
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRationally designed sonocatalyst-enhanced supramolecular ferroptosis inducers for effective cancer therapy
Yida
Pang†
ad,
Yong
Luo†
ac,
Ting
Liu†
e,
Qian
Li†
a,
Longcan
Mei
a,
Junhua
Zhang
a,
Chonglu
Li
 *b,
Junrong
Li
*a and
Yao
Sun
*ad
*b,
Junrong
Li
*a and
Yao
Sun
*ad
aKey Laboratory of Pesticides and Chemical Biology, Ministry of Education, International Joint Research Center for Intelligent Biosensor Technology and Health, College of Chemistry, Central China Normal University, Wuhan 430079, China. E-mail: sunyaogbasp@ccnu.edu.cn
bHubei Province Key Laboratory of Occupational Hazard Identification and Control, School of Public Health, Medical College, Wuhan University of Science and Technology, Wuhan, 430065, China. E-mail: lichonglu@wust.edu.cn
cSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430079, China
dHubei Jiangxia Laboratory, Wuhan 430200, China
eWuhan Jinyintan Hospital, Tongji Medical College of Huazhong University of Science and Technology, Wuhan, 430023, China
First published on 19th September 2025
Abstract
Ferroptosis is a promising strategy against apoptosis-resistant tumors, yet traditional iron-induced approaches face safety issues and unsatisfied efficacy within complex tumor microenvironments, highlighting the need for biocompatible and highly effective ferroptosis inducers. Herein, we rationally constructed a series of supramolecular ferroptosis inducers (Ru1–Ru3) with sonosensitivity and sonocatalytic properties via molecular engineering, designed for cancer treatment through near-infrared fluorescence-guided sonodynamic therapy. Among them, Ru3 exhibited high ultrasound-triggered 1O2 generation efficiency (ΦΔ = 0.89) owing to its extended π-conjugated system and enhanced intramolecular charge transfer effect. Moreover, Ru3 possesses catalase mimic and peroxidase mimic activities, significantly improving ROS generation and diversifying ROS species. Further studies revealed that Ru3 localized predominantly in lysosomes, where it induced lysosomal membrane permeabilization and activated ferritinophagy under US irradiation, leading to the release of iron ions into the cytosol and triggering a Fenton reaction. Furthermore, Ru3 catalyzed the depletion of GSH and the oxidation of NADPH, disrupting redox homeostasis. These effects collectively suppressed GPX4 activity, promoted lipid LPO accumulation, and ultimately enhanced ferroptosis. In vivo experiments confirmed that US-activated Ru3 effectively inhibited 4T1 tumor growth with favorable biosafety. This work provides a research framework for the design of next generation ferroptosis inducers.
Introduction
Despite significant advances in cancer treatment, malignancies with high incidence and mortality rates continue to pose a major threat to human health.1–3 Sonodynamic therapy (SDT) has emerged as a promising anticancer approach, leveraging sonosensitizers to generate cytotoxic singlet oxygen (1O2) upon ultrasound (US) irradiation, effectively eliminating cancer cells. SDT offers several advantages, including precise spatiotemporal control, non-invasive application, and deep tissue penetration (>10 cm).4 The primary mechanism underlying its anticancer effects involves caspase-dependent apoptosis.5,6 However, the overexpression of anti-apoptotic proteins in malignant cells fosters resistance to apoptosis-inducing agents, thus diminishing the therapeutic efficacy of SDT.7–9 This underscores the urgent need to explore novel, non-apoptotic mechanisms of cell death to further enhance the effectiveness of SDT.Ferrous accumulation- and lipid peroxidation (LPO)-mediated ferroptosis, a non-apoptotic cell death pathway, has garnered increasing attention.10–12 However, the direct delivery of iron may induce severe side effects, such as hypersensitivity reactions in normal tissues. Therefore, employing non-iron-dependent agents to induce LPO could be safer for triggering ferroptosis in tumor cells. As the primary ‘executor’ in SDT, reactive oxygen species (ROS) can oxidize polyunsaturated fatty acids to lipid peroxides, ultimately leading to ferroptosis.13–17 The key factor promoting ferroptosis is the elevation of intracellular ROS levels. Recent studies have shown that organic sonosensitizers, with well-defined structures and flexible designs, can effectively generate ROS under US irradiation.18–22 However, most traditional sensitizers (e.g., cyanine and porphyrin) exhibit low ROS generation efficiency and are prone to ROS quenching.23–26 On the other hand, the hypoxic tumor microenvironment (TME) and the overexpression of reductive substances, such as nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione (GSH), can directly or indirectly reduce ROS levels.27 NADPH plays a key role in the recycling of GSH by donating electrons to glutathione reductase, which reduces oxidized glutathione (GSSG) back to GSH. GSH plays a critical role in supporting the antioxidant function of glutathione peroxidase 4 (GPX4), which converts harmful lipid peroxides into benign lipid alcohols, thereby protecting cells. Specifically, by directly affecting NADPH and depleting GSH, the activity of GPX4 can be inhibited, disrupting the redox balance in tumor cells and promoting ROS-mediated ferroptosis.28 Therefore, in the development of efficient sonosensitizers, it is crucial to consider their inherent multifunctionality, including the ability to alleviate hypoxia and disrupt antioxidant defense systems, to more effectively induce ferroptosis.
Recent studies have shown that supramolecular coordination complexes (SCCs), including Pt(II)- or Ru(II)-based SCCs, etc., demonstrate distinct advantages over small molecular precursors in applications such as bioimaging, photodynamic therapy, and SDT.29–33 The formation of SCCs effectively confines sonosensitizers within a rigid structure, thereby minimizing the aggregation-induced ROS quenching effects. Furthermore, the incorporation of heavy metals such as the Ru atom into SCCs enhances intersystem crossing (ISC) and intramolecular charge transfer (ICT) processes, promoting the generation of sonosensitizer-induced ROS.34–36 More importantly, the presence of Ru(II) metal centers in SCCs, with their rich redox, optoelectronic, and sonocatalytic properties, may endow these complexes with significant potential for mimicking enzymatic and sonocatalytic activities, particularly in adapting to the complex tumor microenvironment.37–40 However, the use of SCCs as potential US-activated ferroptosis inducers remains an underexplored area.
In this study, we successfully designed and synthesized a series of supramolecular ferroptosis inducers (Ru1–Ru3) via molecular engineering, which possess sonosensitizing and sonocatalytic properties for near-infrared (NIR) fluorescence-guided SDT. Among them, Ru3 exhibited superior 1O2 generation capability (ΦΔ = 0.89), attributed to its larger π-conjugated system and enhanced ICT effect, compared to homologues Ru1 (ΦΔ = 0.64) and Ru2 (ΦΔ = 0.82). In vitro studies demonstrated that Ru3 triggered a cascade of molecular events under US activation: first, Ru3 displayed catalase (CAT)-mimic and peroxidase (POD)-mimic catalytic activities, significantly enhancing ROS generation and expanding their diversity. And Ru3 mainly accumulated in lysosomes, where the ROS generated increased lysosomes' membrane permeability (LMP), activated ferritinophagy, and released Fe2+ into the cytoplasm, where it catalyzed the Fenton reaction, further amplifying oxidative stress. More importantly, Ru3 significantly disrupted cellular redox homeostasis by catalyzing GSH depletion and promoting NADPH oxidation. These effects collectively suppressed GPX4 activity, promoted lipid LPO accumulation, and ultimately enhanced ferroptosis. Finally, in vivo experiments demonstrated that US-activated Ru3 can effectively and safely ablate tumors in 4T1 tumor-bearing mice under NIR fluorescence-guided assistance. Therefore, this study provides a promising strategy for the development of long-wavelength-emitting supramolecular ferroptosis inducers with potential clinical applications.
