
Linkage of the PAMAM type dendrimer with the gel type resin based on glycidyl methacrylate terpolymer as a method of preparation of the polymer support for the recyclable palladium catalyst for Suzuki–Miyaura cross-coupling reactions†
Agnieszka Bukowska*,
Wiktor Bukowski,
Karol Bester and
Sylwia Flaga
Faculty of Chemistry, Rzeszów University of Technology, al. Powstańców W-wy 6, 35-959 Rzeszów, Poland. E-mail: abuk@prz.edu.pl; Fax: +48 17 854 36 55; Tel: +48 17 865 13 38
First published on 28th May 2015
Abstract
Poly(amidoamine) PAMAM type hyperbranched systems were immobilized on an epoxy functionalized polymer based on glycidyl methacrylate (the GMA resin). Two methods were applied. The first included two-stage synthesis of the zeroth order PAMAM dendrimer (1), starting from tris(2-aminoethyl)amine (TAEA), and its immobilization on the GMA resin. The second relied on multi-stage chemical modification of the GMA resin in turn with TAEA, methyl acrylate and ethylenediamine. The provided multi NH2-functionalyzed polymers (abbreviated respectively 2 and 5) were used for immobilization of palladium(II) ions to obtain two polymer supported palladium catalysts (2-Pd and 5-Pd, respectively). The synthesized polymers and polymer supported Pd catalysts were characterized using different instrumental techniques (FT-IR, DR UV-Vis, ICP-OES, DSC and SEM). Catalyst 2-Pd was found as a clearly more active in the model reaction of p-bromoanisole. It was tested successfully as catalysts in the Suzuki–Miyaura cross-coupling reactions of a series of aryl bromides with phenylboronic acid in water–organic solvent systems. It was also recycled successfully ten times in the coupling reaction of p-bromoanisole with phenylboronic acid carried out in the mixture of isopropyl alcohol and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) at 70 °C.
:1 v/v) at 70 °C.
1. Introduction
Many palladium catalytic systems have been developed for the C–C cross-coupling reactions since the discovery of these transformations. They have been summarized in several reviews.1–8 Both the simple palladium(II) salts, e.g. PdCl2 and (CH3CO)2Pd, and different soluble palladium complexes, e.g. palladium phosphine, pincer, and NCH complexes, were explored to date as potential homogeneous catalysts. Numerous porous and nonporous materials were utilized as supports for immobilization of the palladium complexes and nanoparticles, e.g. carbon, metal oxides, silica, zeolites, soluble and insoluble polymer materials, including different dendrimers.Dendrimers and soluble and insoluble polymers are recognized as particular advantageous supports for stabilizing catalytically active palladium nanoparticles. The use of the typical homogeneous palladium catalysts usually makes their isolation problematical and increases a risk of contamination of the reaction products with this toxic metal. Furthermore, the reuse of homogeneous palladium compounds usually resulted in forming inactive palladium black. Therefore the use of the heterogeneous systems which are able to stabilize palladium nanoparticles seem to be more favorable.
Interesting supports for immobilization of the catalytically active palladium species turn out to be the polymer gels derived from the terpolymers of glycidyl methacrylate, styrene and divinylbenzene (GMA resins). The amine and imine functionalized gels based on the GMA resins, developed in our laboratory, showed relatively high catalytic activity in the Suzuki–Miyaura reactions of aryl bromides and iodides with aryl boronic acids, both when palladium was immobilized in the form of Pd(0) particles and as Pd(II) complexes.9–12 Unfortunately, attempts at catalyst reuse showed that a clear increase in the catalytic activity of the previously developed systems was observed after their reuse. To improve the stability of the polymer supported palladium catalysts in the Suzuki–Miaura reactions we decided to attach PAMAM type dendritic systems to the selected GMA gel, based on our previous works.
PAMAM dendrimers are well known for providing a good environment for stabilizing metal nanoparticles, including palladium.13–18 Encapsulation of metal nanoparticles inside poly(amidoamine) dendrimers was originally demonstrated by Tomalia19 and Crooks13,20 and co-workers. PAMAM dendrimers were previously studied several times as supports for palladium catalysts for C–C coupling reactions, both in a soluble form and after their immobilization of insoluble supports.21–26 The better results were obtained after immobilization of the poly(amidoamine) systems on insoluble supports.23–26
For instance, the Pd catalyst based on the soluble PAMAM G3-OH dendrimer21 was used only for the coupling of the aryl iodides consisting of electron withdrawing substituents with p-tolylboronic acid. The reaction was performed in boiling EtOH within about 2 h (5 examples). This catalyst was inactive under the same conditions in the reaction involving bromobenzene. This halide reacts with p-tolylboronic acid only in DMF at 153 °C within 48 h, providing 70% yield of the product.
Similar catalysts based on G1–G5 DAB-dendrimers (DAB – diaminobutane) described by Astruc et al.27 allowed converting iodobenzene to biphenyl within 1 h in the reaction with phenylboronic acid but at 100 °C using the mixture of CH3CN–H2O (3/1 v/v) and sodium acetate as a base. In this case, a decrease in the catalytic activity was observed when the dendrimer generation increased above G2. Furthermore, the catalyst based on G5 DAB-dendrimers was marked by the relatively low activity.
The catalysts obtained by Trzeciak and co-workers,22 based also on the soluble PAMAM–NH2 dendrimers (G2, G3 and G4 generation) allowed to convert 4-bromotoluene within 3 h in the reaction with phenylboronic acid performed at 80 °C in the i-PrOH–H2O mixture using 2 eq. Cs2CO3. In this case, the yield of 4-methylbiphenyl diminished slightly with an increase in the dendrimer generation.
