.182(6) and 2.155(6) Å in 6, as well as 2.1654(15) and 2.2137(15) Å in 9. The solid state structure thus reveals an elongation of the Mo–N bond lengths in the N,N isomeric structure found in 2 which is likely caused by the trans influence of the oxido ligands. A comparison of the Mo–O (phenolate) bond lengths in these complexes shows a comparable but opposite trend, i.e. an elongation of the Mo–O (phenolate) bonds in the molecular structures of 5, 6 and 9, again likely caused by the oxido trans influence. It can be thus further reasoned that the isomeric arrangement compared to the oxidation state of the metal center has a more significant impact on the metal–ligand bonding situation in these complexes.
Conclusions
The reaction of functionalized iminophenolate ligands with molybdenum(VI) metal precursors [MoO2Cl2] and [MoO2Cl2(DME)] yielded disubstituted complexes 1 and 2, whereas the use of [MoO2(acac)2] as a metal source led to monosubstituted complex 3 coordinated by one iminophenolate and one acac− moiety. Subsequent oxygen atom transfer from 1 or 2 to phosphanes gave rise to distinct molybdenum(IV) and molybdenum(V) complexes. The products obtained from the reactions of 1 and 2 were dependent on the nature of the phosphane (PMe3 or PPh3) as well as the employed stoichiometry. The use of excess of PMe3 yielded molybdenum(IV) oxido phosphane complexes 4 and 5. Upon the use of stoichiometric amounts of PMe3 or PPh3 molybdenum(V) μ-oxido complexes 6 and 7 were obtained. Compounds 4 and 5 readily reacted with molecular oxygen under ambient conditions to give oxido peroxido complexes 8 and 9, while 6 and 7 did not show similar reactivity. Complexes 1, 2, 4, 5 and 9 exist as isomeric mixtures in solution, but for 8 only one isomer is observed.Furthermore 1 and 2 adopt a different ligand arrangement than 5, 6, 8 and 9 in the solid state. A comparison of Mo–N and Mo–O bond lengths within the solid state structures of 1, 2, 5, 6, 8 and 9 reveals a significant impact of the oxido trans influence on the bonding situation in these complexes. Dioxido molybdenum(VI) complexes 1 and 2 have been found to catalyze the aerobic oxidation of PMe3 and PPh3 with noteworthy reactivity differences depending on the nature of the ligand. In the oxidation of PMe3, compound 1 was significantly more active and selective in comparison with 2, however 2 marked the first example of a catalyst capable of transferring both peroxido oxygen atoms to one substrate molecule and thus partially oxidizing trimethyl phosphane to methyl dimethylphosphinate (OP(OMe)Me2). In the catalytic aerobic oxidation of PPh3, complex 1 proved to be virtually unreactive whereas complex 2 gave significant yields of OPPh3. In the case of PMe3 oxidation, the active site is coordinated by a substrate molecule. Thus, prior elimination of PMe3 is necessary, occurring readily under O2 which we have recently demonstrated.16 This is not possible with the more bulky PPh3 as evidenced by the formation of dinuclear μ-oxido bridged complexes in stoichiometric reactions. However catalysis experiments using these dimeric compounds 6 and 7 as catalysts in the oxidation of PPh3 showed the phenyl-based dinuclear compound 7 exhibiting catalytic activity whereas 6 was completely inactive. This indicates lower activation energy for the disproportionation of 7, which is required for the reaction with molecular O2, in comparison with compound 6. The results presented herein provide further understanding of the species involved in OAT reactions as well as oxygen activation with molybdenum(IV)/(VI) systems and also insight into the diverse effects of ligand functionalization on the reactivity of such systems.
Experimental section
General
Unless specified otherwise, experiments were performed under inert conditions using standard Schlenk equipment. Commercially available chemicals were purchased from Sigma-Aldrich and used as received. No further purification or drying operations have been performed. The metal precursors [MoO2(acac)2]20 and [MoO2Cl2(DME)]21 were synthesized according to known procedures. Solvents were purified via a Pure-Solv MD-4-EN solvent purification system from Innovative Technology, Inc. Methanol was refluxed over activated magnesium for at least 24 h and then distilled prior to use. The 1H, 13C and 31P NMR spectra were recorded on a Bruker Optics instrument at 300/75/121 MHz. The peaks are denoted as singlet (s), broad singlet (bs), doublet (d), doublet of doublets (dd), triplet (t), pseudo-doublet (“d”), pseudo-triplet (“t”) and multiplet (m). The used solvents and peak assignment are mentioned in the specific data sets. Resonances in 31P NMR were referenced to phosphoric acid as an external standard. Electron impact mass spectroscopy (EI-MS) measurements have been performed with an Agilent 5973 MSD mass spectrometer with a push rod. ESI-MS as well as HR-MS (ESI+) measurements were performed at the University of Graz, Department of Analytical Chemistry. ESI measurements were performed using an Agilent 1100 Series LCMSD (SL type), HR-MS (ESI+) measurements were performed using a Thermo Scientific Q-Exactive mass spectrometer in positive ion mode, and the used solvent was acetonitrile. The peaks are denoted as cationic mass peaks, and the unit is the according ion mass/charge ratio. Solid samples for infrared spectroscopy were measured on a Bruker Optics ALPHA FT-IR Spectrometer. Liquid samples were recorded in benzene on a Bruker FT-MIR matrix MF in situ spectrometer using a glass fiber optic probe. The IR bands are reported with wavenumber (cm−1) and intensities (s, strong; m, medium; w, weak). All elemental analyses were performed at the Technical University of Graz, Institute of Inorganic Chemistry using a Heraeus Vario Elementar automatic analyzer.
X-ray diffraction analyses
Single-crystal X-ray diffraction analyses were performed on a BRUKER-AXS SMART APEX II diffractometer equipped with a CCD detector. All measurements were performed using monochromatized Mo Kα radiation from an Incoatec microfocus sealed tube at 100 K (cf. Table S1†). Absorption corrections were performed semi-empirical from equivalents. The structures were solved by direct methods (SHELXS-97)27 and refined by full-matrix least-squares techniques against F2 (SHELXL-2014/6).27 CCDC 1476035–1476040 and 1486756 contain the supplementary crystallographic data for this paper. Full experimental details for single-crystal X-ray diffraction analyses of all compounds are provided in the ESI.†Ligand synthesis
All ligands are stable towards air and moderately stable towards moisture. They can be stored in a desiccator over P2O5 for several weeks without decomposition.N), 7.53 (d, 1H, ArH), 6.97 (d, 1H, ArH), 3.36 (t, 2H, CH2), 3.17 (t, 2H, CH2), 3.06 (s, 3H, OMe), 1.69 (m, 2H, CH2), 1.62 (s, 9H, tBu), 1.32 (s, 9H, tBu) ppm; IR (ATR, cm−1) ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) : 1631 (s), 1440 (s), 1390 (m), 1361 (m), 1251 (m), 1173 (m), 1120 (s), 876 (w).
N), 7.64 (d, 1H, ArH), 7.11–6.90 (m, 6H, ArH), 1.68 (s, 9H, tBu), 1.34 (s, 9H, tBu) ppm; IR (ATR, cm−1) : 1605 (m), 1437 (m), 1361 (m), 929 (m), 901 (s), 878 (m), 794 (s), 568 (m), 448 (m).
: 1631 (s), 1440 (s), 1390 (m), 1361 (m), 1251 (m), 1173 (m), 1120 (s), 876 (w).
N), 7.64 (d, 1H, ArH), 7.11–6.90 (m, 6H, ArH), 1.68 (s, 9H, tBu), 1.34 (s, 9H, tBu) ppm; IR (ATR, cm−1) : 1605 (m), 1437 (m), 1361 (m), 929 (m), 901 (s), 878 (m), 794 (s), 568 (m), 448 (m).