Results and discussion
Design, synthesis, and characterization of Ru(II) metallacycles
To develop supramolecular ferroptosis inducers with high ROS generation efficiency and sonocatalytic capability, a series of novel sonosensitizers were synthesized through rational molecular design and structural modifications. In this design, thienothiadiazole (TTD) was selected as an electron-deficient core to construct a donor–acceptor–donor (D–A–D) conjugated structure, considering that the strong push–pull electron effect would favor enhanced ROS generation.41,42 First, L-1 was developed by using sterically hindered 3-hexylthienyl-substituted pyridine as the electron donor. Based on thienyl donor engineering, L-2 was obtained by introducing a phenyl ring structure to further expand the π-conjugation. To further enhance the ICT effect, L-3 was synthesized by incorporating a piperazine structure with stronger electron-donating ability. The chemical structures of L-1 to L-3 were comprehensively characterized using 1H NMR, 13C NMR, and MALDI-TOF MS (Fig. S1–S24). Furthermore, density functional theory (DFT) calculations were performed using Gaussian 09 to determine the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of L-1, L-2, and L-3 (Fig. S25). The results showed that the energy gaps of L-1 (1.74 eV), L-2 (1.66 eV), and L-3 (1.59 eV) gradually decreased, validating their potential as sonosensitizers.43,44 To further enhance the ROS generation efficiency and impart catalytic properties to these sonosensitizers, we chose a half-sandwich Ru(II) acceptor, with low dark toxicity and high ROS yield,45,46 to self-assemble with L-1, L-2, and L-3 at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio to form SCCs, named Ru1, Ru2, and Ru3 (Scheme 1). Compared to the individual small-molecule precursors, the rigid structure and incorporation of heavy metal Ru in SCCs may enhance ROS yield under US activation. More critically, the presence of Ru(II) metal centers in SCCs, with their rich redox, optoelectronic, and catalytic capabilities, may endow these SCCs with significant potential for mimicking enzymatic and sonocatalytic activities, particularly in adapting to the complex tumor microenvironment.
:1 molar ratio to form SCCs, named Ru1, Ru2, and Ru3 (Scheme 1). Compared to the individual small-molecule precursors, the rigid structure and incorporation of heavy metal Ru in SCCs may enhance ROS yield under US activation. More critically, the presence of Ru(II) metal centers in SCCs, with their rich redox, optoelectronic, and catalytic capabilities, may endow these SCCs with significant potential for mimicking enzymatic and sonocatalytic activities, particularly in adapting to the complex tumor microenvironment.
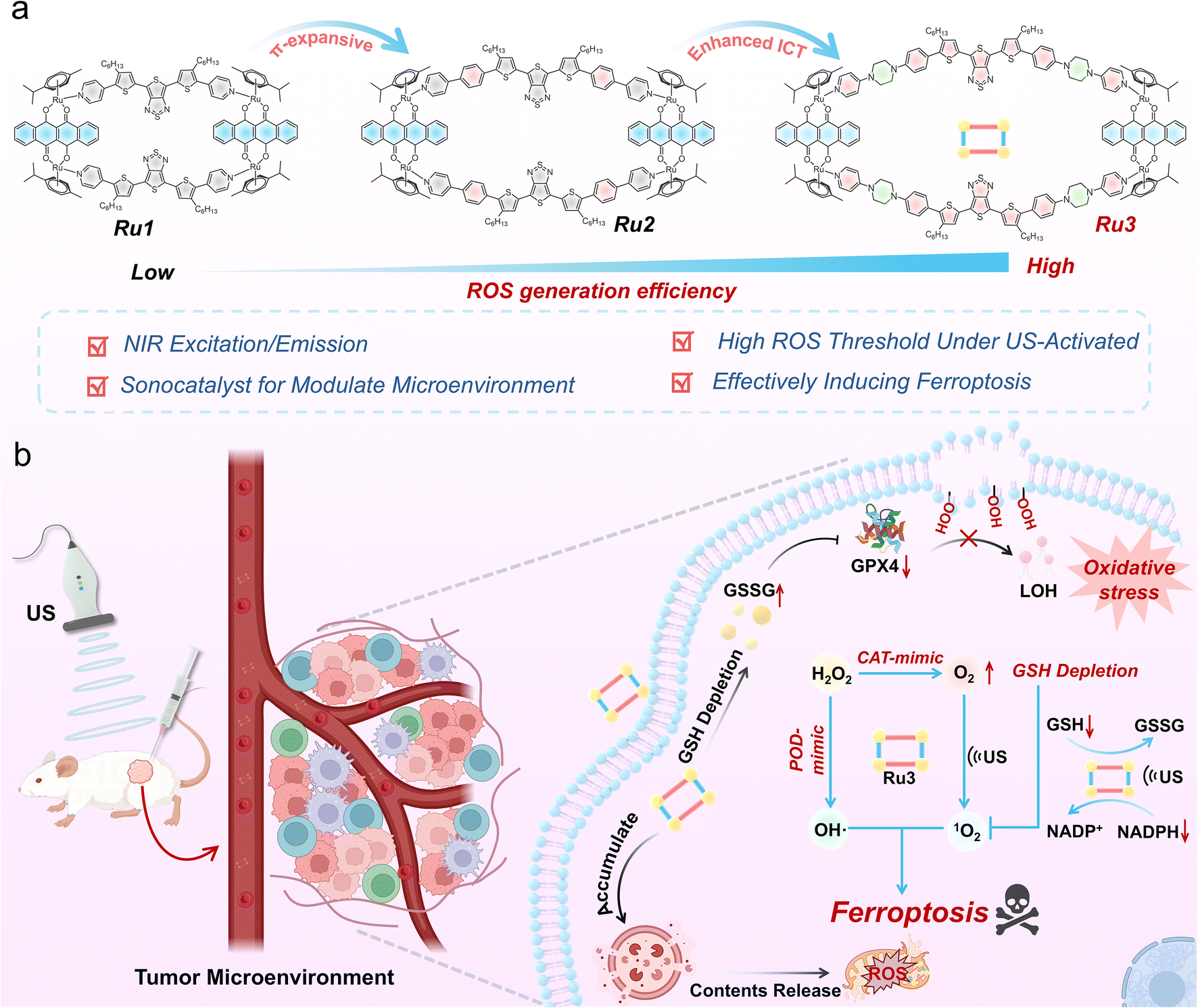 | ||
| Scheme 1 Schematic illustration of the supramolecular ferroptosis inducer Ru3 modulating the tumor microenvironment under US irradiation for tumor therapy. (a) Molecular engineering strategy used for the synthesis of Ru3. (b) The potential anti-tumor mechanisms of Ru3. | ||
The SCCs, Ru1, Ru2, and Ru3, were synthesized through the reaction of L-1, L-2, and L-3 with a Ru(II) acceptor in a 1:1 methanol/chloroform mixture, stirred at room temperature for 12 hours, affording total yields of 6.48% (Ru1), 4.10% (Ru2), and 2.43% (Ru3), respectively, as detailed in the SI. To characterize the synthesized SCCs, we initially employed 1H NMR and 2D COSY (Fig. S26–S37). As shown in Fig. 1a and b, a downfield shift of the pyridine proton peak (Ha) was observed compared to the corresponding ligands, with chemical shifts of 8.69–8.83 ppm for Ru1, 8.74–8.87 ppm for Ru2, and 8.35–8.71 ppm for Ru3. For Ru1, Ru2, and Ru3, the proton H1 of the SCCs was upfield-shifted by approximately 0.20, 0.40, and 0.24 ppm, respectively, relative to the proton H1 of the free Ru(II) receptor, while proton H2 was upfield-shifted by approximately 0.16, 0.17, and 0.21 ppm, respectively. These observed chemical shift changes during the self-assembly process are primarily attributed to the decrease in electron density when the TTD-based precursors coordinate to the electron-deficient Ru(II) centers. The 2D COSY spectra further confirmed the correct assignment of each proton in Ru1, Ru2, and Ru3. The clear spectra and distinctive NMR signals from both 1H NMR and 2D COSY spectra support the formation of these discrete SCCs. To further confirm the assembly of the SCCs, electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS) was carried out (Fig. 1c). Characteristic peaks corresponding to the elimination of trifluoromethanesulfonate (OTf−) counterions were observed, indicating successful formation of the [2 + 2] rectangular SCCs, with m/z values of 975.21 for [Ru1-3OTf]3+, 1076.35 for [Ru2-3OTf]3+, and 1189.69 for [Ru3-3OTf]3+. And all assigned isotope peaks closely matched the theoretical distribution, confirming that Ru1, Ru2, and Ru3 have the anticipated [2 + 2] assembly structure (Fig. S29, S33 and S37). Finally, DFT calculations were performed to optimize the most stable conformations of Ru1, Ru2, and Ru3. These calculations revealed that all three SCCs adopt a planar rectangular geometry (Fig. 1d and S38), with approximate cavity dimensions of 7.84/21.64 Å, 7.78/29.36 Å, and 7.75/37.31 Å (width/length) for Ru1, Ru2, and Ru3, respectively. These combined results confirm the successful self-assembly of the designed SCCs, Ru1, Ru2, and Ru3.
 | ||
| Fig. 1 The characterization of SCCs Ru1–Ru3. (a) The chemical structure of Ru1, Ru2 and Ru3. (b) The partial 1H NMR (400 MHz, 298 K) spectra of metallacycles Ru1–Ru3. (top: acceptor, middle: SCCs, and bottom: ligand) (c) Calculated and experimental ESI-TOF-MS spectra of Ru1 ([Ru1-3OTf]3+), Ru2 ([Ru2-3OTf]3+), and Ru3 ([Ru3-3OTf]3+). (d) Optimized molecular model of Ru3, top view (left), side view (right). For clarity, counterions and hydrogen atoms are omitted. | ||
Photophysical properties, sonodynamic performance, and sonocatalytic activity studies
Based on these molecules, we subsequently investigated the photophysical and sonodynamic properties of Ru1, Ru2, and Ru3. Ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectroscopy were employed to study the optical properties of these molecules in dimethyl sulfoxide (DMSO). Ru1, Ru2, and Ru3 exhibited maximum absorption wavelengths at 632 nm, 664 and 712 nm, respectively, which were similar to the absorption bands of precursors L-1 to L-3 (Fig. 2a and S39). Additionally, the maximum emission wavelengths of these molecules were observed at 944 nm, 959 nm and 1032 nm (with 808 nm as the excitation wavelength), indicating their potential application in NIR fluorescence imaging. Notably, compared to Ru1 and Ru2, Ru3 exhibited a significant redshift in both absorption and emission wavelengths, ascribed to the expanded π-conjugated system and enhanced ICT in the molecular backbone. | ||
| Fig. 2 In vitro photophysical properties, sonodynamic performance, and sonocatalytic activity of SCCs Ru3. (a) Normalized absorption spectra and the emission spectra (λex = 808 nm) of Ru1–Ru3 in DMSO. (b) Normalized absorption spectra of Ru1–Ru3 after incubation in PBS for 1–6 d. (c) Normalized absorption spectra of Ru1–Ru3 in DMF under US irradiation (1 W cm−2). (d) Fluorescent images of DCFH for detecting ROS generation by Ru1–Ru3 in different ratios of DMF and H2O during US irradiation. (e) Average fluorescence intensity of RDPP induced by Ru3 and Rubpy. (f) The DO (ΔO2) production after H2O2 co-incubation with Ru3. (g) UV/vis absorption spectra of MB after co-incubation with/or H2O2 and Ru3. (h) Time-dependent GSH consumption perform after co-incubation with Ru3 or H2O2 using DTNB as an indicator (n = 3, mean ± SD). (i) Average absorption spectra of NADPH after co-incubation with Ru3 at different time points in PBS solution. (j) Schematic illustration of the sonocatalytic capability of Ru3. | ||
Next, the chemical stability and sonostability of Ru1, Ru2, and Ru3 were evaluated by monitoring changes in their absorption spectra. As shown in Fig. 2b, S40 and S41, after incubating in phosphate-buffered saline (PBS) or 10% fetal bovine serum (FBS) for 6 days, no significant degradation in their absorption spectra was observed, indicating strong stability under physiological conditions. To assess the sonodynamic performance of these sonosensitizers, the ROS generation efficiency of Ru1, Ru2, and Ru3 was evaluated using 2′,7′-dichlorofluorescein (DCFH) as an indicator. Previous literature reports indicate that SDT commonly employs low-intensity focused US in a frequency range of 0.5–2 MHz. Among these, 1 MHz offers a suitable balance between spatial resolution, controllability and ROS production efficiency, providing sufficient tissue penetration depth while minimizing tissue damage.4,7–9 Upon US irradiation (1 W cm−2, 1 MHz), DCFH alone did not produce fluorescence. Interestingly, after 5 minutes of irradiation, the DCF fluorescence intensity increased by 7.21-fold, 10.14-fold, and 14.31-fold for Ru1, Ru2, and Ru3, respectively (Fig. 2c). These results demonstrated that Ru3 has significantly higher ROS generation efficiency than Ru1 and Ru2. Furthermore, electron paramagnetic resonance (EPR) spectroscopy was conducted to assess the types of ROS generated by Ru1, Ru2, and Ru3 under US irradiation. Among them, 1O2 was detected using 2,2,6,6-tetramethylpiperidine (TEMP) as the indicator, while hydroxyl radicals (˙OH) and superoxide anions (O2˙−) were detected using 5,5-dimethyl-1-pyrroline N-oxide (DMPO). As shown in Fig. S42, under US irradiation, Ru1, Ru2, and Ru3 primarily generated 1O2, with Ru3 exhibiting the highest efficiency. The 1O2 quantum yields (ΦΔ) of Ru1, Ru2, and Ru3 under US irradiation were determined to be 0.64, 0.82, and 0.89, respectively, using methylene blue (MB, ΦΔ = 0.52) as a reference sonosensitizer (Fig. S43). These three sonosensitizers produced negligible amounts of ˙OH and O2˙− (Fig. S44 and S45).
Previous studies have demonstrated that the incorporation of heavy atoms such as Ru in SCCs enhances ISC, thereby promoting ROS generation under external energy stimuli.47 To explore the underlying mechanisms, we performed geometry optimization and molecular orbital analysis of Ru1 to Ru3 using Gaussian 09. The results indicate that, compared to Ru1 and Ru2, Ru3 has the smallest singlet-triplet energy gap (ΔEST) (Ru1 = 0.46 eV, Ru2 = 0.40 eV, and Ru3 = 0.31 eV, Fig. S46). Additionally, the formation of SCCs effectively confines the sonosensitizer within a rigid structure, minimizing the aggregation-induced ROS quenching effect. To evaluate the ROS quenching resistance, we assessed the ROS generation of Ru1, Ru2, and Ru3 in various DMF/water mixtures (Fig. 2d). The results showed that, with increasing water content in the DMF/water mixture, the ROS generation of Rubpy decreased dramatically due to strong intermolecular π–π stacking interactions. In contrast, Ru3 exhibited superior ROS quenching resistance, attributed to its rigid structure and the steric hindrance of the Ru acceptor.29–35 Furthermore, to investigate whether SCCs can effectively generate ROS in deeper tissues, we studied the ROS penetration depth of Ru3 in 1% lipid (solidified by 1% agarose) using a tissue model. The results revealed that Ru3 achieved a penetration depth exceeding 10 cm (Fig. S47). In addition to the excellent sonodynamic performance exhibited by Ru3 under US, its sonothermal properties have also been further evaluated (Fig. S48). Overall, Ru3 demonstrated efficient ROS generation under US conditions, deep tissue ROS penetration, and excellent resistance to ROS quenching, underscoring its potential as a promising ferroptosis inducer.