The polymer supported Pd catalysts described by Sreekumar et al.,23 based on the commercial gel-type chloromethyl polystyrene (1% DVB) which was converted to NH2 form and used for the immobilization the PAMAM dendritic systems up to 3rd generation, involving Michael addition of methyl acrylate to the amino groups of the polymer followed by amidization with ethylenediamine, allowed to convert activated 4-nitroiodobenzene with phenylboronic acid in the dioxane–water mixture (4/1 v/v) at 100 °C using Na2CO3 as a base.
Another catalysts synthesized by Murugan et al.,24 consisting of the poly(amidoamine) of 1st, 2nd and 3rd generation immobilized on the chloromethyl polystyrene cross-linked with 2% divinylbenzene, used in the reaction of bromobenzene with phenylboronic acids performed in the boiling EtOH–H2O mixture using Na2CO3 as a base, yielded about 74.8–88.5% biphenyl within 12 h. In this case it was found that the catalyst activity increased slightly with an increase in poly(amidoamine) generation.
Quite recently, the synthesis of dendrimer encapsulated Pd nanoparticles (Pd DENs) anchored onto the inner walls of flow microreactors was presented.25 The catalytic platform formed in this way was successfully used in a test Suzuki–Miyaura cross-coupling reaction of iodobenzene with p-tolylboronic acid in ethanol. The system consisting of the G3 PAMAM dendrimer attached covalently to the inner walls of the microreactor exhibited the best activity as compared to higher dendrimer generations (G4 and G5). This catalytic system exhibited not only very high activity but also good stability. It could be used in the model Suzuki–Miyaura reaction for more than 7 days with a low Pd leaching of 1.2 ppm. This system was also tested successfully in the reaction of a series of para-substituted aryl halides and boronic acids and in the Heck–Cassar coupling reaction (copper-free Sonogashira reaction).26
2. Experimental
2.1 Materials
The chemicals used for preparation of the polymer supported palladium(II) catalytic systems and in the performed catalytic studies were commercially available from Aldrich, Merck or Fluka and were used as received, unless otherwise stated. The GMA resin (an epoxy group loading of ∼1.40 mmol g−1) used as a matrix for preparation of the polymer supported poly(amidoamines) was synthesized from the mixture of glycidyl methacrylate (GMA, 20 mol%), styrene (S, 77 mol%) and divinylbenzene (DVB, 3 mol%) using the procedure described previously.282.2 Synthesis of polymer supported dendrimers
The polymer supports for palladium(II) ion immobilization (resins 2 and 5) were obtained using two methods. The first method included the synthesis of PAMAM dendrimer 1 according to the procedure described in ref. 29 and its immobilization on the GMA resin to obtain resin 2. The second method consisted in converting the GMA resin in turn on the reaction with an excess of tris(2-aminoethyl)amine (TAEA) (similarly to ref. 30), methyl acrylate and ethylenediamine (EDA) (for detail see ESI†).2.3 Immobilization of palladium(II) ions – general procedure
Resin 2 or 5 (0.5 g) was swelled in 5 ml THF in a glass reactor. Pd(OAc)2 (0.18 g) dissolved in the mixture of THF and H2O (20:1 v/v) was then added. The reactor was sealed and placed on a vibrational shaker. The mixture has shaken for 24 hours at room temperature. The polymer supported palladium complex (2-Pd or 5-Pd, respectively) was transferred to a polypropylene syringe equipped with a Luer valve and a cotton filter and washed many times with the THF–H2O mixture to remove physically absorbed palladium(II) ion residuals. The beads of the final products were dried under reduced pressure at 40 °C.
2.4 Resin and catalyst characterization
The percentage of nitrogen in the synthesized polymers was determined using the classic Kjeldahl method. The Pd loadings in the polymer supported complexes was determined using a ICP-OES Horiba Jobin Yvon Optima 2 spectrometer (λ = 340.458 nm). The FT-IR spectra of the synthesized polymer materials were recorded using a Thermo Scientific Nicolet 8700 spectrometer and KBr pellets. The polymer supported palladium complexes were also studied by a single bead technique using a FTIR Nicolet iN10MX microscope and a micro-compression diamond cell. The DR UV-Vis spectra of the synthesized polymer materials were recorded using a Jasco V-670 spectrophotometer equipped with a Jasco ISN-723 UV-Vis NIR 60 mm integrating sphere and BaSO4 as a standard. The morphology of the beads of the polymer supported catalysts was studied by means of a VEGA3 TESCAN scanning electron microscope equipped with a X-ray energy dispersive (EDS) system. The images were recorded in two modes using a secondary electron detector (SEI images) and a back scattered electron detector (BSE). Glass-transition temperature (Tg) measurements were performed via differential scanning calorimetry with a Toledo 822e calorimeter (Mettler Toledo, Switzerland) with Stare System software. The resins were heated from 30 to 300 °C and then cooled to 30 °C. This procedure was repeated twice. The rate of heating and cooling was adjusted to 10 °C min−1.The swelling of the resins was determined in the following way: 0.08 g of an appropriate dry resin was added to a 1 ml graduated syringe that was equipped with a polypropylene filter. An appropriate solvent was added. After swelling equilibrium was achieved, the excess of the solvent was removed with the plunger.
The GC and GCMS analyses were performed using Agilent 7890 instruments equipped with split/splitless injectors, autosamplers, a flame ionization or mass detector, and HP-5+ or HP-5MS capillary column, respectively.
2.5 Catalytic tests
3. Results and discussion
3.1 Synthesis of polymer supports
We reported previously that the amine12 and imine11 functionalized polymers derived from the GMA gels and a series of aliphatic amines and aldehydes can be used successfully as supports for palladium species. The polymer supported palladium catalysts provided in this way turned out to be active in the Suzuki–Miyaura cross-coupling of a series of aryl bromides and iodides with phenylboronic acid under relatively mild conditions. Developing our studies in the area of the preparation of the new polymer supports for the catalytic purposes we decided to attach a dendritic poly(amidoamine) system to the selected GMA resin. It was expected that the presence of numerous nitrogen donor centers will influence advantageously the distribution of the catalytically active palladium species within the polymer beads and improve the catalyst stability.To achieve the goals mentioned above two strategies were considered. The first method included the synthesis of dendrimer 1 based on tris(2-aminoethyl)amine (TAEA) (Scheme S1†). The synthesis of 1 was performed according the procedure described in ref. 29. The obtained PAMAM type dendrimer was then applied for modification of the GMA resin according to Scheme 1. The reaction was performed in the mixture of DMF and MeOH (3:1 v/v). This solvent system ensured good solubility of 1. Furthermore, it swelled well both the starting resin and that formed as a result of immobilization of dendrimer moieties on the GMA resin. Several attempts were performed to optimize the reaction conditions (Table S1†). The effectiveness of the performed reactions was concluded based on the percentage of nitrogen in the final products (Table S1†), on the one hand, and comparing the FTIR spectra recorded for the GMA resin and the products of its modification, on the other hand (Fig. S2†). Independently of the molar ratio of 1 to epoxy groups of the starting resin the identical yields of modification after 24 hour reaction at 60 °C were observed. When the temperature was increased by 10 °C, an increase of 5% in the yield could be noted. Treating the GMA resin with the equimolar quantity of 1 at room temperature for 7 days resulted in 32% modification of the epoxy groups. Based on the obtained results the temperature of 70 °C was acknowledged as optimal for the preparation of polymer supported dendrimer 2.
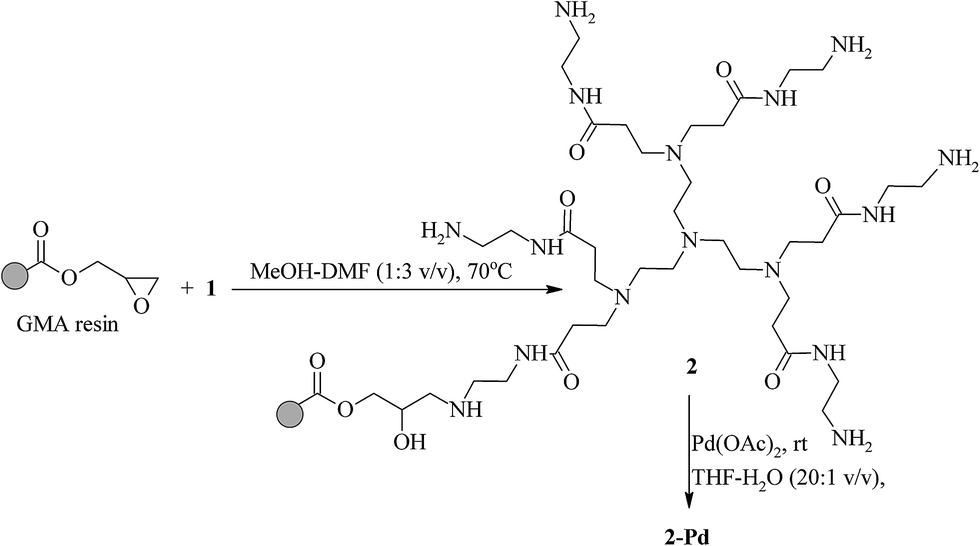 | ||
| Scheme 1 Synthesis of polymer supported catalyst 2-Pd. | ||
Immobilization of 1 on the GMA resins resulted in changes in the FTIR spectra of the polymer beads (see Fig. S2†). The intensity of the bands at 840 cm−1 and 900 cm−1 (the epoxy ring stretches) diminished and strong absorption in the range of 3600–2500 cm−1 (OH and N–H stretching vibrations) was detected. Furthermore, the new bands at 1650 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration) and 1550 cm−1 (N–H deformation and C–N stretching vibrations characteristic of amides) appeared. These findings prove the immobilization of 1 according to Scheme 1.
O stretching vibration) and 1550 cm−1 (N–H deformation and C–N stretching vibrations characteristic of amides) appeared. These findings prove the immobilization of 1 according to Scheme 1.
The final beads of polymer supported dendrimer 2 were yellowish in color and transparent. The modified polymer showed different swelling ability compared with GMA resin (Table S2†). The swelling of 2 improved in methanol, water and the mixtures of MeOH–CH2Cl2 (1:1 v/v), H2O–THF (1:1 v/v), DMF–MeOH (1:3 v/v), i-PrOH–H2O (1:1 v/v), EtOH–H2O (1:1 v/v). It produced poorer results in methylene chloride, THF, THF–H2O (20:1 v/v), DMF, DMF–MeOH (3:1 and 1:1 v/v) and the mixture of MeOH–CH2Cl2 (5:1 v/v).