Complex syntheses
All complexes are sensitive towards moisture; complexes 4 and 5 are additionally highly sensitive towards air. They can be stored in an N2-filled glovebox for several weeks without decomposition.N), 7.71 (d, 2H, ArH), 7.09 (d, 2H, ArH), 3.63–3.58 (m, 4H, CH2), 3.16–3.09 (m, 2H, CH2), 303–2.99 (m, 2H, CH2), 2.92 (s, 6H, OMe), 2.00–1.92 (m, 4H, CH2), 1.29 (s, 18H, tBu), 1.27 (s, 18H, tBu) ppm; 13C NMR (75 MHz, C6D6, 25 °C, N,N isomer) δ: 168.04 (CHN), 160.98 (Ar–O), 142.08, 139.58, 129.74, 128.19 (HSQC), 122.19 (Ar), 69.48 (CH2), 58.11 (OMe), 57.36 (CH2), 35.65, 34.38 (q-tBu), 31.60, 31.56 (tBu), 31.30 (CH2) ppm; IR (ATR, cm−1) : 1628 (s), 1559 (w), 1460 (w), 1247 (s), 1108 (s), 1047 (w), 913 (m), 902 (s), 841 (s), 751 (s), 549 (s); EI-MS (70 eV) m/z: 738.6 [M]+; Anal. calcd for C38H60MoN2O6: C, 61.94; H, 8.21; N, 3.80; found: C, 61.62, H, 8.09; N, 3.78%.
N), 7.44–7.41 (m, 2H, ArH), 7.41 (s, 1H, CHN), 7.20–7.16 (m, 1H, ArH), 7.09–7.00 (m, 1H, ArH), 6.90–6.75 (m, 7H, ArH), 6.44 (d, 1H, ArH), 1.70 (s, 9H, tBu), 1.49 (s, 9H, tBu), 1.26 (s, 9H, tBu), 1.23 (s, 9H, tBu); 1H NMR (300 MHz, C6D6, N,N isomer) δ: 7.88 (s, 2H, CHN), 7.58 (d, 2H, ArH), 7.09–7.00 (m, 4H, ArH), 6.90–6.75 (m, 8H, ArH), 1.37 (s, 18H, tBu), 1.24 (s, 18H, tBu); 13C NMR (HSQC 300/75 MHz, C6D6, 25 °C, O,N isomer, q-C obscured) δ: 170.13, 165.54 (CHN), 131.70, 129.59, 129.26, 128.97, 128.08, 127.95, 126.55, 126.00, 125.72, 125.67, 125.54, 123.26, 123.18, 123.06 (Ar), 31.21, 31.20, 29.85, 29.69 (tBu); 13C NMR (HSQC 300/75 MHz, C6D6, 25 °C, N,N isomer, q-C obscured) δ: 169.00 (CHN), 130.20, 128.91, 128.73, 128.58, 128.48, 123.33, 123.18 (Ar), 31.21, 29.82 (tBu); IR (ATR, cm−1) : 2956 (s), 1611 (s), 1592 (m), 1559 (w), 1486 (w), 1437 (w), 1272 (m), 1253 (s), 1177 (s), 928 (w), 910 (s), 897 (s), 845 (m), 765 (m), 705 (w), 554 (w); EI-MS (70 eV) m/z: 746.6 [M]+; Anal. calcd for C38H60MoN2O6·CH3CN: C, 67.25; H, 7.05; N, 5.35; found: C, 67.03, H, 7.01; N, 5.47%.
N), 7.15–7.12 (m, 2H, ArH), 7.01–6.88 (m, 4H, ArH), 4.83 (s, 1H, CH), 1.65 (s, 9H, tBu), 1.44 (s, 3H, Me), 1.26 (s, 9H, tBu), 1.13 (s, 3H, Me) ppm; 13C NMR (300 MHz, C6D6, 25 °C) δ: 195.26, 185.83 (CO), 166.81 (CHN), 159.17 (Ar–O), 152.24, 142.66, 140.51, 130.47, 129.36, 128.45, 126.23, 123.91, 121.96 (Ar), 103.55 (CH), 35.75, 34.42 (q-tBu), 31.53, 30.09 (tBu), 27.27, 24.90 (Me) ppm; IR (ATR, cm−1) : 2962 (m), 1605 (m), 1518 (m), 1362 (m), 1275 (m), 1250 (m), 1177 (m), 928 (s), 901 (s), 848 (s), 794 (m), 762 (s), 695 (m), 568 (s), 450 (s); EI-MS (70 eV) m/z: 537.3 [M]+; Anal. calcd for C26H35MoNO5: C, 58.32; H, 6.21; N, 2.62; found: C, 58.15, H, 6.04; N, 2.68%.
N), 8.01 (s, 1H, CHN), 7.51 (d, 1H, ArH), 7.40 (d, 1H, ArH), 7.13 (d, 1H, ArH), 7.02 (d, 1H, ArH), 4.39–4.14 (m, 3H, CH2), 4.08–3.99 (m, 1H, CH2), 3.49–3.26 (m, 4H, CH2), 3.17 (s, 3H, OMe), 3.04 (s, 3H, OMe), 2.89–2.76 (m, 1H, CH2), 2.63–2.34 (m, 2H, CH2), 2.29–2.16 (m, 1H, CH2), 1.36 (s, 9H, tBu), 1.35 (s, 9H, tBu), 1.34 (s, 9H, tBu), 1.29 (s, 9H, tBu), 0.92 (d, 9H, PMe3) ppm; 13C NMR (75 MHz, C6D6, 25 °C, putative O,O isomer) δ: 168.92 (CHN), 166.04 (Ar–O), 163.00 (CHN), 162.09 (Ar–O), 140.20, 138.49, 136.50, 136.34, 131.20, 129.96, 129.38, 127.45 (HSQC), 121.25, 121.17 (Ar), 71.06, 69.49, 68.81, 66.68, (CH2), 58.33 (2× OMe), 35.62, 35.45, 34.24, 33.99 (q-tBu), 33.30 (CH2), 31.86, 31.77 (tBu), 30.86 (CH2), 30.10, 29.91 (tBu), 16.53 (d, PMe3) ppm; 31P NMR (121 MHz, C6D6, 25 °C) δ: −3.17 (putative O,N isomer, Mo-PMe3), −3.48 (putative O,O isomer, Mo–PMe3) ppm; IR (FT-IR, benzene, cm−1) : 1612 (s), 1435 (s), 1311 (m), 1256 (s), 1120 (s), 952 (m), 918 (s), 837 (s), 746 (m). ESI-MS (50 V) m/z: 722.3 [M − PMe3]+; Anal. calcd for C41H69MoN2O5P: C, 61.79; H, 8.73; N, 3.52; found: C, 62.14; H, 8.76; N, 3.51%.
N), 8.04 (s, 1H, CHN), 7.84–7.81 (m, 2H, ArH), 7.69–7.66 (m, 2H, ArH), 7.58 (d, 1H, ArH), 7.42 (d, 1H, ArH), 7.23–7.18 (m, 3H, ArH), 7.11–7.05 (m, 3H, ArH), 6.95–6.90 (m, 2H, ArH), 1.42 (s, 9H, tBu), 1.34 (s, 18H, tBu), 1.33 (s, 9H, tBu), 0.45 (d, 9H, PMe3) ppm; 13C NMR (75 MHz, C6D6, 25 °C, O,O isomer) δ: 170.14 (CHN), 166.47 (Ar–O), 163.63 (CHN), 163.08 (Ar–O), 160.20, 157.97, 140.84, 138.73, 136.91, 136.67, 131.89, 131.16, 130.59, 129.28 (2×), 128.65, 128.03 (2×, HSQC), 126.57, 125.92, 124.85 (2×), 123.69 (2×), 121.44, 120.73 (Ar), 35.67, 35.53, 34.28, 34.04 (q-tBu), 31.82, 31.68, 30.15, 29.99 (tBu), 15.39 (d, PMe3) ppm; 31P NMR (121 MHz, C6D6, 25 °C) δ: −0.77 (O,O isomer, Mo-PMe3), −1.99 (putative O,N isomer, Mo-PMe3) ppm; IR (ATR, cm−1) : 2949 (m), 1601 (m), 1587 (m), 1527 (m), 1485 (m), 1254 (m), 1165 (s), 953 (w), 927 (s), 866 (m), 764 (m), 704 (m), 694 (m), 637 (w), 523 (s), 448 (w); HR-MS (ESI+) m/z: [M]+ calcd for C45H61MoN2O3P: 806.3474, found: 806.3474; Anal. calcd for C45H61MoN2O3P: C, 67.15; H, 7.64; N, 3.48; found: C, 67.60; H, 7.43; N, 3.35%.
:1) as the eluent was performed and the first red band was collected. Evaporation of the solvent gave 6 as a dark red solid (68%, 69 mg). For elemental analyses, the complex was recrystallized from dry CH2Cl2. Alternatively, complex 6 is accessible by OAT from complex 1 (100 mg, 0.14 mmol, 1 equiv.) to PPh3 (37 mg, 0.14 mmol, 1 equiv.) in dry toluene (5 mL). After stirring for 24 h at room temperature, purification was followed the procedure described above. Residual traces of PPh3 were first eluted from the basic aluminum oxide column with pentane. The use of polymer-bound PPh3 led to insoluble by-products and thus a simplified work-up procedure. Crystals suitable for single-crystal X-ray diffraction analysis were obtained by slow evaporation of a concentrated MeCN solution at −35 °C. 1H NMR (300 MHz, C6D6, 25 °C) δ: 8.26 (s, 2H, CHN), 8.10 (s, 2H, CHN), 7.57 (d, 2H, ArH), 7.46 (d, 2H, ArH), 7.02 (d, 2H, ArH), 6.84 (d, 2H, ArH), 5.04–4.97 (m, 2H, CH2), 4.62–4.52 (m, 2H, CH2), 4.39–4.30 (m, 2H, CH2), 3.69–3.60 (m, 2H, CH2), 3.42–3.24 (m, 4H, CH2), 3.11–3.05 (m, 2H, CH2), 3.02 (s, 6H, OMe). 2.92 (s, 6H, OMe), 2.78–2.47 (m, 8H, CH2), 2.10–1.92 (m, 2H, CH2), 1.41 (s, 18H, tBu), 1.31 (s, 18H, tBu), 1.22 (s, 18H, tBu), 1.19 (s, 18H, tBu) ppm; 13C NMR (75 MHz, C6D6, 25 °C) δ: 169.91, 169.31 (CHN), 166.94, 162.57 (Ar–O), 140.01, 139.28, 138.48, 137.47, 131.62, 129.96, 129.69, 128.85, 121.95, 121.87 (Ar), 70.26, 70.08, 61.30, 60.77 (CH2), 58.35, 58.20 (OMe), 35.51, 35.23, 34.15, 33.99 (q-tBu), 33.25, 31.95 (CH2), 31.72, 31.44, 29.81, 29.46 (tBu) ppm; IR (ATR, cm−1) : 1611 (s), 1597 (s), 1256 (s), 1104 (br, s), 1014 (br, s), 928 (s), 798 (br, s), 529 (s); EI-MS (70 eV) m/z: 722.6 [MoO(L1)2]+; Anal. calcd for C76H124Mo2N4O10·CH2Cl2: C, 59.80; H, 8.21; N, 3.62; found: C, 59.94; H, 8.40; N, 3.33%.
N), 7.63 (d, 2H, ArH), 7.55 (d, 2H, ArH), 7.43 (s, 2H, CHN), 7.41–7.36 (m, 6H, ArH), 7.09–6.99 (m, 10H, ArH), 6.88 (d, 2H, ArH), 6.55 (d, 2H, ArH), 1.36 (s, 18H, tBu), 1.26 (“d”, 36H, tBu), 1.20 (s, 18H, tBu) ppm; 13C NMR (75 MHz, C6D6, 25 °C) δ: 172.09, 169.54 (CHN), 162.91, 161.76 (Ar–O), 157.25, 155.04, 140.88, 140.52, 139.39, 139.37, 131.00, 130.72, 130.43, 130.16, 128.09 (3×, HSQC), 127.97 (2×, HSQC), 126.82, 126.31 (2×), 126.05 (2×), 122.76, 120.69 (Ar), 35.70, 35.49, 34.27, 34.01 (q-tBu), 31.79, 31.50, 30.52, 30.11 (tBu) ppm; IR (ATR, cm−1) : 1618 (m), 1434 (m), 1250 (s), 1168 (s), 928 (m), 835 (s), 703 (s), 693 (s), 532 (vs); HR-MS (ESI+) m/z: [M]+ calcd for C84H104Mo2N4O7: 1476.6013, found: 1476.6056; Anal. calcd for C84H104Mo2N4O7: C, 68.46; H, 7.11; N, 3.80; found: C, 68.25; H, 6.80; N, 3.71%.
N), 8.18 (s, 1H, CHN), 7.63 (d, 1H, ArH), 7.47 (d, 1H, ArH), 7.03 (d, 1H, ArH), 6.98 (d, 1H, ArH), 5.25–5.17 (m, 1H, CH2), 5.01–5.94 (m, 1H, CH2), 4.22–4.08 (m, 2H, CH2), 3.59–3.52 (m, 1H, CH2), 3.35–3.19 (m, 3H, CH2), 3.04 (s, 3H, OMe), 3.01 (s, 3H, OMe), 2.60–2.29 (m, 4H, CH2), 1.32 (s, 9H, tBu), 1.24 (s, 18H, tBu), 1.15 (s, 9H, tBu) ppm; 13C NMR (75 MHz, C6D6, 25 °C) δ: 169.00, 168.35 (CHN), 162.21, 160.64 (Ar–O), 139.70, 139.26, 139.06, 132.20, 130.67, 129.61, 129.28, 129.00, 121.93, 121.66 (Ar), 69.47, 69.39, 63.76, 59.68 (CH2), 58.22, 58.19 (OMe), 35.06, 34.94 (q-tBu), 34.15 (2× q-tBu), 31.77 (CH2), 31.60 (tBu), 31.53 (CH2), 31.48, 29.73, 29.56 (tBu) ppm; IR (ATR, cm−1) : 1612 (s), 1256 (s), 1108 (br, s), 1089 (br, s), 914 (s), 839 (s), 800 (s), 779 (s), 748 (m), 539 (s); ESI-MS (100 V) m/z : 755.3 [M]+; Anal. calcd for C38H60MoN2O7·0.2CH2Cl2: C, 59.60; H, 7.91; N, 3.64; found: C, 59.26; H, 7.99; N, 4.00%.
N), 8.10 (s, 1H, CHN), 7.89–7.85 (m, 2H, ArH), 7.70 (d, 1H, ArH), 7.61–7.56 (m, 2H, ArH), 7.54 (d, 1H, ArH), 7.24–7.18 (m, 2H, ArH), 7.13–6.99 (m, 4H, ArH), 6.90 (d, 1H, ArH), 6.86 (d, 1H, ArH), 1.37 (s, 9H, tBu), 1.26 (s, 9H, tBu), 1.25 (s, 9H, tBu), 1.23 (s, 9H, tBu) ppm; 13C NMR (HSQC 300/75 MHz, C6D6, 25 °C, q-C obscured) δ: 169.10, 168.74 (CHN), 133.20, 131.41, 130.24, 129.74, 128.81, 128.48, 128.46, 128.09, 127.15, 126.90, 125.44, 125.26 (Ar), 124.74 (2× Ar), 31.19, 31.09, 29.64, 29.54 (tBu); IR (ATR, cm−1) : 2953 (m), 1611 (s), 1589 (s), 1540 (m), 1252 (s), 1172 (s), 927 (s), 838 (s), 765 (m), 704 (m), 692 (m), 538 (br s); HR-MS (ESI+) m/z: [M + Na]+ calcd for C42H52MoN2NaO5: 785.2828, found: 785.2820; Anal. calcd for C42H52MoN2O5: C, 66.30; H, 6.89; N, 3.68; found: C, 65.81; H, 7.05; N, 3.66%.