Considering the redox enzyme-like activity demonstrated by Ru3-based agents, we next examined the catalytic activity of Ru3 under conditions mimicking the intracellular environment. To evaluate the CAT-mimic activity of Ru3, we assessed its ability to decompose H2O2 into O2. Initially, Ru(dpp)3Cl2 (RDPP) was used as an O2 indicator to probe its CAT-mimic performance. RDPP is a well-known luminescent O2 probe, widely used for O2 detection and quantification.48 The fluorescence of RDPP (with an emission maximum of 613 nm) was strongly quenched by molecular O2 due to dynamic quenching (λmax at 455 nm). As shown in Fig. 2e, under US conditions, Ru3 induced a decrease in the fluorescence intensity of RDPP in buffer solution. Similarly, the increase in dissolved O2 and the formation of gas bubbles supported substantial O2 generation (Fig. 2f and S49). We also confirmed this by observing an increase in ROS levels upon the addition of H2O2 (Fig. S50). To assess the POD-mimic activity of Ru3, we measured its ability to induce ˙OH generation. As illustrated in Fig. 2g and S51, the characteristic absorption peak of MB at 665 nm underwent a pronounced reduction, coupled with visible color alterations following the addition of Ru3, thereby confirming the generation of ˙OH. Similarly, by using tetramethylbenzidine (TMB) as a probe, which reacted with ˙OH to generated blue oxidized TMB with a characteristic absorption peak at 652 nm,49 we observed a clear stronger absorption upon the addition of Ru3 (Fig. S52).
GSH is a major endogenous antioxidant that plays a key role in maintaining redox homeostasis, capable of scavenging potential ROS bursts that may subsequently impair therapeutic outcomes.50,51 Given the redox properties of Ru(II), we further investigated the depletion of GSH in the presence of Ru3. To evaluate Ru3's ability to consume GSH, we used 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) as a GSH indicator. DTNB can react with GSH to form 5-thio-2-nitrobenzoic acid (TNB).52 As shown in Fig. 2h, Ru3 could induce a considerable amount of GSH depletion over time, fully demonstrating the effective depletion effect of Ru3 on GSH. Then, 1H NMR spectroscopy further verified the successful conversion of GSH to its oxidized form GSSG (Fig. S53). Also, cellular GSH can be regenerated from oxidized GSSG through a key NADPH-dependent process.53 Therefore, we proceeded to investigate the sonocatalytic oxidation of NADPH by Ru3. The results showed that, upon addition of Ru3, the NADPH absorption at ∼336 nm was significantly reduced with extended US exposure, and the NADPH oxidation turnover frequency (TOF) was 35.34 h−1 (Fig. 2i and S54). Similarly, 1H NMR spectroscopy further validated the transformation of NADPH into its oxidized form, NADP+ (Fig. S55). These observations suggested that Ru3 exhibited CAT/POD-mimic multi-enzyme activity and sonocatalyst-enhanced GSH and NADPH depletion, highlighting its significant potential for sonocatalyst-enhanced SDT in tumor cells ferroptosis (Fig. 2j).
In vitro cell uptake, localization, cytotoxicity and sonocatalytic activity studies
Given the excellent SDT performance demonstrated by the physicochemical properties of supramolecular sonosensitizers, we subsequently conducted in vitro experiments in 4T1 cells. First, inspired by the high-resolution imaging capabilities of Ru3, we evaluated its cellular uptake and localization. As shown in Fig. 3a and S56, after incubation with Ru3, 4T1 cells exhibited NIR-II fluorescence, which increased with incubation time and peaked at 24 hours. And the NIR-II fluorescence intensity of Ru3 remained high during the 48-hour monitoring period. After determining the cellular uptake and retention efficiency of Ru3, we examined its subcellular localization to reveal its distribution within organelles. Incubation of 4T1 cells with Ru3 and the LysoSensor Red probe (a commercial probe for lysosome imaging) for 30 min revealed that the NIR fluorescence signals from Ru3 overlapped with the red fluorescence from LysoTracker Red, showing a Pearson correlation coefficient (PCC) of 0.81 (Fig. 3b). Likewise, Ru3 fluorescence could well overlap with a commercial MitoTracker Red imaging probe (PCC = 0.72). These results suggested that Ru3 effectively entered 4T1 cells and primarily concentrated in lysosomes and mitochondria, which is critical for its anticancer efficacy.54 Then, we used the MTT assay to assess the cell viability of 4T1 cells after treatment with Ru1–Ru3. As shown in Fig. 3c and S57, the results indicated that Ru3 exhibited negligible dark toxicity at low concentrations in the absence of US irradiation. When incubated with 10 μM of Rubpy, Ru1, Ru2, and Ru3, and conducted US irradiation, the cell viability of Rubpy, Ru1, Ru2, and Ru3 decreased to 90%, 78%, 57%, and 35%, respectively, confirming that Ru3 demonstrated significantly better performance in SDT compared to the other treatment groups. | ||
| Fig. 3 In vitro cellular uptake, localization, cytotoxicity and sonocatalytic activity of SCC Ru3 in 4T1 cells. (a) Fluorescence images of 4T1 cells treated with Ru3 (10 μM) at different time points (0 h and 24 h). Scale bar: 10 μm. (b) Colocalization assay of Ru3 (10 μM) in 4T1 cells by using LysoTracker Red and MitoTracker Red. Scale bar: 10 μm. (c) Cell viabilities of 4T1 cells after incubation with different concentrations of Ru3 under US irradiation (1 W cm−2). Error bars represent mean ± SD (n = 3). (d) Fluorescence images of 4T1 cells co-incubated with RDPP under different treatments (Rubpy: 10 μM, Ru3: 10 μM, and US irradiation: 1 W cm−2). Scale bar: 10 μm. (e) Fluorescence images of 4T1 cells co-incubated with DCF under different treatments. Scale bar: 25 μm. (f) Fluorescence images of 4T1 cells stained with HPF, incubated with Ru3 (10 μM) and irradiated with US (1 W cm−2). Scale bar, 10 μm. (g) Calcein-AM and PI-stained images of 4T1 cells after incubation with different treatments (Rubpy: 10 μM, Ru3: 10 μM, and US power: 1 W cm−2). Scale bar, 25 μm. (h) Fluorescence images of GSH levels within the 4T1 cells after co-incubation with different treatment groups. (Rubpy: 10 μM, Ru3: 10 μM, and US power: 1 W cm−2). Scale bar, 10 μm. (i) Relative NADPH activity in 4T1 cell under various treatments. Error bars represent mean ± SD (n = 3). (j) Cell viabilities of 4T1 cells co-incubated (24 h) with Ru3 (10 μM) and various inhibitors, Fer-1 (10 μM), 3-MA (10 μM), Z-VAD-FMK (10 μM) and Nec-1 (10 μM) under US irradiation (1 W cm−2). Error bars represent mean ± SD (n = 3). | ||
To assess the potential of Ru3 as a sonosensitizer adaptable to the tumor microenvironment, we then evaluated the O2 levels within 4T1 cells using the RDPP staining method. As shown in Fig. 3d and S58, in the Ru3-treated group, a significant reduction in the red fluorescence of RDPP was observed, confirming an increase in O2 levels within the 4T1 cells. Conversely, no substantial change in RDPP fluorescence was detected after Rubpy treatment, suggesting its limited O2 production ability. These results indicated that Ru3 may generate O2 through the CAT-mimic process, alleviating hypoxia in 4T1 cells. Then, we incubated the SCC Ru3 together with the DCFH probe to validate the effective ROS generation by Ru3 in 4T1 cells. As shown in Fig. 3e, after incubation with Ru3 and 4T1 cells without irradiation, a visible green fluorescence was observed, which may suggest that Ru3-mediated POD-mimic catalysis induced the generation of ˙OH within the cells. Upon US treatment, Ru3 displayed a marked enhancement in green fluorescence compared to the Rubpy group, indicating US-triggered ROS generation. We further evaluated the POD-mimic activity by employing HPF as a ˙OH probe, and the results revealed a notable increase in HPF fluorescence intensity after Ru3 incubation, indicating the production of ˙OH via the POD-mimic mechanism in 4T1 cells (Fig. 3f). Finally, we used calcein AM and propidium iodide (PI) staining to distinguish live cells (green) from dead cells (red) (Fig. 3g). These results demonstrate that the Ru3 plus US group effectively generated ROS in 4T1 cells, showing enhanced sonodynamic performance.
Next, we investigated the consumption of GSH and the sonocatalytic oxidation of NADPH by Ru3 in 4T1 cells. ThiolTrace Violet 500 was used as an indicator of intracellular GSH levels. As shown in Fig. 3h and S59, the green fluorescence of ThiolTrace Violet 500 in the Ru3-treated group was significantly diminished, indicating a reduction in GSH levels in 4T1 cells, which confirmed Ru3's consumption of intracellular GSH. In contrast, no significant change in fluorescence was observed in the Rubpy-treated group, suggesting a weaker GSH depletion. After US irradiation, the green fluorescence in the Ru3 group almost completely disappeared, indicating that US-triggered ROS generation further depleted intracellular GSH. Subsequently, the sonocatalytic oxidation of NADPH by Ru3 in 4T1 cells was studied (Fig. 3i). After Ru3 treatment and US irradiation, significant oxidation of NADPH was observed, consistent with our sonocatalytic studies.
Cell death mechanism studies
To explore the potential mechanisms of cell death under US irradiation, we assessed the cell viability of 4T1 cells in the presence of various cell death pathway inhibitors (Fig. 3j), including ferroptosis, autophagy, apoptosis and necrosis. After treatment with z-VAD-fmk (an apoptosis inhibitor) or necrostatin-1 (Nec-1, a necrosis inhibitor), cell viability remained almost unchanged, indicating non-apoptotic and non-necrotic cell death. In contrast, the addition of the ferroptosis inhibitor ferrostatin-1 (Fer-1) effectively increased the survival of 4T1 cells, suggesting that US-activated Ru3 may induce ferroptosis. Furthermore, the autophagy inhibitor 3-methyladenine (3-MA) was found to partially improve cell viability, indicating that autophagic processes in lysosomes possibly contribute to this cell death mode.Increasing evidence suggests that lysosome-targeting sonodynamic materials exhibit ferroptosis effects in tumor cells, which are more prominent than ROS-induced cell damage. Lysosomal membrane permeabilization (LMP) induces the release of various substances into the cytoplasm, such as protons and iron, which activate or amplify cell death signaling under iron depletion conditions.55–57 Similarly, mitochondrial dysfunction is a hallmark of ferroptosis. Cells undergoing ferroptosis often show dissipation of mitochondrial membrane potential (MMP), increased mitochondrial membrane permeability, and structural damage to mitochondria. MMP is essential for maintaining mitochondrial function and energy metabolism.58–61 Given that Ru3 was primarily concentrated in both lysosomes and mitochondria, we then investigated the sonodynamic damage to lysosomes and mitochondria in situ. The JC-1 probe was used as a probe to detect the changes in MMP in cells. As shown in Fig. 4a, the Ru3 plus US treatment group exhibited the strongest green fluorescence, indicating synergistic depolarization and mitochondrial membrane damage. Subsequently, using acridine orange (AO) as an indicator, the red fluorescence of AO in 4T1 cells treated with Ru3 plus US disappeared dramatically compared to other treatments, indicating a loss of lysosomal integrity and dysfunction (Fig. 4b).
 | ||
| Fig. 4 Ferroptosis mechanism of 4T1 cells under Ru3 plus US treatment. Fluorescence images of 4T1 cells co-incubated with Rubpy/Ru3 (10 μM) and probes (a) JC-1 and (b) AO, with or without US irradiation. Scale bar JC-1: 25 μm; scale bar AO: 10 μm. (c) Fluorescence images of C11-BODIPY-stained 4T1 cells with the red and green channels indicating reduced C11-BODIPY and oxidized C11-BODIPY, respectively (top) and the mechanism of C11-BODIPY oxidation/reduction. Scale bars: 10 μm. (d) Relative MDA activity in 4T1 cells under various treatments. Error bars represent mean ± SD (n = 3). (e) Bio-TEM images showing ferroptosis after various treatments. Scale bar (top image): 5 μm; scale bar (below image): 1 μm. (f) Western blot assay of GPX4, ACSL4, ATG5 and NCOA4 levels in 4T1 cells after various treatments. (g) Cartoon illustration of the ferroptosis mechanism induced by Ru3 under US irradiation in 4T1 cells. | ||
Previous studies have shown GSH depletion can inactivate GPX4, thereby upregulating ROS-dependent LPO, which is a crucial factor in ferroptosis-induced cell death.10–13,62 Considering the oxidative stress imbalance and GPX4 inactivation, we assessed the changes in LPO levels during the US-induced tumor cell death process. As shown in Fig. 4c and S60, the Ru3 plus US treatment group exhibited significant LPO accumulation, as evidenced by the red fluorescence in the control group and the decrease in red fluorescence and an increase in green fluorescence in the Ru3 plus US treatment group. Moreover, since malondialdehyde (MDA) is a key end product of LPO, we measured the MDA concentration in 4T1 cells treated under different conditions. The results showed that the Ru3 plus US group exhibited the highest MDA levels compared to other control groups (Fig. 4d). Additionally, transmission electron microscopy (TEM) analysis revealed morphological changes in 4T1 cell mitochondria, including mitochondrial shrinkage, increased membrane density, and decreased cristae, which are typical of ferroptosis-induced mitochondrial dysfunction (Fig. 4e).
To further investigate the ferroptosis induced by Ru3 under US irradiation in 4T1 cells, we performed western blot (WB) analysis to detect the expression of ferroptosis-related proteins. As shown in Fig. 4f and S61, GPX4 expression was visibly reduced, which can be attributed to Ru3's ability to deplete GSH, while US irradiation further exacerbated this depletion. These results suggest that the generation of multiple ROS, including ˙OH and 1O2, and the depletion of GSH may together lead to irreversible GPX4 inactivation, consistent with the classical hallmark of ferroptosis. Additionally, we examined ferroptosis-related pathway markers, including Acyl-CoA synthetase long-chain family member 4 (ACSL4), which facilitates the esterification of polyunsaturated fatty acids (PUFAs) into PUFA-phospholipids (PUFA-PLs), which are prone to lipid peroxidation.63–66 WB analysis revealed that Ru3 plus US treatment significantly upregulated the expression of ACSL4, further confirming that Ru3 can induce ferroptosis in tumor cells. Previous reports have shown that oxidative stress can induce ferritinophagy and increase intracellular iron levels. This prompted us to explore whether ferritinophagy occurs in the cells. Nuclear receptor coactivator 4 (NCOA4) is known to be a key regulator of ferritinophagy, as it binds to ferritin and facilitates its transport to lysosomes for degradation.67–72 Notably, WB results showed a significant increase in NCOA4 expression, as well as in the LC3II/LC3I ratio and autophagy-related protein 5 (ATG5) following Ru3 plus US treatment, which could be reversed by NAC treatment. Additionally, changes in the levels of the relevant proteins GPX4, ASCL4, and ACOA4 were further monitored through Raman signal variations, and the results were consistent with the ferroptosis outcomes (Fig. S62). Overall, the enzyme-like activity and sonocatalytic performance of Ru3 enhanced ROS generation, induced ferritinophagy, depleted GSH and NADPH, and ultimately inhibited GPX4 activity, significantly promoting ferroptosis in tumor cells (Fig. 4g).
In vivo anticancer application
Given the excellent in vitro synergistic therapeutic effects of Ru3, we evaluated its in vivo antitumor efficacy using a 4T1 tumor-bearing mouse model. Before conducting in vivo experiments, we first performed hemolysis tests to ensure the biocompatibility of Ru3 in biological systems (Fig. S63). Next, a total of 30 mice were randomly divided into 6 groups (5 mice per group): (1) control group, (2) US group, (3) Rubpy group, (4) Rubpy + US, (5) Ru3 group, and (6) Ru3 + US group. Mice in the Rubpy and Ru3 groups were intratumorally injected with Rubpy (1 mg Ru per kg) and Ru3 (1 mg Ru per kg), respectively, while the other groups received PBS. Following intratumoral injection of PBS, Rubpy or Ru3, the mice were subjected to US irradiation (1.0 MHz, 1.0 W cm−2, 50% duty cycle, 5 min) at 12 hours post-injection (US, Rubpy + US, and Ru3 + US groups), or left untreated (PBS, Rubpy, and Ru3 groups) (Fig. 5a). Mouse body weight and tumor volume were recorded every other day. We then visualized the tumor after injection with Ru3 by using NIR fluorescence imaging, which guided subsequent sonodynamic therapy (Fig. S64). After US irradiation, the results showed complete tumor eradication in the Ru3 plus US group after 14 days of treatment. In contrast, the Rubpy plus US group exhibited only mild tumor suppression (Fig. 5b and c). Furthermore, to assess the acute systemic toxicity of Ru3, we monitored body weight changes in the mice (Fig. 5d). The results indicated that the weight loss observed during treatment was negligible. Finally, on day 20, normal organs from each group of mice were collected for hematoxylin and eosin (H&E) staining. After 20 days of treatment, no significant organ damage was observed in any of the treatment groups (Fig. S65). It is noteworthy that the tumor tissue from the Ru3 + US group exhibited a significantly lower cancer cell density compared to the other control groups, indicating the strong effectiveness of Ru3-mediated sonodynamic therapy. GPX4 immunofluorescence staining showed a marked downregulation in the Ru3 + US group, suggesting that Ru3-induced ferroptosis plays a key role in its antitumor activity (Fig. 5d). These preliminary but comprehensive evaluations suggest that the sonosensitizer Ru3 exhibits excellent biocompatibility and is suitable for potential in vivo therapeutic applications. | ||
| Fig. 5 In vivo antitumor experiments. (a) The protocol of the experiment. (b) Representative image of 4T1 tumor-bearing mice under different treatments at day 1 and day 14. (c) The linear curve depicts the 4T1 tumor suppression in different treatment groups. Error bars represent mean ± SD (n = 5). (d) Representative image depicts the body weight change of mice after various treatments. Error bars represent mean ± SD (n = 5). (e) H&E and GPX4 staining of tumor slices collected from tumor mouse models. Scale bar: H&E, 100 μm; GPX4, 50 μm. | ||
Conclusions
In conclusion, we successfully developed a series of supramolecular ferroptosis inducers (Ru1–Ru3) through a molecular engineering approach for NIR fluorescence-guided ferroptosis-enhanced SDT. Under US irradiation, Ru3, with its extended π-conjugated system and enhanced ICT effect, demonstrated superior ROS generation. In vitro studies revealed that Ru3 exhibited both CAT-mimic and POD-mimic catalytic activities, further boosting ROS production and broadening their diversity. Furthermore, Ru3 catalyzed the depletion of GSH and promoted NADPH oxidation, disrupting cellular redox homeostasis, inducing autophagy, and triggering ferroptosis in 4T1 tumor cells. In vivo, Ru3 effectively inhibited 4T1 tumor growth upon US activation with negligible side effects. This work offers a promising strategy for developing long-wavelength-emitting supramolecular sonosensitizers, particularly for catalytically enhanced ferroptosis therapy with potential clinical applications.Author contributions
Y. Sun conceived the project and designed the experiments. Y. Pang, C. Li and J. Zhang designed, synthesized and characterized the materials. L. Mei carried out theoretical calculations. Y. Luo and Q. Li performed the in vitro and in vivo studies. Y. Sun, Y. Pang, C. Li and J. Li wrote the manuscript. All authors analyzed and discussed the results and have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
All the data supporting this article have been included in the main text and the SI. Supplementary information includes characterization data and synthesis steps of Ru1–Ru3; NMR spectra and MS spectra of Ru1–Ru3; cell imaging and therapeutic data, as well as in vivo imaging data. See DOI: https://doi.org/10.1039/d5sc05712d.Acknowledgements
We thank the National Natural Science Foundation of China (22374055, 22022404, 22474047, and 22204055), the National Natural Science Foundation of Hubei Province (2022CFA033), and the Fundamental Research Funds for the Central Universities (2662025SYPY005).References
- Y. D. Guo, P. Hu and J. L. Shi, J. Am. Chem. Soc., 2024, 146, 10217–10233 CrossRef CAS PubMed.
- X. R. Wang, X. J. Song, Q. L. Wei, W. J. Wang, H. E. Xu and X. C. Dong, Coord. Chem. Rev., 2024, 521, 216189 CrossRef CAS.
- G. Lei, L. Zhuang and B. Y. Gan, Cancer Cell, 2024, 42, 513–534 CrossRef CAS PubMed.
- Y. Zhang, X. Q. Zhang, H. C. Yang, L. Yu, Y. Xu, A. Sharma, P. Yin, X. Y. Li, J. S. Kim and Y. Sun, Chem. Soc. Rev., 2021, 50, 11227–11248 RSC.
- H. C. Yang, L. Tu, J. Li, S. Y. Bai, Z. X. Hu, P. Yin, H. Y. Lin, Q. Yu, H. D. Zhu and Y. Sun, Coord. Chem. Rev., 2022, 453, 214333 CrossRef CAS.
- X. J. Xing, S. J. Zhao, T. Xu, L. Huang, Y. Zhang, M. H. Lan, C. W. Lin, X. L. Zheng and P. F. Wang, Coord. Chem. Rev., 2021, 445, 214087 CrossRef CAS.
- Y. A. Martins, T. Z. Pavan and R. F. V. Lopez, Int. J. Pharm., 2021, 610, 121243 CrossRef PubMed.
- D. Li, Y. J. Zhu, W. W. Yin, X. T. Lin, G. Kim, Z. Y. Liu, S. W. Jung, J. Seo, S. Kim, J. S. Kim, H. Y. Huang and P. Y. Zhang, Chem. Soc. Rev., 2025, 54, 7610–7653 RSC.
- S. Son, J. H. Kim, X. W. Wang, C. L. Zhang, S. A. Yoon, J. Shin, A. Sharma, M. H. Lee, L. Cheng, J. S. Wu and J. S. Kim, Chem. Soc. Rev., 2020, 49, 3244–3261 RSC.
- F. T. Zeng, S. Nijiati, L. G. Tang, J. M. Ye, Z. J. Zhou and X. Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300379 CrossRef CAS PubMed.
- N. Kang, S. Son, S. H. Min, H. Hong, C. Kim, J. An, J. Kim and H. Kang, Chem. Soc. Rev., 2023, 52, 3955–3972 RSC.
