
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Correlating single-molecule and ensemble-average measurements of peptide adsorption onto different inorganic materials
Seong-Oh
Kim
a,
Joshua A.
Jackman
a,
Masahito
Mochizuki
b,
Bo Kyeong
Yoon
a,
Tomohiro
Hayashi
*bc and
Nam-Joon
Cho
*ad
aSchool of Materials Science and Engineering and Centre for Biomimetic Sensor Science, Nanyang Technological University, 50 Nanyang Drive 637553, Singapore. E-mail: njcho@ntu.edu.sg
bDepartment of Electronic Chemistry, Interdisciplinary Graduate School of Science and Engineering, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8502, Japan. E-mail: hayashi@echem.titech.ac.jp
cSurface and Interface Science Laboratory, RIKEN, Wako, 2-1 Hirosawa, Saitama 351-0198, Japan
dSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive 637459, Singapore
First published on 9th May 2016
Abstract
The coating of solid-binding peptides (SBPs) on inorganic material surfaces holds significant potential for improved surface functionalization at nano-bio interfaces. In most related studies, the goal has been to engineer peptides with selective and high binding affinity for a target material. The role of the material substrate itself in modulating the adsorption behavior of a peptide molecule remains less explored and there are few studies that compare the interaction of one peptide with different inorganic substrates. Herein, using a combination of two experimental techniques, we investigated the adsorption of a 16 amino acid-long random coil peptide to various inorganic substrates – gold, silicon oxide, titanium oxide and aluminum oxide. Quartz crystal microbalance-dissipation (QCM-D) experiments were performed in order to measure the peptide binding affinity for inorganic solid supports at the ensemble average level, and atomic force microscopy (AFM) experiments were conducted in order to determine the adhesion force of a single peptide molecule. A positive trend was observed between the total mass uptake of attached peptide and the single-molecule adhesion force on each substrate. Peptide affinity for gold was appreciably greater than for the oxide substrates. Collectively, the results obtained in this study offer insight into the ways in which inorganic materials can differentially influence and modulate the adhesion of SBPs.
Introduction
Solid-binding peptides (SBPs) are a versatile class of biomacromolecules which are widely employed as molecular building blocks to functionalize various types of inorganic and organic solid materials.1–3 One of the most intriguing aspects of SBPs is the possibility of utilizing short amino acid sequences which selectively bind with high affinity to a target substrate. Indeed, SBPs are a useful tool for surface functionalization, offering a simple approach to improve the biocompatibility of a material surface or act as molecular ligands to facilitate the conjugation of other biomolecules.4,5 Among the scope of applications, SBPs enable the fine placement of biomolecules on solid surfaces6–9 and the fabrication of self-assembled nanostructures7,10 towards nanoarchitectonics-based design strategies.11,12 For all these reasons, there is significant interest in understanding the factors which determine how SBPs selectively bind to target substrates.The interaction between SBPs and material surfaces is governed by the interplay of various noncovalent intermolecular forces with different physical origins, including the double-layer electrostatic force, van der Waals force, hydration force, steric interactions, and the entropic force.13–16 The binding affinity and selectivity of peptide attachment are difficult to predict on the basis of substrate properties and peptide characteristics (e.g., amino acid composition, electrical charge, polarity) alone. Empirical investigations are necessary in order to directly address these questions, and typically focus on comparing the attachment of sequence-related peptides in a library to one target substrate or the attachment of one peptide to multiple substrates. In either case, a combination of different experimental techniques and theoretical analysis can help to establish correlations between material properties and peptide binding affinity.17–20 So far, a major emphasis in the field has been on engineering SBPs with optimal binding properties to a target material, and a wealth of knowledge remains to be learned about how material substrates differentially modulate the adsorption properties of SBPs, with a few studies comparing the binding affinity of peptides on two substrates.21–23
The goal of the present study is to investigate the adsorption responses of one SBP candidate upon attachment to a series of inorganic substrates – gold, silicon oxide, titanium oxide, and aluminum oxide. In particular, we discuss a comparative analysis of the peptide attachment kinetics at the single-molecule and ensemble-average levels for the first time. The SBP is a 16-amino acid amphipathic peptide (AGSWLRDIWTWLQSAL) engineered from a longer 27-amino-acid sequence which has demonstrated virucidal activity in solution24 and can be noncovalently immobilized onto hydrophilic solid supports for patterning applications.25 The engineered peptide investigated here possesses one negatively and one positively charged residues (electrically neutral) as well as neutral hydrophilic and hydrophobic residues with an amphipathic disposition (Fig. 1). Systematic experiments are performed based on adsorption kinetic measurements of peptide binding affinity by quartz crystal microbalance-dissipation (QCM-D) and single-molecule force measurements of peptide attachment by atomic force microscopy (AFM). The combination of experimental techniques allows us to correlate changes in the peptide binding affinity and the corresponding attachment/detachment kinetics.