Furthermore, the modification of the GMA resin with 1 resulted in an increase in the glass temperature (Tg) of the polymer beads of ∼17 °C (Tg,GMA resin = 113 °C). It might be caused by the hydrogen interactions between the functional groups introduced to the starting material with 1 and those formed as a consequence of the epoxy ring opening reaction or by the side cross-linking of the resin beads performing as a result of the reaction of the NH2 groups of 1 to two or more epoxy groups attached to neighboring polymer chains.
In the view of the relatively low yield of the direct immobilization of 1 (32–39%) compared with the similar transformations including the reaction of the epoxy groups with tris(2-aminoethyl)amine,30 the multi-stage path of immobilization of the dendritic PAMAM type system on the GMA resin was also examined (Scheme 2). The starting resin bearing epoxy groups was first reacted with TAEA using the procedure described previously in ref. 27. Provided in this way the GMA–TAEA resin (3) was then treated with an excess of methyl acrylate in MeOH–CH2Cl2 (3:1 v/v) at 0 °C to room temperature for 48 hours. Finally, the resin with methyl ester functionalities (4) prepared in the reaction of 3 with methyl acrylate was reacted with an excess of EDA (150 eq.) in MeOH–CH2Cl2 (3:1 v/v) at −30 °C and rested at room temperature for 48 hours. The effectiveness of the procedure was proved using FTIR spectroscopy and elementary analysis.
 | ||
| Scheme 2 Synthesis of polymer supported palladium catalyst 5-Pd. | ||
The conversion of the amine groups under the influence of methyl acrylate resulted in changes in absorption in the ranges characteristic of the N–H vibrations (Fig. S3†). The broad absorption bands observed initially for 3 in the range of from 3600 to 2200 cm−1 (N–H stretching vibrations) underwent narrowing in the case of 4. In addition, an increase in the intensity of the band at 1750 cm−1 was noted for 4 as a consequence of increasing the loading of CO groups. Finally, the bands at 1650 and 1560 cm−1 which appeared in the spectra of 5 proved the presence of the secondary amide functionalities in the final product. Moreover, the introducing amide (NH) and amine (NH2) groups into the polymer beads, as a consequence of the substitution of CH3O groups by EDA moieties, resulted in a re-increase in absorption intensity in the range of N–H stretching vibrations.
The elemental analysis showed 11.8% N loading in the final product (5). It corresponds to 8.4 mmol N per gram of the resin beads (Table 1). Given these values the yield of the multi-stage modification of the GMA resin was assessed. It amounted to about 42%.
| Resin | Nitrogen loading | Number of N atoms in the dendritic moiety | Catalyst | Pd loading | N/Pd ratio | ||
|---|---|---|---|---|---|---|---|
| % | mmol g−1 | % | mmol g−1 | ||||
| 2 | 5.7 | 4.1 | 16 | 2-Pd | 16.3 | 1.5 | 2.7 |
| 5 | 11.8 | 8.4 | 14 | 5-Pd | 8.1 | 0.8 | 10.5 |
Thus, one can easily notice that although the multi-stage procedure of the immobilization of the PAMAM like hyperbranched system provided a loading of nitrogen about twice higher than the direct immobilization of 1, the final effectiveness of immobilization of the poly(amidoamine) hyperbranched system also in this case was relatively low. The higher nitrogen loading in the beads of 5 compared with the loading of 2 resulted from the high efficiency of the first stage of modification – the addition of tris(2-aminoethyl)amine to the GMA resin. The yield of TAEA addition amounted to about 88% while the direct immobilization of 1 gave only about 32%.
The chemical transformations performed according to the multi-stage path also resulted in changes in the values of Tg of the provided polymer beads compared with the GMA resin. The addition of TAEA led to an increase in Tg of ∼20 °C. Probably, this finding, similarly to that observed in the case of the immobilization of 1, is caused by the hydrogen interactions including the polar groups introduced with TAEA molecules. The cross-linking of the beads as a results of the side reactions of NH2 groups of the immobilized TAEA moieties of with the epoxy groups attached to the neighboring polymer chains can be another reason of increasing Tg in this case. The addition of methyl acrylate to amine groups of the immobilized TAEA moieties resulted in lowering Tg of the resin to 125 °C. The glass temperature of the polymer beads increased once more, to 140 °C, after treating the polymer supported methyl ester with an excess of ethylenediamine. It is probably caused again by the escalation of the hydrogen interactions including NH2 amine groups of EDA moieties and the further cross-linking of the polymer beads resulting from the side reactions of the diamine moieties with more than one methoxy group.
The hydrogen interaction and side cross-linking seem to be also responsible for a dramatic decrease in the swelling ability of 5 in the most explored solvent systems (Table S2†).
3.2 Palladium(II) ion immobilization
Resins 2 and 5 bearing attached dendritic systems were used for immobilization of palladium(II) ions to obtain the new catalysts for Suzuki–Miyaura reactions. The polymer supports were shaken with the solution of palladium(II) acetate for 24 hours at room temperature. Equimolar quantities of Pd(II) ions were used in the performed experiments. THF and H2O in a ratio of 20:1 v/v were used as a reaction medium. As a result, polymer supported palladium systems 2-Pd and 5-Pd consisting of 16.3 and 8.1% Pd(II) ions, respectively, were obtained. Interestingly, resin 2 bearing the moieties of dendrimer 1 was able to bind about twice as much Pd(II) ion than resin 5 with the hyperbranched system obtained using the multi-stage procedure, although the loading of nitrogen was about half higher in the case of 5 than the loading of 2. Probably, some of the donor groups within the beads of 5 were inaccessible for palladium ions under the applied reaction conditions due to the lesser ability of this resin to swell in the mixture of THF–H2O (20:1 v/v) which was used as a medium for Pd(II) ion complexing.