- N. Singh, D. Kim, S. Min, E. Kim, S. Kim, Y. S. Zhang, H. Kang and J. S. Kim, Coord. Chem. Rev., 2025, 522, 216236 CrossRef CAS.
- S. K. Yao, F. W. Xu, Y. Wang, J. Z. Shang, S. M. Li, X. Y. Xu, Z. P. Liu, W. J. He, Z. J. Guo and Y. C. Chen, J. Am. Chem. Soc., 2025, 147, 11132–11144 CrossRef CAS PubMed.
- L. Zhu, X. Y. Wang, T. Tian, Y. Y. Chen, W. J. Du, W. Wei, J. Zhao, Z. J. Guo and X. X. Wang, Chem. Sci., 2024, 15, 10499–10507 RSC.
- T. Xiong, Y. C. Chen, Q. Peng, S. Lu, S. R. Long, M. L. Li, H. Wang, S. Lu, X. Q. Chen, J. L. Fan, L. Wang and X. J. Peng, Adv. Mater., 2024, 36, 2309711 CrossRef CAS PubMed.
- Q. Hu, W. J. Zhu, J. J. Du, H. Y. Ge, J. Z. Zheng, S. R. Long, J. L. Fan and X. J. Peng, Chem. Sci., 2023, 14, 9095–9100 RSC.
- W. Li, S. L. Yin, Y. Shen, H. Y. Li, L. Yuan and X. B. Zhang, J. Am. Chem. Soc., 2023, 145, 3736–3747 CrossRef CAS PubMed.
- X. Q. Zhang, C. L. Li, Y. Zhang, X. F. Guan, L. C. Mei, H. L. Feng, J. Li, L. Tu, G. Q. Feng, G. Z. Deng and Y. Sun, Adv. Funct. Mater., 2022, 32, 2207259 CrossRef CAS.
- Z. R. Gong and Z. F. Dai, Adv. Sci., 2021, 8, 2002178 CrossRef CAS PubMed.
- C. Zhang and K. Y. Pu, Adv. Mater., 2023, 35, 2303059 CrossRef CAS PubMed.
- Y. D. Pang, F. Zhao, Y. T. Wang, Q. Song, Q. Wang, J. R. Li, R. P. Zhang and Y. Sun, Coord. Chem. Rev., 2025, 535, 216664 CrossRef CAS.
- Y. L. Wang, L. S. Luo, T. T. Zhang, J. R. Hu, H. L. Wang, F. Bao, C. L. Li, Y. Sun and J. R. Li, ACS Appl. Mater. Interfaces, 2024, 16, 52068–52079 CrossRef CAS PubMed.
- J. Zhu, C. C. Chu, D. S. Li, X. Pang, H. L. Zheng, J. Q. Wang, Y. S. Shi, Y. Zhang, Y. Cheng, E. Ren, J. L. Cheng, X. Y. Chen and G. Liu, Adv. Funct. Mater., 2019, 29, 1904056 CrossRef.
- Z. G. Chen, L. Sang, Y. J. Liu and Z. Q. Bai, Adv. Sci., 2025, 12, 2417439 CrossRef CAS PubMed.
- S. Liang, J. J. Yao, D. Liu, L. Rao, X. Y. Chen and Z. H. Wang, Adv. Mater., 2023, 35, 2211130 CrossRef CAS PubMed.
- Y. R. Yang, J. Huang, M. Liu, Y. G. Qiu, Q. H. Chen, T. J. Zhao, Z. X. Xiao, Y. Q. Yang, Y. T. Jiang, Q. Huang and K. L. Ai, Adv. Sci., 2023, 10, 2204365 CrossRef CAS PubMed.
- X. M. Yao, W. Li, D. Fang, C. Y. Xiao, X. Wu, M. H. Li and Z. Luo, Adv. Sci., 2021, 8, 2100997 CrossRef CAS PubMed.
- H. C. Yang, X. M. Yao, Y. Q. Liu, X. K. Shen, M. H. Li and Z. Luo, ACS Nano, 2023, 17, 15328–15353 CrossRef CAS PubMed.
- H. Sepehrpour, W. X. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS PubMed.
- C. L. Li, Y. D. Pang, Y. L. Xu, M. J. Lu, L. Tu, Q. Li, A. Sharma, Z. Z. Guo, X. Y. Li and Y. Sun, Chem. Soc. Rev., 2023, 52, 5340–5342 RSC.
- D. D. Xu, Y. Li, S. C. Yin and F. H. Huang, Chem. Soc. Rev., 2024, 53, 3167–3204 RSC.
- Y. Qin, X. H. Chen, Y. X. Gui, H. Wang, B. Z. Tang and D. Wang, J. Am. Chem. Soc., 2022, 144, 12825–12833 CrossRef CAS PubMed.
- Y. L. Xu, C. L. Li, S. Lu, Z. Z. Wang, S. Liu, X. J. Yu, X. P. Li and Y. Sun, Nat. Commun., 2022, 13, 2009 CrossRef CAS PubMed.
- Y. D. Pang, Q. Li, J. L. Wang, S. Y. Wang, A. Sharma, Y. L. Xu, H. Y. Hu, J. R. Li, S. Liu and Y. Sun, Angew. Chem., Int. Ed., 2025, 64, e202415802 CrossRef CAS PubMed.
- Y. Ding, Z. R. Tong, L. L. Jin, B. L. Ye, J. Zhou, Z. Q. Sun, H. Yang, L. J. Hong, F. H. Huang, W. L. Wang and Z. W. Mao, Adv. Mater., 2022, 34, 2106388 CrossRef CAS PubMed.
- B. X. Huang, X. Liu, G. L. Yang, J. Tian, Z. Y. Liu, Y. C. Zhu, X. P. Li, G. Q. Yin, W. Zheng, L. Xu and W. A. Zhang, CCS Chem., 2022, 4, 2090–2101 CrossRef CAS.
- K. Li, L. Y. Zhang, C. Yan, S. C. Wei, M. Pan, L. Zhang and C. Y. Su, J. Am. Chem. Soc., 2014, 136, 4456–4459 CrossRef CAS PubMed.
- S. Chen, K. Li, F. Zhao, L. Zhang, M. Pan, Y. Z. Fan, J. Guo, J. Y. Shi and C. Y. Su, Nat. Commun., 2016, 7, 13169 CrossRef CAS PubMed.
- C. Liang, J. E. Xie, S. L. Luo, C. Huang, Q. L. Zhang, H. Y. Huang and P. Y. Zhang, Nat. Commun., 2021, 12, 5001 CrossRef CAS PubMed.
- Y. L. Xu, Y. D. Pang, L. S. Luo, A. Sharma, J. F. Yang, C. L. Li, S. Liu, J. B. Zhan and Y. Sun, Angew. Chem., Int. Ed., 2024, 63, e202319966 CrossRef CAS PubMed.
- L. Yang, J. C. Huang, Y. N. Liao, D. P. Hu, Y. K. He, N. Feng, R. T. K. Kwok, J. W. Y. Lam, J. Zhang, B. Z. He and B. Z. Tang, Adv. Healthcare Mater., 2025, 14, 2500513 CrossRef CAS PubMed.
- J. Zhu, Y. Q. Zhu, J. H. Huang, W. J. Zhang, J. Q. Hou, B. Z. Tang and D. Wang, Adv. Mater., 2025, 37, 2502452 CrossRef CAS PubMed.
- C. Fu, W. Zhao, X. L. Wang, X. He, Y. T. Yin, J. Y. Li, Q. Y. Deng, C. H. Yan, Y. L. Yin, Z. M. Wang and R. Hu, Aggregate, 2025, 6, e70058 CrossRef CAS.
