
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A focus on substituents effect in the force-promoted disrotatory ring-opening of cis-cyclobutenes
Lei
Chen
 and
Guillaume
De Bo
*
and
Guillaume
De Bo
*
Department of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: Guillaume.debo@manchester.ac.uk
First published on 15th April 2025
Abstract
Symmetry-forbidden reactions are notoriously difficult to investigate as they are typically overshadowed by the corresponding symmetry-allowed pathway. Mechanical activation allows access to reaction pathways disfavoured using other methods of activation, such as the symmetry-forbidden disrotatory ring-opening of substituted cis-cyclobutenes. In a recent publication, Bowser, et al. have studied the effects of various substituents on this reaction using atomic force microscopy and computational analysis (B. H. Bowser, C. L. Brown, J. Meisner, T. B. Kouznetsova, T. J. Martínez and S. L. Craig, Chem. Sci., 2025, https://doi.org/10.1039/D5SC00253B). The largest effect is observed with substituents close to the scissile bond having the ability to stabilise the diradical character of the disrotatory ring-opening reaction pathway.
In polymer mechanochemistry, polymers are used to activate force-sensitive molecules (mechanophores),1 sometimes along unusual reaction pathways. The most striking example of such unique reactivity is perhaps the symmetry-forbidden disrotatory ring-opening reaction of cis cyclobutene (CBE) and benzocyclobutene (BCB) mechanophores (Fig. 1). The first example was reported by Moore and coworkers in a landmark paper demonstrating the anti-Woodward-Hoffman ring-opening of BCB.2 This seminal report was quickly followed by computational and single-molecule studies of BCB and CBE by the groups of Marx, Martínez, and Craig.3–8
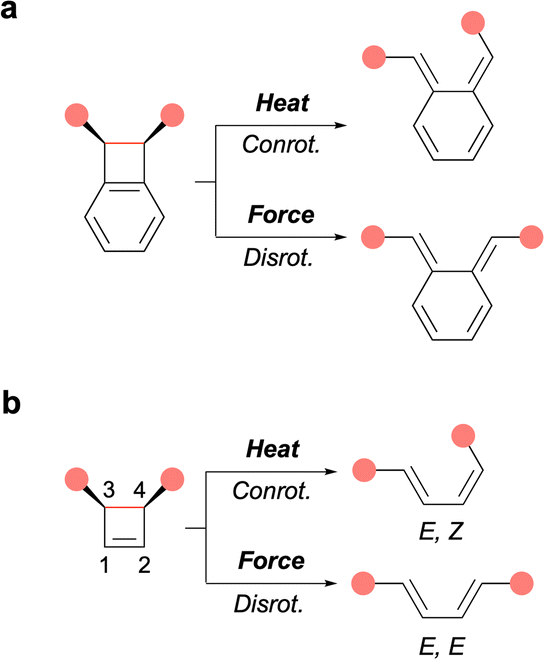 | ||
| Fig. 1 Thermal and mechanical activation of cis-BCB (a) and cis-CBE (b) mechanophores, leading to conrotatory and disrotatory ring-opening reactions, respectively. Pink disks indicate the position of the pulling points. Scissile bonds shown in red. | ||
Mechanical activation offers a unique opportunity to study the disrotatory reaction pathway in more detail, for example to investigate substituent effects, as it is mostly inaccessible to thermal activation. Previously, Craig and Martínez have compared the forbidden/allowed ring-opening of cis/trans BCB and CBE mechanophores, differing by the presence of a fused (BCB) or connected (CBE) phenyl ring(s) at positions 1 and 2 of the 4-membered ring core (Fig. 1). They found that BCB and CBE respond in contrasting ways to mechanical force as the forbidden ring-opening of cis-BCB occurs at lower force than the allowed ring-opening of trans-BCB, while the opposite is true for cis- and trans-CBE.8
In a recent article published in this journal, the Craig and Martínez groups teamed up again (https://doi.org/10.1039/D5SC00253B) to investigate the effect of substituents in positions 1,2 and 3,4 of various CBE mechanophores using atomic force microscopy (AFM) in concert with computational methods (Fig. 2).9
 | ||
| Fig. 2 Copolymers containing CBE mechanophores with various substituents and epoxides for adhesion to the tip/surface of the AFM setup, and their activation force (f*) determined by AFM. | ||
First, the effect of substitution at positions 1 and 2, which are remote from the scissile bond, was investigated on multimechanophore polymers 1Ph–X possessing phenyl groups of varying electronic density (X = H, OMe, F) at these positions. All three polymers exhibit a similar activation force (f*) as measured by AFM (Fig. 2). The low level of influence from substituents at these positions was further confirmed with 1Me, which displays an activation force in the same range. In previously investigated systems, the activation of a CBE mechanophore is believed to proceed via both disrotatory and conrotatory pathways at low force, with the disrotatory reaction channel being predominant at higher forces.8 This picture was confirmed in the present study from the activation energy calculated for the range of forces recorded by AFM. This means that the ring-opening could theoretically proceed via a symmetry-allowed thermal conrotatory process, leading to an E,Z butadiene, or via the concurrent force-driven disrotatory pathway to afford the E,E product (Fig. 1). The subtle difference in contour length between the 2 butadiene isomers was matched with the elongation plateaus recorded in the AFM profiles to conclude that the disrotatory pathway was predominant in these experiments.
