Improving the esterification activity of Pseudomonas fluorescens and Burkholderia cepacia lipases via cross-linked cyclodextrin immobilization†
Ivaldo I. Juniora,
Emanuela Calcio Gaudinob,
Katia Martinab,
Giancarlo Cravottob,
Rafael Luquec and
Rodrigo O. M. A. de Souza*a
aDepartamento de Química Orgânica, Federal University of Rio de Janeiro, Brazil. E-mail: souzarod21@gmail.com
bDipartimento di Scienza e Tecnologia del Farmaco, University of Turin, Via P. Giuria 9, 10125 Torino, Italy
cDepartamento de Química Orgánica Universidad de Córdoba, Edificio Marie Curie (C-3), Campus de Rabanales, Ctra Nnal IV-A, Km 396, E14014, Cordoba, Spain. E-mail: q62alsor@uco.es
First published on 1st July 2014
Abstract
The search for a new, efficient and sustainable matrix for biocatalyst immobilization is a growing area in biotechnology. Our proposed approach deals with the utilization of solid cross-linked β-cyclodextrin as supports for enzyme immobilization. Results obtained in terms of enzyme activity and thermal stability of novel immobilised materials have been found to remarkably improve those obtained using commercial immobilized enzymes in esterification reactions (e.g., monostearin synthesis).
Introduction
Lipases are triacylglycerol acyl hydrolytic enzymes that have found applications as hydrolytic, esterification and transesterification biocatalysts.1 Upon immobilization, supported enzymes can further provide an easily separable and reusable system (together with enhanced product recovery) which boasts of enhanced resistance to deactivation as compared to free enzymes.2–4 Immobilization has several implications when generating increasingly stable biocatalysts compatible with continuous processing technologies.5 Various strategies to immobilize enzymes on a number of supports have been reported. These range from the more extended and widely employed physical methods (e.g., adsorption, entrapping and/or electrostatic immobilization) to chemical protocols including covalent immobilization.4Cyclodextrins (CDs) are a class of macrocyclic structures comprising (α-1,4)-linked β-D-glucopyranose units that possess a relatively non-polar cavity. The internal hydrophobic cavity and the external hydrophilic rim of CDs render them as ideal candidates for modelling host–guest interactions,6–8 drug delivery,9 catalysis,10,11 chiral separation12 and molecular recognition in self-assembled monolayers.12 β-CD has proven to be a good enzyme support, with a number of contributions reporting significant efficiencies in promoting catalytic processes both in water and organic solvents. The addition of β-CD to solutions containing lipases has been reported to enhance reaction rates as well as enantioselectivity and lipase stability.13,14 Furthermore, CD immobilized Candida rugosa lipase offered important advantages (e.g., thermal stability) with respect to its free enzyme counterparts.14–17
Biodiesel comprising alkyl esters of long chain fatty acids has been proposed as a suitable bio-derived replacement for petroleum diesel as a means to reduce gaseous pollutant emissions including CO, SOx, and organic compounds.18 The properties of biodiesel are similar to those of petroleum-based diesel, allowing its use either as a substitute for diesel fuel or more commonly in fuel blends. Several strategies for biodiesel production have been reported in recent years and include homogeneous/heterogeneous19 and biocatalytic triacylglyceride transesterification protocols, with most commonly extended low molecular weight alcohols.20
Mono- and diacylglycerols (MAG and DAG) are well-known biodegradable, biocompatible, nontoxic and nonionic surfactants widely used in food, pharmaceutical and industrial applications.21 Constituted of a hydrophobic and hydrophilic part, the hydrophobic part consists of fatty acids (i.e., lauric, myristic, palmitic, oleic and stearic acid), whereas the hydrophilic part can be formed of glycerol or one of its ester derivatives of organic acids including lactic, citric, acetic or tartaric acid. MAG and DAGs are commonly produced on the basis of the batch alkaline catalyzed chemical glycerolysis of natural oil and fats at high temperatures (220–250 °C) and elevated pressures under nitrogen atmosphere. Besides the high energy consumption of their preparation, high temperatures are responsible for low yields (<50%) and poor product quality which leads to dark-coloured and burned-tasting product formation, thus requiring costly and extensive purification steps.
Biocatalysis can comparably overcome these issues and lead to an environmentally friendly approach for MAG synthesis via selective hydrolysis or alcoholysis using 1,3-regiospecific lipases,22 esterification of glycerol with fatty acids,23,24 and glycerolysis of fats or oils.17,19 Monostearin stands out as one of most relevant available monoacylglycerols due to its numerous applications as an additive in foodstuffs (e.g., candies, ice creams, cakes and bread), due to its important role as an emulsifier, disperser, anti-frothing agent and preservative enhancer.25–27
Following recent research of the groups into green chemical protocols and innovative biotechnological strategies,28,29 we herein report an easy and reproducible one-pot sonochemical β-CD reticulation in the presence of lipases from Bulkhorderea cepaceae and Pseudomonas fluorescens. Ultrasound accelerated the cross-linking reaction while preserving the biocatalytic activity of CD-hybrids. The promising biocatalytic activities of these newly designed immobilized enzymes have been illustrated in the esterification of free fatty acids for biodiesel production and monostearin synthesis.
Experimental section
Materials
The free and immobilized lipases from Bulkhorderea cepaceae (Amano PS) and Pseudomonas fluorescens (AK Amano) were obtained from Amano. n-Heptane and oleic acid were purchased from Tedia Co., while (R,S)-1,2-isopropylidene glycerol and all chromatographic standards were purchased from Sigma-Aldrich. Stearic acid (>98%) and ethanol were purchased from Vetec Ltda. All other chemicals were purchased from Alfa-Aesar Italy and used without further purification. β-CD was kindly provided by Wacker Chemie (Germany). Ultrasound-assisted cross-linking reactions were carried out in an ultrasonic bath (35 kHz, Transonic 460, Elma).GC-MS analysis
All GC-MS analyses were performed using the EN 14105 modified method. Free fatty acids and (R,S)-1,2-isopropylidene glycerol were transformed into more volatile silylated derivatives in the presence of pyridine and N-methyl-N-trimethysilyltrifluoroacetamide (MSTFA). All GC-MS measurements were carried out in duplicate using a DB 5-HT (Agilent, J & W. Scientific, USA) capillary column (10 m × 0.32 mm × 0.1 μm). Quantification was conducted on the basis of calibration curves with internal standards. GC-MS samples were prepared by dissolving 0.1 g of the final product in 1 mL of n-heptane. 100 μL of this solution and pyridine solutions of butanetriol (1 mg mL−1) and tricaprine (8 mg mL−1), used as internal standards, were added to a flask that held 100 μL of MSTFA. After 15 min, these reactants were dissolved in 8 mL n-heptane. One micro-litre of this sample was then injected into Shimadzu CG2010 equipment. The oven was heated at 60 °C during 1 min and then, the temperature was increased until 260 °C at 10 °C min−1.Lowry–Tinsley analysis
Esterification measurements were performed using a modification of the Lowry–Tinsley30 assay. Fatty acid depletion was monitored as follows: 0.30 mL of the reaction solution, including buffer solutions, was added to a tube containing 0.6 mL of n-heptane and 1 mL of cupric acetate–pyridine (5% w/v, pH 6.0). The final solutions were vigorously mixed for 30 s in a vortex shaker and the upper organic phase was measured on a UV/visible spectrophotometer at 715 nm. Each reaction was analyzed in triplicate and content conversion was calculated according to the absorbance percentage difference shown by the stock solution.Enzyme immobilization on HDI cross linked β-CD
β-CD reticulation was performed using 1,6-diisocyanatohexane (HDI) as an efficient cross-linker in the presence of PS-AMANO or AK-AMANO enzymes (10% w/w). β-CD (500 mg) was dissolved in DMF (7 mL) in a 100 mL three-necked round bottom flask and the addition of the corresponding enzyme was preceded by the addition of HDI (800 μL). The mixture was magnetically stirred for 12 h at room temperature and then sonicated at 30 °C for 2 h in an ultrasonic bath (35 kHz, 30 W). The resulting reticulation product was transferred to a mortar, mildly grinded and washed twice with acetone (100 mL), MeOH (100 mL), and water (100 mL). The obtained product was lyophilized and stored as a white powder under a nitrogen atmosphere.Esterification activity assay
Oleic acid and ethanol were used as substrates for the esterification reaction. Every mL of n-heptane solution contained an equimolar mixture of substrates (0.1 mol−1) as well as the free or immobilized lipase (10 mg mL−1). The reaction mixture (40 °C) was stirred at 200 rpm in 1 mL vials. 300 μL aliquots were taken at intervals and residual fatty acid levels were analyzed using the previously described Lowry–Tinsley method. Specific esterification activity (mmol min−1 mg) was determined by calculating the conversion of fatty acid to ester and defined as micromoles per hour per milligram of protein, according to Cao et al.29Results and discussion
Highly cross-linked CDs are insoluble polymers obtained from the reaction of CDs with a series of bi-functional reagents.31,32 These include diphenylcarbonate, epichlorohydrin, diisocyanates and the ultrasound-promoted copper-catalyzed azide–alkyne cycloaddition of a randomly propargylated β-CD with 1,3-bis(azidomethyl)benzene.33 Our proposed approach deals with the utilization of solid cross-linked β-CDs as enzyme supports which can in principle be obtained at room temperature under mild sonication via reticulation using hexamethylene diisocyanate (HDI).A range of typical solvents was screened in the esterification experiments (Table 1). The esterification activities of free and commercial immobilized lipases from Pseudomonas fluorescens and Bulkholderia cepaceae with AKβCD and PSβCD were initially compared. Results clearly demonstrate that all types of immobilized lipases exhibited improved esterification activity with respect to their free enzymes counterparts regardless of the utilized solvents. Interestingly, both AK and PSβCD showed superior activity in hydrocarbon solvents (e.g., n-hexane and n-heptane) as well as significantly superior esterification activities as compared to that of commercial immobilized lipases (Table 1). These results prove the remarkable advantages of the proposed immobilization protocol.
| Organic solvents | Esterification activity (mmol min−1 mg) | |||||
|---|---|---|---|---|---|---|
| AKa | AKIMa | AKβCDa | PSb | PSIMb | PSβCDb | |
a AK: Pseudomonas fluorescens; AKIM immobilized AK, AKβCD: AK immobilized on crosslinked β-CD.b PS: Bulkholderia cepaceae; PSIM immobilized PS, PSβCD: PS immobilized on crosslinked β-CD. Reaction conditions: oleic acid and butanol 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (0.1 mol−1) in n-heptane, 40 °C and 200 rpm, with 10 mg mL−1 of biocatalyst. :1 (0.1 mol−1) in n-heptane, 40 °C and 200 rpm, with 10 mg mL−1 of biocatalyst. |
||||||
| n-Hexane | 120.22 | 139.34 | 191.56 | 145.71 | 204.44 | 223.45 |
| n-Heptane | 119.56 | 150.32 | 198.72 | 155.23 | 208.33 | 221.97 |
| Iso-octane | 99.43 | 125.34 | 155.44 | 111.19 | 165.46 | 201.75 |
| MTBE | 98.51 | 112.5 | 121.33 | 100.06 | 128.19 | 178.68 |
| Cyclohexane | 110.65 | 137.29 | 165.87 | 134.12 | 173.08 | 203.65 |
The thermal stability of immobilized lipases AKIM, PSIM, AKβCD and PSβCD was subsequently investigated. Reactions were conducted under identical conditions to those selected for the esterification activity, although numerous temperatures, varying from 30 to 70 °C, were used (Fig. 1).
 | ||
| Fig. 1 Thermal stability of immobilized lipases. | ||
Results depicted in Fig. 1 illustrate the improved stability of immobilized AKβCD and PSβCD with respect to commercial immobilized enzymes. Firstly, the commercial AKIM exhibited a significant loss in activity at 60 °C (e.g., over 25% total activity loss). Comparably, AKβCD maintained almost quantitative esterification activity under comparable temperatures, with a clearly noticeable improved thermo-resistance. Similar profiles were found for PSβCD. Interestingly, the commercial immobilized PSIM starts losing activity sharply at 45 °C, while PSβCD exhibited a 100% activity at the same temperature. In this preparation, a substantial decrease in esterification activity was only observed above 55 °C.