The immobilization of Pd(II) ions on 2 and 5 led to a further decrease in the swelling ability of the polymer beads (Table S2†). However the changes of the swelling were clearly less in the case of the pair of 5 and 5-Pd.
The immobilization of Pd(II) ions on 2 and 5 resulted in changes in the FTIR spectra of the polymer beads, particularly in the ranges characteristic of NH and carboxylic group vibrations.31 These findings indicated that amine groups are involved in coordinating palladium(II) ions bound to acetate ions (Fig. S4†).
The immobilization of palladium(II) acetate on 2 and 5 resulted in changes of the DR UV-Vis spectra of the obtained materials as well (Fig. S5 and S6†). The red shift of the maximum absorption characteristic of d–d transitions of Pd(II), observed at 400 nm for Pd(AcO)2, proves that the donor groups of 2 and 5 are employed in coordinating the metal ions. The shift was higher in the case of the spectrum recorded for 2-Pd. It results probably from its higher Pd loading.
3.3 Catalytic activity of the polymer supported palladium catalysts
Polymer supported Pd(II) complexes 2-Pd and 5-Pd were explored as catalysts in the Suzuki–Miyaura reactions (Table 2).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Ar-Br | T, °C | mol% 2-Pd | Solvent | Base | Time, min | Conversion of ArBrb, % |
|
|||||||
| R |
|||||||
| a 0.9 mmol ArBr, 1.08 PhB(OH)2, 1.8 mmol base.b Determined by GC. | |||||||
| 1 | H | 25 | 0.5 | i-Pr:H2O (1:1 v/v) |
KOH | 600 | 100 |
| 2 | H | 50 | 0.5 | i-Pr:H2O (1:1 v/v) |
KOH | 90 | 99 |
| 3 | H | 70 | 0.5 | i-Pr:H2O (1:1 v/v) |
KOH | 80 | 100 |
| 4 | H | 70 | 0.5 | i-Pr:H2O (1:1 v/v) |
K2CO3 | 80 | 99.5 |
| 5 | H | 70 | 0.5 | i-Pr:H2O (1:1 v/v) |
K3PO4 | 80 | 99 |
| 6 | H | 70 | 0.5 | i-Pr:H2O (1:1 v/v) |
Cs2CO3 | 80 | 100 |
| 7 | H | 70 | 0.5 | Diglym:H2O (1:1 v/v) |
KOH | 240 | 100 |
| 8 | H | 70 | 0.5 | EtOH:H2O (1:1 v/v) |
KOH | 25 | 100 |
| 9 | H | 70 | 0.5 | DMF:H2O (1:1 v/v) |
KOH | 240 | 100 |
| 10 | OCH3 | 70 | 0.5 | i-Pr:H2O (1:1 v/v) |
KOH | 30 | 100 |
| 11 | CH3 | 70 | 0.5 | i-Pr:H2O (1:1 v/v) |
KOH | 40 | 100 |
| 12 | OCH3 | 70 | 1 | i-Pr:H2O (1:1 v/v) |
KOH | 15 | 100 (1st run) |
| 13 | OCH3 | 70 | 1 | i-Pr:H2O (1:1 v/v) |
KOH | 25 | 100 (2nd run) |
| 14 | OCH3 | 70 | 1 | i-Pr:H2O (1:1 v/v) |
KOH | 40 | 99 (3rd run) |
| 15 | OCH3 | 70 | 1 | i-Pr:H2O (1:1 v/v) |
KOH | 40 | 100 (4th run) |
| 16 | OCH3 | 70 | 1 | i-Pr:H2O (1:1 v/v) |
KOH | 40 | 99 (5th run) |
| 17 | OCH3 | 70 | 1 | i-Pr:H2O (1:1 v/v) |
KOH | 40 | 99 (6th run) |
| 18 | OCH3 | 70 | 1 | i-Pr:H2O (1:1 v/v) |
KOH | 40 | 99 (7th run) |
| 19 | OCH3 | 70 | 1 | i-Pr:H2O (1:1 v/v) |
KOH | 40 | 98 (8th run) |
| 20 | OCH3 | 70 | 1 | i-Pr:H2O (1:1 v/v) |
KOH | 40 | 100 (9th run) |
| 21 | OCH3 | 70 | 1 | i-Pr:H2O (1:1 v/v) |
KOH | 40 | 99 (10th run) |
| 22 | OH | 70 | 0.5 | i-Pr:H2O (1:1 v/v) |
KOH | 150 | 90 |
| 23 | F | 70 | 0.5 | i-Pr:H2O (1:1 v/v) |
KOH | 20 | 99 |
| 24 | Naphtyl | 70 | 0.5 | i-Pr:H2O (1:1 v/v) |
KOH | 240 | 99 |


Catalyst 2-Pd (0.5 mol%) was selected to study the effect of temperature on the rate of the coupling reaction of bromobenzene with phenylboronic acid. Similarly to our previous work, the coupling was performed in the presence of KOH (2 eq.), at 25, 50 or 70 °C, using the mixture of i-PrOH and H2O (1:1 v/v) as a reaction medium. It was found that under applied conditions the complete or nearly complete conversion of bromobenzene was obtained respectively within about 600, 120 and 80 min. However, lower reaction temperatures caused longer induction periods (Fig. 1). The s-shaped character of the kinetic curves proved that 2-Pd could be recognized as a precursor of the catalytic species which form within the beads under reaction conditions as a result of reduction of Pd(II) ions to Pd(0) nanoparticles. SEM EDS analysis showed the presence of Pd nanoparticles on the surface of the beads recovered after completing the reactions.
