
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZn(II)-driven impact of monomeric transthyretin on amyloid-β amyloidogenesis†
Yelim
Yi
a,
Bokyung
Kim
b,
Mingeun
Kim
 a,
Young Ho
Ko
c,
Jin Hae
Kim
*b and
Mi Hee
Lim
*a
a,
Young Ho
Ko
c,
Jin Hae
Kim
*b and
Mi Hee
Lim
*a
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: miheelim@kaist.ac.kr
bDepartment of New Biology, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu 42988, Republic of Korea. E-mail: jinhaekim@dgist.ac.kr
cCenter for van der Waals Quantum Solids, Institute for Basic Science, Pohang 37673, Republic of Korea
First published on 4th February 2025
Abstract
Extracellular accumulation of amyloid-β (Aβ) peptides in the brain plays a significant role in the development of Alzheimer's disease (AD). While the co-localization and interaction of proteins and metal ions with Aβ in extracellular milieu are established, their precise pathological associations remain unclear. Here we report the impact of Zn(II) on the anti-amyloidogenic properties of monomeric transthyretin (M-TTR), which coexists spatially with Aβ and Zn(II) in extracellular fluids. Our findings demonstrate the Zn(II)-promoted ternary complex formation involving M-TTR, Aβ, and Zn(II) as well as M-TTR's proteolytic activity towards Aβ. These interactions decrease the inhibitory effect of M-TTR on the primary nucleation process of Aβ as well as its ability to improve cell viability upon treatment of Aβ. This study unveils the variable activities of M-TTR towards Aβ, driven by Zn(II), providing insights into how metal ions influence the entanglement of M-TTR in the Aβ-related pathology linked to AD.
Introduction
Alzheimer's disease (AD), the most common form of dementia, is a leading cause of death and currently lacks a cure.1 In the brains of AD patients, the formation of extracellular deposits occurs as a consequence of the conversion of amyloid-β (Aβ) peptides into toxic oligomers and fibrils.2,3 To release the burden of these aggregates in the brain, proteins available in extracellular fluids sequester cerebral Aβ species, compensating for the pathological cascade of Aβ by protein–protein interactions.4,5 Transthyretin (TTR) is a major extracellular protein identified as an Aβ-binding protein in the cerebrospinal fluid (CSF) and serves as a carrier of Aβ from the brain to the blood.6–10 Previous studies have demonstrated the ability of TTR to ameliorate neuropathologic manifestations of AD in vivo,6 and the decrease in its levels in extracellular fluids from AD patients further supports its neuroprotective function in the disease pathology.11–13Additionally, highly concentrated metal ions have been detected in Aβ aggregate deposits.14–19 These metal ions bind to Aβ peptides, forming metal–Aβ complexes, which alters the aggregation and toxicity of Aβ.14–26 In the case of TTR, the diseased states lead to the dissociation of its native homotetrameric structure into aggregation-prone monomers (M-TTR), rendering it vulnerable to metal coordination by exposing additional surfaces.27–31Ex vivo observations have denoted the prevalence of M-TTR in the CSF and its aggregates enriched with Zn(II), implying the capacity of M-TTR to interact with Zn(II).32–34 Despite these findings, the connection of this interactive network involving Aβ, M-TTR, and Zn(II) to the pathology of AD remains elusive. Herein, we questioned whether M-TTR exhibits anti-amyloidogenic activity against Aβ in the presence of Zn(II). Thus, we determined the direct binding between M-TTR and Aβ under Zn(II)-present conditions, with the subsequent variation in the structure of Aβ. Furthermore, the influence of M-TTR on the aggregation and toxicity profiles of Aβ in the presence of Zn(II) was probed.
Results and discussion
Using a TTR mutant, Phe87Met/Leu110Met TTR, which mainly remains monomeric (M-TTR) in an aqueous solution at neutral pH and room temperature,35,36 we explored the impact of M-TTR on the aggregation of Aβ in the presence of Zn(II), as illustrated in Fig. 1a and b. We first monitored the aggregation kinetics of Aβ40, which is the most prevalent Aβ in the CSF,37 in the presence of Zn(II) upon treatment of various concentrations of M-TTR by the thioflavin-T (ThT) assay that enables to quantify the amount of β-sheet-containing aggregates.21,38 As shown in Fig. 1c, the ThT fluorescence intensity for the entire aggregation process of Aβ40 treated with Zn(II) remarkably lowered with the addition of M-TTR. M-TTR incubated with or without Zn(II) did not produce ThT-detectable aggregates (Fig. S1†). This manifests the inhibition of M-TTR on the formation of ThT-reactive Aβ40 aggregates in the presence of Zn(II). Given the sub-equimolar, equimolar, and supra-equimolar ratios of M-TTR to Zn(II) under physiological conditions,32,33,39 these results suggest that the inhibitory effect of M-TTR on the assembly of Zn(II)–Aβ40 may vary in biological environments. Such modulative effect of M-TTR on Aβ40 aggregation with Zn(II) was further confirmed by transmission electron microscopy (TEM). As illustrated in Fig. 1d, Aβ40 treated with M-TTR in the presence of Zn(II) exhibited a mixture of smaller amorphous aggregates and chopped fibrils, distinct from larger aggregates observed for Zn(II)-bound Aβ40 [Zn(II)–Aβ40]. For the samples of M-TTR, no aggregates were visible under Zn(II)-treated conditions. Clearly, these results indicate that M-TTR can significantly redirect the aggregation of Aβ40 in the presence of Zn(II). The toxicity of the resultant samples in human neuroblastoma SH-SY5Y (5Y) cells was then evaluated by the MTT assay [MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]. Aβ40 aggregates produced with M-TTR and Zn(II) were observed to be less cytotoxic by ca. 10%, compared to those formed only with Zn(II), under our experimental conditions (Fig. 1e). The Zn(II)-treated M-TTR sample was not significantly toxic. Collectively, M-TTR in the presence of Zn(II) reduces the formation of toxic Aβ40 aggregates. It should be noted that the hydrolytic products of Aβ40 generated in the presence of M-TTR and Zn(II) may contribute to the cell viability (vide infra). The effects of M-TTR without Zn(II) on Aβ40 aggregation and toxicity were consistent with those observed in the previous reports,8,41–43 showing more discernible anti-amyloidogenic activity relative to that under Zn(II)-treated conditions (Fig. 1). Overall, our results indicate that Zn(II) impairs M-TTR's ability to control the aggregation of Aβ40 and improve the cell viability upon treatment of Aβ40. Specifically, a ca. 4% decrease in cell viability was observed when cells were treated with Zn(II)–Aβ40 incubated with M-TTR, compared to those added with Aβ40 and M-TTR without Zn(II). | ||
| Fig. 1 Anti-amyloidogenic activity of M-TTR against Aβ40 with and without Zn(II). (a) Amino acid sequences and structures of M-TTR and Aβ40 determined by NMR spectroscopy (PDB 2NBO36 for M-TTR; PDB 2LFM40 for Aβ40). (b) Scheme of the aggregation studies. (c) Aggregation kinetics of Aβ40 in the presence of M-TTR with (top) and without (bottom) Zn(II) monitored by the ThT assay. Conditions: [Aβ40] = 25 μM; [M-TTR] = 0.125–37.5 μM; [ZnCl2] = 25 μM; 20 mM HEPES, pH 7.4, 150 mM NaCl; 37 °C; quiescent conditions; λex = 440 nm; λem = 490 nm. The error bars denote s.e.m. for n = 9 examined over three independent experiments. (d) Morphologies of the resultant peptide and protein aggregates detected by TEM. Conditions: [Aβ40] = 25 μM; [M-TTR] = 25 μM; [ZnCl2] = 25 μM; 20 mM HEPES, pH 7.4, 150 mM NaCl; 37 °C; 24 h incubation; quiescent conditions. Scale bars, 200 nm. (e) Survival of the cells upon treatment of Aβ40 incubated with M-TTR in the presence (top) and absence (bottom) of Zn(II). The viability of 5Y cells, analyzed by the MTT assay, was calculated, compared to that with an equivalent amount of the buffered solution. Conditions: [Aβ40] = 25 μM; [M-TTR] = 25 μM; [ZnCl2] = 25 μM. The error bars denote s.e.m. for n = 6 examined over three independent experiments. All statistical significance was determined by a two-sided unpaired Student's t-test. *P < 0.05. | ||
Confirming the decrease in anti-amyloidogenic activity of M-TTR in the presence of Zn(II), we identified the specific microscopic step in the assembly of Aβ40 influenced by M-TTR under Zn(II)-added conditions. We initially analyzed the microscopic processes in the aggregation mechanisms of Zn(II)–Aβ40 without M-TTR by global fitting of previously delineated mathematical models for the protein assembly44 to experimental data sets. As shown in Fig. S2,† macroscopic aggregation curves for various concentrations of Aβ40 with equimolar Zn(II) were obtained by the ThT assay. Based on their normalized curves depicted in Fig. 2a, the values of half-time (t1/2), the time at which the ThT fluorescence intensity reaches half of its maximum value, were plotted against the peptide concentration on a double logarithmic scale. A resultant linear plot denotes that a single aggregation mechanism is dominant44,45 for the self-assembly of Zn(II)–Aβ40 under our experimental conditions. Global fitting of various kinetic models to the curves revealed that an aggregation model involving primary nucleation and elongation steps (i.e., nucleation–elongation model)44 well-described the experimental data (Fig. 2a and Table S1†). A reasonable global fit to the data was also acquired if the secondary nucleation step was included in the aforementioned model (Fig. S3†). To evaluate whether the secondary nucleation process is entailed in the assembly of Zn(II)–Aβ40, we performed additional experiments with seeds, preformed Aβ40 aggregates generated in the presence of Zn(II) (Fig. S4†). According to previously reported studies, the addition of a relatively small amount of seeds (e.g., up to 5% v/v) can dramatically increase the aggregation rate of Aβ, representing the assembly with substantial intermediation of the secondary nucleation step.44,46,47 Our results, however, showed similar t1/2 values of Zn(II)–Aβ40 aggregation with various quantities of seeds up to 5% v/v. Therefore, the primary nucleation and elongation processes predominantly participate in the aggregation of Zn(II)–Aβ40, without significant involvement of the secondary nucleation step.