In order to obtain better insights into the observed improved activities and stabilities of β-CD immobilized enzymes, a series of characterization studies were conducted. These included XRD, TG-DTA and XPS. Results have been depicted in Fig. 2 and 3 (see also ESI†). XRD data point to β-CD material amorphisation upon cross-linking and enzyme immobilization (see ESI†). Important differences were also observed in TG-DTA experiments (Fig. 2). These clearly indicate two distinctive steps of mass loss associated with β-CD decomposition (present in all systems at ca. 330 °C) with an additional significant mass loss in the 450–550 °C range, which seemed to be associated with enzyme removal. The high decomposition temperature may be indicative of a strong enzyme–β-CD interaction.23
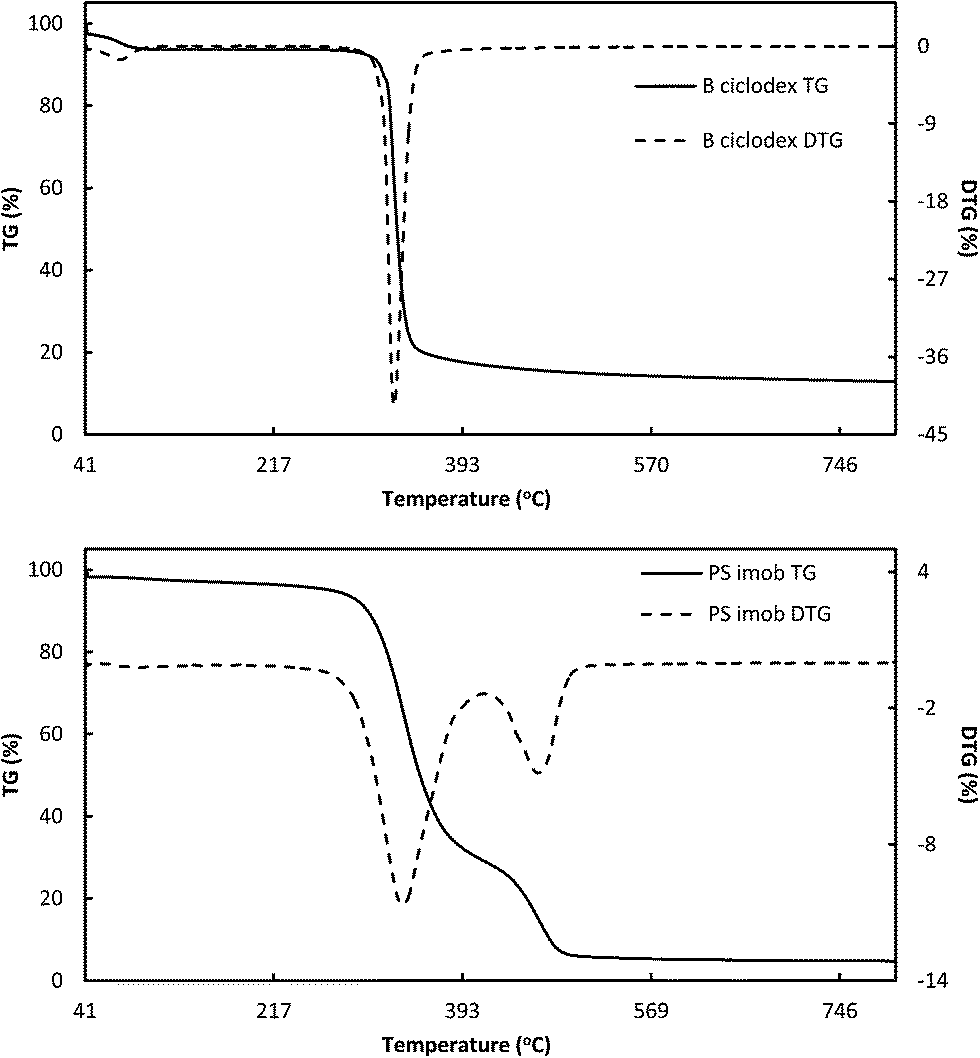 | ||
| Fig. 2 TG profiles of β-CD as compared to PS β-CD. | ||
 | ||
| Fig. 3 C1s spectra of AKBCD (top) and BCD (bottom) showing significantly different C contributions. | ||
This possible strong enzyme–β-CD interaction supports activity data obtained for β-CD biocatalytic hybrids (Table 1 and Fig. 1). XPS results were also in good agreement with TG-DTA data and confirmed the presence of the enzyme in β-CD cross-linked systems as indicated by the number and proportion of C species in the material (Fig. 3). N could also be detected in AK and PSβCD materials (as compared to blank β-CD samples, see ESI†). Detected N derives from peptide bonds present in the protein, while no N was comparably detected in β-CD.
Biocatalytic applications
The observed enzymatic activity enhancement under the proposed immobilization methodology was subsequently extended to previously optimized processes.29 Firstly, the esterification of oleic acid and ethanol was conducted according to previously optimised conditions by Costa et al. 2011 (Fig. 4).34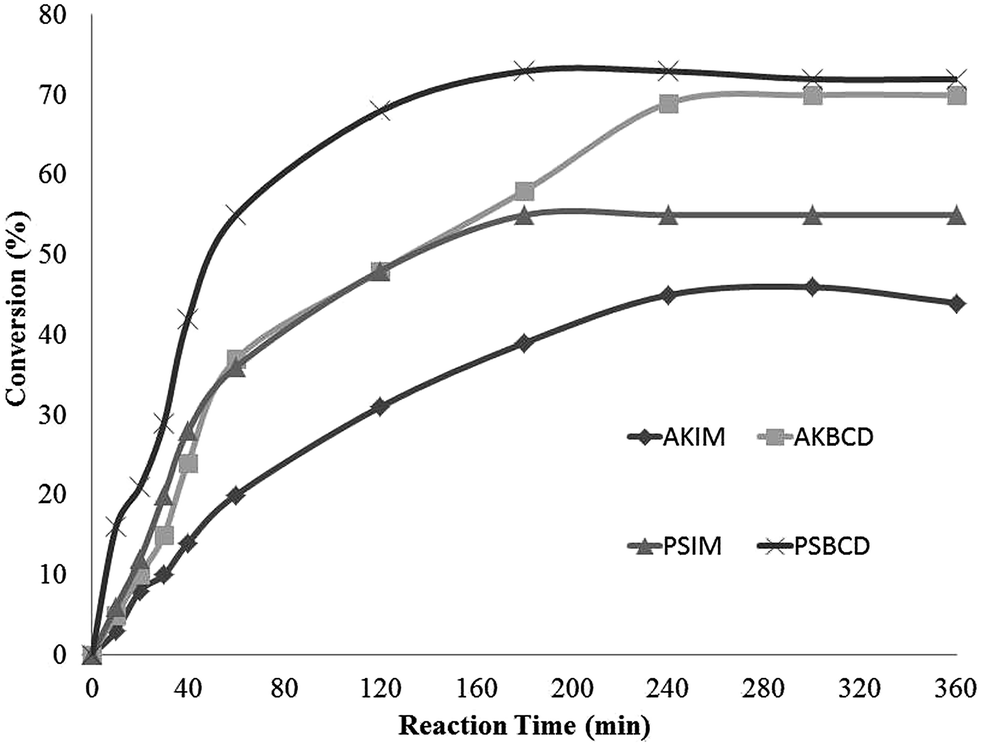 | ||
| Fig. 4 Ethyl oleate synthesis catalyzed by immobilized lipases. Reaction conditions: 0.1 mol−1 conc. oleic acid–ethanol, 10 mg lipase, 1 mL n-heptane, 200 rpm, 40 °C. | ||
Results from Fig. 4 point to a significantly improved performance of immobilized lipases AKβCD and PSβCD with respect to commercial immobilized PSIM and AKIM enzymes from Amano. The latter two exhibited regular conversions after 4 h (39% for AKIM and 51% for PSIM). Interestingly, reaction equilibrium was achieved after 3 h, with a 72% conversion rate when this reaction was catalyzed by PSβCD. Similar results were achieved only after 4 h with AKβCD, which in any case predate those observed for AKIM.
In another application, monostearin synthesis was attempted following previously optimized conditions by Junior et al. 2011.35
As a result, the novel immobilised biocatalysts were able to provide moderate to good conversions after 4 hours reaction, 62% by AKβCD and 53% by PSβCD, which are significant improvements as compared to PSIM and AKIM lipases (Fig. 5).
 | ||
| Fig. 5 Monostearin synthesis catalysed by immobilized lipases. | ||
In conclusion, the development of highly active and stable novel immobilised enzymes using a simple and efficient cross-linking protocol with beta-cyclodextrins was successfully accomplished, rendering versatile biocatalysts for different applications including esterifications. We envisage this methodology to be extended to a number of related chemistries of relevance to waste valorisation to a range of valuable products that will be reported in due course.
The reusability of the biocatalyst was also evaluated and expressed in terms of monoacylglycerol conversion as depicted in Fig. 6.
 | ||
| Fig. 6 Recycles of new immobilized biocatalysts. (A) New immobilized lipases. (B) Commercial lipases. | ||
As observed, both biocatalysts were able to maintain a high performance and stability during 9 cycles. Comparably, a significant decrease in conversion was observed for AKBCD with respect to PSBCD after 9 uses. Interestingly, commercial lipases showed both reduced activities and a gradual decrease in activity after 6–7 reuses.
Acknowledgements
ROMAS and IJ gratefully acknowledge CAPES, FAPERJ, CNPq and FINEP for financial support. RL gratefully acknowledges Ministerio de Ciencia e Innovación, Gobierno de España for the concession of a Ramon y Cajal contract (ref. RYC-2009-04199) and funding under project no. CTQ2011-28954-C02-02 as well as Consejeria de Ciencia e Innovación, Junta de Andalucía for funding under project no. P10-FQM-6711. GC and KM acknowledge the University of Turin (Finanziamento ricerca locale 2013 ex-60%).References
- Lipases and Phospholipases, Methods in Molecular Biology, ed. G. Sandoval, Humana Press, Springer, Netherlands, 2012, vol. 861 Search PubMed.
- P. Torres-Salas, A. del Monte-Martinez, B. Cutiño-Avila, B. Rodriguez-Colinas, M. Alcalde, A. O. Ballesteros and F. Plou, Adv. Mater., 2011, 23, 5275–5282 CrossRef CAS PubMed.
- F. Pion, A. F. Reano, P.-H. Ducrot and F. Allais, RSC Adv., 2013, 3, 8988–8997 RSC.
- K. Viswanathan, G. Li and R. A. Gross, Macromolecules, 2010, 43, 5245–5255 CrossRef CAS.
- K. M. Polizzi, A. S. Bommarius, J. M. Broering and J. F. Chaparro-Riggers, Curr. Opin. Chem. Biol., 2007, 11, 220–225 CrossRef CAS PubMed.
- D. I. Fried, F. J. Brieler and M. Fröba, ChemCatChem, 2013, 5, 862–884 CrossRef CAS.
- (a) M. E. Davis, J. E. Zuckerman, C. H. J. Choi, D. Seligson, A. Tolcher, C. A. Alabi, Y. Yen, J. D. Heidel and A. Ribas, Nature, 2010, 464, 1067–1070 CrossRef CAS PubMed.
- Y. Zhu, L. Che, H. He, Y. Jia, J. Zhang and X. Li, J. Controlled Release, 2011, 152, 317–324 CrossRef CAS PubMed.
- M. Adeli, F. Hakimpoor, M. Parsamanesh, M. Kalantari, Z. Sobhani and F. Attyabi, Polymer, 2011, 52, 2401–2413 CrossRef CAS.
- J. Zhang and P. X. Ma, Nano Today, 2010, 5, 337–350 CrossRef CAS PubMed.
- Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890–7908 RSC.
- L. Marinescu and M. Bols, Curr. Org. Chem., 2010, 14, 1380–1398 CrossRef CAS.
- L. Szente and J. Szeman, Anal. Chem., 2013, 85, 8024–8030 CrossRef CAS PubMed.
- C. Yong and L. Yu, Chem. Soc. Rev., 2010, 39, 495–505 RSC.
- A. Ghanem and V. Schurig, Org. Biomol. Chem., 2003, 1, 1282–1291 CAS.