- W. Zhao, C. Fu, H. Y. Gao, Y. Z. Zhou, C. H. Yan, Y. L. Yin, R. Hu and B. Z. Tang, Mater. Chem. Front., 2023, 7, 6229–6235 RSC.
- C. L. Li, L. Tu, J. F. Yang, C. Liu, Y. L. Xu, J. R. Li, W. Tuo, B. Olenyuk, Y. Sun, P. J. Stang and Y. Sun, Chem. Sci., 2023, 14, 2901–2909 RSC.
- L. Tu, C. L. Li, X. X. Xiong, J. H. Kim, Q. Li, L. C. Mei, J. R. Li, S. Liu, J. S. Kim and Y. Sun, Angew. Chem., Int. Ed., 2023, 62, e202301560 CrossRef CAS PubMed.
- Y. Li, F. H. Huang, P. J. Stang and S. C. Yin, Acc. Chem. Res., 2024, 57, 1174–1187 CrossRef CAS PubMed.
- B. J. Geng, J. Y. Hu, X. L. He, Z. L. Zhang, J. M. Cai, D. Y. Pan and L. X. Shen, Adv. Mater., 2024, 36, 2313670 CrossRef CAS PubMed.
- H. X. Zhang, J. Z. Lv, H. Wu, Y. H. He, M. Y. Li, C. H. Wu, D. Lv, Y. Y. Liu and H. Yang, Sci. Adv., 2025, 11, eadq3870 CrossRef CAS PubMed.
- Y. Zheng, W. J. Wang, J. X. Chen, K. Peng, X. X. Chen, Q. H. Shen, B. B. Liang, Z. W. Mao and C. P. Tan, Adv. Sci., 2024, 12, 2411629 CrossRef PubMed.
- D. Wen, J. Feng, R. P. Deng, K. Li and H. J. Zhang, Nat. Commun., 2024, 15, 9359 CrossRef CAS PubMed.
- D. Li, M. H. Fan, H. B. Wang, Y. J. Zhu, B. L. Yu, P. Y. Zhang and H. Y. Huang, Chem. Sci., 2024, 15, 10027–10035 RSC.
- P. P. Xue, H. L. Zhuang, S. J. Shao, T. J. Bai, X. M. Zeng and S. Q. Yan, ACS Nano, 2024, 18, 25795–25812 CrossRef CAS PubMed.
- J. Zhou, Y. Z. Zhang, G. C. Yu, M. R. Crawley, C. R. P. Fulong, A. E. Friedman, S. Sengupta, J. F. Sun, Q. Li, F. H. Huang and T. R. Cook, J. Am. Chem. Soc., 2018, 140, 7730–7736 CrossRef CAS PubMed.
- Y. Li, B. Liu, Y. Zheng, M. Hu, L. Y. Liu, C. R. Li, W. Zhang, Y. X. Lai and Z. W. Mao, J. Med. Chem., 2024, 67, 16235–16247 CrossRef CAS PubMed.
- T. Feng, Z. X. Tang, J. Karges, J. Shu, K. Xiong, C. Z. Jin, Y. Chen, G. Gasser, L. N. Ji and H. Chao, Chem. Sci., 2024, 15, 6752–6762 RSC.
- A. M. Wu, M. Li, Y. Y. Chen, W. Zhang, H. R. Li, J. Z. Chen, K. Gu and X. X. Wang, Adv. Healthcare Mater., 2024, 13, 2302556 CrossRef CAS PubMed.
- K. Q. Ma, H. Yang, T. R. Shen, Y. K. Yue, L. L. Zhao, X. G. Liu, F. J. Huo and C. X. Yin, Chem. Sci., 2022, 13, 3706–3712 RSC.
- S. Wang, C. Chen, J. M. Wu, J. Y. Zhang, J. W. Y. Lam, H. Y. Wang, L. Chen and B. Z. Tang, Sci. China:Chem., 2022, 65, 870–876 CAS.
- N. Montesdeoca, L. Johannknecht, E. Efanova, J. Heinen-Weiler and J. Karges, Angew. Chem., Int. Ed., 2024, 63, e202412585 CrossRef CAS PubMed.
- X. Tian, J. Y. Cheng, L. Yang, Z. X. Li and M. M. Yu, Chem. Biomed. Imaging, 2024, 2, 518–525 CrossRef CAS PubMed.
- Y. S. Yang, X. L. Liu, D. Y. Xi, Y. B. Zhang, X. C. Gao, K. Xu, H. Y. Liu and M. X. Fang, Chem. Biomed. Imaging, 2024, 3, 169–179 CrossRef PubMed.
- S. Doll, B. Proneth, Y. Y. Tyurina, E. Panzilius, S. Kobayashi, I. IngoId, M. Irmler, J. Beckers, M. Aichler, A. Walch, H. Prokisch, D. Trümbach, G. W. Mao, F. Qu, H. Bayir, J. Füllekrug, C. H. Scheel, W. Wurst, J. A. Schick, V. E. Kagan, J. P. F. Angeli and M. Conrad, Nat. Chem. Biol., 2017, 13, 91–98 CrossRef CAS PubMed.
- R. Li, X. J. Yan, C. C. Xiao, T. T. Wang, X. J. Li, Z. Y. Hu, J. L. Liang, J. B. Zhang, J. Y. Cai, X. Sui, Q. L. Liu, M. L. Wu, J. Q. Xiao, H. T. Chen, Y. S. Liu, C. H. Jiang, G. Lv, G. H. Chen, Y. C. Zhang, J. Yao, J. Zheng and Y. Yang, Nat. Commun., 2024, 15, 4760 CrossRef CAS PubMed.
- Y. Q. Wang, M. Z. Hu, J. Cao, F. X. Wang, J. R. Han, T. W. Wu, L. X. Li, J. S. Yu, Y. J. Fan, G. L. Xie, H. Y. Lian, Y. Y. Cao, N. Naowarojna, X. Wang and Y. L. Zou, Cell, 2025, 188, 412–429 CrossRef CAS PubMed.
- X. Tian, J. Y. Cheng, L. Yang, Z. X. Li and M. M. Yu, Chem. Biomed. Imaging, 2024, 2, 518–525 CrossRef CAS PubMed.
- Y. Y. Fang, X. C. Chen, Q. Y. Tan, H. H. Zhou, J. Xu and Q. Gu, ACS Cent. Sci., 2021, 7, 980–989 CrossRef CAS PubMed.
- A. Anandhan, M. Dodson, A. Shakya, J. J. Chen, P. F. Liu, Y. Y. Wei, H. Tan, Q. Wang, Z. Y. Jiang, K. Yang, J. G. N. Garcia, S. K. Chambers, E. Chapman, A. Ooi, Y. Yang-Hartwich, B. R. Stockwell and D. D. Zhang, Sci. Adv., 2023, 9, eade9585 CrossRef CAS PubMed.
- A. T. Ouyang, T. Chen, Y. Feng, J. H. Zou, S. Y. Tu, M. J. Jiang, H. M. Sun and H. B. Zhou, Adv. Sci., 2024, 11, 2404365 CrossRef CAS PubMed.
- C. Wang, F. Yuan, C. Fu, Y. Li, G. Dai, H. S. Kim, S. Xia, L. Yu, S. Debnath, W. X. Ren, J. Shu, M. Qiu and J. S. Kim, J. Am. Chem. Soc., 2025, 147, 136–148 CrossRef PubMed.
- Q. Ding, C. Wang, H. Wang, C. Xiang, Z. Wang, Y. Wang, L. Zhao, M. Vendrell and J. S. Kim, J. Am. Chem. Soc., 2025, 148, 16661–16671 CrossRef PubMed.
- J. Kim, Y. Xu, J. H. Lim, J. Y. Lee, M. Li, J. M. Fox, M. Vendrell and J. S. Kim, J. Am. Chem. Soc., 2025, 147, 701–712 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |