 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing excess electron transport in DNA†
Fazel
Fakhari‡
,
Yun-Yun K.
Chen
and
Steven E.
Rokita‡
*
Department of Chemistry and Biochemistry, University of Maryland, College Park, Maryland 20742, USA
First published on 27th June 2013
Abstract
The efficiency of excess electron transport in duplex DNA can be enhanced by limiting the pathways available for migration and using a donor of moderate strength that suppresses radical recombination through selective electron transfer to distal pyrimidines rather than proximal purines.
Charge transport (CT) in DNA contributes to a range of natural processes and has recently been integrated into nanostructures and sensing devices.1,2 However, challenges still remain before DNA may be widely applied to nanoelectronics, and much effort has focused on identifying and optimizing parameters that limit CT efficiency between defined donors and acceptors. The intervening nucleotide sequence, helical structure and conformational dynamics are all crucial in optimizing long distance CT.3 The selection of appropriate donors and acceptors is also equally important for successful CT.4–6
The number of pathways available for charge migration in DNA is another variable that is expected to influence the apparent efficiency of transport between a specific donor–acceptor pair, but direct study of this has been minimal. Most model systems are based on DNA with a charge donor appended to the 5′-terminus of a duplex strand. For example, hairpin systems are typically capped with a terminal donor.7 Likewise, most double-stranded systems using organic and organometallic donors rely on their attachment near to a helical terminus as well.2,8 Alternative placement of a donor at an internal position within helical DNA has received less attention and offers an intrinsic competition between charge migration in two directions (Fig. 1).9–11 The efficiency of delivering an electron from a donor in the middle of a duplex to an acceptor (BrdU) has now been compared directly to an equivalent system with the donor at the duplex terminus in order to restrict electron migration.
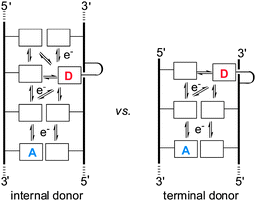 | ||
| Fig. 1 Freedom of charge migration may influence the efficiency of its transport from a donor (D) to an acceptor (A) in DNA. | ||
Transport of charge from a donor to acceptor also competes against rapid recombination of the proximal ion pair formed by initial charge injection into DNA.12 Both experimental and theoretical investigations have demonstrated that increasing the distance between these counter ions will suppress their non-productive recombination.13 Judicious choice of donors and their surrounding nucleotide sequence may reduce this back electron transfer as illustrated previously with hole transport.4,14 This same strategy has now been tested as described below for enhancing excess electron transfer (EET) initiated by single electron transfer from a photoexcited donor to DNA.
Our model system for comparing donor placement and electron injection relied on BrdU as an electron acceptor. This is easily incorporated into DNA by standard phosphoramidite chemistry and acts with a trapping efficiency near unity by rapidly eliminating bromide and generating a transient uridinyl radical that oxidizes its 5′ neighboring deoxyribose and creates a site of alkaline lability.10,15,16 A methylated derivative of the chosen diaminonaphthyl donor (DN) had originally been synthesized using a norbornyl protection strategy,10 but the parent donor lacking the methyl groups could only be isolated in low yield under equivalent conditions. Substituting the norbornyl group with a phthalimide group facilitated deprotection and provided high yields of the desired donor (a, Chart 1 and ESI†). The donor was easily coupled through its hydroxylamine group to abasic sites in oligonucleotides that resulted from deglycosylation of the uridine-containing parents (ESI†).17
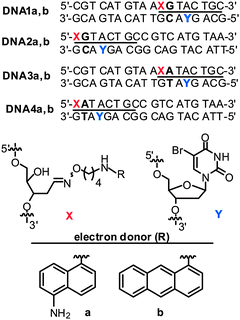 | ||
| Chart 1 Duplex DNA alternatively containing donors of (a) diaminonaphthalene and (b) aminoanthracene for EET studies. | ||
Restricting the potential migration of an excess electron in DNA was expected to enhance the efficiency of CT from donor to acceptor. This was confirmed after reaction was initiated by irradiation of the donor DN (λ > 335 nm) in duplexes DNA1a and DNA2a that contained identical nucleotide sequences but alternative placement of the donor DN and acceptor BrdU within the duplex as indicated in Chart 1. However, the sequence and the 5′ to 3′ orientation between these redox active centers remained constant. The characteristic strand scission resulting from electron trapping by BrdU was quantified after gel electrophoresis and monitored over time of irradiation (Fig. 2). Initial rates of scission were indeed enhanced 2-fold for the duplex with DN at a terminal versus an internal position (Fig. 3). However, the signal-to-noise ratio for this first comparison was only moderate due to the relatively low efficiency of CT.