Substituents at positions 3 and 4, adjacent to the scissile bond, have a much greater effect as determined from polymers 2Me, possessing Me groups in all four positions, and 2TMS, in which one of the Me group is replaced by a trimethylsilylacetylene (TMSA) moiety. The same contour length analysis was used to confirm that these polymers also react predominantly via a disrotatory mechanism to produce the longer E,E isomer. However, these ring openings occur at much lower forces (f*) than in polymers 1x (Fig. 2), indicating a lower activation barrier for this disrotatory process. A mechanophore’s reactivity can be assessed from its mechanochemical coupling (i.e. how efficiently force affects the activation energy) and its intrinsic (i.e. force-free) reactivity along a specific reaction pathway. Despite their varied f* values, all the CBE mechanophores show a similar mechanochemical coupling, which is inherent to the cis geometry of the core CBE structure. In contrast, the presence of Me, and even more of TMSA, increases the intrinsic reactivity of these CBE mechanophores compared to their unsubstituted counterpart (1Me). As the disrotatory pathway displays a significant diradical character,8 the difference in reactivity is mainly attributed to the ability of these groups to stabilise the developing radicals in positions 3 and 4. Computational analysis even allowed the weighing of the stabilising contribution of each group (1.5–2 and 4.5–6.5 kcal per mol per Me and TMSA groups respectively). Interestingly, 2TMS proved to be mechanochromic as the TMSA group imparts the corresponding ring-opened product with fluorescence–emissive properties due to the conjugation of TMSA with the butadiene backbone, as demonstrated by sonication.
By combining single force spectroscopy and computational techniques, the authors were able to investigate the symmetry-forbidden disrotatory ring-opening of CBE mechanophores in great detail. Notably, they were able to identify the mechanical signature of the disrotatory process and isolate the effect of substituents on the ring-opening, which supports the diradical character of the disrotatory mechanism. The competition between concerted and radical mechanisms has been invoked in many force-promoted pericyclic reactions and the latter is often considered dominant in the nN regime.10 However, even though it’s been shown computationally,10,11 there are few experimental validations of the radical mechanism.12 The possibility of probing these competing mechanisms, and underlying effects, by AFM is an exciting prospect and would help shed light on the reactivity of mechanophores at low force and strain rate, and near the mechanistic cross over point. This study further highlights the unique position occupied by the field of polymer mechanochemistry in the study of reaction pathways inaccessible to other methods of activation.
Author contributions
L. C. and G. D. B. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC; EP/X023788/1), and the European Research Council (ERC Consolidator selected by the ERC, funded by EPSRC) for funding.Notes and references
- G. De Bo, Macromolecules, 2020, 53, 7615–7617 CrossRef CAS.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423–427 CrossRef CAS PubMed.
- J. Ribas-Arino, M. Shiga and D. Marx, Chem.–Eur. J., 2009, 15, 13331–13335 CrossRef CAS PubMed.
- M. T. Ong, J. Leiding, H. Tao, A. M. Virshup and T. J. Martínez, J. Am. Chem. Soc., 2009, 131, 6377–6379 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova, Z. Niu, M. T. Ong, H. M. Klukovich, A. L. Rheingold, T. J. Martínez and S. L. Craig, Nat. Chem., 2015, 7, 323–327 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova, Z. Niu, A. L. Rheingold and S. L. Craig, J. Org. Chem., 2015, 80, 11895–11898 CrossRef CAS PubMed.
- B. H. Bowser, S. Wang, T. B. Kouznetsova, H. K. Beech, B. D. Olsen, M. Rubinstein and S. L. Craig, J. Am. Chem. Soc., 2021, 143, 5269–5276 CrossRef CAS PubMed.
- C. L. Brown, B. H. Bowser, J. Meisner, T. B. Kouznetsova, S. Seritan, T. J. Martínez and S. L. Craig, J. Am. Chem. Soc., 2021, 143, 3846–3855 CrossRef CAS PubMed.
- B. H. Bowser, C. L. Brown, J. Meisner, T. B. Kouznetsova, T. J. Martínez and S. L. Craig, Chem. Sci., 2025 10.1039/D5SC00253B.
- S. Akbulatov and R. Boulatov, ChemPhysChem, 2017, 18, 1422–1450 CrossRef CAS PubMed.
- M. Cardosa-Gutierrez, G. De Bo, A.-S. Duwez and F. Remacle, Chem. Sci., 2023, 14, 1263–1271 RSC.
- Z. S. Kean, Z. Niu, G. B. Hewage, A. L. Rheingold and S. L. Craig, J. Am. Chem. Soc., 2013, 135, 13598–13604 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |