
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnergy migration, charge transfer, and charge dissociation in self-assembling nonfullerene acceptor aggregates with zincporphyrin-nonfullerene acceptor dyads†
Yasunari
Tamai
 *ab,
Midori
Akiyama
c,
Lorenzo
Vallan
c,
Daiki
Sasada
c,
Katsuaki
Suzuki
d,
Hironori
Kaji
*d,
Takumi
Urakami
c,
Hirofumi
Sato
c,
Masahiro
Higashi
*e,
Seiichiro
Izawa
f,
Motohisa
Kubota
c,
Tomokazu
Umeyama
g and
Hiroshi
Imahori
*chi
*ab,
Midori
Akiyama
c,
Lorenzo
Vallan
c,
Daiki
Sasada
c,
Katsuaki
Suzuki
d,
Hironori
Kaji
*d,
Takumi
Urakami
c,
Hirofumi
Sato
c,
Masahiro
Higashi
*e,
Seiichiro
Izawa
f,
Motohisa
Kubota
c,
Tomokazu
Umeyama
g and
Hiroshi
Imahori
*chi
aDepartment of Advanced Materials Science, Graduate School of Frontier Sciences, The University of Tokyo, Kashiwanoha 5-1-5, Kashiwa, 277-8561, Japan. E-mail: tamai@edu.k.u-tokyo.ac.jp
bJapan Science and Technology Agency (JST), PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama, 332-0012, Japan
cDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyoto 615-8510, Japan. E-mail: imahori@scl.kyoto-u.ac.jp
dInstitute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: kaji@scl.kyoto-u.ac.jp
eDepartment of Complex Systems Science, Graduate School of Informatics, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8601, Japan. E-mail: higashi@nagoya-u.jp
fMaterials and Structures Laboratory, Institute of Science Tokyo, 4259 Nagatsuta-cho, Midori-ku, Yokohama, Kanagawa 226-8503, Japan
gDepartment of Applied Chemistry, Graduate School of Engineering, University of Hyogo, Himeji, Hyogo 671-2280, Japan
hInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Kyoto 606-8501, Japan
iInstitute for Liberal Arts and Sciences (ILAS), Kyoto University, Kyoto 606-8316, Japan
First published on 29th July 2025
Abstract
The synergy between self-assembling donor–acceptor–donor type nonfullerene acceptors (TACIC-Br) and zincporphyrin-nonfullerene acceptor linked molecules (ZnP-TACIC) provides a compelling model for examining key multi-step processes, including energy migration, charge transfer (CT), and charge dissociation (CD) in photosynthesis and organic photovoltaics (OPVs). Remarkably, TACIC-Br molecules exhibited a strong tendency to aggregate, even in the good solvent CHCl3. However, when the proportion of the poor solvent (MeOH) exceeded 40% in a CHCl3/MeOH mixture (v/v), these aggregates displayed an unusually prolonged excited singlet-state lifetime, comparable to TACICs in thin films. Solid-state NMR spectroscopy and theoretical calculations revealed that within the TACIC aggregates, a slipped or T-shaped dimeric π–π packing arrangement is favored, positioning the thienoazacoronene donor unit and the 1,1-dicyanomethylene-3-indanone acceptor unit in close proximity. This supramolecular packing effectively suppresses both nonradiative and radiative decay processes in CHCl3/MeOH mixtures and thin films, contrasting sharply with typical self-quenching observed in conventional dye aggregates. Time-resolved transient absorption measurements showed efficient energy migration, CT, and CD within these composite aggregates. With an extremely long singlet excited-state diffusion length (LD) of 45.6 nm, facilitated by the prolonged excited singlet-state lifetime, TACICs are well-suited for efficient energy migration. Notably, after quantitative CT at the ZnP-TACIC molecule, 35% of the CT states in the aggregates dissociated to form free ion pairs. This integrated supramolecular approach adeptly emulates both light-harvesting and CT and CD processes in photosynthesis and OPVs, thereby offering potential applications in solar energy conversion.
Introduction
Supramolecular chemistry investigates intermolecular bonds such as hydrogen bonding, π–π interactions, and electrostatic forces. This field focuses on understanding the structures and functions of the assemblies formed through interactions between two or more complex chemical species.1 The overarching goal is to develop molecular and supramolecular devices, which are integrated chemical systems with organized structures and specialized functions built upon supramolecular architectures.2–14 However, constructing supramolecular systems with enhanced, multifaceted functionality poses a challenge due to the inherent weakness and complexity of these interactions. When self-assembling units are deliberately introduced into molecules, they may promote organization, yet this often compromises the intended functions due to interference from the rather bulky self-assembling units. Therefore, the ideal approach involves embedding self-assembling units directly into molecular frameworks without hindering functional properties.A compelling area within supramolecular chemistry is light energy conversion.15–30 Photosynthesis serves as inspiration, where sunlight is captured by precisely arranged chromophores in light-harvesting complexes. The collected energy is directed to reaction centers for charge separation (CS), transforming light into chemical energy. Mimicking this natural multi-step processes, supramolecular strategies have been employed to integrate energy transduction and CS in both solutions and electrodes.15–37 These methods have demonstrated potential for efficiently converting light energy into chemical energy in solar fuels generation and electricity in organic photovoltaics (OPVs).
Nonfullerene acceptors (NFAs) have gathered considerable attention due to their high light-harvesting properties in the visible and near-infrared region, facile HOMO–LUMO level tuning, and self charge transfer (CT) and charge dissociation (CD) at interfaces of OPVs, improving power conversion efficiencies (PCEs) of up to 20%.38–40 Recently, we introduced a new class of NFAs, termed TACIC (X = H in Fig. 1), specifically designed for OPVs.41–43 The OPV device with PBDB-T donor polymer and TACIC showed a PCE of 9.92%, which was comparable to a PCE (9.71%) of the OPV device with PBDB-T and the representative NFA, ITIC (vide infra).41 Given the self-assembling characteristics of thienoazacoronene through π–π interactions,44–46 we hypothesized that integrating this moiety into the donor moiety of acceptor–donor–acceptor (A–D–A) type NFAs would strengthen intermolecular interactions, positively impacting photodynamics. Indeed, TACIC stands apart from conventional NFAs including ITIC, exhibiting an extended excited singlet-state lifetime in thin films (τ = 1.3–2.3 ns) compared to chloroform solution (≤0.22 ns). This distinct behavior stems from its intrinsic self-assembly, which effectively reduces both radiative (kr) and nonradiative (knr) decay rate constants in films. Importantly, this self-assembly is an inherent feature of its molecular structure, preserving its core photophysical properties despite the yet-to-be-elucidated supramolecular structures of TACIC due to its amorphous state. It should be emphasized here that the prolonged excited singlet state of NFAs enables a reduced driving force for photoinduced charge transfer (CT) from donor polymers to NFAs, thereby minimizing open-circuit voltage loss and enhancing PCE in OPVs.38–40 We hypothesized that if TACIC aggregates in solutions could form structures akin to those in films, efficient energy migration within the aggregates would ensure. More importantly, integrating D–A molecules capable of CS within these aggregates would enable energy migration to facilitate CT and CD.
 | ||
| Fig. 1 Molecular structures of ZnP-TACIC, ZnP-ref, TACIC (X = H), and TACIC-Br (X = Br). | ||
In this study, we designed a TACIC-Br (X = Br in Fig. 1) and a zincporphyrin (ZnP)-TACIC dyad (Fig. 1). TACIC-Br is expected to aggregate in solution due to its structural similarity to TACIC. Meanwhile, ZnP acts as an excellent electron donor for photoinduced electron transfer (ET),17,29,30 and ET from ZnP to the excited singlet state (S1) of TACIC is anticipated based on their optical and electrochemical properties.30,41 First, we fully characterized the supramolecular aggregates of TACIC-Br using spectroscopic measurements and theoretical calculations. Subsequently, ZnP-TACIC molecules were incorporated into TACIC-Br aggregates in solution, and their photodynamics were analyzed using time-resolved transient absorption (TA) spectroscopy. Our results revealed efficient energy migration, CT, and CD within the aggregates. This is highlighted by an extraordinary singlet excited-state diffusion length (LD) of 45.6 nm originating from the long-lived excited singlet state in the aggregates, surpassing those of typical organic semiconductors (5–10 nm).47–50
Results and discussion
Synthesis
The synthetic routes to TACIC-Br and ZnP-TACIC are illustrated in Schemes S1 and S2, and details of the synthesis are provided in the ESI.† Briefly, TACIC-Br was obtained by Knoevenagel condensation between the corresponding aldehyde41 and (6-bromo-2,3-dihydro-3-oxo-1H-inden-1-ylidene)propanedinitrile.51ZnP-TACIC was synthesized by Stille coupling between the corresponding ZnP tin reagent51,52 and TACIC-Br. ZnP-ref was also prepared.53 The molecular structures were characterized by solution 1H NMR spectroscopy (Fig. S1†), Fourier transfer infrared (FT-IR) spectroscopy (Fig. S2†), and high-resolution mass spectrometry (HRMS) (Fig. S3†).Aggregation behavior of TACIC-Br
The aggregation behavior of TACIC-Br was examined in a mixture of CHCl3 and MeOH (Fig. 2a). In the good solvent, CHCl3, TACIC-Br displayed an absorption peak at 700 nm with a shoulder at 650 nm, which is similar to that of TACIC.41 As the ratio of the poor solvent, MeOH, increased, the band at 700 nm decreased, and a new peak emerged at 750 nm. Further increasing the MeOH ratio led to the formation of precipitates. Correspondingly, the fluorescence also exhibited a redshift with decreasing intensity (Fig. 2b). These behaviors suggest the formation of TACIC-Br aggregates in the mixed solvent. | ||
Fig. 2 (a) UV-vis-NIR absorption spectra and (b) fluorescence spectra of TACIC-Br in a mixture of CHCl3 and MeOH (1 × 10−5 M). The inset depicts the ratio of CHCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (v/v). :MeOH (v/v). | ||
The fluorescence lifetime of the TACIC-Br aggregates was measured in a mixture of CHCl3 and MeOH (Table S1†). The sample was excited at 636 nm, with emission monitored at 750 nm. When the MeOH ratio ranged from 0–30%, the fluorescence lifetime (τ) remained nearly constant at 0.4–0.5 ns, displaying a single short-lived component (Fig. S4†). However, with MeOH ratios exceeding 40%, the average lifetime considerably increased due to the emergence of a long-lived component. Dynamic light scattering measurements revealed the formation of TACIC-Br aggregates with mean diameters (DM) between 10–15 nm regardless of the CHCl3 to MeOH ratio (Fig. S5†). Based on trends in the UV-visible-NIR absorption spectra, steady-state fluorescence spectra, fluorescence lifetimes, and aggregate sizes as a function of the MeOH ratio, we proposed a plausible formation mechanism for the TACIC-Br aggregates. Within the CHCl3:MeOH ratios of 100:0 to 60:40 (v/v), aggregates of DM = 10–15 nm with a fluorescence lifetime of 0.4–0.5 ns are formed, but the aggregated states are largely comparable. As the MeOH ratio increases further (CHCl3:MeOH = 60:40 to 40:60), a dramatic change in the aggregated state occurs, while maintaining a similar size, thereby prolonging the average fluorescence lifetime from 0.4–0.5 ns to 2.3 ns, which is close to the aggregated states of TACIC (τ = 1.6 ns),41TACIC-Br (τ = 1.9 ns), and TACIC derivatives (τ = 1.3–2.3 ns)42 in films. For the subsequent time resolved TA experiments, we fixed the solvent mixed ratio at 50:50, reflecting the representative TACIC-Br aggregates with an extended fluorescence lifetime (vide infra).