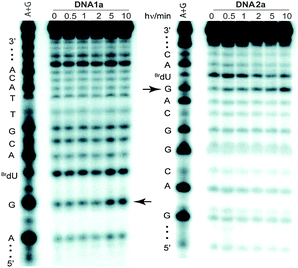 | ||
| Fig. 2 Phosphoimagery of 20% denaturing polyacrylamide gels showing strand scission of DNA1a and DNA2b after UV irradiation (λ > 335 nm) and piperidine treatment. Arrows indicate the scission sites caused by electron trapping by the BrdU residue. | ||
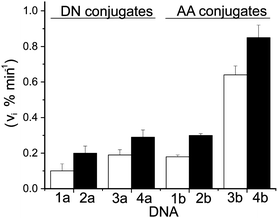 | ||
| Fig. 3 EET from donors (DN and AA) to BrdU in DNA1–DNA4 as indicated by strand scission after UV irradiation and piperidine treatment. Initial scission rates (vi) were obtained by linear fitting of % scission vs. time (ESI†). Each analysis was repeated a minimum of three times and error bars represent their standard deviation. | ||
A second set of duplexes (DNA3a and DNA4a, Chart 1) was subsequently prepared with an expectation of increasing the signal-to-noise for an additional test of donor placement and electron migration. For these duplexes, the G/C base pair adjacent to the donor was replaced with an A/T to avoid the likely suppression of electron migration encountered from an intermediate cytosine radical that is stabilized by proton transfer from its hydrogen bonding partner guanine.5,11,18 This change of sequence was sufficient to enhance the efficiency of CT to the acceptor BrdU by more than 100% (Fig. 3), and the trend of efficiency versus donor position remained constant. Each example illustrates the benefit of limiting the migration of the injected electron by placing the donor at a helix terminus.
The reducing potential of the electron donor is another variable that may affect the efficiency of delivering charge to its acceptor within DNA. Initial selection of these donors was guided by the original uncertainty that excess electron transport was even possible in DNA. Thus, a very strong photoexcited donor (Eox* below −3 V vs. SCE) was selected at first for injecting an electron indiscriminately into any of the nucleobases.10 This strategy became unnecessary and unsuitable once back electron transfer from the initial charge-separated species was found to limit long distance transport.12 Alternative application of a more selective donor provided an opportunity to suppress radical recombination by placing an unreactive nucleobase between the initial pair of charges.4,14 In order to investigate this strategy, a derivative of 1-aminoanthracene (AA) was chosen as a close analog of DN for comparison. Unlike the excited state of DN, the excited state of AA is capable of only reducing pyrimidines and not purines due to its moderate potential of −2.25 V vs. SCE.19,20 Consequently, surrounding AA with purines enforced a distance between charged species that should enhance excess electron transport between donor and acceptor in DNA (Fig. 4).
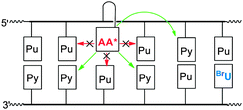 | ||
| Fig. 4 Photoexcited 1-aminoanthracene (AA*) conjugates are capable of injecting electrons to pyrimidines (Py) but not purines (Pu). | ||
The donor AA was prepared and conjugated to DNA by the same methods used for the DN derivative (ESI†). Transport efficiency was then measured within identical nucleotide sequences (Chart 1) to establish direct comparisons between AA, DN and the trends already established by DN. In each example, the AA conjugates supported a greater level of transport than their equivalent DN conjugates (Fig. 3, ESI†). The enhancement ranged from a low of approximately 50% for DNA2 to a high of 250% for DNA4. In addition, preferential transport from donors placed at a terminal rather than central region of a duplex was confirmed by comparing AA-containing DNA1b and DNA3b with DNA2b and DNA4b, respectively. The sensitivity of excess electron migration through A/T base pairs vs. G/C base pairs is even more pronounced for the selective AA donor as evident from a comparison between DNA2b and DNA4b (Fig. 3). When all parameters are optimized – donor placement, transport sequence and redox potential of the excited-state donor – the efficiency of CT increases by more than 8-fold as represented by DNA1a and DNA4b (Fig. 3).