 | ||
| Fig. 2 Aggregation kinetics of Zn(II)-added Aβ40 with and without M-TTR analyzed by the ThT assay. (a) Normalized aggregation curves of Aβ40 in the presence of equimolar Zn(II) (top) and the t1/2 value of each curve plotted against [Aβ40] for obtaining scaling exponent (γ) (bottom). The nucleation–elongation model was well-fitted to the data (top; solid lines). (b) Normalized aggregation curves of Zn(II)–Aβ40 in the presence of M-TTR (top) and the combined rate constant, k+kn, plotted against [M-TTR] (bottom). The nucleation–elongation model with the variations in k+kn values was well-fitted to the data (top; solid lines). (c) Normalized aggregation curves of Zn(II)–Aβ40 with seeds in the absence and presence of M-TTR (top; mean residual error, 0.0078) and the relative k+ values plotted against [M-TTR] (bottom). Solid lines are the fits of Zn(II)–Aβ40 aggregation profiles upon treatment of various [M-TTR] with the alteration in k+. (d) Schematic representation of Aβ40 aggregation with and without Zn(II) (black lines) affected by M-TTR (red lines). (e) Change in the relative k+kn values of Zn(II)-added and -unadded Aβ40 aggregation plotted as a function of [M-TTR]. | ||
Next, as presented in Fig. 2b, the inhibitory effect of M-TTR on the aggregation of Aβ40 in the presence of Zn(II) was quantitatively analyzed through global fitting of the nucleation–elongation model44 to the experimental data shown in Fig. 1c. As a result, the combined rate constants of primary nucleation (kn) and elongation (k+), k+kn values, gradually decreased with increasing the concentration of M-TTR, showing a reduction by ca. 90% with 1.5 equiv. of M-TTR. This implies the ability of M-TTR to impede either primary nucleation, elongation, or both of Zn(II)–Aβ40 assembly. To probe which of the microscopic steps is mainly influenced by M-TTR, Zn(II)–Aβ40 aggregation with M-TTR was observed in the presence of 35% v/v seeds, which can induce elongation-dominant aggregation (Fig. S5†).44,46,47Fig. 2c illustrates that M-TTR did not noticeably affect the aggregation kinetics of Zn(II)–Aβ40 with seeds exhibiting similar k+ values. This demonstrates that M-TTR predominantly hampers the primary nucleation process, rather than the elongation step, in the self-assembly of Zn(II)–Aβ40. Collectively, as summarized in Fig. 2d, in the presence of Zn(II), Aβ40 assembles via the combination of primary nucleation and elongation processes. Intriguingly, M-TTR can dominantly retard the primary nucleation of Zn(II)–Aβ40.
Similar experiments were conducted with Zn(II)-untreated samples (Fig. S6–S10 and Table S1†). Metal-free Aβ40 aggregation involves primary nucleation, elongation, and secondary nucleation catalyzed by the surface of preformed aggregates that can be saturated by incoming peptide species (Fig. 2d), consistent with previously reported results.48 The nucleation steps of metal-free Aβ40 (particularly, primary nucleation), following previous reports,8,43 can be primarily affected by the introduction of M-TTR. Since the elongation steps for both Zn(II)-treated and metal-free Aβ40 were not considerably perturbed by M-TTR, the change in k+kn values upon addition of M-TTR can be interpreted as M-TTR-triggered alteration in the primary nucleation of Aβ40. As shown in Fig. 2e, the k+kn value of Zn(II)-added Aβ40 was lowered by M-TTR, with an even greater reduction observed under Zn(II)-unadded conditions. In particular, the addition of M-TTR (2.5 μM) reduced the k+kn value of Zn(II)–Aβ40 by ca. 60%, whereas the value was lowered by ca. 100% in the presence of M-TTR (1.5 μM) under Zn(II)-untreated conditions. Overall studies manifest that while Zn(II) diminishes the inhibitory effect of M-TTR on the formation of primary nuclei of Aβ40, it does not completely eliminate the effect.
To elucidate how Zn(II) decreases the anti-amyloidogenic activity of M-TTR towards Aβ40, we investigated Zn(II)-binding properties of M-TTR in the absence and presence of Aβ40. Given the limited spectral changes of M-TTR mixed with Zn(II), the dissociation constant (Kd) was estimated through competitive Zn(II) titration experiments using Zincon (2-carboxy-2′-hydroxy-5′-sulfoformazyl-benzene monosodium salt), an indicator that shows electronic absorption (Abs) at 618 nm upon Zn(II) coordination with a Kd value in the micromolar range (Fig. S11†).49 As shown in Fig. S12a,† electronic Abs spectra exhibited no noticeable variation in Abs at 618 nm when the solution of M-TTR and Zincon was titrated with sub-equimolar Zn(II). In contrast, the addition of supra-equimolar Zn(II) notably elevated the Abs at 618 nm. These observations indicate that M-TTR can bind to 1 equiv. of Zn(II) with a binding affinity similar to or greater than that of Zincon. Considering that M-TTR includes three Zn(II)-binding sites,33 the affinity of M-TTR binding to Zn(II) greater than 1 equiv. could be weaker than that of Zincon. Based on the spectral changes at 618 nm, the apparent Kd value for Zn(II)–M-TTR was calculated to be in a micromolar range. Given the Hill coefficient (n) of 0.74 (±0.04), as depicted in Fig. S12b,† this apparent Kd value indicates the involvement of at least two Zn(II)-binding events in M-TTR.