 | ||
| Fig. 1 Effect of temperature on the coupling of bromobenzene with phenylboronic acid in the presence of 2-Pd (0.9 mmol ArBr, 1.08 PhB(OH)2, 1.8 mmol KOH, 4.5 ml i-PrOH–4.5 ml H2O, 0.5 mol% cat). | ||
K2CO3, Cs2CO3 and K3PO4, besides KOH, were also explored as bases in the reaction of PhBr with PhB(OH)2 carried out in the presence of 2-Pd. All these bases allowed to complete the coupling within about 60 minutes at 70 °C (Fig. S7†). However, the use of the basic salts shortened clearly an induction period of the reaction.
Some other solvent systems such as the mixtures of diglyme–H2O and DMF–H2O and EtOH–H2O (1:1 v/v) could be also used as a medium for performing the coupling of PhBr with PhB(OH)2 in the presence of 2-Pd (Fig. S8†). The use of the first two mixtures, however, turned out to be clearly less advantageous than the use of the i-PrOH–H2O mixture. In these solvent systems the coupling was 4–5 times slower. The third of the solvents mixture mentioned above, containing the primary alcohol, facilitated the coupling about 2.5 faster. Probably, the presence of alcohols facilitate the formation of catalytically active Pd(0) nanoparticles under reaction conditions.
Complex 2-Pd facilitated complete coupling PhBr with PhB(OH)2 about 3 times faster than 5-Pd (Fig. 2). Therefore, it was used for the further catalytic studies.
 | ||
| Fig. 2 Kinetics plots obtained for the reaction of bromobenzene with phenylboronic acid performed in the presence of 2-Pd and 5-Pd (0.9 mmol ArBr, 1.08 PhB(OH)2, 1.8 mmol KOH, 4.5 ml i-PrOH–4.5 ml H2O, 0.5 mol% cat., 70 °C). | ||
The studies were performed for a series aryl bromides which were reacted with phenylboronic acid in the medium of the i-PrOH–H2O mixture, at 70 °C, using KOH as a base and 0.5% 2-Pd. It was found that under the applied conditions the coupling of the most of the explored bromides were complete within 40–240 minutes. An incomplete conversion (about 90%) was noted only in the case of p-bromophenol (Table 2).
In the presence of 2-Pd fluorobromobenzene turned out to be the most reactive. It reacted quantitatively with PhB(OH)2 within 20 minutes. The reactivity of Me- and MeO-substituted bromobenzenes was very similar. These bromides were converted completely within about 25–30 minutes. Two unsubstituted bromoarenes (bromobenzene and bromonaphthalene) reacted with phenylboronic acid clearly slower. The complete or nearly complete conversion of bromobenzene occurred within 60 min. 1-Bromonaphtalene was converted within about 240 minutes. p-Bromophenol and bromobenzene reacted with phenylboronic acid similarly at the initial stage of the reaction but in the case of the first the reaction slowed down after achieving about 60% conversion.
Attempts of the use of catalyst 2-Pd in the coupling reactions of aryl chlorides and phenylboronic acid were also performed. However, they were failed.
It has been shown that numerous factors (e.g. the nature of reactants and basic reagents, solvents and ligands, reaction temperature, reagent concentrations) can influence the activity of palladium(0) species in the Suzuki–Miyaura coupling reactions. Thus, it was very difficult to make comparisons of the activity of the catalyst described in this work with the activity of different palladium catalytic systems published previously. That is why we limited to the systems developed in our labs and the catalysts based on soluble and insoluble poly(amidoamine) PAMAM dendritic systems used in batch conditions by other authors.
Compared the activity of 2-Pd (and 5-Pd) with the activity of the polymer supported Pd catalysts based on imine functionalized gels described in ref. 12 we could conclude that the former showed clearly higher activity than the latter. The activity of 2-Pd and 5-Pd is similar to the amine functionalized systems described in ref. 11.
The catalysts described in this work seems be more active than those described by other authors in ref. 21–26, based on both the soluble and insoluble supports.
Catalysts 2-Pd and 5-Pd differed from the polymer supported dendritic poly(amidoamine) systems reported previously with the clear higher loading of palladium in the polymer beads. Furthermore, catalyst 2-Pd, which showed clearly higher activity, was prepared based on the support obtained by the direct immobilization of the poly(amidoamine) dendrimer on the polymer resin. Another difference between previously used catalysts and the catalysts described herein is that 2-Pd and 5-Pd were applied for the coupling reactions in the form of Pd(II) complexes. It caused that the catalytically active Pd(0) species indispensable for performing the oxidative addition of aryl halides had to form in situ under reaction conditions. Maybe, this factor contributed to their better distribution within the polymer beads increasing the activity of the Pd(0) nanoparticles encapsulated in the beads of 2-Pd and 5-Pd. In our previous work, it was showed using SEM EDS analysis that the reduction of the Pd(II) ions immobilized on the gel-type polymer conducting under Suzuki–Miyaura reaction conditions results in distributing Pd(0) nanoparticles within the hole beads.12
The polymer supported catalysts described by Sreekumar23 and Murugan24 and co-workers were prepared including the stage of reduction of the Pd(II) ions, initially coordinating with dendritic poly(amidoamine) moieties immobilized on the polystyrene core, with hydrazine23 or NaBH4.24 Pd(0) nanoparticles which formed as a result of the quick reduction could be stabilized by dendritically functionalized polymer first of all within an external part of the nonporous glassy polymer beads (the polymers applied as catalyst supports had the gel-type nature) due to incompatibility of the hydrophobic beads (cross-linked polystyrene was used as a starting material) with polar solvents which had to be use as a reaction medium.
3.4 Catalyst recycling
The possibility of the reuse of catalyst 2-Pd was also studied. The experiments were performed for the reaction of p-bromoanisole with phenylboronic acid. The following reaction conditions were applied: 1 mol% of Pd in relation to bromoanisole, 70 °C, 2 eq. KOH, i-Pr–H2O (1:1 v/v). The catalyst beads were reused nine times without regeneration. It was observed that the color of the polymer beads became darker and darker after each next reuse of the catalyst. This finding confirmed the gradual reduction of Pd(II) ions to Pd(0) species conducting within the polymer beads under reaction conditions.
Based on the FTIR spectra of the flattened beads recorded for the fresh and recovered catalyst using a FTIR microscope (Fig. 3) we concluded that Pd(II) ion reduction was accompanied with the migration of acetate ions from the beads. A clear decrease in the intensity of the absorption bands at 1550 and 1320 cm−1 characteristic of acetate ions was observed in the spectra of the recovered beads. The similar findings we observed in our two previous work.11,12
 | ||
| Fig. 3 FTIR spectra of 2-Pd recorded for the fresh and recovered catalyst. | ||
Using the ICP-OES analysis it was established that there were not observed the distinct changes in the Pd loading for the catalyst beads recovered after the first run (Table 3). However, the loading of palladium diminished in about 9% after 5th run and about 12.5% after 10th run. The gradual leaching of palladium from the support seems to prove the mechanisms proposed for the C–C coupling reactions in the presence of DENs.32
| Catalyst 2-Pd | Pd content 2-Pd, wt% |
|---|---|
| Fresh | 16.34 |
| After 1st run | 16.55 |
| After 5th run | 14.89 |
| After 10th run | 14.30 |
The changes of the Pd loading did not influence the efficiency of 2-Pd. It provided the complete or nearly complete conversion of p-bromoanisole even after the 10th use although its activity changed in the first three runs (Table 3). After the 3rd run the recovered catalyst demonstrated the unchangeable activity during the reuse up to the 10th run, although some differences in the initial periods of the reaction could be still noted (Fig. 4); the conversion of p-bromoanisole observed after the first 10 minutes of the reaction increased from 5% in the 2nd, 8% in the 3rd, 25% in the 5th to 78% in the 9th run. This finding seems to prove the developing the catalytically active palladium nanoparticles within the polymer supports under reaction conditions.
 | ||
| Fig. 4 Recycling of 2-Pd in the reaction of p-bromoanisole with phenylboronic acid (0.9 mmol p-CH3OArBr, 1.08 PhB(OH)2, 1.8 mmol KOH, 4.5 ml i-PrOH–4.5 ml H2O 1 mol% cat., 70 °C). | ||
To exclude the presence of the free homogenous catalytic species in the reaction mixture the Sheldon test was performed. Catalyst 2-Pd was filtered off from the hot reaction mixture after attaining 60% conversion of p-bromoanisole. Lack of the further conversion of aryl bromide even after continuing the reaction for 4 hours at 70 °C was observed.
The beads of the fresh and recovered catalyst were also studied using scanning electron microscopy. The representative microphotographs taken for the fresh catalyst and the beads recovered after their the 1st and 10th use are presented in Fig. 5. The SEM images combined with EDS analysis proved the presence of palladium nanoparticles on the surface of the recovered beads. A number and size of the particles increased in an consequence of the subsequent use of 2-Pd. It was also noted that the surface of the beads had become rougher and rougher due to the partial mechanical destruction of the beads under reaction conditions. The last might also influence advantageously the distribution of Pd(0) nanoparticles on the surface of the polymer beads and their catalytic activity.
 | ||
| Fig. 5 SEM images of 2-Pd; A – fresh catalyst, B – after 1st reuse, C – after 10th reuse. | ||
Comparing the results of the catalytic studies described in this work with the results obtained previously by our group for the polymer supported catalysts consisting of the simple polyamine moieties and their imine derivatives immobilized on the same GMA resins11,12 we can conclude that catalyst 2-Pd comprising the moieties of PAMAM dendrimer 1 shows clearly better stability than the latter. The catalysts developed previously dramatically lost the activity after each their reuse in the model Suzuki–Miyaura reaction.
Catalyst 2-Pd seems to be also more stable than the catalysts bearing dendritic poly(amidoamine) moieties reported previously.22–24 The catalyst developed by Trzeciak and co-workers22 based on the soluble PAMAM dendrimers lost quickly the activity during attempts of their reuse. The heterogeneous polymer supported Pd catalysts obtained using the PAMAM dendritic systems immobilized on the polystyrenes23,24 could be recycled 3–5 times without considerable loss of activity but their activity gradually decreased on further recycling stages. The catalyst presented in this work seem to stabilize its activity after the first three uses.