Alternative explanations for this enhanced efficiency of excess electron transport were also considered, but none were consistent with the data or literature precedent. The integrated absorptivities above 335 nm for both donors were nearly identical (ESI†) and could not account for differences in electron injection into DNA. Conformational differences between AA and DN linked to DNA are also expected to be negligible. Their conjugation to the abasic parent duplexes increased the thermal stability almost equivalently with an average of +5 °C for DN and +6 °C for AA (ESI†). Typically, aromatic compounds conjugated to the 5′-terminus stack on top of the helix rather than unwind and intercalate into the duplex.6,21 The similarity in stabilization detected for terminal and internal conjugates indicates similar binding modes, namely stacking rather than intercalation. A solvatochromic derivative of DN also recently confirmed its accomodation within an abasic site.22 Finally, reduction of BrdU through interduplex transfer of an electron would not have exhibited the sensitivity to the internal nucleobase sequence as evident with DNA1–DNA4. Moreover, our original studies with a related DN conjugate were insensitive to conditions that suppress intermolecular processes.10
Limiting the available paths for electron migration and suppressing back electron transfer from the initial charge-separated states measurably promote excess electron transport in duplex DNA. These parameters represent only two of the many that affect transport, but their influence is significant enough to support a large increase in its efficiency from the aromatic donor to the BrdU electron trap. Combined with the known preference of excess electron transport for A/T vs. G/C base pairing,5,11,18 the efficiency of CT can be controlled by judicious choice of donor potential and DNA sequence. Such considerations should be incorporated into future systems that rely on excess electron transport through DNA to convey a signal, trigger an actuator or initiate other related applications of nanosystems based on DNA.
Authors would like to thank Dr Neil P. Campbell, Timothy J. Simons and Angel Li for initial studies and useful discussions. Financial support from NSF (CHE-0517498) and the Howard Hughes Medical Institute Undergraduate Science Education Program are also gratefully acknowledged.
Notes and references
- (a) F. Boussicault and M. Robert, Chem. Rev., 2008, 108, 2622–2645 CrossRef CAS; (b) E. J. Merino, A. K. Boal and J. K. Barton, Curr. Opin. Chem. Biol., 2008, 12, 229–237 CrossRef CAS; (c) W. Su, V. Bonnard and G. A. Burley, Chem.–Eur. J., 2011, 17, 7982–7991 CrossRef CAS; (d) Y. W. Kwon, C. H. Lee, D. H. Choi and J. I. Jin, J. Mater. Chem., 2009, 19, 1353–1380 RSC; (e) B. Ge, Y. C. Huang, D. Sen and H.-Z. Yu, Angew. Chem., Int. Ed., 2010, 49, 9965–9967 CrossRef CAS.
- J. C. Genereux, A. K. Boal and J. K. Barton, J. Am. Chem. Soc., 2010, 132, 891–905 CrossRef CAS.
- (a) B. Giese and A. Biland, Chem. Commun., 2002, 667–672 RSC; (b) Top. Curr Chem., ed. G. B. Schuster, Springer, New York, 2004, vol. 236 & 237 Search PubMed; (c) Charge Transfer in DNA: From Mechanism to Application, ed. H.-A. Wagenknecht, Wiley-VCH Verlag GmbH & Co., Weinheim, 2005 Search PubMed; (d) L. Valis, Q. Wang, M. Raytchev, I. Buchvarov, H.-A. Wagenknecht and T. Fiebig, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10192–10195 CrossRef CAS; (e) F. C. Grozema, S. Tonzani, Y. A. Berlin, G. C. Schatz, L. D. A. Siebbeles and M. A. Ratner, J. Am. Chem. Soc., 2008, 130, 5157–5166 CrossRef CAS.
- T. T. Williams, C. Dohno, E. D. A. Stemp and J. K. Barton, J. Am. Chem. Soc., 2004, 126, 8148–8158 CrossRef CAS.
- A. Manetto, S. Breeger, C. Chatgilialoglu and T. Carell, Angew. Chem., Int. Ed., 2006, 45, 318–321 CrossRef CAS.
- K. Kawai, Y. Osakada, E. Matsutani and T. Majima, J. Phys. Chem. B, 2010, 114, 10195–10199 CrossRef CAS.
- (a) S. M. M. Conron, A. K. Thazhathveetil, M. R. Wasielewski, A. L. Burin and F. D. Lewis, J. Am. Chem. Soc., 2010, 132, 14388–14390 CrossRef CAS; (b) M. J. Park, M. Fujitsuka, K. Kawai and T. Majima, J. Am. Chem. Soc., 2011, 133, 15320–15323 CrossRef CAS.
- C.-S. Liu, R. Hernandez and G. B. Schuster, J. Am. Chem. Soc., 2004, 126, 2877–2884 CrossRef CAS.
- (a) A. Schwögler, L. T. Burgdorf and T. Carell, Angew. Chem., Int. Ed., 2000, 39, 3918–3920 CrossRef; (b) B. Giese, B. Carl, T. Carl, T. Carell, C. Behrens, U. Hennecke, O. Schiemann and E. Feresin, Angew. Chem., Int. Ed., 2004, 43, 1848–1851 CrossRef CAS; (c) M. Raytchev, E. Mayer, N. Amann, H.-A. Wagenknecht and T. Fiebig, ChemPhysChem, 2004, 5, 706–712 CrossRef CAS.