Furthermore, we probed the interaction of Aβ40 with M-TTR in the presence of Zn(II) by two-dimensional 1H–15N heteronuclear single quantum coherence nuclear magnetic resonance (2D 1H–15N HSQC NMR) spectroscopy, as presented in Fig. 3a and S13.† The discernible alteration in the NMR signals of amino acid residues in or near the N-terminal region of 15N-labeled Aβ40 was observed upon addition of Zn(II) (Fig. 3a, top). Specifically, the peaks of some amino acid residues (e.g., Arg5, Asp7, Ser8, and Val12) adjacent to the Zn(II)-binding site in Aβ14–19 disappeared, suggestive of Zn(II) binding to Aβ40.50,51 Interestingly, mixing M-TTR with the solution of 15N-labeled Aβ40 with Zn(II) restored the vanished peaks to certain extents. This reveals that M-TTR can partially disrupt Zn(II) binding to Aβ40. The comparable Zn(II)-binding affinities of Aβ14–18 and M-TTR (Fig. S12†) in the micromolar range support this competitive Zn(II) binding of M-TTR with Aβ40. Prior studies described that Zn(II) coordination to M-TTR increases the structural flexibility of the region flanking α-helix (Fig. 1a).33 Given that the helical region of M-TTR can be involved in the association with Aβ,8,42,52,53 when it interacts with Zn(II) from Zn(II)–Aβ40, its subsequent structural perturbation could impact the binding and reactivity with Aβ40. The spectra further displayed that Zn(II) moderately induced the chemical shift perturbations (CSPs) of peaks corresponding to Ser8, Glu11, Val12, Gln15, Phe20, and Val24 in 15N-labeled Aβ40 treated with M-TTR (Fig. 3a, middle). Without Zn(II), the modest CSPs for 15N-labeled Aβ40 at Gln15, Val24, and Lys28 were revealed upon treatment of M-TTR (Fig. 3a, bottom). These may indicate the Zn(II)-directed ternary complexation among M-TTR, Zn(II), and Aβ40.
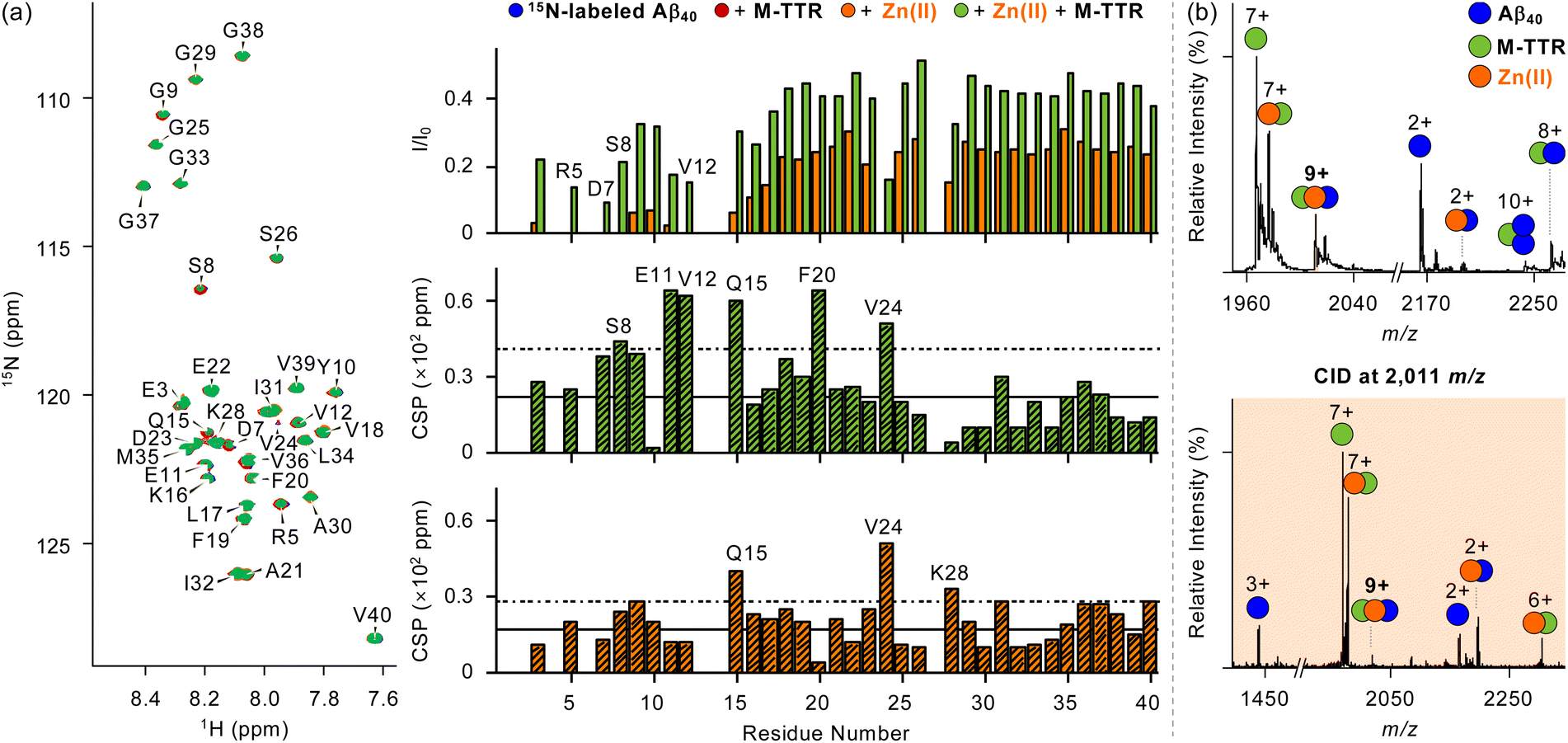 | ||
| Fig. 3 Interaction of M-TTR with Aβ40 in the presence of Zn(II). (a) 2D 1H–15N HSQC NMR (850 MHz) spectra of 15N-labeled Aβ40 treated with either M-TTR, Zn(II), or both. (Top) The peak intensity ratios (I/I0) of amino acid residues in 15N-labeled Aβ40 upon treatment of Zn(II) with (green) and without (orange) M-TTR were calculated. The CSPs of amino acid residues in (middle) M-TTR-added 15N-labeled Aβ40 upon addition of Zn(II) and (bottom) 15N-labeled Aβ40 upon treatment of M-TTR were obtained. The average of CSPs and the sum of the average and one standard deviation are indicated with solid and dashed lines, respectively. Conditions: [15N-labeled Aβ40] = 35 μM; [M-TTR] = 35 μM; [ZnCl2] = 35 μM; 50 mM HEPES, pH 7.4; 7% v/v D2O; 10 °C. (b) M-TTR binding to Aβ40 in the presence of Zn(II) monitored by ESI–MS and ESI–MS2. Charge states are marked above the peaks in the spectra. Conditions: [Aβ40] = 100 μM; [M-TTR] = 100 μM; [ZnCl2] = 100 μM; 20 mM ammonium acetate, pH 7.4; 37 °C; 3 h incubation; quiescent conditions. The samples were diluted 10-fold before injection into the mass spectrometer. The measurements were conducted in triplicate. | ||
Investigations employing electrospray ionization-mass spectrometry (ESI–MS) further corroborate these findings. As depicted in Fig. 3b, a peak at ca. 2011 m/z, assigned to be a 9+-charged ion, appeared when Aβ40 was incubated with M-TTR and Zn(II). Tandem MS (ESI–MS2) analysis with applying collision-induced dissociation (CID) energy to this peak resulted in multiply charged peaks corresponding to [M-TTR + Zn(II)]7+ and [Aβ40 + Zn(II)]2+, indicating a ternary complexation of Aβ40 with M-TTR and Zn(II). This metal-assisted ternary complexation associated with Aβ was proposed to alter the aggregation.54 This may support the contribution of Zn(II) to the suppressive effect of M-TTR on the assembly of Aβ40 accompanied by the perturbation in Aβ40's primary nucleation (Fig. 2d). It should be noted that the Zn(II)-binding affinity of M-TTR in the presence of Aβ could not be accurately determined under our experimental conditions, likely due to the formation of a ternary complex. Without Zn(II) treatment, 8+-charged (ca. 2263 m/z) and 10+-charged (ca. 2243 m/z) peaks appeared (Fig. S14†), corresponding to heterodimers and heterotrimers comprised of M-TTR with monomeric and dimeric Aβ40, respectively.41 The direct interaction between the aggregates of Aβ40 and M-TTR was also monitored by cross-linking experiments. Glutaraldehyde, a well-known cross-linking reagent, covalently conjugated the peptide and protein aggregates generated from the aggregation studies (Fig. 1b).42,55 The size distribution of the resultant samples was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) with western blotting using anti-Aβ (6E10) and anti-TTR antibodies (Fig. S15†). As shown in Fig. S16,† different from the samples of either Aβ40 or M-TTR with and without Zn(II), new bands between 35 kDa and 48 kDa were displayed for Aβ40 samples incubated with M-TTR under both Zn(II)-treated and -untreated41 conditions. These observations substantiate that dimeric Aβ40 can form heterotetramers with dimeric M-TTR with and without Zn(II).
Moving forward, the impact of the direct interaction between Aβ40 and M-TTR in the presence of Zn(II) on the structure of Aβ40 was assessed through ESI–MS studies. As depicted in Fig. 4, after incubating Aβ40 with M-TTR and Zn(II) for 24 h, new peaks at ca. 850 m/z and ca. 1325 m/z were detected, distinguishing them from the spectra obtained without Zn(II) treatment (Fig. S17†). ESI–MS2 in conjunction with CID characterized these peaks as Aβ1–142+ and Aβ15–402+ fragments, respectively. These fragments were not generated with Aβ40 samples relatively shortly incubated with M-TTR and Zn(II) for 3 h (Fig. S18†) and, thus, they could not be detected in the 2D 1H–15N HSQC NMR spectrum of Aβ40 with M-TTR in the presence of Zn(II) (Fig. 3). According to prior studies regarding the proteolytic competence of TTR against peptides or proteins induced by Zn(II),56,57 these fragmentations could result from the proteolytic activity of M-TTR in the presence of Zn(II). The binding of truncated Aβ with full-length ones can accelerate the generation of primary nuclei.58 Thus, proteolytic reactions catalyzed by M-TTR with Zn(II) could decrease the inhibitory effect of M-TTR on the primary nucleation process of Aβ40 (Fig. 2d). In particular, Aβ15–40 was suggested as an amyloid core segment capable of interacting with itself and the full-length of Aβ. While Aβ15–40 has intrinsic amyloidogenic properties to form toxic aggregates, it can redirect the assembly of full-length Aβ40 into non-fibrillar and less toxic aggregates.59 These aggregates may contribute to the observed cell viability upon treatment with the Aβ40 sample incubated with M-TTR and Zn(II) (Fig. 1e). In the case of Aβ1–14, as the least hydrophobic domain of Aβ40,60 it is less likely to interact with itself or Aβ40 to form amyloids; however, previous studies reported that the N-terminal regions of Aβ, including Aβ1–14, could interact with cell receptors (e.g., glutamate or N-methyl-D-aspartate receptors), potentially leading to cytotoxicity.60 Thus, the generation of Aβ1–14 may contribute to the observed reduction in cell viability when cells were treated with the Aβ40 sample incubated with M-TTR and Zn(II), compared to the sample without Zn(II) (Fig. 1e). It is worth noting that additional hydrolytic products of Aβ (e.g., Aβ1–203+, Aβ21–402+, Aβ1–223+, Aβ23–403+, and Aβ34–40+) were observed under both Zn(II)-treated and -untreated conditions (Fig. S19†), denoting that a minute quantity of Zn(II) remaining in the samples due to experimental limitations in purification processes may cause peptide hydrolysis. It is noteworthy that liquid chromatography coupled with MS (LC–MS) could not distinguish Aβ fragments detected under Zn(II)-treated conditions, possibly due to their similar retention time61 and the detection limit of LC–MS under our experimental conditions (Fig. S20†). Overall results illustrate that M-TTR in the presence of Zn(II) promotes the proteolysis of Aβ40 between His14 and Gln15, which potentially influences the aggregation and toxicity of Aβ40.
 | ||
| Fig. 4 Proteolytic activity of M-TTR against Aβ40 in the presence of Zn(II). Aβ fragments [Aβ1–142+ (850 m/z) and Aβ15–402+ (1325 m/z)] were investigated by ESI–MS and ESI–MS2. Charge states are marked above the peaks in the spectra. Conditions: [Aβ40] = 100 μM; [M-TTR] = 100 μM; [ZnCl2] = 100 μM; 20 mM ammonium acetate, pH 7.4; 37 °C; 24 h incubation; quiescent conditions. The samples were diluted 10-fold before injection into the mass spectrometer. The measurements were conducted in triplicate. | ||
Conclusions
Our studies shed light on the intricate interplay among M-TTR, Zn(II), and Aβ40 in the context of AD pathology. We demonstrate that Zn(II) retains the anti-amyloidogenic ability of M-TTR, which spatially overlaps with Aβ in extracellular fluids,6–9 against Aβ40, albeit to a lesser extent. Through comprehensive investigations, we manifest the formation of a Zn(II)-promoted ternary complex involving M-TTR, Aβ40, and Zn(II), which can tune the suppressive effect of M-TTR on the primary nucleation process and cytotoxicity of Aβ40. Such binding also fosters the proteolytic cleavage of Aβ40 and further redirects its aggregation and toxicity profiles. These interactions among M-TTR, Aβ40, and Zn(II) reduce M-TTR's anti-amyloidogenic activity against Aβ40 but allow it to maintain some inhibitory effect in the presence of Zn(II). Collectively, our findings offer a perspective on the collaborative roles of M-TTR and Zn(II) in modulating Aβ aggregation and toxicity. In the CSF, the presence of various metal ions and proteins capable of coordinating metal ions, in addition to M-TTR and Zn(II), suggests that dynamic interactions involved in Aβ-related pathology associated with AD may also extend to other metal ions (e.g., copper)41,62 and extracellular proteins (e.g., human serum albumin).63–66 Therefore, these findings contribute to our understanding of potential therapeutic targets and interventions for AD.67–69Data availability
All experimental details and data supporting the findings of this study are available within the paper and its ESI.† The data are also available from the corresponding authors upon reasonable request.Author contributions
Y. Y., J. H. K., and M. H. L. designed the research. Y. Y. performed biochemical and cell studies (cross-linking experiments with gel/western blot, ThT assay, and MTT assay) and the measurements of TEM, ESI–MS, and electronic Abs spectroscopy with data analyses and analyzed the LC–MS data. B. K. and J. H. K. prepared, purified, and characterized M-TTR. Y. H. K. and J. H. K. conducted 2D NMR studies with data analysis. M. K. conducted the ThT assay and the LC–MS analysis. Y. Y. and M. H. L. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grants {Creative Research Initiative [RS-2022-NR070709] (M. H. L.); NRF-2020R1I1A2074335 (J. H. K.)} and the Institute for Basic Science of Korea [IBS-R034-D1 (Y. H. K.)], funded by the Korean government. M. K. thanks the Sejong Science Fellowship Grants (RS-2023-00214034).Notes and references
- L. Traxler, R. Lucciola, J. R. Herdy, J. R. Jones, J. Mertens and F. H. Gage, Nat. Rev. Neurol., 2023, 19, 434 CrossRef CAS PubMed.
- D. M. Walsh and D. J. Selkoe, Nat. Rev. Neurosci., 2016, 17, 251 CrossRef CAS PubMed.
- E. Karran and B. De Strooper, Nat. Rev. Drug Discovery, 2022, 21, 306 CrossRef CAS PubMed.
- S. H. Han, J. C. Park and I. Mook-Jung, Prog. Neurobiol., 2016, 137, 17 CrossRef CAS PubMed.
- Y. Yi, J. Lee and M. H. Lim, Trends Chem., 2024, 6, 128 CrossRef CAS.
- J. N. Buxbaum, Z. Ye, N. Reixach, L. Friske, C. Levy, P. Das, T. Golde, E. Masliah, A. R. Roberts and T. Bartfai, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2681 CrossRef CAS PubMed.
- X. Li, E. Masliah, N. Reixach and J. N. Buxbaum, J. Neurosci., 2011, 31, 12483 CrossRef CAS.
- X. Li, X. Zhang, A. R. A. Ladiwala, D. Du, J. K. Yadav, P. M. Tessier, P. E. Wright, J. W. Kelly and J. N. Buxbaum, J. Neurosci., 2013, 33, 19423 CrossRef CAS PubMed.
- A. L. Schwarzman, L. Gregori, M. P. Vitek, S. Lyubski, W. J. Strittmatter, J. J. Enghilde, R. Bhasin, J. Silverman, K. H. Weisgraber, P. K. Coyle, M. G. Zagorski, J. Talafous, M. Eisenberg, A. M. Saunders, A. D. Roses and D. Goldgaber, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8368 CrossRef CAS PubMed.
- M. Alemi, C. Gaiteiro, C. A. Ribeiro, L. M. Santos, J. R. Gomes, S. M. Oliveira, P.-O. Couraud, B. Weksler, I. Romero, M. J. Saraiva and I. Cardoso, Sci. Rep., 2016, 6, 20164 CrossRef CAS PubMed.
- S. F. Gloeckner, F. Meyne, F. Wagner, U. Heinemann, A. Krasnianski, B. Meissner and I. Zerr, J. Alzheimer's Dis., 2008, 14, 17 CAS.
- I. Elovaara, C. P. Maury and J. Palo, Acta Neurol. Scand., 1986, 74, 245 CrossRef CAS PubMed.
- E. M. Castano, A. E. Roher, C. L. Esh, T. A. Kokjohn and T. Beach, Neurol. Res., 2006, 28, 155 CrossRef CAS PubMed.
- M. G. Savelieff, G. Nam, J. Kang, H. J. Lee, M. Lee and M. H. Lim, Chem. Rev., 2019, 119, 1221 CrossRef CAS PubMed.
- J.-M. Suh, M. Kim, J. Yoo, J. Han, C. Paulina and M. H. Lim, Coord. Chem. Rev., 2023, 478, 214978 CrossRef CAS.
- K. P. Kepp, Coord. Chem. Rev., 2017, 351, 127 CrossRef CAS.
- S. Ayala, P. Genevaux, C. Hureau and P. Faller, ACS Chem. Neurosci., 2019, 10, 3366 CrossRef CAS PubMed.
- P. Faller and C. Hureau, Dalton Trans., 2009, 1080 RSC.
- Y. Miller, B. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9490 CrossRef CAS PubMed.
- Y. Yi and M. H. Lim, RSC Chem. Biol., 2023, 4, 121 RSC.
- S. Park, C. Na, J. Han and M. H. Lim, Metallomics, 2023, 15, mfac102 CrossRef PubMed.
- J. Han, Z. Du and M. H. Lim, Acc. Chem. Res., 2021, 54, 3930 CrossRef CAS PubMed.
- E. Nam, Y. Lin, J. Park, H. Do, J. Han, B. Jeong, S. Park, D. Y. Lee, M. Kim, J. Han, M.-H. Baik, Y.-H. Lee and M. H. Lim, Adv. Sci., 2023, 11, 2307182 CrossRef PubMed.
- Z. Du, E. Nam, Y. Lin, M. Hong, T. Molnár, I. Kondo, K. Ishimori, M.-H. Baik, Y.-H. Lee and M. H. Lim, Chem. Sci., 2023, 14, 5340 RSC.
- J. Han, J. Yoon, J. Shin, E. Nam, T. Qian, Y. Li, K. Park, S.-H. Lee and M. H. Lim, Nat. Chem., 2022, 14, 1021 CrossRef CAS PubMed.
- A. Abelein, A. Gräslund and J. Danielsson, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5407 CrossRef CAS PubMed.
- L. Saelices, L. M. Johnson, W. Y. Liang, M. R. Sawaya, D. Cascio, P. Ruchala, J. Whitelegge, L. Jiang, R. Riek and D. S. Eisenberg, J. Biol. Chem., 2015, 290, 28932 CrossRef CAS PubMed.
- W. Colon and J. W. Kelly, Biochemistry, 1992, 31, 8654 CrossRef CAS PubMed.
- T. R. Foss, R. L. Wiseman and J. W. Kelly, Biochemistry, 2005, 44, 15525 CrossRef CAS PubMed.
- Z. H. Lai, W. Colon and J. W. Kelly, Biochemistry, 1996, 35, 6470 CrossRef CAS PubMed.
- A. Quintas, D. C. Vaz, I. Cardoso, M. J. M. Saraiva and R. M. M. Brito, J. Biol. Chem., 2001, 276, 27207 CrossRef CAS PubMed.
- S. Susuki, Y. Ando, T. Sato, M. Nishiyama, M. Miyata, M. A. Suico, T. Shuto and H. Kai, Amyloid, 2008, 15, 108 CrossRef CAS PubMed.
- L. d. C. Palmieri, L. M. T. R. Lima, J. B. B. Freire, L. Bleicher, I. Polikarpov, F. C. L. Almeida and D. Foguel, J. Biol. Chem., 2010, 285, 31731 CrossRef CAS PubMed.
- N. Marchi, V. Fazio, L. Cucullo, K. Kight, T. Masaryk, G. Barnett, M. Volgelbaum, M. Kinter, P. Rasmussen, M. R. Mayberg and D. Janigro, J. Neurosci., 2003, 23, 1949 CrossRef CAS PubMed.
- X. Jiang, C. S. Smith, H. M. Petrassi, P. Hammarstrom, J. T. White, J. C. Sacchettini and J. W. Kelly, Biochemistry, 2001, 40, 11442 CrossRef CAS.
- J. Oroz, J. H. Kim, B. J. Chang and M. Zweckstetter, Nat. Struct. Mol. Biol., 2017, 24, 407 CrossRef CAS.
- R. Whelan, F. M. Barbey, M. R. Cominetti, C. M. Gillan and A. M. Rosická, Transl. Psychiatry, 2022, 12, 473 CrossRef PubMed.
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, 2010, 1804, 1405 CrossRef CAS PubMed.
- H. Tamano, R. Nishio, Y. Shakushi, M. Sasaki, Y. Koike, M. Osawa and A. Takeda, Sci. Rep., 2017, 7, 42897 CrossRef CAS PubMed.
- S. Vivekanandan, J. R. Brender, S. Y. Lee and A. Ramamoorthy, Biochem. Biophys. Res. Commun., 2011, 411, 312 CrossRef CAS PubMed.
- Y. Yi, W. Ryu, B. Kim, S. Muniyappan, K. Park, J. H. Kim and M. H. Lim, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-cx1zb.
- J. Du and R. M. Murphy, Biochemistry, 2010, 49, 8276 CrossRef CAS PubMed.
- S. A. Ghadami, S. Chia, F. S. Ruggeri, G. Meisl, F. Bemporad, J. Habchi, R. Cascella, C. M. Dobson, M. Vendruscolo, T. P. J. Knowles and F. Chiti, Biomacromolecules, 2020, 21, 1112 CrossRef CAS PubMed.
- G. Meisl, J. B. Kirkegaard, P. Arosio, T. C. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. J. Knowles, Nat. Protoc., 2016, 11, 252 CrossRef CAS PubMed.
- T. E. R. Werner, D. Bernson, E. K. Esbjorner, S. Rocha and P. Wittung-Stafshede, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 27997 CrossRef CAS.
- S. Linse, Pure Appl. Chem., 2019, 91, 211 CrossRef CAS.
- P. Arosio, R. Cukalevski, B. Frohm, T. P. J. Knowles and S. Linse, J. Am. Chem. Soc., 2014, 136, 219 CrossRef CAS PubMed.
- G. Meisl, X. Yang, E. Hellstrand, B. Frohm, J. B. Kirkegaard, S. I. Cohen, C. M. Dobson, S. Linse and T. P. J. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9384 CrossRef CAS PubMed.
- C. E. Sabel, J. M. Neureuther and S. Siemann, Anal. Biochem., 2010, 397, 218 CrossRef CAS PubMed.
- Y. Yi, Y. Lin, J. Han, H. J. Lee, N. Park, G. Nam, Y. S. Park, Y. H. Lee and M. H. Lim, Chem. Sci., 2021, 12, 2456 RSC.
- N. Rezaei-Ghaleh, K. Giller, S. Becker and M. Zweckstetter, Biophys. J., 2011, 101, 1202 CrossRef CAS PubMed.
- J. Du, P. Y. Cho, D. T. Yang and R. M. Murphy, Protein Eng., Des. Sel., 2012, 25, 337 CrossRef CAS PubMed.
- D. T. Yang, G. Joshi, P. Y. Cho, J. A. Johnson and R. M. Murphy, Biochemistry, 2013, 52, 2849 CrossRef CAS PubMed.
- G. D. Natale, G. Sabatino, M. F. M. Sciacca, R. Tosto, D. Milardi and G. Pappalardo, Molecules, 2022, 27, 5066 CrossRef PubMed.
- F. Lopez-Gallego, J. M. Guisan and L. Betancor, Methods Mol. Biol., 2013, 1051, 33 CrossRef CAS PubMed.
- M. A. Liz, S. C. Leite, L. Juliano, M. J. Saraiva, A. M. Damas, D. Bur and M. M. Sousa, Biochem. J., 2012, 443, 769 CrossRef CAS PubMed.
- I. E. Gouvea, M. Y. Kondo, D. M. Assis, F. M. Alves, M. A. Liz, M. A. Juliano and L. Juliano, Biochimie, 2013, 95, 215 CrossRef CAS PubMed.
- C. Dammers, M. Schwarten, A. K. Buell and D. Willbold, Chem. Sci., 2017, 8, 4996 RSC.
- K. Taş, B. D. Volta, C. Lindner, O. El Bounkari, K. Hille, Y. Tian, X. Puig-Bosch, M. Ballmann, S. Hornung, M. Ortner, S. Prem, L. Meier, G. Rammes, M. Haslbeck, C. Weber, R. T. A. Megens, J. Bernhagen and A. Kapurniotu, Nat. Commun., 2022, 13, 5004 CrossRef PubMed.
- B. Murray, B. Sharma and G. Belfort, ACS Chem. Neurosci., 2017, 8, 432 CrossRef CAS PubMed.
- S. Du, E. R. Readel, M. Wey and D. W. Armstrong, Chem. Commun., 2020, 56, 1537 RSC.
- L. Ciccone, C. Fruchart-Gaillard, G. Mourier, M. Savko, S. Nencetti, E. Orlandini, D. Servent, E. A. Stura and W. Shepard, Sci. Rep., 2018, 8, 13744 CrossRef PubMed.
- T. S. Choi, H. J. Lee, J. Y. Han, M. H. Lim and H. I. Kim, J. Am. Chem. Soc., 2017, 139, 15437 CrossRef CAS PubMed.
- D. A. Bushinsky and R. D. Monk, Lancet, 1998, 352, 305 CrossRef.
- P. C. Johnson, W. O. Smith and B. Wulff, J. Appl. Physiol., 1959, 14, 859 CrossRef CAS PubMed.
- C. Cantarutti, M. C. Mimmi, G. Verona, W. Mandaliti, G. W. Taylor, P. P. Mangione, S. Giorgetti, V. Bellotti and A. Corazza, Biomolecules, 2022, 12, 1066 CrossRef CAS PubMed.
- M. Ramesh, C. Balachandra, P. Andhare and T. Govindaraju, ACS Chem. Neurosci., 2022, 13, 2209 CrossRef CAS PubMed.
- Q. Cao, D. H. Anderson, W. Y. Liang, J. Chou and L. Saelices, J. Biol. Chem., 2020, 295, 14015 CrossRef CAS PubMed.
- M. Ramesh and T. Govindaraju, Chem. Sci., 2022, 13, 13657 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Table S1 and Fig. S1–S20. See DOI: https://doi.org/10.1039/d4sc08771b |
| This journal is © The Royal Society of Chemistry 2025 |