To further examine the intermolecular interactions within the aggregates, solid-state NMR (ssNMR) spectroscopy was performed on TACIC (X = H) as a reference for TACIC-Br. One-dimensional (1D) 1H magic angle spinning (MAS) and 13C cross-polarization (CP)/MAS spectra of TACIC are presented in Fig. S6 and S7,† respectively. In the 1D 1H MAS spectrum, peaks observed at −1 to 3 ppm, 5 ppm, and 6–10 ppm were attributed to protons in different environments: alkyl and alkoxy side chains (H_aliphatic), α-carbon of the alkoxy side chain on the thienoazacoronene unit (H1), and the π-conjugated unit (H2–H11) within the aggregates, respectively. A detailed assignment is provided in Fig. S6.† While the 1D 13C CP/MAS spectra exhibited significant signal overlap, some peaks (C1, C2, C3, C4, and C5) could be distinctly assigned with the aid of DFT chemical shift calculations, as shown in Fig. S7.†
Further insight into the intermolecular packing was gained from a two-dimensional (2D) 1H–13C heteronuclear correlation (HETCOR) spectrum which identifies spatially proximate 1H–13C spin pairs. Weak correlations were detected between the 13C peak at 68.5 ppm and 115 ppm and the 1H peak near 5.0 ppm, corresponding to H1/C3 and H1/C4 pairs (Fig. 3a, red circles). Given that the intramolecular distances of H1/C3 and H1/C4 exceed 10 Å in the DFT-optimized structure, these cross peaks were attributed to intermolecular correlation. These findings suggest that within the amorphous TACIC aggregate, a slipped or T-shaped dimeric π–π packing arrangement is preferred (see the next theoretical calculation section), wherein the thienoazacoronene unit and the 1,1-dicyanomethylene-3-indanone acceptor unit are positioned in close proximity (Fig. 3b). Such π–π packing rationalizes the suppressed nonradiative decay of TACIC in the aggregates formed within the mixed solvents and in thin films (knr = 5.8 × 108 s−1) compared to its behavior in pure chloroform (knr = 4.2 × 109 s−1).41
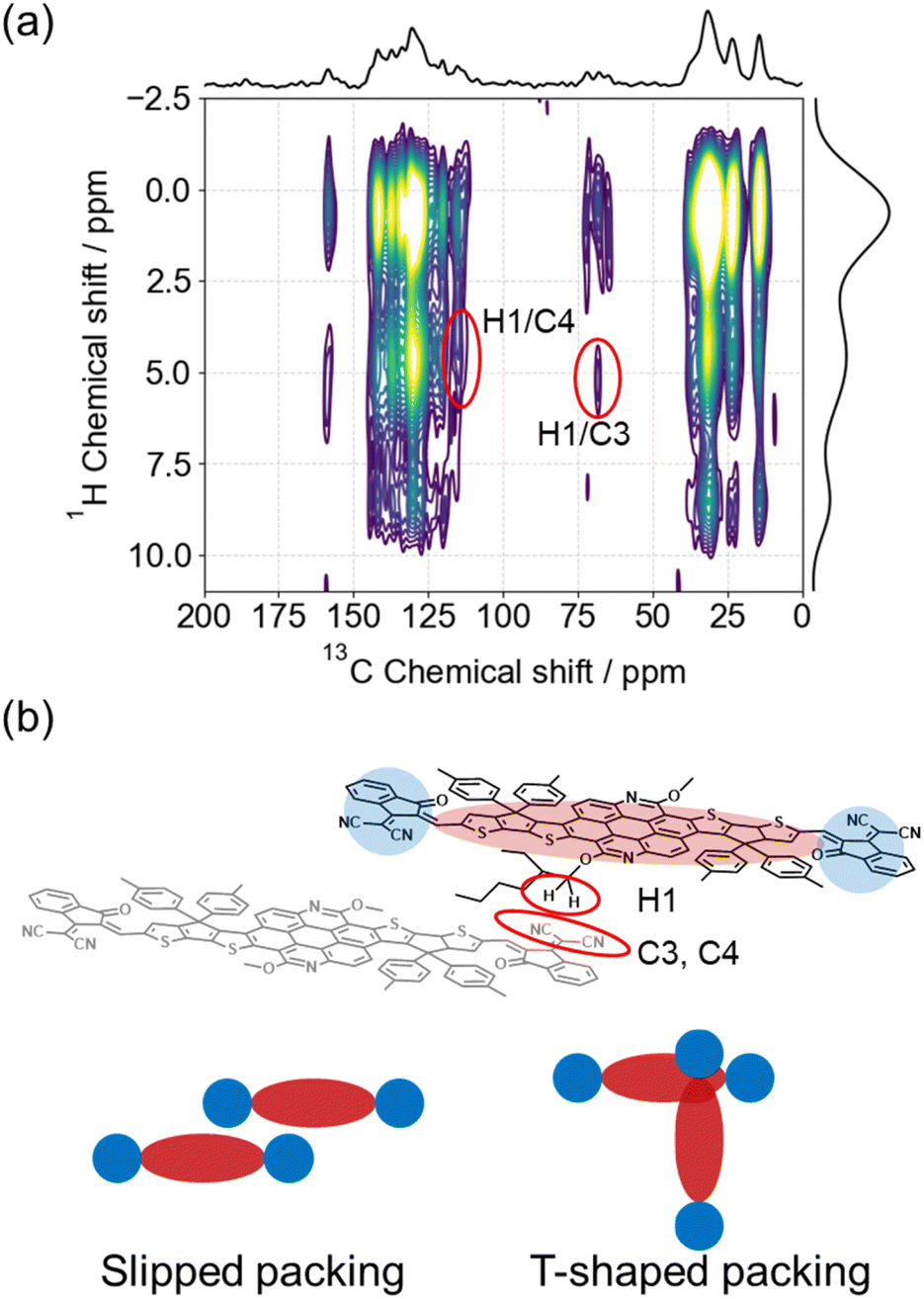 | ||
| Fig. 3 (a) 2D 1H–13C HETCOR ssNMR spectrum of TACIC and (b) expected aggregated structures. | ||
To further explore intermolecular interactions, theoretical calculations were conducted, with computational details provided in the ESI.† First, molecular dynamics (MD) simulations were performed to investigate the stacking structures of TACIC in the film state. The results revealed that two π–π packing conformations are nearly equally dominant: the slipped structure, where the acceptor moieties overlap, and the T-shaped structure, where the acceptor and donor moieties overlap (Fig. S8†). This finding aligns with the ssNMR results discussed earlier. Notably, this contrasts with ITIC, a representative NFA, where only the slipped structure is predominant.39