Probably, the higher stability of 2-Pd results from the covalent linking the moieties of dendrimer 1 with the gel type GMA resin. The support provided in this way is less hydrophobic than classic polystyrene resins. It contain high local concentration of donor groups (NH2, NH, CO, C–O, and OH; the last groups form after ring opening reaction upon 1) which might participate in stabilization of palladium particles. The benefits of covalent immobilization of dendritic PAMAM systems was also noted by Ricciardi et al. in published recently works.25,26
4. Conclusion
To sum up, the results of the performed catalytic studies led us to conclusion that the immobilization of Pd(II) ions on the gel consisting of the moieties of the PAMAM type dendrimer provides the polymer supported Pd(II) complexes which can be used as effective catalysts in the Suzuki–Miyaura cross-coupling reactions of aryl bromides with phenylboronic acid (Scheme 3). | ||
| Scheme 3 Suzuki–Miyaura cross-coupling reactions. | ||
It was concluded that 2-Pd bearing the moieties of dendrimer 1 stands out from the similar “dendritic” catalytic systems described previously in open literature.21–24 The developed catalyst retained its catalytic activity up to the 10th reuse in batch conditions.
Acknowledgements
The authors thank Piotr Sagan from University of Rzeszów for the SEM measurements.References
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176–4211 CrossRef CAS.
- V. Farina, Adv. Synth. Catal., 2004, 346, 1553–1582 CrossRef CAS PubMed.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS PubMed.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS PubMed.
- Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181–5203 RSC.
- N. Selander and K. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed.
- Á. Molnár, Chem. Rev., 2011, 111, 2251–2320 CrossRef PubMed.
- A. M. Trzeciak, E. Mieczyńska, J. J. Ziółkowski, W. Bukowski, A. Bukowska, J. Noworól and J. Okal, New J. Chem., 2008, 32, 1124–1130 RSC.
- T. Borkowski, A. M. Trzeciak, W. Bukowski, A. Bukowska, W. Tylus and L. Kępiński, Appl. Catal., A, 2010, 378, 83–89 CrossRef CAS PubMed.
- K. Bester, A. Bukowska and W. Bukowski, Appl. Catal., A, 2012, 443–444, 181–190 CrossRef CAS PubMed.
- K. Bester, A. Bukowska and W. Bukowski, J. Mol. Catal. A: Chem., 2013, 378, 124–134 CrossRef CAS PubMed.
- R. M. Crooks, M. Zhao, L. Sun, V. Chechlik and L. K. Yeung, Acc. Chem. Res., 2001, 34(3), 181–190 CrossRef CAS PubMed.
- R. Andrés, E. de Jesús and J. C. Flores, New J. Chem., 2007, 31, 1161–1191 RSC.
- X. Peng, Q. Pan and G. L. Rempel, Chem. Soc. Rev., 2008, 37, 1619–1628 RSC.
- M. Pittelkow, T. Brock-Nannestad, K. Moth-Poulsen and J. B. Christensen, Chem. Commun., 2008, 2358–2360 RSC.
- V. S. Myers, M. G. Weir, E. V. Carino, D. F. Yancey, S. Pande and R. M. Crooks, Chem. Sci., 2011, 2, 1632–1646 RSC.
- Q. M. Kainz and O. Reiser, Acc. Chem. Res., 2014, 47(2), 667–677 CrossRef CAS PubMed.
- L. Balogh and D. A. Tomalia, J. Am. Chem. Soc., 1998, 120, 7355–7356 CrossRef CAS.
- M. Zhao, L. Sun and R. M. Crooks, J. Am. Chem. Soc., 1998, 120, 4877–4878 CrossRef CAS.
- M. Pittelkow, K. Moth-Poulsen, U. Boas and J. B. Christensen, Langmuir, 2003, 19, 7682–7684 CrossRef CAS.
- T. Borkowski, P. Subik, A. M. Trzeciak and S. Wołowiec, Molecules, 2011, 16, 427–441 CrossRef CAS PubMed.
- G. R. Krishnan and K. Sreekumar, Soft Mater., 2010, 8(2), 114–129 CrossRef CAS PubMed.
- E. Murugan, J. N. Jebaranjitham and A. Usha, Appl. Nanosci., 2012, 2, 211–222 CrossRef CAS PubMed.
- R. Ricciardi, J. Huskens, M. Holtkamp, U. Karst and W. Verboom, ChemCatChem, 2015, 7, 936–942 CrossRef CAS PubMed.
- R. Ricciardi, J. Huskens and W. Verboom, Org. Biomol. Chem., 2015, 13, 4953–4959 CAS.
- J. Lemo, K. Heuzé and D. Astruc, Inorg. Chim. Acta, 2006, 359, 4909–4911 CrossRef CAS PubMed.
- A. Bukowska, W. Bukowski and J. Noworól, J. Appl. Polym. Sci., 2007, 106, 3800–3807 CrossRef CAS PubMed.
- M. X. Tang, C. T. Redemann and F. C. Szoka Jr, Bioconjugate Chem., 1996, 7, 703–714 CrossRef CAS PubMed.
- A. Bukowska and W. Bukowski, J. Appl. Polym. Sci., 2012, 124(2), 904–912 CrossRef CAS PubMed.
- M. Badertscher, P. Bühlmann and E. Pretsch, in Structure Determination of Organic Compounds, Springer-Verlag, Berlin, Heidelberg, 2009 Search PubMed.
- A. K. Diallo, C. Ornelas, L. Salmon, J. Ruiz Aranzaes and D. Astruc, Angew. Chem., Int. Ed., 2007, 46, 8644–8648 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04637h |
| This journal is © The Royal Society of Chemistry 2015 |