- (a) E. Y. Ozmen, M. Sezgin and M. Yilmaz, J. Mol. Catal. B: Enzym., 2009, 57, 109–114 CrossRef CAS.
- B. Boscolo, F. Trotta and E. Ghibaudi, J. Mol. Catal. B: Enzym., 2010, 62, 155–161 CrossRef CAS.
- K. P. Dhakea, A. H. Karoyo, M. H. Mohamed, L. D. Wilson and B. M. Bhanage, J. Mol. Catal. B: Enzym., 2013, 87, 105–112 CrossRef.
- R. Luque, J. Lovett, B. Datta, J. Clancy, J. M. Campelo and A. A. Romero, Energy Environ. Sci., 2010, 3, 1706–1721 CAS.
- P. Cintas, S. Mantegna, E. Calcio Gaudino and G. Cravotto, Ultrason. Sonochem., 2010, 17, 985–989 CrossRef CAS PubMed.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–564 CAS.
- (a) S. Saadi, A. A. Ariffin, H. M. Ghazali, M. S. Miskandar, S. M. Abdulkarin and H. C. Boo, J. Food Sci., 2011, 76, C21–C31 CrossRef CAS PubMed; (b) P. Pacivova, I. Buresova and H. Bilkova, J. Sci. Food Agric., 2010, 90, 2282–2288 CrossRef PubMed; (c) H. Szelag and W. Zwierzykowski, Colloids Surf., A, 1999, 155, 349–357 CrossRef CAS; (d) P. Kohler and W. Grosch, J. Agric. Food Chem., 1999, 47, 1863–1869 CrossRef CAS PubMed.
- E. Martin, Trav. Chim. Aliment. Hyg., 1988, 79, 406–410 Search PubMed.
- C. G. Frost and L. Mutton, Green Chem., 2010, 12, 1687–1703 RSC.
- A. Pohar and I. Plazl, Chem. Biochem. Eng. Q., 2009, 23, 537–544 CAS.
- V. Caballero, F. M. Bautista, J. M. Campelo, D. Luna, J. M. Marinas, A. A. Romero, J. M. Hidalgo, R. Luque, A. Macario and G. Giordano, Process Biochem., 2009, 44, 334–342 CrossRef CAS.
- M. Kotwal, S. S. Deshpande and D. Srinivas, Catal. Commun., 2011, 12, 1302–1306 CrossRef CAS.
- (a) P. Cintas, G. Cravotto, E. Calcio Gaudino, L. Orio and L. Boffa, Catal. Sci. Technol., 2012, 2, 85–87 RSC; (b) G. Cravotto, E. Calcio Gaudino, S. Tagliapietra, D. Carnaroglio and A. Procopio, Green Process. Synth., 2012, 1, 269–273 CAS.
- X. Cao, J. Yang, L. Shu, B. Yu and Y. Yan, Process Biochem., 2009, 44, 177–182 CrossRef CAS.
- R. R. Lowry and I. J. Tinsley, J. Am. Oil Chem. Soc., 1976, 53, 470 CrossRef CAS PubMed.
- A. Binello, B. Robaldo, A. Barge, R. Cavalli and G. Cravotto, J. Appl. Polym. Sci., 2008, 107, 2549–2557 CrossRef CAS.
- G. Cravotto, V. V. Fokin, D. Garella, A. Binello, L. Boffa and A. Barge, J. Comb. Chem., 2010, 12, 13–15 CrossRef CAS PubMed.
- R. M. Blanco, P. Terreros, M. Fernandez-Perez, C. Otero and G. Diaz-Gonzalez, J. Mol. Catal. B: Enzym., 2004, 30, 83–93 CrossRef CAS.
- I. C. R. Costa, S. G. F. Leite, I. C. R. Leal, L. S. M. Miranda and R. O. M. A. de Souza, J. Braz. Chem. Soc., 2011, 22, 1993–1998 CrossRef CAS.
- I. I. Junior, F. K. Sutili, L. S. M. Miranda, I. C. R. Leal and R. O. M. A. de Souza, J. Mol. Catal. B: Enzym., 2011, 72, 313–318 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra03797a |
| This journal is © The Royal Society of Chemistry 2014 |