Next, the time-dependent density functional theory (TDDFT) calculations were employed to examine the fluorescent properties of the TACIC monomer and its slipped and T-shaped π–π packing dimers. The S1 fluorescent states of the TACIC monomer and dimers are primarily characterized by the HOMO–LUMO transitions. In the monomer, the HOMO and LUMO are delocalized across the entire molecule, resulting in a large transition dipole strength (Fig. 4). However, in TACIC dimers, the transition dipole strengths are significantly reduced because the HOMO and LUMO are separated and localized significantly on the overlapped moieties of respective monomers. The same trend was found for TACIC-Br (Fig. S9†). In contrast, the HOMO and LUMO of ITIC dimers are delocalized throughout the dimer, leading to an enhanced transition dipole moment strength (Fig. S10†). These theoretical results are consistent with the experimental results of radiative rate constants (kr); the kr of TACIC in the film state (4.8 × 107 s−1) is smaller than in pure chloroform (3.0 × 108 s−1), whereas the kr of ITIC in the film state (7.5 × 108 s−1) is larger than in the solution (3.6 × 108 s−1).41
 | ||
| Fig. 4 Calculated transition dipole strength (μ), oscillator strength (f), HOMO and LUMO (isovalue = 0.02) of (a) TACIC monomer, (b) slipped dimer, and (c) T-shaped dimer at the S1 optimized geometry. The phenyl sidechains are omitted for simplicity. | ||
Optical properties of ZnP-TACIC
The UV-vis-NIR absorption spectra of ZnP-TACIC, TACIC-Br, and ZnP-ref were measured in benzonitrile (PhCN), which has a dielectric constant of 26, close to the estimated value of the CHCl3:MeOH mixed solvent (50:50, v/v) (19). The absorption spectrum of ZnP-TACIC is almost a linear combination of those of ZnP-ref and TACIC-Br, indicating negligible interaction between the ZnP and TACIC units in the ground state (Fig. 5a). The fluorescence spectra of ZnP-TACIC and TACIC-Br were recorded in PhCN, with the TACIC-Br moiety selectively excited at 690 nm (Fig. 5b). The fluorescence of ZnP-TACIC is significantly quenched compared to that of TACIC-Br, suggesting the occurrence of photoinduced ET from ZnP to the TACIC-Br excited singlet-state (S1). This is consistent with the exothermic driving forces for CS (0.42 eV) and charge recombination (CR) (1.28 eV) in ZnP-TACIC in benzonitrile, based on the first oxidation potential of ZnP (0.32 V vs. Fc/Fc+), the first reduction potential of TACIC (−0.96 V vs. Fc/Fc+), and the optical HOMO–LUMO gap (1.70 eV) of TACIC.41
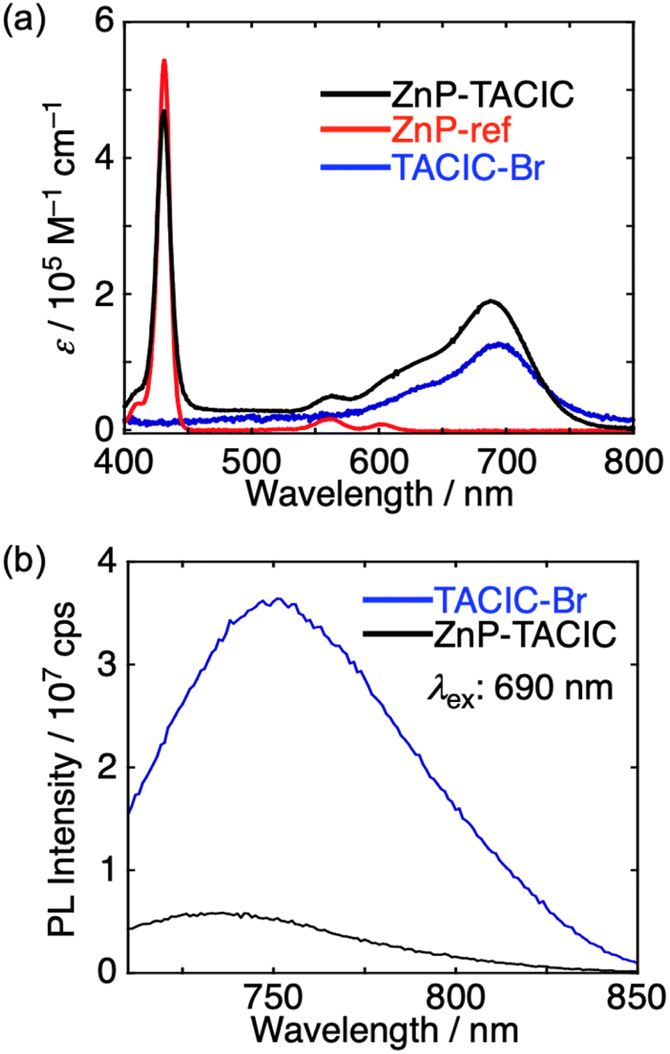 | ||
| Fig. 5 (a) UV-vis-NIR absorption spectra of ZnP-TACIC, ZnP-ref, and TACIC-Br in benzonitrile. (b) Fluorescence spectra of TACIC-Br and ZnP-TACIC in benzonitrile. | ||
Additive effects of ZnP-TACIC
We examined the additive effects of ZnP-TACIC on the optical properties of the TACIC aggregates. The ratio of CHCl3:MeOH (50:50, v/v) was fixed (vide supra), while the molar ratio of TACIC-Br:ZnP-TACIC was varied. Upon adding the ZnP-TACIC solution to the TACIC-Br aggregate solution, a slight decrease in the TACIC-Br absorption at 700 and 750 nm, as well as a slight increase in the ZnP absorption, was observed (Fig. 6a). Meanwhile, the fluorescence of TACIC-Br at 830 nm was significantly quenched with a molar ratio of up to 60:1 when the TACIC-Br moiety was excited solely at 700 nm (Fig. 6b). These results suggest that ZnP-TACIC is incorporated into the TACIC-Br aggregates (denoted as TACIC-Br/ZnP-TACIC), and efficient energy migration in the TACIC-Br/ZnP-TACIC aggregates and subsequent CS in ZnP-TACIC occur when a small molar ratio of ZnP-TACIC is added to the TACIC-Br aggregate solution.
 | ||
| Fig. 6 (a) UV-vis-NIR absorption spectra and (b) fluorescence spectra of TACIC-Br/ZnP-TACIC in a mixture of CHCl3 and MeOH (1 × 10−5 M, 50:50, v/v). The inset depicts the molar ratio of TACIC-Br:ZnP-TACIC. | ||
From the dynamic light scattering experiments, the size of the TACIC-Br aggregates appears to decrease slightly as the MeOH ratio in CHCl3 increases (Fig. S5†). However, this trend is not consistent, and we are therefore unable to evaluate the effect of ZnP-TACIC as an additive. To investigate the impact of mixing order on aggregate formation, we conducted the following experiments: Order A: (1) a CHCl3 solution of ZnP-TACIC was added to a CHCl3 solution of TACIC-Br to achieve a molar ratio of TACIC-Br:ZnP-TACIC = 10:1. (2) MeOH was then added to adjust the solvent ratio to CHCl3:MeOH = 50:50. Order B: (1) MeOH was added to a CHCl3 solution of TACIC-Br to reach a solvent ratio of approximately CHCl3:MeOH = 50:50. (2) A CHCl3 solution of ZnP-TACIC was then added to achieve a final molar ratio of TACIC-Br:ZnP-TACIC = 10:1 while maintaining the CHCl3:MeOH ratio at approximately 50:50. In both cases, selective excitation of TACIC-Br resulted in pronounced fluorescence quenching, indicating that energy migration and charge transfer processes occur efficiently regardless of the ZnP-TACIC incorporation sequence in the TACIC-Br aggregates.
Transient absorption measurements
The time-resolved TA measurements were performed for TACIC-Br and ZnP-TACIC in CHCl3. Upon excitation at 700 nm, positive absorptions arising from the S1 state at 600 and 1100 nm, as well as negative ground-state bleaching at 700 nm, were observed for TACIC-Br (Fig. 7a). A similar absorption profile was noted for TACIC.41 The absorption decayed with a time constant of 410 ps (Fig. S11a†), which agreed with the fluorescence lifetime of TACIC-Br (τ = 438 ps). For ZnP-TACIC, the absorptions decayed faster than those in TACIC-Br, suggesting the occurrence of photoinduced ET from ZnP to the S1 state of TACIC (Fig. 7b). Indeed, as the S1 absorption decayed, characteristic ground-state bleaching at 420 nm, arising from ZnP radical cation (ZnP˙+), and positive absorption at 750 nm, arising from TACIC radical anion (TACIC˙−), emerged.41 Given the spectral overlap between TACIC˙− and the ground-state bleaching and considering the intrinsic synchronization of radical cation and anion dynamics, the charge dynamics were analyzed using the 420 nm signal, which exhibits minimal spectral overlap. From the absorption profile fitting, the CS and CR time constants of 8.8 ps and 140 ps were obtained (Fig. S11b†). Taking into account the fluorescence lifetime of TACIC-Br, the charge separation efficiency approaches unity.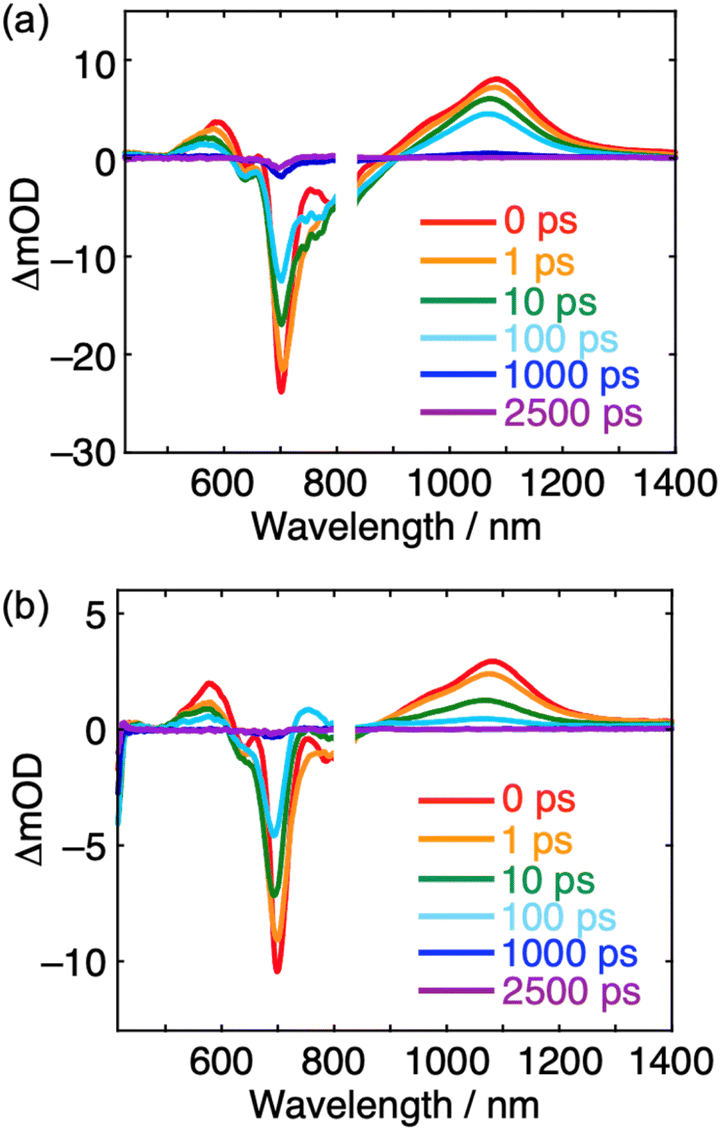 | ||
| Fig. 7 Femtosecond time-resolved transient absorption spectra of (a) TACIC-Br and (b) ZnP-TACIC in CHCl3 (3 × 10−5 M). The inset depicts the delay time after excitation at 700 nm. | ||
The time-resolved TA measurements were also conducted for the TACIC-Br/ZnP-TACIC aggregates with a molar ratio of TACIC-Br:ZnP-TACIC (10:1) in a mixture of CHCl3 and MeOH (50:50, v/v). In the subsequent measurements, we focused on the visible region due to its information richness. Upon excitation at 700 nm, positive absorption arising from the S1 state of TACIC-Br at 600 nm was observed for the TACIC-Br aggregates in both the absence and presence of ZnP-TACIC (Fig. 8). In the aggregated state, the ground-state bleaching (as well as the steady-state absorption) of TACIC-Br exhibits a red shift relative to that in solution, leading to increased spectral overlap with TACIC˙−. Consequently, the relatively weak absorption of TACIC˙− is masked by the intense ground-state bleaching. However, in the presence of ZnP-TACIC, a positive absorption at 800 nm, attributable to the absorption tail of TACIC˙−, and the characteristic ground-state bleaching at 420 nm, arising from ZnP˙+, were detected. This supports the formation of the charge-separated state for the TACIC-Br/ZnP-TACIC aggregates. As in the solution systems, the ground-state bleaching signal at 420 nm, derived from ZnP˙+ with minimal spectral overlap, was employed in the subsequent decay analysis.
 | ||
| Fig. 8 Femtosecond time-resolved transient absorption spectra of (a) TACIC-Br and (b) TACIC-Br/ZnP-TACIC with a molar ratio of 10:1 in a mixture of CHCl3 and MeOH (50:50, v/v, 1 × 10−5 M of TACIC-Br). The inset depicts the delay time after excitation at 700 nm. Strong scattering of the excitation pump pulse by aggregates obscures the wavelength region around 700 nm. The vertical axis has been expanded to better visualize the S1 state and TACIC˙−. The complete intense ground-state bleaching signal up to its peak is presented in Fig. S12.† | ||
To determine whether energy transfer from TACIC-Br molecules near ZnP-TACIC or energy migration among the TACIC-Br aggregate contributes to CS, the excitation intensity dependence of TA spectra was investigated. As the excitation intensity at 700 nm increased, CS took place more rapidly in the TACIC-Br/ZnP-TACIC aggregates with a molar ratio of TACIC-Br:ZnP-TACIC (10:1) in a mixture of CHCl3 and MeOH (50:50, v/v) (Fig. 9 and S12†). This suggests that the S1 state of TACIC-Br far from ZnP-TACIC does not contribute to CS due to the fast relaxation of the S1 state by singlet-singlet annihilation. However, under the weak excitation intensity (<5 μJ cm−2), the CS rate constant slows down, exceeding the CS rate constant of ZnP-TACIC in CHCl3 (8.8 ps). These results demonstrate that CS is regulated by diffusion-controlled singlet excited-state migration. Under the excitation intensity of 18.7 μJ cm−2, the CR rate constant of 260 ps is longer than that of ZnP-TACIC in CHCl3 (140 ps). We underscore that approximately 35% of the charge-separated state in the aggregates persisted on a nanosecond timescale, in stark contrast to those in ZnP-TACIC in CHCl3, where the charge-separated state decayed completely within 1 ns. These results indicate that a fraction of TACIC˙− diffuse through the aggregates opposing the Coulomb attraction, thereby preventing CR.
 | ||
| Fig. 9 (a) Excitation intensity dependence of ground-state bleaching formation corresponding to ZnP˙+ (averaged over 420–430 nm) and (b) plot of CS rate constant as a function of excitation fluence at 700 nm. The molar ratio of TACIC-Br/ZnP-TACIC aggregates is 10:1 and the ratio of CHCl3 and MeOH is 50:50 (v/v). The inset depicts the excitation intensity. The black lines in panel (a) represent the best fitting curves of the transient absorption data with bi-exponential functions and a constant offset. The horizontal line in panel (b) represents the CS rate constant of ZnP-TACIC in CHCl3 (8.8 ps) as a guide for the eye. | ||
The singlet excited-state diffusion length (LD) of TACIC-Br was determined to be 45.6 nm (Fig. S13†). Given the LD value and the singlet excited-state lifetime of 2.2 ns, diffusion coefficient (D) was calculated to be 9.5 × 10−3 cm2 s−1. This value is lower than that of ITIC-Cl (2.7 × 10−2 cm2 s−1),54 which we previously determined using the same methodology. Considering τ = 140 ps for ITIC-Cl, the LD value of TACIC-Br is 2.4 times higher than that of ITIC-Cl, demonstrating the potential utility of the TACIC structure for efficient energy migration.
Experimental
Experimental details including materials, methods, synthetic procedures, solution 1H NMR spectra, IR spectra, mass spectra, optical measurements, DLS measurements, solid-state 1H and 13C NMR spectra, MD and TDDFT calculations, TA measurements, and the details of determination of LD are provided in the ESI.†Conclusions
The dynamic interplay between the self-assembling nonfullerene acceptor TACIC-Br and the zincporphyrin (ZnP)-TACIC dyad presents a fascinating model for emulating energy transduction and charge separation, akin to natural photosynthesis and organic photovoltaics. Notably, TACIC-Br molecules exhibit a strong tendency to aggregate even in the favorable solvent CHCl3. However, when the proportion of the poor solvent (MeOH) surpasses 40% in a CHCl3/MeOH mixture (v/v), these aggregates exhibit an extended singlet-state lifetime, comparable to TACIC thin films. Solid-state NMR spectroscopy and theoretical calculations showed for the first time the importance of dimeric π–π packing structures for the unusual photophysical properties of TACIC aggregates: within these aggregates, a slipped or T-shaped dimeric π–π packing arrangement is favored, bringing the thienoazacoronene donor unit and the 1,1-dicyanomethylene-3-indanone acceptor unit into close proximity. This supramolecular π–π packing effectively minimizes both nonradiative and radiative decay processes, shaping the unique photophysical behavior of TACIC in chloroform/MeOH mixtures and thin films, in contrast to its behavior in pure chloroform. Such relationship between molecular structure, packing structure, and photophysical properties of NFAs are pivotal for rational molecular design of photoactive organic functional molecules in solid states.In CHCl3/MeOH mixtures exceeding 40% MeOH, TACIC-Br/ZnP-TACIC aggregates exhibit substantial fluorescence quenching of TACIC-Br by ZnP-TACIC, even at a TACIC-Br:ZnP-TACIC molar ratio of up to 30:1. Time-resolved transient absorption measurements highlighted efficient energy migration and charge separation within these aggregates, demonstrating an extraordinarily long singlet excited-state diffusion length (LD) of 45.6 nm due to the prolonged excited singlet state lifetime—far exceeding the typical 5–10 nm range observed in organic semiconductors and ranking among the longest diffusion lengths reported for recently developed novel NFAs. Remarkably, after quantitative charge-transfer at the charge separation molecule, approximately 35% of the charge-separated states within these aggregates successfully dissociate, forming free ion pairs as the TACIC radical anion fraction diffuses and overcomes Coulomb attraction. This integrated supramolecular strategy adeptly replicates both light-harvesting and charge separation mechanisms (including charge transfer and charge dissociation) observed in photosynthesis and organic photovoltaics. It holds promise for driving future advancements in artificial photosynthesis, solar fuels, and organic photovoltaics.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. Imahori conceived and designed this work. D. Sasada, M. Kubota, and T. Umeyama conducted the synthesis and characterization of the products. M. Akiyama and L. Vallan performed spectroscopic measurements. Y. Tamai and S. Izawa conducted TA and diffusion length measurements. K. Suzuki and H. Kaji performed solid-sate NMR measurements. T. Urakami, H. Sato, and M. Higashi carried out theoretical calculations. Y. Tamai, M. Akiyama, and H. Imahori co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Transformative Research Areas (A) (Dynamic Exciton: 20H05831, 20H05832, 20H05837, 20H05840, 21H05394, 23H03951), and Scientific Research (A) (24H00485), and JST PRESTO program (JPMJPR23J7), JSPS Core-to-Core Program (JPJSCCA20220004), and JST CREST (JPMJCR2431).Notes and references
- J.-M. Lehn, Angew. Chem., Int. Ed., 1988, 27, 89 CrossRef.
- K. Ariga, Y. Yamauchi, G. Rydzek, Q. Ji, Y. Yonamine, K. C.-W. Wu and J. P. Hill, Chem. Lett., 2014, 43, 36 CrossRef CAS.
- A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320 CrossRef CAS PubMed.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729 CrossRef CAS PubMed.
- J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080 CrossRef CAS PubMed.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459 RSC.
- I. V. Kolesnichenko and E. V. Anslyn, Chem. Soc. Rev., 2017, 46, 2385 RSC.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861 RSC.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404 RSC.
- T. Ogoshi, T. Kakuta and T. Yamagishi, Angew. Chem., Int. Ed., 2019, 58, 2197 CrossRef CAS PubMed.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144 RSC.
- C. Guo, A. C. Sedgwick, T. Hirao and J. L. Sessler, Coord. Chem. Rev., 2021, 427, 213560 CrossRef CAS.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS.
- J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802 CrossRef CAS PubMed.
- H. Imahori, Bull. Chem. Soc. Jpn., 2007, 80, 621 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS PubMed.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjö, S. Styring, V. Sundström and L. Hammarström, Acc. Chem. Res., 2009, 42, 1899 CrossRef CAS PubMed.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS.
- N. Aratani, D. Kim and A. Osuka, Acc. Chem. Res., 2009, 42, 1922 CrossRef CAS.
- P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847 RSC.
- S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059 RSC.
- M. Rudolf, S. V. Kirner and D. M. Guldi, Chem. Soc. Rev., 2016, 45, 612 RSC.
- C. B. KC and F. D'Souza, Coord. Chem. Rev., 2016, 322, 104 CrossRef CAS.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283 CrossRef CAS.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216 RSC.
- H. Kumagai, Y. Tamaki and O. Ishitani, Acc. Chem. Res., 2022, 55, 978 CrossRef CAS PubMed.
- H. Imahori, Bull. Chem. Soc. Jpn., 2023, 96, 339 CrossRef CAS.
- H. Imahori and M. Akiyama, J. Porphyrins Phthalocyanines, 2024, 28, 319 CrossRef CAS.
- H. Imahori, H. Norieda, H. Yamada, Y. Nishimura, I. Yamazaki, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 100 CrossRef CAS PubMed.
- W.-S. Li, K. S. Kim, D.-L. Jian, H. Tanaka, T. Kawai, J. H. Kwon, D. Kim and T. Aida, J. Am. Chem. Soc., 2006, 128, 10527 CrossRef CAS PubMed.
- Y. Kuramochi, A. S. D. Sandanayaka, A. Satake, Y. Araki, K. Ogawa, O. Ito and Y. Kobuke, Chem.–Eur. J., 2009, 15, 2317 CrossRef CAS PubMed.
- K. Börjesson, J. Tumpane, T. Ljungdahl, L. M. Wilhelmsson, B. Nordén, T. Brown, J. Mårtensson and B. Albinsson, J. Am. Chem. Soc., 2009, 131, 2831 CrossRef.
- T. Miura, R. Tao, S. Shibata, T. Umeyama, T. Tachikawa, H. Imahori and Y. Kobori, J. Am. Chem. Soc., 2016, 138, 5879 CrossRef CAS PubMed.
- J. Otsuki, T. Okumura, K. Sugawa, S. Kawano, K. Tanaka, T. Hirao, T. Haino, Y. J. Lee, S. Kang and D. A. Kim, Chem.–Eur. J., 2021, 27, 4053 CrossRef.
- J. Royakkers, H. Yang, A. J. Gillett, F. Eisner, P. Ghosh, D. G. Congrave, M. Azzouzi, Z. Andaji-Garmaroudi, A. Leventis, A. Rao, J. M. Frost, J. Nelson and H. Bronstein, Nat. Chem., 2024, 16, 1453 CrossRef CAS PubMed.
- G. C. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H. J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Y. Jen and H. L. Yip, Chem. Rev., 2022, 122, 14180 CrossRef CAS PubMed.
- A. Yamakata, K. Kato, T. Urakami, S. Tsujimura, K. Murayama, M. Higashi, H. Sato, Y. Kobori, T. Umeyama and H. Imahori, Chem. Sci., 2024, 15, 12686 RSC.
- H. Imahori, Y. Kobori and H. Kaji, Acc. Mater. Res., 2021, 2, 501 CrossRef CAS.
- T. Umeyama, K. Igarashi, D. Sasada, Y. Tamai, K. Ishida, T. Koganezawa, S. Ohtani, K. Tanaka, H. Ohkita and H. Imahori, Chem. Sci., 2020, 11, 3250 RSC.
- T. Umeyama, K. Igarashi, D. Sasada, K. Ishida, T. Koganezawa, S. Ohtani, K. Tanaka and H. Imahori, ACS Appl. Mater. Interfaces, 2020, 12, 39236 CrossRef CAS.
- M. Kubota, T. Umeyama, W. Suzuki, T. Koganezawa, M. Akiyama and H. Imahori, Bull. Chem. Soc. Jpn., 2025, 98, uoae147 CrossRef CAS.
- B. He, A. B. Pun, L. M. Klivansky, A. M. McGough, Y. Ye, J. Zhu, J. Guo, S. J. Teat and Y. Liu, Chem. Mater., 2014, 26, 3920 CrossRef CAS.
- B. He, B. A. Zhang, F. Liu, A. Navarro, M. P. Fernández-Liencres, R. Lu, K. Lo, T. L. Chen, T. P. Russell and Y. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 20034 CrossRef CAS PubMed.
- B. He, J. Dai, D. Zherebetskyy, T. L. Chen, B. A. Zhang, S. J. Teat, Q. Zhang, L. Wang and Y. A. Liu, Chem. Sci., 2015, 6, 3180 RSC.
- O. V. Mikhnenko, P. W. M. Blom and T.-Q. Nguyen, Energy Environ. Sci., 2015, 8, 1867 RSC.
- Y. Firdaus, V. M. Le Corre, S. Karuthedath, W. Liu, A. Markina, W. Huang, S. Chattopadhyay, M. M. Nahid, M. I. Nugraha, Y. Lin, A. Seitkhan, A. Basu, W. Zhang, I. McCulloch, H. Ade, J. Labram, F. Laquai, D. Andrienko, L. J. A. Koster and T. D. Anthopoulos, Nat. Commun., 2020, 11, 5220 CrossRef CAS PubMed.
- H. Wang, H. Chen, W. Xie, H. Lai, T. Zhao, Y. Zhu, L. Chen, C. Ke, N. Zheng and F. He, Adv. Funct. Mater., 2021, 31, 2100877 CrossRef CAS.
- Y. Tamai, Aggregate, 2022, 6, e280 CrossRef.
- D. M. Knoll, T. B. Wiesner, S. M. Marschner, Z. Hasssan, P. Weis, M. Kappes, M. Nieger and S. Bräse, RSC Adv., 2019, 9, 30541 RSC.
- T. Umeyama, T. Hanaoka, J. Baek, T. Higashino, F. Abou-Chahine, N. V. Tkachenko and H. Imahori, J. Phys. Chem. C, 2016, 120, 28337 CrossRef CAS.
- S. Takagi, T. K. Miyamoto and Y. Sasaki, Bull. Chem. Soc. Jpn., 1986, 59, 2371 CrossRef CAS.
- Y. Sakamoto, S. Izawa, H. Ohkita, M. Hiramoto and Y. Tamai, Commun. Mater., 2022, 3, 76 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc03828f |
| This journal is © The Royal Society of Chemistry 2025 |