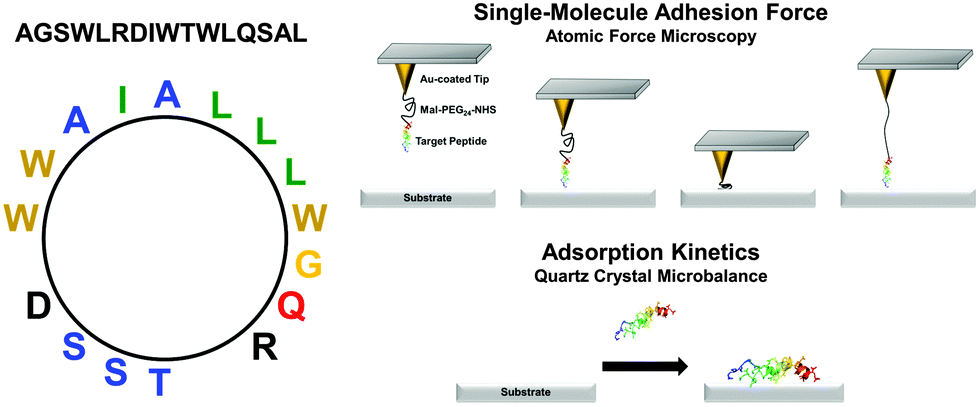 | ||
| Fig. 1 Schematic of experimental design. Helix net wheel plot of the 16-amino acid long peptide is presented. By combining multiple experimental techniques, it is possible to investigate peptide adsorption kinetics and binding affinity for various inorganic substrates. | ||
Materials and methods
Reagents
Phosphate-buffered saline (PBS) (Life Technologies, Carlsbad, CA), ethanol (Merck Millipore, Billerica, MA), dehydrated toluene (Fisher Scientific, Waltham, MA), 11-amino-1-undecanethiol hydrochloride (Sigma-Aldrich, St Louis, Mo), N-hydroxysuccinimide–PEG24-maleimide ester (NHS–PEG–MAL) (Product No. 10314, Quanta Biodesign, Plain City, OH) and polyoxyethylene sorbitan monolaurate (Tween-20, Bio-RAD, Hercules, CA) were used in the experiments. The PBS buffer solution contained 137 mM NaCl and 2.7 mM KCl salt concentrations. All aqueous solutions were prepared using Milli-Q-treated water (18.7 MΩ cm) (Millipore, Billerica, MA).Peptides
High purity peptides (>95%) were synthesized by Anaspec Corporation (San Jose, CA, USA). The sequence of the template peptide is H-Ala-Gly-Ser-Trp-Leu-Arg-Asp-Ile-Trp-Thr-Trp-Leu-Gln-Ser-Ala-Leu-NH2. For AFM experiments, an additional Cys residue was added to the N-terminus or C-terminus. The as-supplied lyophilized peptides were solubilized in deionized water at a stock concentration of 2 mg mL−1 peptide. The exact molar concentration of peptide in solution was determined by standard absorbance measurements at 280 nm and diluted accordingly before experiments.Quartz crystal microbalance-dissipation (QCM-D)
QCM-D experiments were conducted using a Q-Sense E4 instrument (Q-Sense AB, Gothenburg, Sweden) in order to measure the binding affinity of peptides for the solid supports. The QCM-D technique monitors changes in the resonance frequency (Δf) and energy dissipation (ΔD) of an oscillating, piezoelectric quartz crystal sensor chip as functions of time, which reflect the acoustic mass and viscoelastic properties, respectively, of an adsorbate on the surface.26 The sensor chips had a fundamental frequency of 5 MHz and were obtained as-supplied with gold, silicon oxide, titanium oxide, or aluminum oxide coatings. Before experiments, the sensor chips were sequentially rinsed with water and ethanol, dried under nitrogen gas, and were subjected to oxygen plasma treatment for 1 min using an Expanded Plasma Cleaner (model no. PDC-002, Harrick Plasma, Ithaca, NY). A baseline signal was recorded in PBS solution [pH 7.2] and then 7 μM peptide in the same buffer was added. A washing step with PBS solution completed the procedure. In all steps, a peristaltic pump (Reglo Digital, Ismatec, Glattbrugg, Switzerland) was used to inject liquid samples into the measurement chamber under continuous flow conditions at a flow rate of 50 μL min−1. During the experiments, the temperature in the measurement cell was maintained at 25.0 ± 0.5 °C. The experimental data were collected at the 3rd (n = 3), 5th (n = 5), and 7th (n = 7) odd overtones using the QSoft (Q-Sense AB) software package, and the data were normalized according to the overtone number. Data processing was performed using the QTools (Q-Sense AB) and OriginPro 8.5 (OriginLab, Northampton, MA) software packages. All presented data were collected at the 5th overtone.Probe functionalization
In order to measure the peptide binding interaction, silicon AFM probes (CSG-01, NT-MDT, Moscow, Russia) with a spring constant of around 0.03 N m−1 were employed. The probes were coated with a 2 nm thick germanium adhesion layer, followed by a 20 nm thick gold layer by thermal evaporation in a vacuum (base pressure, 1.0 × 10−4 Pa). In order to attach the peptide to the probe tip, the gold-coated probes were first submerged into an ethanol solution with 10 M 11-amino-1-undecanethiol hydrochloride for 2 hours and were then washed with pure ethanol solution. The probes were next immersed in dehydrate toluene solution with N-hydroxysuccinimide–PEG24–maleimide ester (1 mM) for 2 hours. After immersion, the probes were rinsed with pure toluene solution and were allowed to air dry. The dried probes were incubated with 1 mg mL−1 of peptide in deionized water. Before AFM experiments, the functionalized probes were rinsed with PBS buffer solution.Atomic force microscopy (AFM)
A commercial AFM system (NX-Bio, Park Systems, Suwon, South Korea) was employed for the force spectroscopy measurements. Before force measurements, the AFM probes were calibrated using the thermal method, which monitors thermal vibration of the cantilever. All force spectroscopy experiments were performed at room temperature in PBS buffer solution. The probe approach speed and maximum loading force were set at 2 μm s−1 and 1 nN, respectively. Among various possible approaches, autocorrelation analysis is one of the major statistical approaches to evaluate single-molecule adhesion forces.27–29 Under our conditions for the preparation of AFM probes, the histograms exhibited quantized features consisting of multiple peaks originated from events of single and multiple bond attachments. Therefore, the single-molecule adhesion force corresponds to the gap between the peaks. To evaluate the periodicity of the peaks (the gap), we analyzed the autocorrelation of the histograms. In this work, the curves were differentiated to precisely evaluate the periodicity. Error bars were calculated from the standard deviation of the periodicity observed in the differentiated autocorrelation plots.Results and discussion
Peptide binding affinity for solid supports
In order to compare the binding affinity of the peptide for different substrates, quartz crystal microbalance-dissipation (QCM-D) experiments were performed. An initial baseline signal was recorded in aqueous buffer solution, followed by addition of 7 μM peptide at t = 20 min under continuous flow conditions. Peptide adsorption caused a significant decrease in the frequency and a minor increase in the energy dissipation shifts (Fig. 2A and B). The low energy dissipation shift indicates that the adsorbed peptides are rigidly attached to the substrates,30 and also permit the quantitative analysis of the bound adsorbate mass by using the Sauerbrey relationship. The bound mass was in the range of 350 to 450 ng cm−2 which is consistent with a closely-packed peptide adlayer.31 After reaching near-saturation values, a buffer washing step was performed and peptide desorption was monitored, as detected by a positive frequency shift and negligible change in the energy dissipation. More appreciable desorption was observed on the oxide films than on the gold substrate. In general, the peptide affinity for gold was appreciably greater than to the oxide films, as judged on the basis of a larger frequency shift due to adsorption and smaller frequency shift due to desorption.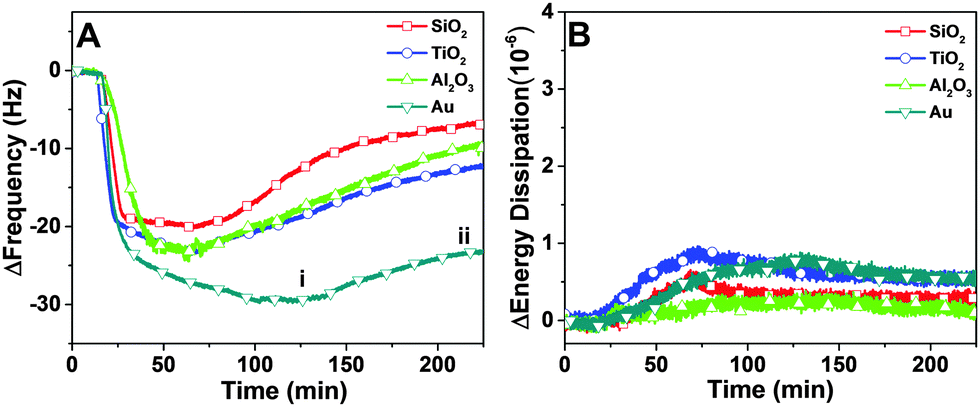 | ||
| Fig. 2 Representative QCM-D kinetic data for peptide adsorption onto solid supports. Changes in (A) frequency and (B) energy dissipation are presented as functions of time. Peptide was added at t = 20 min. On panel A, the labels i and ii represent the measurement values at the saturation and post-washing stages, respectively. | ||
Depending on the substrate, there were quantitative differences in the frequency shift of the adsorption step (Fig. 3A and B). On gold, the average frequency shift was around −30 Hz, while the shifts were around −24 Hz on aluminum oxide and titanium oxide. The smallest shift was around −20 Hz on silicon oxide. In all cases, the average energy dissipation shifts were less than 1 × 10−6. After buffer washing, the final frequency and energy dissipation shifts were also recorded and take into account the effects of peptide adsorption and subsequent desorption (Fig. 3C and D). The final shift was around −27 Hz on gold, and smaller on the oxide substrates, including −12 Hz on titanium oxide, −10 Hz on silicon oxide, and −7 Hz on aluminum oxide.
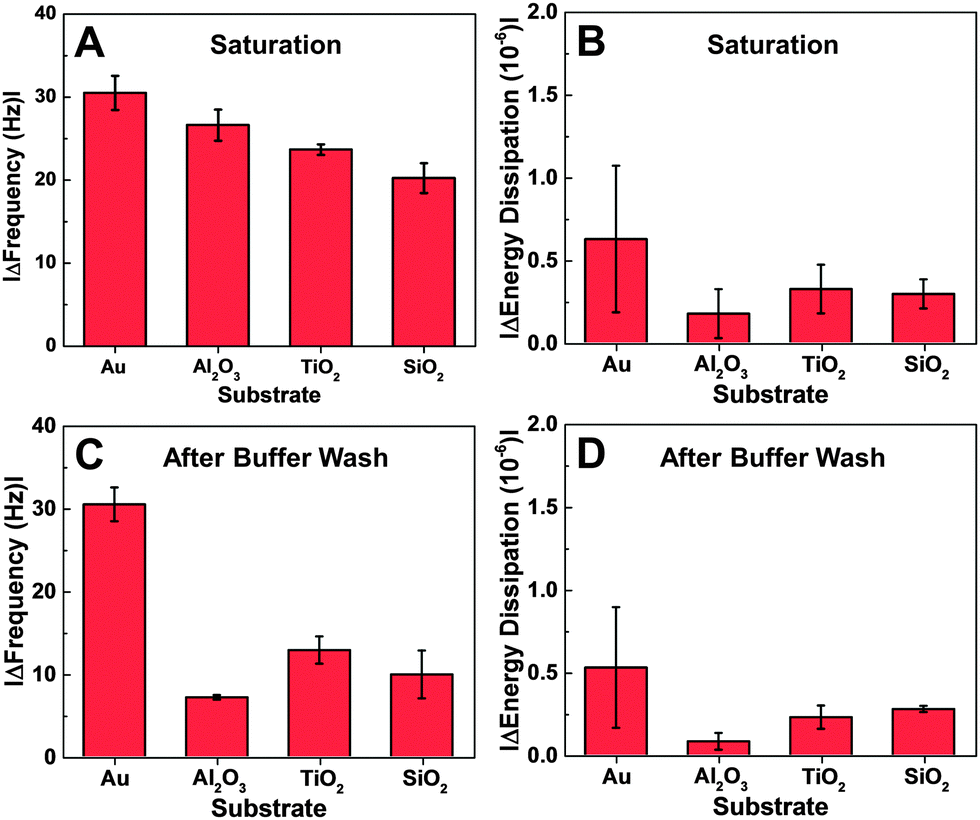 | ||
| Fig. 3 Comparison of QCM-D measurement responses. Maximum changes in (A) frequency and (B) energy dissipation are reported for peptide adsorption on the different substrates. Final changes in (C) frequency and (D) energy dissipation after a washing step are also reported. All reported values are the average and standard deviation for n = 4 measurements. | ||
Taken together, the QCM-D experiments indicate that the peptide affinity for gold is the greatest among the tested cases. The results obtained with the oxide film substrates were more nuanced because there was no general trend between the adsorption and desorption behaviors. The adsorption data support that the peptide has higher binding affinity for titanium oxide and aluminum oxide over silicon oxide. However, despite the observed adsorption behavior, peptide attachment to aluminum oxide was relatively weaker than to the other two substrates.
Adhesion force analysis
Adhesion force analyses were next performed using atomic force microscopy (AFM) in order to evaluate the interaction between the peptide and solid surfaces. First, the AFM probe tip was coated with an N-hydroxysuccinimide–PEG24–maleimide ester that served as a linker for peptide functionalization. The peptide contained an additional cysteine amino acid at either its C- or N-terminus in order to permit covalent binding to the maleimide functional group. Representative AFM measurement data characterizing the adhesion force of peptide attachment onto gold are presented in Fig. 4. Adhesion peaks in the force–distance curves were always observed after the stretching of the PEG chain (around the separation distance of 10 nm) (Fig. 4A), indicating that the adhesion is due to the peptide–substrate interaction and not due to other non-specific interactions. As presented in Fig. 4B, the histogram of collected adhesion forces consists of several peaks due to the sum of the events for the detachment of one or more bound peptides from the substrate. In order to determine the adhesion force of an individual bound peptide, autocorrelation function analysis was conducted in order to identify periodic trends in the adhesion force histogram (Fig. 4C). An autocorrelation function, G(x), of the histogram data was calculated according to the following equation:where
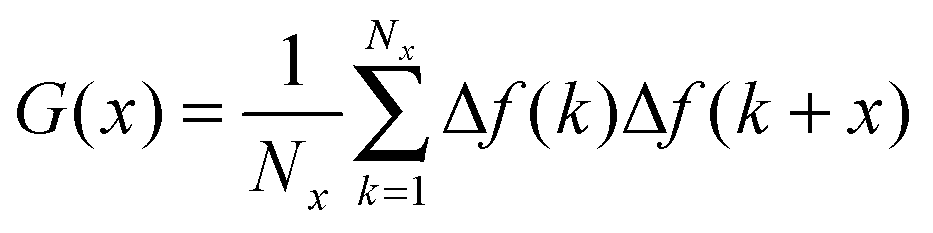
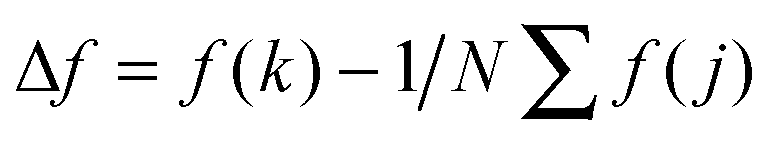 for integer j from 1 to N, and Δf corresponds to the unit binding force. The calculations assume that the histogram has period P such that f(x) = f(x + P) = f(x − P). Indeed, periodicities were observed in the autocorrelation function, as indicated by arrows and there was a unit adhesion force of around 120 pN for this particular case. Similar periodicity was also seen in the autocorrelation functions for peptides on the other substrates.
for integer j from 1 to N, and Δf corresponds to the unit binding force. The calculations assume that the histogram has period P such that f(x) = f(x + P) = f(x − P). Indeed, periodicities were observed in the autocorrelation function, as indicated by arrows and there was a unit adhesion force of around 120 pN for this particular case. Similar periodicity was also seen in the autocorrelation functions for peptides on the other substrates.
 | ||
| Fig. 4 Single-molecule AFM force spectroscopy. (A) Representative force curve obtained using the P8C–Au system. Adhesion force is indicated with a double-sided arrow. The dots reflect the individual data points collected as a function of separation distance and the illustrative line connects the dots. (B) Representative histogram for adhesion forces observed for the interaction between the peptide and the gold surface. The peptide is attached to the tether via a cysteine residue at the C-terminus. (C) Autocorrelation-lag plot in order to determine the single-molecule adhesion force. | ||
Hence, the AFM measurements enabled the comparative measurements of the mean adhesion force for peptide attachment to the different substrates as well as the adhesion force of an individual peptide molecule on each substrate.
Table 1 summarizes the mean adhesion force, which was experimentally measured for peptide attachment on each substrate. On gold, the mean adhesion force was around 290 pN, whereas lower adhesion forces of around 120–140 pN were observed on titanium oxide and aluminum oxide. The smallest adhesion forces were recorded on silicon oxide, with a value of around 70 pN. Peptides with cysteine residues at either the C- or the N-terminus were both evaluated, and demonstrated similar adhesion forces. The AFM results indicate that it is possible to evaluate the affinity of peptide for solid supports regardless of the choice of which terminus is chosen for fixation to the AFM probe.
| Peptide tethering site | SiO2 (pN) | TiO2 (pN) | Al2O3 (pN) | Gold (pN) |
|---|---|---|---|---|
| C-terminus | 76 ± 44 (101) | 135 ± 81 (104) | 120 ± 66 (104) | 291 ± 86 (109) |
| N-terminus | 72 ± 39 (103) | 143 ± 64 (103) | 118 ± 67 (96) | 292 ± 71 (112) |
Based on the autocorrelation analysis of the adhesion force histogram for each histogram, the evaluated single-molecule adhesion forces obtained in each system are all presented in Fig. 5. The data agree well with the mean adhesion force, with both types of peptides exhibiting the strongest adhesion to gold among the tested substrates. Moreover, the peptides interacted more weakly with the other oxide solid supports, which is consistent with the QCM-D measurement results. Therefore, the peptide binding affinity for the solid supports in both the unlabeled and tethered states was similar and demonstrates the complementarity of the two measurement techniques. While both techniques can evaluate the binding affinity based on different physical principles (total uptake vs. adhesion force), they do so in different coverage regimes and the QCM-D technique carries the added advantage of probing the mass and viscoelastic properties of the adlayer, including the corresponding desorption behavior and related conformational aspects.
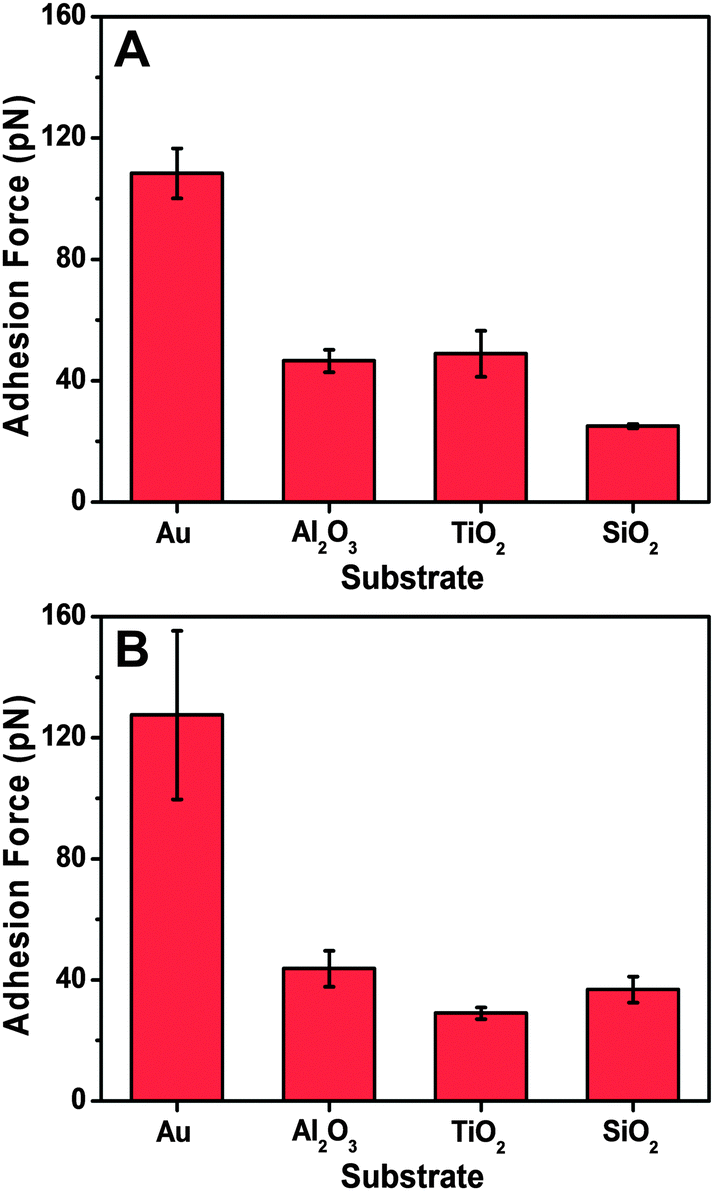 | ||
| Fig. 5 Comparison of AFM single-molecule adhesion force analysis on different substrates. Evaluated single-molecule adhesion forces are presented for the peptide tethered to the NHS–PEG–MAL linker via the (A) C-terminus or (B) N-terminus. | ||
Of note, the bound peptides to aluminum oxide were sensitive to buffer washing and exhibited the most significant desorption behavior, as reflected in the final QCM-D measurement values (cf.Fig. 4C). Hence, the data support that peptide adsorption onto the substrates strongly depends on the peptide–substrate interaction while peptide–peptide interactions are also important for adlayer stabilization.
Finally, we discuss the underlying mechanism of the strong binding between the peptide and gold. The results obtained clearly support the greater peptide attachment to gold versus the other three oxides, which is in good agreement with our previous result for another peptide.20 In that work, the gold-binding peptide (MHGKTQATSGTIQS) had a large population of polar residues in its sequence, similar to the SBP in this work. In both cases, electrostatic attractions are an important factor in governing the interaction between the peptide and different surfaces (see, e.g., ref. 32 for preferential adsorption of charged amino acids on titanium oxide). In the particular case of gold, the strong interaction is likely due to the electrostatic interaction of charged and polar groups of the peptide with their image charges. Compared with the electrostatic interactions between charged amino acid groups on the peptide and ionized functional groups on the oxide surface, the interaction between the polar and charged groups with their image charges is stronger because of the larger number of interaction- and position-independent bonds formed due to the homogenous distribution of free electrons in gold.33–37 The charged and polar groups on the oxide surfaces are fixed. Also, the peptide in the current study (AGSWLRDIWTWLQSAL) has a large number of polar amino acids (especially T, R, W) that are known to have particularly strong binding to gold surfaces (see ref. 38 and references therein). In such cases, the peptides need to optimize their conformations and orientations so as to maximize the interaction energy, resulting in the lower affinities. Indeed, it should be emphasized that not all peptides prefer binding to gold over silica-based materials, as evidenced by at least one peptide that binds to quartz but not gold.39 In that case, it was suggested that conformationally flexible peptides have greater propensity to bind to gold surfaces than more rigid peptides with similar amino acid sequences.38 From these findings, there is strong motivation to explore quantitative measurement methods in the context of probing peptide binding affinity and selectivity on different surfaces.
Conclusion
In this work, we have investigated the adsorption of a short peptide to four inorganic material substrates. A combination of experimental approaches allowed us to compare the adsorption kinetics and binding affinity of peptide attachment for the different substrates through measurements conducted at the single-molecule and ensemble-average levels. A general trend in the total mass uptake and binding affinity was observed, with the greatest values obtained for bound peptides on gold. These findings open the door to integrating experimental measurements conducted at the single-molecule and ensemble-average levels in order to understand the adsorption behavior of peptides on solid supports.Acknowledgements
The authors wish to acknowledge support from the National Research Foundation (NRF-NRFF2011-01) and Nanyang Technological University to N.J.C. T.H. acknowledges the support of MEXT KAKENHI Grant Number 26282118. Dr Jeongeun Seo is gratefully acknowledged for assistance with the figure illustrations.References
- A. Care, P. L. Bergquist and A. Sunna, Trends Biotechnol., 2015, 33, 259–268 CrossRef CAS PubMed.
- C. Tamerler, T. Kacar, D. Sahin, H. Fong and M. Sarikaya, Mater. Sci. Eng., C, 2007, 27, 558–564 CrossRef CAS.
- M. Sarikaya and C. Tamerler, Abstr. Pap. Am. Chem. Soc., 2009, 238 Search PubMed.
- S. R. Meyers, X. Khoo, X. Huang, E. B. Walsh, M. W. Grinstaff and D. J. Kenan, Biomaterials, 2009, 30, 277–286 CrossRef CAS PubMed.
- D. Khatayevich, M. Gungormus, H. Yazici, C. So, S. Cetinel, H. Ma, A. Jen, C. Tamerler and M. Sarikaya, Acta Biomater., 2010, 6, 4634–4641 CrossRef CAS PubMed.
- Y. Yamashita, H. Kirimura, M. Okuda, K. Nishio, K. I. Sano, K. Shiba, T. Hayashi, M. Hara and Y. Mishima, Small, 2006, 2, 1148–1152 CrossRef PubMed.
- K. Sano, H. Sasaki and K. Shiba, J. Am. Chem. Soc., 2006, 128, 1717–1722 CrossRef CAS PubMed.
- N. Matsukawa, K. Nishio, K. Sano, K. Shiba and I. Yamashita, Langmuir, 2009, 25, 3327–3330 CrossRef CAS PubMed.
- N. Matsukawa and I. Yamashita, Appl. Phys. Express, 2011, 4, 057001 CrossRef.
- C. R. So, J. L. Kulp, E. E. Oren, H. Zareie, C. Tamerler, J. S. Evans and M. Sarikaya, ACS Nano, 2009, 3, 1525–1531 CrossRef CAS PubMed.
- K. Ariga, Q. Ji, W. Nakanishi, J. P. Hill and M. Aono, Mater. Horiz., 2015, 2, 406–413 RSC.
- K. Ariga, J. Li, J. Fei, Q. Ji and J. P. Hill, Adv. Mater., 2016, 28, 1251–1286 CrossRef CAS PubMed.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 1992 Search PubMed.
- D. Leckband and J. Israelachvili, Q. Rev. Biophys., 2001, 34, 105–267 CrossRef CAS PubMed.
- R. A. Latour and L. L. Hench, Biomaterials, 2002, 23, 4633–4648 CrossRef CAS PubMed.
- R. A. Latour and C. J. Rini, J. Biomed. Mater. Res., 2002, 60, 564–577 CrossRef CAS PubMed.
- T. Hayashi, K. Sano, K. Shiba, Y. Kumashiro, K. Iwahori, I. Yamashita and M. Hara, Nano Lett., 2006, 6, 515–519 CrossRef CAS PubMed.
- T. Hayashi, K. I. Sano, K. Shiba, K. Iwahori, I. Yamashita and M. Hara, Langmuir, 2009, 25, 10901–10906 CrossRef CAS PubMed.
- Y. Arai, K. I. Okabe, H. Sekiguchi, T. Hayashi and M. Hara, Langmuir, 2011, 27, 2478–2483 CrossRef CAS PubMed.
- M. Mochizuki, M. Oguchi, S. O. Kim, J. A. Jackman, T. Ogawa, G. Lkhamsuren, N. J. Cho and T. Hayashi, Langmuir, 2015, 31, 8006–8012 CrossRef CAS PubMed.
- Y. Fang, N. Poulsen, M. B. Dickerson, Y. Cai, S. E. Jones, R. R. Naik, N. Kröger and K. H. Sandhage, J. Mater. Chem., 2008, 18, 3871–3875 RSC.
- M. Hnilova, C. R. So, E. E. Oren, B. R. Wilson, T. Kacar, C. Tamerler and M. Sarikaya, Soft Matter, 2012, 8, 4327–4334 RSC.
- J. P. Palafox-Hernandez, Z. Tang, Z. E. Hughes, Y. Li, M. T. Swihart, P. N. Prasad, T. R. Walsh and M. R. Knecht, Chem. Mater., 2014, 26, 4960–4969 CrossRef CAS.
- N.-J. Cho, H. Dvory-Sobol, A. Xiong, S.-J. Cho, C. W. Frank and J. S. Glenn, ACS Chem. Biol., 2009, 4, 1061–1067 CrossRef CAS PubMed.
- N.-J. Cho, S.-J. Cho, K. H. Cheong, J. S. Glenn and C. W. Frank, J. Am. Chem. Soc., 2007, 129, 10050–10051 CrossRef CAS PubMed.
- M. Rodahl, F. Höök, A. Krozer, P. Brzezinski and B. Kasemo, Rev. Sci. Instrum., 1995, 66, 3924–3930 CrossRef CAS.
- E. L. Florin, V. T. Moy and H. E. Gaub, Science, 1994, 264, 415–417 CAS.
- Y. Iiguni and H. Watarai, Analyst, 2010, 135, 1426–1427 RSC.
- S. Kado, K. Yamada, T. Murakami and K. Kimura, J. Am. Chem. Soc., 2005, 127, 3026–3030 CrossRef CAS PubMed.
- O. Mermut, D. C. Phillips, R. L. York, K. R. McCrea, R. S. Ward and G. A. Somorjai, J. Am. Chem. Soc., 2006, 128, 3598–3607 CrossRef CAS PubMed.
- C. Keller and B. Kasemo, Biophys. J., 1998, 75, 1397–1402 CrossRef CAS PubMed.
- A. M. Sultan, Z. E. Hughes and T. R. Walsh, Langmuir, 2014, 30, 13321–13329 CrossRef CAS PubMed.
- M. Hoefling, F. Iori, S. Corni and K.-E. Gottschalk, Langmuir, 2010, 26, 8347–8351 CrossRef CAS PubMed.
- F. Iori and S. Corni, J. Comput. Chem., 2008, 29, 1656–1666 CrossRef CAS PubMed.
- F. Iori, R. Di Felice, E. Molinari and S. Corni, J. Comput. Chem., 2009, 30, 1465–1476 CrossRef CAS PubMed.
- P. V. C. Medeiros, G. K. Gueorguiev and S. Stafström, Carbon, 2015, 81, 620–628 CrossRef CAS.
- Z. Zhong, S. Patskovskyy, P. Bouvrette, J. H. T. Luong and A. Gedanken, J. Phys. Chem. B, 2004, 108, 4046–4052 CrossRef CAS.
- S. Corni, M. Hnilova, C. Tamerler and M. Sarikaya, J. Phys. Chem. C, 2013, 117, 16990–17003 CAS.
- T. Kacar, J. Ray, M. Gungormus, E. E. Oren, C. Tamerler and M. Sarikaya, Adv. Mater., 2009, 21, 295–299 CrossRef CAS.
| This journal is © the Owner Societies 2016 |