Catalytic aerobic oxidation of phosphanes
In a representative reaction, 5 mg of the respective catalyst (approx. 0.007 mmol) was dissolved in dry C6D6 under a N2 atmosphere in a 50 mL Schlenk flask. Subsequently 100 equiv. of the respective phosphane (0.7 mmol PMe3: 53 mg; PPh3: 184 mg) were added to the solution. The Schlenk flask was then flushed with dry O2 and placed under O2 pressure (1.5 atm) whereupon the reaction solution was stirred for 24 h at room temperature. Subsequently the Schlenk flask was cooled to 0 °C and twice evacuated and backfilled with dry N2. After addition of 0.2 mL of dry CD3CN to ensure complete dissolution of all formed products, the reaction was monitored via1H and 31P NMR spectroscopy, phosphane oxide yields were calculated via integration of the respective peak areas in the 1H NMR spectra, and conversion included the peak areas for the formed side product (OP(OMe)Me21H NMR in C6D6δ: 3.34 (d, 3H, OMe), 1.08 (d, 6H, 2× Me); 31P NMR in C6D6δ: 49.93). Blank experiments without catalysts were included for both phosphanes, where yields of <5% were observed for PMe3 and PPh3.Acknowledgements
Financial support by the Austrian Science Fund (FWF) (grant number P26264) and NAWI Graz is gratefully acknowledged.References
- (a) C. G. Young, in Biomimetic Oxidations Catalyzed by Transition Metal Complexes, ed. B. Meunier, World Scientific, Singapore, 2000, pp. 415–459 Search PubMed; (b) S. Groysman and R. H. Holm, Biochemistry, 2009, 48, 2310–2320 CrossRef CAS PubMed.
- (a) F. J. Hine, A. J. Taylor and C. D. Garner, Coord. Chem. Rev., 2010, 254, 1570–1579 CrossRef CAS; (b) P. Basu and S. J. Burgmayer, Coord. Chem. Rev., 2011, 255, 1016–1038 CrossRef CAS PubMed; (c) R. Hille, J. Hall and P. Basu, Chem. Rev., 2014, 114, 3963–4038 CrossRef CAS PubMed.
- K. Heinze, Coord. Chem. Rev., 2015, 300, 121–141 CrossRef CAS.
- (a) R. H. Holm, E. I. Solomon, A. Majumdar and A. Tenderholt, Coord. Chem. Rev., 2011, 255, 993–1015 CrossRef CAS; (b) A. Majumdar and S. Sarkar, Coord. Chem. Rev., 2011, 255, 1039–1054 CrossRef CAS.
- S. A. Hauser, M. Cokoja and F. E. Kühn, Catal. Sci. Technol., 2013, 3, 552–561 CAS.
- G. Lyashenko, G. Saischek, M. E. Judmaier, M. Volpe, J. Baumgartner, F. Belaj, V. Jancik, R. Herbst-Irmer and N. C. Mösch-Zanetti, Dalton Trans., 2009, 5655–5665 RSC.
- M. E. Judmaier, C. Holzer, M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2012, 51, 9956–9966 CrossRef CAS PubMed.
- M. Volpe and N. C. Mösch-Zanetti, Inorg. Chem., 2012, 51, 1440–1449 CrossRef CAS PubMed.
- (a) J. A. Schachner, P. Traar, C. Sala, M. Melcher, B. N. Harum, A. F. Sax, M. Volpe, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2012, 51, 7642–7649 CrossRef CAS PubMed; (b) M. E. Judmaier, C. H. Sala, F. Belaj, M. Volpe and N. C. Mösch-Zanetti, New J. Chem., 2013, 37, 2139 RSC; (c) A. Dupé, M. K. Hossain, J. A. Schachner, F. Belaj, A. Lehtonen, E. Nordlander and N. C. Mösch-Zanetti, Eur. J. Inorg. Chem., 2015, 3572–3579 CrossRef; (d) N. C. Mösch-Zanetti, D. Wurm, M. Volpe, G. Lyashenko, B. Harum, F. Belaj and J. Baumgartner, Inorg. Chem., 2010, 49, 8914–8921 CrossRef PubMed.
- N. Zwettler, A. Dupé, J. A. Schachner, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2015, 54, 11969–11976 CrossRef CAS PubMed.
- Modern Oxidation Methods, ed. J.-E. Bäckvall, Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, Germany, 2010 Search PubMed.