- T. Ito and S. E. Rokita, J. Am. Chem. Soc., 2003, 125, 11480–11481 CrossRef CAS.
- C. Wagner and H.-A. Wagenknecht, Chem.–Eur. J., 2005, 11, 1871–1876 CrossRef CAS.
- (a) P. Kaden, E. Mayer-Enthart, A. Trifonov, T. Fiebig and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2005, 44, 1636–1639 CrossRef CAS; (b) T. Takada, C. Lin and T. Majima, Angew. Chem., Int. Ed., 2007, 46, 6681–6683 CrossRef CAS; (c) J. C. Genereux, K. E. Augustyn, M. L. Davis, F. Shao and J. K. Barton, J. Am. Chem. Soc., 2008, 130, 15150–15156 CrossRef CAS; (d) P. Daublain, A. K. Thazhathveetil, V. Shafirovich, Q. Wang, A. Trifonov, T. Fiebig and F. D. Lewis, J. Phys. Chem. B, 2010, 114, 14265–14272 CrossRef CAS; (e) T. Ito, T. Uchida, K. Tanabe, H. Yamada and S. Nishimoto, J. Photochem. Photobiol., B, 2011, 219, 115–121 CrossRef CAS.
- (a) F. D. Lewis, P. Daublain, B. Cohen, J. Vura-Weis and M. R. Wasielewski, Angew. Chem., Int. Ed., 2008, 47, 3798–3800 CrossRef CAS; (b) F. C. Grozema, S. Tonzani, Y. Berlin, G. C. Schatz, L. D. A. Siebbeles and M. A. Ratner, J. Am. Chem. Soc., 2009, 131, 14204–14205 CrossRef CAS.
- K. Kawai, Y. Osakada, M. Fujitsuka and T. Majima, J. Phys. Chem. B, 2008, 112, 2144–2149 CrossRef CAS.
- T. Ito, A. Kondo, S. Terada and S. Nishimoto, J. Am. Chem. Soc., 2006, 128, 10934–10942 CrossRef CAS.
- (a) H. Sugiyama, Y. Tsutsumi and I. Saito, J. Am. Chem. Soc., 1990, 112, 6720–6721 CrossRef CAS; (b) G. P. Cook and M. M. Greenberg, J. Am. Chem. Soc., 1996, 118, 10025–10030 CrossRef CAS.
- (a) T. Lindahl, S. Ljungquist, W. Siegert, B. Nyberg and B. Sperens, J. Biol. Chem., 1977, 252, 3286–3294 CAS; (b) K. Kubo, H. Ide, S. S. Wallace and Y. W. Kow, Biochemistry, 1992, 31, 3703–3708 CrossRef CAS.
- (a) Z. Cai, X. Li and M. D. Sevilla, J. Phys. Chem. B, 2002, 106, 2755–2762 CrossRef CAS; (b) T. Ito and S. E. Rokita, Angew. Chem., Int. Ed., 2004, 43, 1839–1842 CrossRef CAS.
- N. P. Campbell, A. S. Finch and S. E. Rokita, ChemPhysChem, 2010, 11, 1768–1773 CrossRef CAS.
- (a) H.-A. Wagenknecht, Nat. Prod. Rep., 2006, 23, 973–1006 RSC; (b) A. S. Finch, PhD thesis, University of Maryland, College Park, 2008.
- (a) W. C. Ho, C. Steinbeck and C. Richert, Biochemistry, 1999, 38, 12597–12606 CrossRef CAS; (b) S. Narayanan, J. Gall and C. Richert, Nucleic Acids Res., 2004, 32, 2901–2911 CrossRef CAS; (c) E. K. Y. Leung and D. Sen, Chem. Biol., 2007, 14, 41–51 CrossRef CAS; (d) F. D. Lewis, A. K. Thazhathveetil, T. A. Zeidan, J. Vura-Weis and M. R. Wasielewski, J. Am. Chem. Soc., 2010, 132, 444–445 CrossRef CAS; (e) F. Shao, K. Augustyn and J. K. Barton, J. Am. Chem. Soc., 2005, 127, 17445–17452 CrossRef CAS.
- A. Fakhari M and S. E. Rokita, Chem. Commun., 2011, 47, 4222–4224 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and oligonucleotide conjugation of electron donors, HPLC and absorption spectra of conjugates, thermal denaturation of duplex DNA, initial rate of strand scission induced by excess electron transfer. See DOI: 10.1039/c3cc43887b |
| ‡ Current address: Department of Chemistry, Johns Hopkins University, Baltimore, MD 21218, USA. E-mail: rokita@jhu.edu; Tel: +1 410-516-5793. |
| This journal is © The Royal Society of Chemistry 2013 |