- (a) C. E. MacBeth, A. P. Golombek, V. G. Young Jr., C. Yang, K. Kuczera, M. P. Hendrich and A. S. Borovik, Science, 2000, 289, 938–941 CrossRef CAS PubMed; (b) R. L. Shook, S. M. Peterson, J. Greaves, C. Moore, A. L. Rheingold and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 5810–5817 CrossRef CAS PubMed; (c) W. Nam, Acc. Chem. Res., 2015, 48, 2415–2423 CrossRef CAS PubMed; (d) K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS PubMed.
- H. Arzoumanian, J. F. Petrignani, M. Pierrot, F. Ridouane and J. Sanchez, Inorg. Chem., 1988, 27, 3377–3381 CrossRef CAS.
- (a) J. Tachibana, T. Imamura and Y. Sasaki, J. Chem. Soc., Chem. Commun., 1993, 1436–1438 RSC; (b) M. Minato, D.-Y. Zhou, K.-i. Sumiura, Y. Oshima, S. Mine, T. Ito, M. Kakeya, K. Hoshino, T. Asaeda, T. Nakada and K. Osakada, Organometallics, 2012, 31, 4941–4949 CrossRef CAS.
- G. Lyashenko, G. Saischek, A. Pal, R. Herbst-Irmer and N. C. Mösch-Zanetti, Chem. Commun., 2007, 701–703 RSC.
- A. Dupé, M. E. Judmaier, F. Belaj, K. Zangger and N. C. Mösch-Zanetti, Dalton Trans., 2015, 44, 20514–20522 RSC.
- (a) C. Y. Lorber, S. P. Smidt and J. A. Osborn, Eur. J. Inorg. Chem., 2000, 655–658 CrossRef CAS; (b) S. N. Rao, N. Kathale, N. N. Rao and K. N. Munshi, Inorg. Chim. Acta, 2007, 360, 4010–4016 CrossRef CAS; (c) M. A. Katkar, S. N. Rao and H. D. Juneja, RSC Adv., 2012, 2, 8071 RSC.
- H. Audouin, R. Bellini, L. Magna, N. Mézailles and H. Olivier-Bourbigou, Eur. J. Inorg. Chem., 2015, 2015, 5272–5280 CrossRef CAS.
- G. Alesso, M. Sanz, M. E. G. Mosquera and T. Cuenca, Eur. J. Inorg. Chem., 2008, 2008, 4638–4649 CrossRef.
- H. Gehrke and J. Veal, Inorg. Chim. Acta, 1969, 3, 623–627 CrossRef CAS.
- T. Robin, F. Montilla, A. Galindo, C. Ruiz and J. Hartmann, Polyhedron, 1999, 18, 1485–1490 CrossRef CAS.
- N. Zwettler, J. A. Schachner, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2016, 55, 5973–5982 CrossRef CAS PubMed.
- J. Leppin, C. Förster and K. Heinze, Inorg. Chem., 2014, 53, 1039–1047 CrossRef CAS PubMed.
- J. M. Mayer, Inorg. Chem., 1988, 27, 3899–3903 CrossRef CAS.
- (a) R. D. Rogers, E. Carmona, A. Galindo, J. L. Atwood and L. G. Canada, J. Organomet. Chem., 1984, 277, 403–415 CrossRef CAS; (b) K. Most, J. Hoßbach, D. Vidović, J. Magull and N. C. Mösch-Zanetti, Adv. Synth. Catal., 2005, 347, 463–472 CrossRef CAS; (c) K. Hüttinger, C. Förster, T. Bund, D. Hinderberger and K. Heinze, Inorg. Chem., 2012, 51, 4180–4192 CrossRef PubMed.
- (a) A. B. Blake, F. A. Cotton and J. S. Wood, J. Am. Chem. Soc., 1964, 86, 3024–3031 CrossRef CAS; (b) B. Modec, M. Šala and R. Clérac, Eur. J. Inorg. Chem., 2010, 2010, 542–553 CrossRef; (c) M. M. El-Essawi, F. Weller, K. Stahl, M. Kersting and K. Dehnicke, Z. Anorg. Allg. Chem., 1986, 542, 175–181 CrossRef CAS; (d) J. Leppin, C. Förster and K. Heinze, Inorg. Chem., 2014, 53, 12416–12427 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1476035–1476040 and 1486756. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01692h |
| This journal is © The Royal Society of Chemistry 2016 |