DOI:
10.1039/D5RA00879D
(Paper)
RSC Adv., 2025,
15, 17290-17301
Synthesis and characterization of novel N-(2-(pyrazin-2-yl-oxy)ethyl)-4-(trifluoromethoxy)benzamide scaffolds, and biological evaluation and molecular docking studies†
Received
6th February 2025
, Accepted 15th May 2025
First published on 23rd May 2025
Abstract
A new series of biologically potent N-(2-(6-substituted-1H-pyrazin-2-yloxy)ethyl)-4-(trifluoromethoxy)benzamide scaffolds was synthesized, and their structures were confirmed by 1H NMR, 13C NMR, and mass spectrometry. All the synthesized molecules were tested against antibacterial activity against various pathogenic microorganisms and exhibited remarkable activity. Compounds 12a and 13a exhibited good antibacterial activity against pathogenic cell lines, Staphylococcus aureus and Escherichia coli. Additionally, synthesized molecules 12a and 13a were screened for anticancer activity against the A549 (lung cancer) cell line. These compounds displayed excellent anticancer activity with IC50 values of 19 + 0.50 μM, 17 ± 0.5 μM, A549 (lung cancer). Molecular docking studies results were well supported by strong intercalative interactions of the synthesized compounds with target proteins.
Introduction
Pyrazines are heterocyclic compounds characterized by a six-membered ring with two nitrogen atoms. These compounds serve as versatile scaffolds in medicinal chemistry due to their electronic properties and ability to participate in diverse chemical interactions. Structural modifications of the pyrazine ring allow for fine-tuning of biological activity and specificity. Nitrogen atoms in the ring system facilitate hydrogen bonding and metal coordination, broadening their biological applications.1–5 Pyrazine derivatives have garnered attention in medicinal chemistry due to their diverse biological activities.6–9 Recent studies highlight their potential as antimicrobial agents and their promising cytotoxicity against lung cancer cells.10–12 In recent years, the quest for novel bioactive compounds has driven significant research into diverse chemical classes with potential therapeutic applications. Among these, pyrazine derivatives have emerged as a prominent area of interest due to their versatile biological activities.13,14 The antimicrobial properties of pyrazine derivatives have garnered particular attention as the global rise in antimicrobial resistance challenges conventional treatment strategies.15 These derivatives have demonstrated significant efficacy against various pathogenic microorganisms, including bacteria and fungi.16 Their ability to inhibit microbial growth and potentially overcome resistance mechanisms makes them valuable candidates for developing new antimicrobial agents.17 Over the years, numerous drugs and bioactive molecules have been developed based on the pyrazine core moiety; among them, some medicinally potent pyrazine drug derivatives are shown in Fig. 1.
 |
| | Fig. 1 Chemical structure of biologically potent pyrazine drugs. | |
In addition to their antimicrobial potential, pyrazine derivatives have also shown promise in oncology. Cancer remains a leading cause of morbidity and mortality worldwide, and the search for effective, targeted therapies is crucial. Pyrazine-based compounds have exhibited cytotoxic effects against various cancer cell lines, including those of lung cancer,18–23 which is one of the most prevalent and aggressive types of cancer. The ability of these compounds to induce selective toxicity in cancer cells while sparing normal cells is of particular interest in designing more effective and less toxic cancer therapies.
In this manuscript, we explore the synthesis and evaluation of new N-(2-(6-substituted1-yl)pyrazin-2-yloxy)ethyl)-4-(trifluoromethoxy)benzamide derivatives, focusing on their antimicrobial activities and cytotoxic effects against human lung cancer cell lines. By investigating these dual properties, we aim to contribute to developing new therapeutic agents that address both infectious diseases and cancer, potentially offering new avenues for treatment in these critical areas. In this manuscript, the present work describes the synthesis of a novel series of N-(2-((6-substituted pyrazin-2-yloxy)ethyl)-4-(trifluoromethoxy)benzamide derivatives. These compounds were synthesized using a key intermediate, N-(2-((6-chloropyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (5), which was further derivatized through nucleophilic substitution and palladium-catalyzed cross-coupling reactions.
Results and discussions
The synthetic route of newly prepared compounds (6a–20a) was represented in Scheme 1. Initially, the starting material N-(2-hydroxyethyl)-4-(trifluoromethoxy)benzamide (3) was easily synthesized from 4-(trifluoromethoxy)benzoic acid (1) and 2-aminoethanol (2), in the presence of N,N-diisopropylethylamine (DIPEA), 1-hydroxybenzotriazole (HOBt) (1.5 eq.), and N,N-ethyl carbodiimide hydrochloride (EDC·HCl) (1.5 eq.) was added at 0 °C and stirred for 12 h at 25 °C, furnishing compound 3 with an excellent yield. Then, compound N-(2-hydroxyethyl)-4-(trifluoromethoxy)benzamide reacted with 2,6-dichloro pyrazine (4) in the presence of potassium tert-butoxide (1.1 eq.) in THF (20 mL) at 0 °C and stirred for 2 h at 25 °C obtained reaction intermediate N-(2-((6-chloropyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (5) good yield. In this study, a pyrazine-based intermediate, N-(2-((6-chloropyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (compound 5), was employed as a key scaffold for the synthesis of a series of novel target derivatives. The synthesis strategy involved the formation of a new carbon–nitrogen (C–N) bond, enabling the development of N-(2-((6-substituted pyrazin-2-yloxy)ethyl)-4-(trifluoromethoxy)benzamide derivatives, labeled as 6a–20a. For derivatives (6a–16a), condensation reactions were carried out between compound 5 and various amine-containing reagents in dimethyl sulfoxide (DMSO) under optimized temperature conditions. This route allowed efficient nucleophilic substitution at the 6-chloropyrazine moiety. In contrast, compounds (17a–20a) were synthesized via palladium-catalyzed amination reactions. These reactions were conducted in 1,4-dioxane, employing cesium carbonate (Cs2CO3) as a base and Brett Phos Pd G3 as the catalyst, under conventional heating conditions. This protocol facilitated the formation of C–N bonds with amines that required more reactive or sterically hindered systems.
 |
| | Scheme 1 Synthesis of substituted novel N-(2-(pyrazin-2-yl-oxy)ethyl)-4-(trifluoromethoxy)benzamide scaffolds derivatives (6a–20a). | |
All desired compounds were obtained in moderate to high yields, ranging from 53% to 90%, indicating the efficiency and versatility of the developed synthetic methodologies. The structures of the synthesized compounds were unambiguously confirmed using liquid chromatography-mass spectrometry (LC-MS), 1H NMR, and 13C NMR spectroscopic techniques. Detailed yields and structural assignments are summarized in Table 1.
Table 1 Synthesis of substituted pyrazine derivatives (6a–20a) under the conventional method in the presence of Brett-Phos Pd G3
| S. No. |
Comp. |
Synthesized final compounds |
M. P. (°C) |
Yield (%) |
| 1 |
6a |
 |
103 |
63 |
| 2 |
7a |
 |
106 |
86 |
| 3 |
8a |
 |
113 |
90 |
| 4 |
9a |
 |
136 |
87 |
| 5 |
10a |
 |
168 |
56 |
| 6 |
11a |
 |
189 |
62 |
| 7 |
12a |
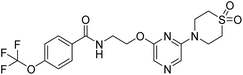 |
196 |
63 |
| 8 |
13a |
 |
188 |
89 |
| 9 |
14a |
 |
150 |
58 |
| 10 |
15a |
 |
165 |
66 |
| 11 |
16a |
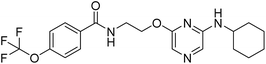 |
180 |
55 |
| 12 |
17a |
 |
110 |
64 |
| 13 |
18a |
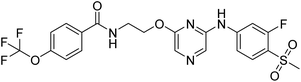 |
97 |
66 |
| 14 |
19a |
 |
125 |
59 |
| 15 |
20a |
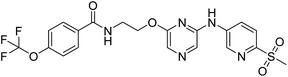 |
120 |
53 |
Biological evaluation
Anti-cancer activity.
Cell culture. The Human lung cancer (A549)24–28 cells were procured from the National Center for Cell Sciences (NCCS), Pune, India. The selected cancer cells were maintained in Dulbecco's modified eagles' medium (DMEM) supplemented with 2 mM l-glutamine and balanced salt solution (BSS) adjusted to contain 1.5 g L−1 Na2CO3, 0.1 mM non-essential amino acids, 1 mM sodium pyruvate, 2 mM l-glutamine, 1.5 g L−1 glucose, 10 mM (4-(2-hydroxyethyl)-1-piperazineethane sulfonic acid) (HEPES) and 10% fetal bovine serum (GIBCO, USA). Penicillin and streptomycin (100 IU/100 μg) were adjusted to 1 mL L−1. The cells were maintained at 37 °C with 5% CO2 in a humidified CO2 atmosphere.
Morphological study. The selected cells that were grown on cover slips (1 × 105 cells per cover slip) were incubated with the complex at different concentrations, and they were then fixed in an ethanol: acetic acid solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The cover slips were gently mounted on glass slides for the morphometric analysis. Three monolayers per experimental group were micrographed. The morphological changes of the cells were analyzed using Nikon (Japan) bright field inverted light microscopy at 10× magnification.
:1, v/v). The cover slips were gently mounted on glass slides for the morphometric analysis. Three monolayers per experimental group were micrographed. The morphological changes of the cells were analyzed using Nikon (Japan) bright field inverted light microscopy at 10× magnification.
Evaluation of cytotoxicity
The inhibitory concentration (IC50) value was evaluated using an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. The cells were grown (1 × 104 cells per well) in a 96-well plate for 48 h to 80% confluence. The medium was replaced with fresh medium containing the serially diluted sample, and the cells were further incubated for 48 h. The culture medium was removed, and 100 μL of the MTT [3-(4,5-dimethylthiozol-2-yl)-3,5-diphenyl tetrazolium bromide] (Hi-Media) solution was added to each well and incubated at 37 °C for 4 h. After removal of the supernatant, 50 μL of DMSO was added to each of the wells and incubated for 10 min to solubilize the formazan crystals. The optical density was measured at 620 nm in an ELISA multi-well plate reader (Thermo Multiskan EX, USA). The OD value was used to calculate the percentage of viability using the following formula.
| % of viability = OD value of experimental sample/OD value of experimental control ×100 |
Fluorescence microscopic analysis of apoptotic cell death
Approximately 1 μL of a dye mixture (100 mg mL−1 acridine orange (AO) and 100 mg mL−1 ethidium bromide (EtBr) in distilled water) was mixed with 0.9 mL of cell suspension (1 × 105 cells per mL) on clean microscope cover slips. The pretreated cancer cells were collected, washed with phosphate-buffered saline (PBS) (pH 7.2), and stained with 10 μL of AO/EtBr. After incubation for 2 min, the cells were washed twice with PBS (5 min each) and visualized under a fluorescence microscope (Nikon Eclipse, Inc., Japan) at 20× magnification with an excitation filter at 580 nm.29–32
Cytotoxic assay. An analysis of the effect of the extract on the cell response of the Human lung cancer cells by using the MTT assay. Fig. 2 shows the in vitro cytotoxicity activity of the extract (up to 100 μg ml−1 concentrations) against selected lung cancer cells. The experimental results demonstrate that the extract inhibited cell proliferation dose-dependently. From Fig. 2, the IC50 values of a sample against cancer cells were calculated, and it was found to be 19 and 17 μg ml−1 CPd12 and CPd13 13 respectively. It can be noticed from the results that the observed IC50 values of the material are low and significantly inhibit the proliferation of Human cancer cells.
 |
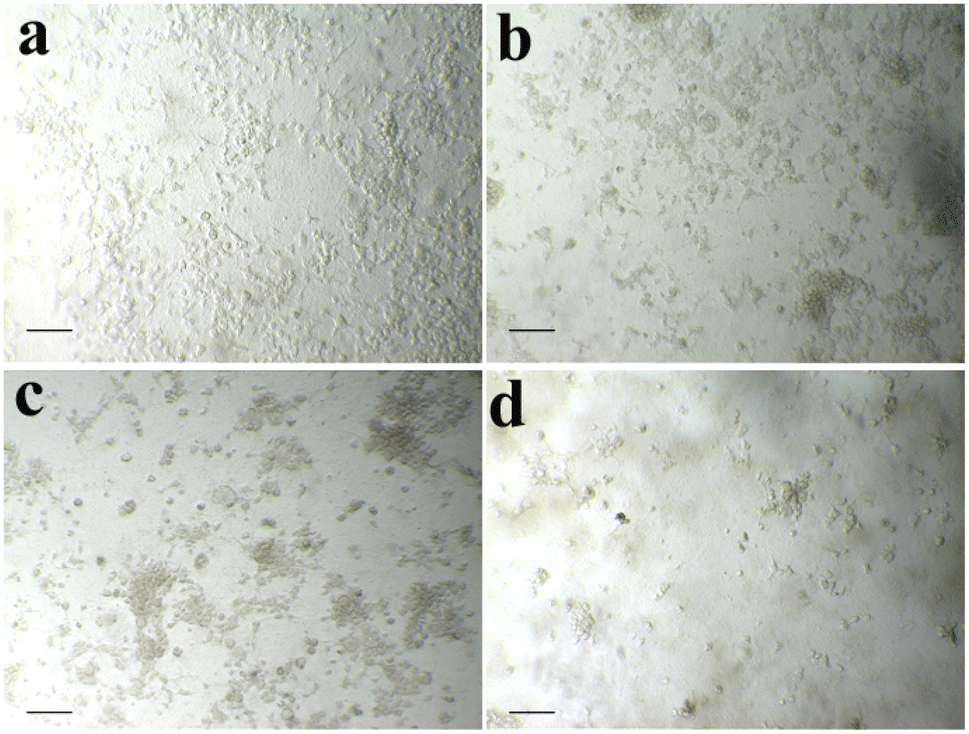
| | Fig. 2 A549 (lung cancer) cells. Control (a), 10 μg ml−1 (b), 25 μg ml−1 (c), 50 μg ml−1 (d), and scale bars – 50 μm. | |
Cell morphology analysis. The morphological changes of selected cancer cells in the absence and presence of samples at various concentrations are shown in Fig. 3 and 4. It could be observed from Fig. 3 and 4, control cells showed no remarkable changes in their morphology. However, in the presence of samples, the cells show improved cell shrinkage and membrane blebbing, and floating cells are formed in a dose-dependent manner. It is well accepted that cytological investigations elucidate the antiproliferative effect mediated through membrane blebbing, membrane instability, and disturbing the cytoskeleton of the cells by the samples.
 |
| | Fig. 3 Morphological changes were observed in A549 cells after treatment with the synthesized compounds at various concentrations. | |
 |
| | Fig. 4 Morphological changes observed in A549 cells exposed to the synthesized compounds at various concentrations compared to the control. | |
Acridine orange/ethidium bromide (AO/EtBr) staining method
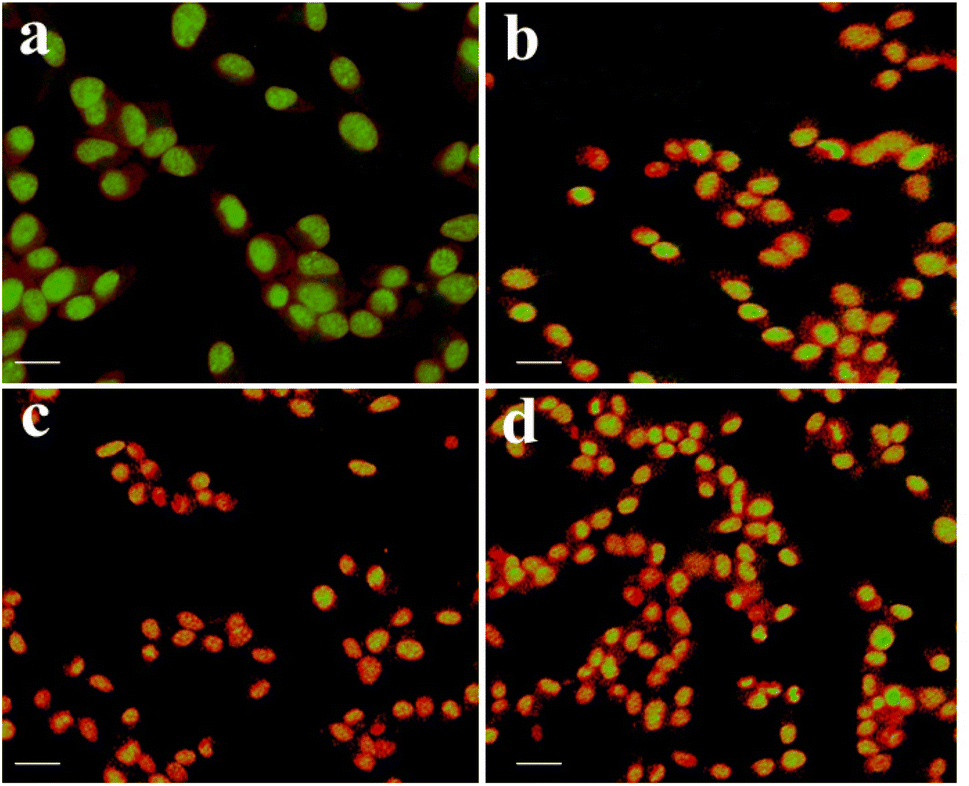
In order to study the effect of apoptogenic activity of the material on cancer cells, we conducted the fluorescence microscopic analysis. Fluorescence microscopy images of A549 cancer cells in the absence of samples (Control) and in the presence of samples are shown in Fig. 5. Fig. 5(a) shows that the untreated Lung cancer cells (control) did not show any significant adverse effect compared to the sample treated cells (Fig. 5(b–d)). From Fig. 5(b–d), it can be observed that with the addition of the sample to the A549 cells (Fig. 6), the green color of the cells is transformed into orange/red color cells, which is due to the induction of apoptosis and the nuclear condensation effect on the cells.33–35
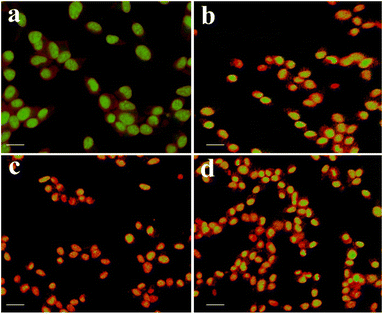 |
| | Fig. 5 Fluorescence microscopy images of A549 cancer cells showing morphological changes in the absence (control) and presence of test samples. | |
 |
| | Fig. 6 Control (a), 10 μg ml−1 (b), 25 μg ml−1 (c), 50 μg ml−1 (d), and scale bars – 50 μm. | |
This choice from the literature to examine the binding mode conformation structure that contributes to the interrelations between synthesized compounds and the protein. The best fitted poses, binding energy scores, and a list of hydrogen bonds were determined. From the docking studies, it was predicted that few compounds would have strong interactions against the active site of the bound protein. The rank of potent binding score compounds are: comp. 20 > comp. 12 > comp. 11 > comp. 15 > comp. 19 = −6.8 > −6.7 > −6.5 > −6.4 > −6.2 kcal mol−1. Remaining compounds binding affinities showed good to moderate.36 Its promising binding affinity suggests potential as an antimicrobial agent. The compounds 12a and 13a showed excellent anticancer activity results, which are summarized in Table 2.
Table 2 Anticancer activity data of the synthesized 2,6-disubstituted pyrazine scaffolds (μg mL−1)a
| Sample |
A549 cells (IC50) |
| IC50 – values of respective compounds (at 24 h) standard used: doxorubicin. |
| Compound 12a |
19 ± 0.5 |
| Compound 13a |
17 ± 0.5 |
| Doxorubicin |
16 ± 1.3 |
Statistics. All the in vitro experiments were done in triplicate and repeated at least three times. The statistical software SPSS version 17.0 was used for the analysis. P value < 0.01 was considered significant.
Antibacterial activity. The antibacterial susceptibility of prepared compounds against Gram-positive and Gram-negative bacterial strains was assessed. S. aureus and E. coli were evaluated by the disk diffusion/Kirby–Bauer method.37–41 Briefly, a 100 μL sample of freshly grown bacterial suspension (∼104 and ∼106 CFU per mL of S. aureus and E. coli, respectively, cultured in LB medium) was spread onto nutrient agar plates. Small sterile paper disks of uniform size (10 mm) were impregnated with as-prepared compounds and colloidal samples and then placed on the nutrient agar plates. A disk impregnated with ciprofloxacin was also placed on nutrient agar for positive control, and PBS acted as a negative control, respectively. Plates were then incubated at 37 °C for 24 h. The resulting bacterial colonies' distance inhibition zones around the disks were then recorded.The antibacterial zone inhibition (mm) of samples against pathogenic microorganisms is given in Table 3. The results revealed that most tested compounds (6a–20a) showed moderate to good inhibitory activity against all the strains. All the synthesized compounds (6a–20a) showed very good activity.
Table 3 Antibacterial zone of inhibition of synthesized benzamide derivatives (6a–20a)
| S. No. |
Compounds |
E. coli |
S. aureus |
| 50 (μg ml−1) |
100 (μg ml−1) |
150 (μg ml−1) |
50 (μg ml−1) |
100 (μg ml−1) |
150 (μg ml−1) |
| 1 |
6a |
1.6 |
2.2 |
2.4 |
1.7 |
1.8 |
2.1 |
| 2 |
7a |
1.6 |
2.1 |
2.3 |
1.4 |
2.0 |
2.2 |
| 3 |
8a |
1.5 |
1.9 |
2.2 |
1.6 |
1.9 |
2.2 |
| 4 |
9a |
1.3 |
1.8 |
2.1 |
1.5 |
1.9 |
2.2 |
| 5 |
10a |
1.6 |
1.8 |
2.0 |
1.7 |
1.8 |
2.3 |
| 6 |
11a |
1.7 |
1.9 |
2.2 |
1.8 |
1.9 |
2.2 |
| 7 |
12a |
1.9 |
2.3 |
2.9 |
1.8 |
2.5 |
3.0 |
| 8 |
13a |
1.7 |
2.2 |
2.7 |
1.8 |
2.1 |
2.8 |
| 9 |
14a |
1.6 |
1.9 |
2.1 |
1.5 |
1.9 |
2.0 |
| 10 |
15a |
1.5 |
1.8 |
2.2 |
1.4 |
1.8 |
2.0 |
| 11 |
16a |
1.5 |
1.8 |
2.2 |
1.4 |
1.8 |
2.0 |
| 12 |
17a |
1.3 |
1.8 |
2.0 |
1.8 |
2.0 |
2.0 |
| 13 |
18a |
1.7 |
2.2 |
2.2 |
1.8 |
2.0 |
2.0 |
| 14 |
19a |
1.7 |
2.2 |
2.3 |
1.7 |
1.9 |
1.9 |
| 15 |
20a |
1.6 |
1.7 |
1.9 |
1.8 |
1.9 |
2.4 |
| 16 |
Ciprofloxacin |
2.0 |
2.5 |
3.0 |
2.0 |
2.5 |
3.0 |
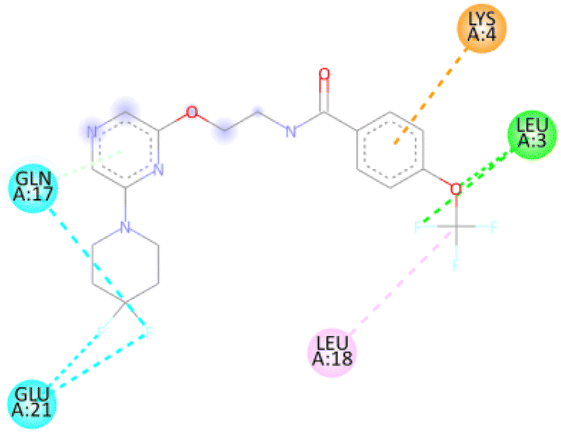
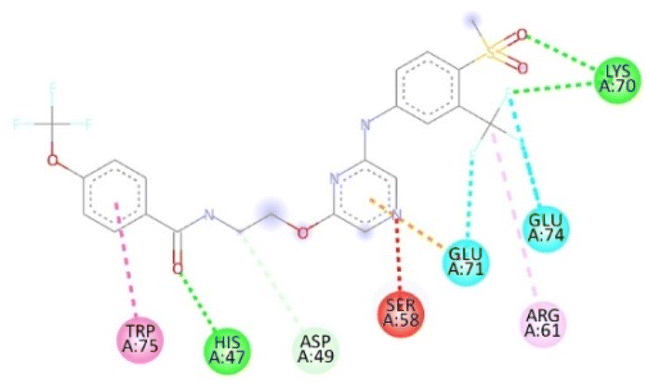
Molecular docking studies. For affected identification and result enhancement, analytical techniques that “dock” organic compounds into the structures of molecular targets and “score” their potential in pertaining to binding sites are widely applied.42 The Chem Draw Ultra 15.0 was used to create the structural model, which was then saved as a PDBQT file for Discovery Studio 2021 to display.43 Using the corresponding 3D structure and the default MOE energy reduction method parameters (gradient: 0.05, Force Field: MMFF94X), the energies of the observed molecule were decreased. The obtained model was then used in the MOE's “Systematic Conformational Search.” Selected protein (PDBID: ICEI) crystal structures were obtained via the Protein Data Bank (http://www.rcsb.org/pdb). The proteins were fixed in a variety of ways before docking, such as by deleting solvent molecules and co-ligands, introducing hydrogen's, altering the chain, and selecting active sites. The novel synthesized modeled compounds (6a to 20a) docked with the active site of the antibacterial protein residual (PDBID: 1CEI). The modeled three-dimensional (3-D) and two-dimensional (2-D) compounds and their distances between atoms (Å), binding interactions, and binding energies are shown in Tables 4 and 5.
Table 4 The molecular docking interaction of compounds with the antibacterial protein (PDBID: 1CEI)
| S. No. |
Compounds |
Interaction residues (PDBID: 1CI) |
Distances (Å) |
H-interactions involved |
Binging energy (kcal mol−1) |
| Ligand |
Protein |
| 1 |
6a |
NH |
O-GLU: A14 |
3.01 |
2 |
−5.9 |
| O |
HN-LEU: A3 |
2.26 |
| 2 |
7a |
O |
HN-LYS: A70 |
2.33 |
3 |
−6.5 |
| N |
HN-LYS: A70 |
2.43 |
| O |
HN-ASP: A62 |
2.05 |
| 3 |
8a |
NH |
O-GLU: A21 |
2.34 |
2 |
−6.2 |
| O |
HN-GLN: A17 |
2.34 |
| 4 |
9a |
NH |
O-ASP: A35 |
2.46 |
2 |
−6.4 |
| O |
HO-THR: A51 |
2.76 |
| 5 |
10a |
N |
HN-LYS: A81 |
2.36 |
1 |
−6.1 |
| 6 |
11a |
O |
HN-LEU; A3 |
2.42 |
2 |
−6.5 |
| F |
HN-LEU; A3 |
2.70 |
| 7 |
12a |
F |
HN-LEU: A3 |
2.84 |
4 |
−6.7 |
| NH |
O-GLN: A17 |
2.15 |
| N |
HO-THR: A11 |
2.21 |
| O |
HN-LYS:85 |
1.94 |
| 8 |
13a |
NH |
O-GLU: A21 |
2.43 |
3 |
−5.7 |
| NH |
O-GLN: A17 |
3.08 |
| F |
HN-LEU: A3 |
2.08 |
| 9 |
14a |
NH |
O-ASN: A60 |
2.31 |
3 |
−5.4 |
| O |
HO-SER: A58 |
2.20 |
| O |
HN-LYS: A70 |
3.30 |
| 10 |
15a |
F |
HN-LYS: A70 |
2.93 |
3 |
−6.4 |
| F |
HN-LYS: A70 |
2.75 |
| N |
HN-HYS: A47 |
2.49 |
| 11 |
16a |
N |
HO-SER: A58 |
2.30 |
3 |
−6.0 |
| N |
HN-ASN: A60 |
2.38 |
| F |
O-HNASN: A79 |
2.95 |
| 12 |
17a |
O |
HN-GLY: A17 |
2.80 |
4 |
−5.5 |
| NH |
O-GLY: A17 |
2.65 |
| NH |
O-GLU: A21 |
2.37 |
| F |
HN-LEU: A3 |
2.42 |
| 13 |
18a |
N |
HN-LEU: A3 |
3.02 |
3 |
−5.4 |
| NH |
O-GLU:21 |
2.31 |
| O |
HN-GLN: A17 |
2.38 |
| 14 |
19a |
O |
HN-LYS: A70 |
2.47 |
3 |
−6.2 |
| F |
O-LYS: A70 |
2.78 |
| O |
HN-HYS: A47 |
2.18 |
| 15 |
20a |
F |
HN-PHE: A84 |
2.53 |
5 |
−6.5 |
| F |
HN-PHE: A84 |
2.57 |
| O |
HN-LYS: A73 |
2.32 |
| NH |
O-GLU: A66 |
2.45 |
| O |
HN-GLY: A87 |
2.33 |
Table 5 3D and 2D molecular docking poses interactions between compounds with the active sites of the bacterial protein (PDBID: 1CEI)
| S. No |
Compounds |
3D structure of a molecule interaction with an enzyme |
2D structure of a molecule interaction with protein |
| 6 |
11a |
 |
 |
| 7 |
12a |
 |
 |
| 10 |
15a |
 |
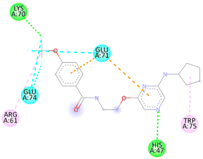 |
| 14 |
19a |
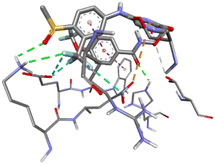 |
 |
| 15 |
20a |
 |
 |
Conclusion
In summary, a new series of biologically potent N-(2-(6-substituted-1H-pyrazin-2-yloxy)ethyl)-4-(trifluoromethoxy)benzamide derivatives was successfully synthesized and characterized by 1H NMR, 13C NMR, and mass spectrometry. The biological screening highlighted the dual therapeutic potential of these compounds, with 12a and 13a exhibiting notable antibacterial activity against Staphylococcus aureus and Escherichia coli. Furthermore, these lead compounds demonstrated significant anticancer activity against the A549 lung cancer cell line, with IC50 values of 19 ± 0.50 μM and 17 ± 0.50 μM, respectively. Molecular docking studies supported the experimental findings, indicating strong intercalative interactions between the synthesized molecules and their respective target proteins. These results underscore the potential of 12a and 13a as promising scaffolds.
Experimental section
All the chemicals used in the present investigation were purchased from Sigma-Aldrich Chemical Company, India. Melting points were determined on a Cintex melting point apparatus and are uncorrected. The purity of the compounds was monitored by TLC on silica gel-G plates (Merk, 60F 254), visualized with ultraviolet light. IR spectra (KBr) were recorded on a PerkinElmer BX series FT-IR spectrophotometer, and 1H and 13C-NMR spectra on a Varian Gemini 400 MHz spectrometer (Chemical shifts in δ ppm) using TMS as internal standard. Mass spectra were recorded using the electron spray ionization technique on the LCMS-2010A mass spectrometer at 70 eV.
General procedure for the construction of N-(2-hydroxyethyl)-4-(trifluoromethoxy)benzamide (3)
To a stirred solution of 4-(trifluoromethoxy)benzoic acid (5 g, 24.26 mmol; 1 eq.) in THF (100 mL) was added 2-aminoethan-1-ol (1.63 g, 26.68 mmol; 1.1 eq.) followed by DIPEA (12.68 mL, 72.77 mmol; 3 eq.), HOBt (4.92 g, 36.39 mmol; 1.5 eq.) and EDC. HCl (6.98 g, 36.39 mmol; 1.5 eq.) was added at 0 °C and stirred for 12 h at 25 °C. After completion of the reaction, the reaction mixture was diluted with EtOAc (200 mL) and washed with water (200 mL) and brine solution (100 mL). The separated organic layer was concentrated and purified by column chromatography using a silica gel column to yield N-(2-hydroxyethyl)-4-(trifluoromethoxy)benzamide (5.1 g, 20.46 mmol; 84.83% Yield), white solid. 1H NMR (400 MHz, DMSO-d6): δ 8.55 (t, J = 5.20 Hz, 1H), 7.98 (dd, J = 2.80, 5.80 Hz, 2H), 7.46 (d, J = 8.00 Hz, 2H), 4.73 (t, J = 5.60 Hz, 1H), 3.54–3.49 (m, 2H), 3.36–3.31 (m, 2H). 13C NMR (101 MHz, DMSO-d6): δ 165.54, 150.63, 134.15, 129.98, 121.72, 121.01, 119.16, 60.11, 42.72. 19F NMR (377 MHz, DMSO-d6): δ −56.69. LCMS: m/z: 250.0 (M + H)+.
General procedure for the synthesis of N-(2-((6-chloropyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (5)
To a stirred solution of N-(2-hydroxyethyl)-4-(trifluoromethoxy)benzamide (5 g, 20.07 mmol; 1 eq.) in THF (80 mL) was added 2,6-dichloropyrazine (2.99 g, 20.07 mmol; 1 eq.) followed by potassium tert-butoxide (2.48 g, 22.07 mmol; 1.1 eq.) in THF (20 mL) at 0 °C and stirred for 2 h at 25 °C. After completion of the reaction, the reaction mixture was diluted with EtOAc (200 mL) and washed with water (100 mL) and brine solution (100 mL). The separated organic layer was concentrated and purified by column chromatography using a silica gel column to yield N-(2-((6-chloropyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (6.2 g, 17.14 mmol; 84% Yield), white solid. 1H-NMR (400 MHz, DMSO-d6): δ 8.80 (s, 1H), 8.33 (d, J = 6.40 Hz, 2H), 7.97 (d, J = 8.40 Hz, 2H), 7.47 (d, J = 8.40 Hz, 2H), 4.48 (t, 2H), 3.69 (m, 2H). 13C NMR (101 MHz, DMSO-d6): δ 165.79, 159.28, 150.76, 144.65, 135.57, 134.10, 133.75, 130.02, 121.71, 121.08, 119.15, 65.81, 40. 62. 19F NMR (377 MHz, DMSO-d6): −56.69. LCMS: m/z: 362.1 (M + H)+.
General method for the synthesis of N-(2-((6-(piperidin-1-yl)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide compounds (6a–13a)
To a stirred solution of N-(2-((6-chloropyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (1 eq.) in DMSO (w/v 20%) was added 2°-amines (6a to 13a) (2 eq.) at room temperature and stirred for 12 h at 80 °C. After completion of the reaction, the reaction mixture was diluted with EtOAc and washed with water and brine solution. The separated organic layer was concentrated and purified by column chromatography using a silica gel column to yield the desired compounds 6a to 13a.
N-(2-((6-(azetidin-1-yl)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (6a). 1H NMR (400 MHz, DMSO-d6): δ 8.80 (t, J = 5.60 Hz, 1H), 7.98–7.95 (m, 2H), 7.47–7.44 (m, 3H), 7.34 (s, 1H), 4.37 (t, J = 5.60 Hz, 2H), 3.98 (t, J = 7.60 Hz, 4H), 3.66–3.61 (m, 2H), 2.37–2.30 (m, 2H). 13C NMR (101 MHz, DMSO-d6): δ 165.69, 158.93, 154.85, 150.74, 133.83, 130.011, 121.701, 121.07, 120.79, 120.12, 119.15, 64.10, 50.94, 17.02. LCMS: m/z: 427.0 (M + H)+.
N-(2-((6-(pyrrolidin-1-yl)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (7a). 1H NMR (400 MHz, DMSO-d6): δ 8.79 (t, J = 5.60 Hz, 1H), 7.96–7.94 (m, 2H), 7.47–7.37 (m, 4H), 4.41 (t, J = 5.60 Hz, 2H), 3.66–3.62 (m, 2H), 3.39–3.34 (m, 4H), 1.92–1.89 (m, 4H). 13C NMR (101 MHz, DMSO-d6): δ 165.70, 158.70, 151.55, 150.71, 133.85, 129.99, 124.26, 121.70, 121.44, 121.05, 119.15, 118.49, 116.6, 63.84, 46.44, 25.24. LCMS: m/z: 441.0 (M + H)+.
N-(2-((6-(piperidin-1-yl)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (8a). 1H NMR (400 MHz, DMSO-d6): δ 8.79 (t, J = 5.60 Hz, 1H), 7.96 (d, J = 8.40 Hz, 2H), 7.78 (s, 1H), 7.46 (d, J = 8.40 Hz, 2H), 7.40 (s, 1H), 4.38 (t, J = 5.60 Hz, 2H), 3.64 (q, J = 5.60 Hz, 2H), 3.53–3.50 (m, 4H), 1.59–1.52 (m, 6H). 13C NMR (101 MHz, DMSO-d6):δ 165.70, 158.24, 153.32, 150.72, 133.84, 130.02, 121.84, 121.07, 119.54, 63.93, 45.45, 25.29, 24.52. LCMS: m/z: 411.2 (M + H)+.
N-(2-((6-morpholinopyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (9a). 1H NMR (400 MHz, DMSO-d6):δ 8.79 (t, J = 5.60 Hz, 1H), 7.97–7.93 (m, 2H), 7.79 (s, 1H), 7.51 (s, 1H), 7.46 (d, J = 8.00 Hz, 2H), 4.42–4.39 (m, 2H), 3.67–3.63 (m, 6H), 3.48–3.45 (m, 4H), 13C NMR (101 MHz, DMSO-d6): δ 165.71, 158.23, 153.40, 150.3, 133.83, 130.01, 121.81, 121.70, 121.08, 119.15 66.16, 64.02, 44.81. LCMS: m/z: 413.0 (M + H)+.
N-(2-((6-(4-cyanopiperidin-1-yl)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (10a). 1H NMR (400 MHz, DMSO-d6): δ 8.79 (t, J = 5.60 Hz, 1H), 7.97–7.94 (m, 2H), 7.83 (s, 1H), 7.48–7.45 (m, 3H), 4.39 (t, J = 5.60 Hz, 2H), 3.84–3.79 (m, 2H), 3.67–3.62 (m, 2H), 3.39–3.36 (m, 2H), 3.15–3.10 (m, 1H), 1.95–1.90 (m, 2H), 1.74–1.66 (m, 2H). 13C NMR (101 MHz, DMSO-d6): 165.72, 158.21, 152.90, 150.74, 133.82, 130.00, 122.46, 122.04, 121.71, 121.05, 120.56, 119.15, 64.03, 43.00, 27.83, 25.88. LCMS: m/z: 436.0 (M + H)+.
N-(2-((6-(4,4-difluoropiperidin-1-yl)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (11a). 1H NMR (400 MHz, DMSO-d6): 1H-NMR (400 MHz, DMSO-d6): δ 8.79 (t, J = 5.60 Hz, 1H), 7.96–7.92 (m, 3H), 7.52 (s, 1H), 7.46 (d, J = 8.00 Hz, 2H), 4.41 (t, J = 5.60 Hz, 2H), 3.70–3.62 (m, 6H), 2.03–1.95 (m, 4H). LCMS: m/z: 447.2 (M + H)+.
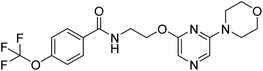
N-(2-((6-(1,1-dioxidothiomorpholino)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (12a). 1H NMR (400 MHz, DMSO-d6): δ 8.80 (t, J = 5.60 Hz, 1H), 7.99–7.95 (m, 3H), 7.58 (s, 1H), 7.47 (d, J = 8.00 Hz, 2H), 4.40 (t, J = 5.60 Hz, 2H), 4.06 (s, 4H), 3.67–3.63 (m, 2H), 3.15 (s, 4H). LCMS: m/z: 461.0 (M + H)+.
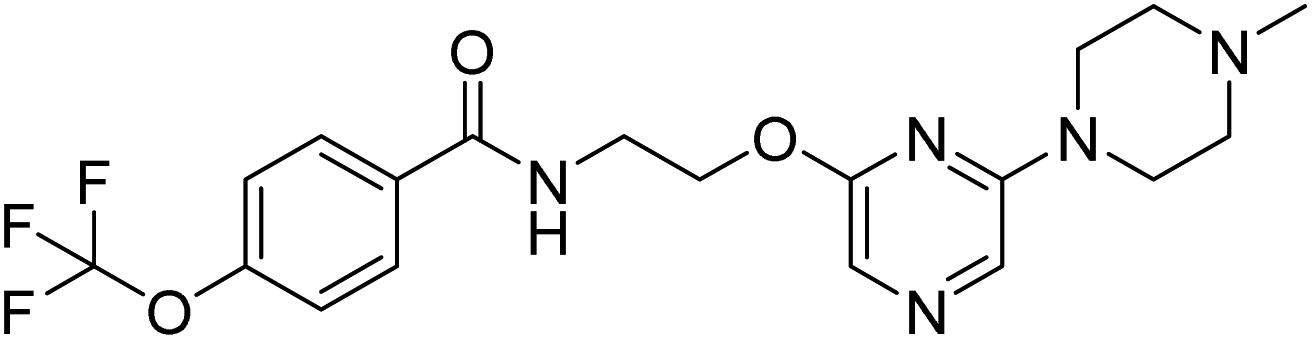
N-(2-((6-(4-methyl piperazine-1-yl)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (13a). 1H NMR (400 MHz, DMSO-d6): δ 8.79 (t, J = 5.20 Hz, 1H), 7.95 (d, J = 8.80 Hz, 2H), 7.79 (s, 1H), 7.47–7.45 (m, 3H), 4.40 (t, J = 5.60 Hz, 2H), 3.64 (q, J = 5.60 Hz, 2H), 3.49 (t, J = 4.80 Hz, 4H), 2.35 (t, J = 4.80 Hz, 4H), 0.19 (t, J = Hz, 3H). 13C NMR (101 MHz, DMSO-d6): 165.72, 158.22, 153.31, 150.72, 133.84, 130.01, 121.87, 121.71, 121.06, 120.50, 119.15, 63.97, 54.54, 46.23, 44.40. LCMS: m/z: 426.2 (M + H)+.
General method for synthesis of N-(2-((6-(cyclobutylamino)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide derivatives (14a to 16a)
To a stirred solution of N-(2-((6-chloropyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (1 eq.) in DMSO (w/v 20%) was added 1°-amines (14a to 16a) (3 eq.) at room temperature and stirred for 12 h at 120 °C. After completion of the reaction, the reaction mixture was diluted with EtOAc and washed with water and brine solution. The separated organic layer was concentrated and purified by column chromatography using a silica gel column to yield the desired compounds 14, 15, and 16.
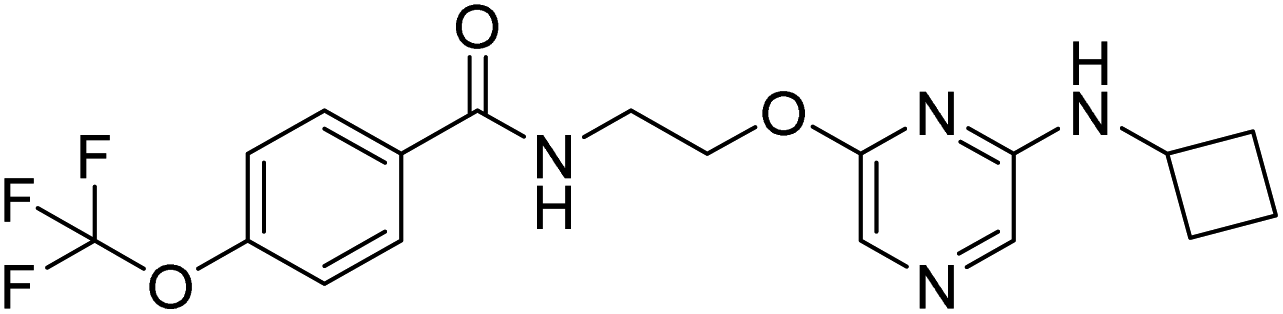
N-(2-((6-(cyclobutylamino)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (14a). 1H NMR (400 MHz, DMSO-d6): δ 8.79 (t, J = 5.60 Hz, 1H), 7.98 (d, J = 8.80 Hz, 2H), 7.47 (d, J = 8.00 Hz, 2H), 7.40 (s, 1H), 7.28 (s, 1H), 7.25 (d, J = 6.80 Hz, 1H), 4.36 (t, J = 5.60 Hz, 2H), 4.24–4.18 (m, 1H), 3.68–3.62 (m, 2H), 2.50–2.28 (m, 2H), 1.92–1.86 (m, 2H), 1.71–1.66 (m, 2H). 13C NMR (101 MHz, DMSO-d6): 165.71, 159.01, 152.94, 150.72, 133.86, 130.02, 123.16, 121.71, 121.08, 119.16, 118.34, 63.98, 46.16, 30.77, 15.30. LCMS: m/z: 397.2 (M + H)+.
N-(2-((6-(cyclopentyl amino)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (15a). 1H NMR (400 MHz, DMSO-d6): δ 8.79 (t, J = 4.80 Hz, 1H), 7.98 (d, J = 8.80 Hz, 2H), 7.47–7.44 (m, 3H), 7.25 (s, 1H), 6.99 (d, J = 6.40 Hz, 1H), 4.37 (t, J = 5.60 Hz, 2H), 4.06–4.01 (m, 1H), 3.66–3.62 (m, 2H), 1.93–1.90 (m, 2H), 1.67–1.62 (m, 2H), 1.59–1.50 (m, 2H), 1.44–1.41 (m, 2H). 13C NMR (101 MHz, DMSO-d6): 165.69, 159.02, 153.77, 150.72, 133.85, 130.01, 123.69, 121.71, 121.06, 119.16, 117.65, 63.97, 52.33, 32.84, 24.00. LCMS: m/z: 411.2 (M + H)+.
N-(2-((6-(cyclohexylamino)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (16a). 1H NMR (400 MHz, DMSO-d6):δ 8.79 (t, J = 5.20 Hz, 1H), 7.98–7.95 (m, 2H), 7.47–7.45 (m, 3H), 7.23 (s, 1H), 6.86 (d, J = 7.60 Hz, 1H), 4.37–4.34 (m, 2H), 3.66–3.57 (m, 3H), 3.66–3.57 (m, 2H), 1.72–1.68 (m, 2H), 1.59–1.56 (m, 1H), 1.36–1.20 (m, 6H); LCMS: m/z: 425.1 (M + H)+.
General method for the construction of N-(2-((6-((2-fluoro-4-(methyl sulfonyl)phenyl)amino)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide derivatives (17a–20a)
To a stirred solution of N-(2-((6-chloropyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (1 eq.) in 1,4-dioxane (w/v 20%) was added amine (17a–20a), Cs2CO3 (2.5 eq.) and degassed with argon for 2 min and to this resulting reaction mixture was added Brett-Phos Pd G3 (0.05 eq.) at room temperature and stirred for 12 h at 110 °C. After completion of the reaction, the reaction mixture was diluted with EtOAc and washed with water and brine solution. The separated organic layer was concentrated and purified by column chromatography using a silica-gel column to yield the desired compounds 17a to 20a.
N-(2-((6-((2-fluoro-4-(methyl sulfonyl)phenyl)amino)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (17a). 1H NMR (400 MHz, DMSO-d6):δ 9.66 (s, 1H), 8.81 (t, J = 5.60 Hz, 1H), 8.53 (t, J = 8.00 Hz, 1H), 8.12 (s, 1H), 7.99–7.91 (m, 2H), 7.81–7.66 (m, 3H), 7.45 (d, J = 8.00 Hz, 2H), 4.48 (t, J = 5.20 Hz, 2H), 3.72–3.68 (m, 2H), 3.23 (s, 3H). LCMS: m/z: 515.0 (M + H)+.
N-(2-((6-((3-fluoro-4-(methyl sulfonyl)phenyl)amino)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (18a). 1H NMR (400 MHz, DMSO-d6): δ 10.23 (s, 1H), 8.83 (t, J = 5.20 Hz, 1H), 7.97–7.95 (m, 1H), 7.89 (s, 1H), 7.80–7.76 (m, 3H), 7.57–7.54 (m, 1H), 7.46 (d, J = 8.00 Hz, 2H), 4.51 (t, J = 5.20 Hz, 2H), 3.75–3.71 (m, 2H), 3.26 (s, 3H). LCMS: m/z: 515.0 (M + H)+.
N-(2-((6-((4-(methyl sulfonyl)-3-(trifluoromethyl)phenyl)amino)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (19a). 1H NMR (400 MHz, DMSO-d6):δ 10.38 (s, 1H), 8.83 (t, J = 5.20 Hz, 1H), 8.39 (d, J = 2.00 Hz, 1H), 8.18–8.15 (m, 1H), 8.09–8.06 (m, 1H), 7.99–7.94 (m, 2H), 7.94 (s, 1H), 7.79 (s, 1H), 7.46–7.44 (m, 2H), 4.50 (t, J = 5.60 Hz, 2H), 3.74–0.70 (m, 2H), 3.23 (s, 3H). LCMS: m/z: 565.0 (M + H)+.
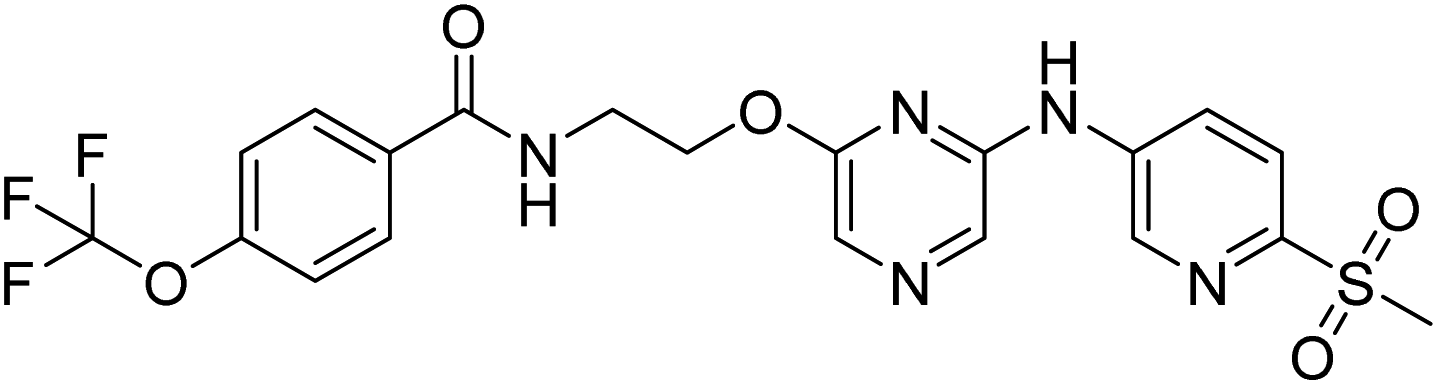
N-(2-((6-((6-(methyl sulfonyl)pyridin-3-yl)amino)pyrazin-2-yl)oxy)ethyl)-4-(trifluoromethoxy)benzamide (20a). 1H NMR (400 MHz, DMSO-d6): δ 10.24 (s, 1H), 8.92 (d, J = 2.40 Hz, 2H), 8.83 (t, J = 5.60 Hz, 1H), 8.43–8.41 (m, 1H), 7.97–7.91 (m, 5H), 7.77 (s, 1H), 7.45 (d, J = 8.00 Hz, 2H), 4.52 (t, J = 5.60 Hz, 2H), 3.74–3.70 (m, 2H), 3.22 (s, 3H). LCMS: m/z: 498.0 (M + H)+.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to acknowledge financial support from Syngeneic International Ltd, under which this research was pursued. We are thankful to Syngene International Ltd, Hyderabad, for providing spectral data and the Department of Biochemistry, Bharathiar University, Coimbatore, for their valuable contribution to doing biological studies.
References
- D. Choudhary, S. Garg, M. Kaur, H. Singh Sohal, D. Singh Malhi, L. Kaur, M. Verma, A. Sharma and V. Mutreja, Polycyclic Aromat. Compd., 2023, 43(5), 4512–4578 CrossRef CAS.
- A. Climova, E. Pivovarova, B. Rogalewicz, A. Raducka, M. Szczesio, I. Korona-Głowniak, A. Korga-Plewko, M. Iwan, K. Gobis and A. Czylkowska, Molecules, 2022, 27(11), 3467 CrossRef CAS PubMed.
- A. Hameed, S. T. Zehra, S. J. A. Shah, K. M. Khan, R. D. Alharthy, N. Furtmann, J. Bajorath, M. N. Tahir and J. Iqbal, Chem. Biol. Drug Des., 2015, 86, 1115–1120 CrossRef CAS PubMed.
- G.-Q. Chen, H.-Y. Guo, Z.-S. Quan, Q.-K. Shen, X. Li and T. Luan, Molecules, 2023, 28(21), 7440 CrossRef CAS PubMed.
- H. Wen, W. Dai, H. Huang, S.-L. Liu, J. Liu, Le-J. Huang, X.-H. Huang, J.-L. Zeng, Z.-W. Gan, Z.-Y. Zhang and J.-X. Lan, Eur. J. Med. Chem., 2023, 258, 115544 CrossRef PubMed.
- L. Peng-Cheng, H.-Q. Li, J. Sun, Z. Yang and H.-L. Zhu, Bioorg. Med. Chem., 2010, 18(13), 4606–4614 CrossRef PubMed.
- M. L. N. Dubuisson and M.-B. Rees, Med. Chem., 2004, 4(15), 421–435 CAS.
- K. Singh*, J. K. Gupta, K. Shah, S. Saha, M. Arockia Babu, S. Kumar and M. Agrawal, Curr. Org. Chem., 2024, 28(3), 176–184 CrossRef CAS.
- H.-M. Wu, K. Zhou, T. Wu and Y.-G. Cao, Chem. Biol. Drug Des., 2016, 88(3), 411–421 CrossRef CAS PubMed.
- K. Zurbonsen, A. Michel, P.-A. Bonnet, L. Gannoun-Zaki, M.-N. Mathieu and C. Chevillard, Eur. J. Pharmacol., 1997, 320(2–3), 215–221 CrossRef CAS PubMed.
- J. Hu, S. Chen, R. Mao, C. Liao, H. Yang and J. Zhao, J. Inorg. Biochem., 2018, 186, 246–256 CrossRef CAS PubMed.
- R. Sahu, K. Shah, Y. Gautam and K. Sahu, Curr. Org. Chem., 2023, 27(23), 821–843 CrossRef CAS.
- H. Zhang, Y. Wang, P. Zhu, J. Liu, S. Xu, H. Yao, J. Jiang, W. Ye, X. Wu and J. Xu, Eur. J. Med. Chem., 2015, 97, 235–244 CrossRef CAS PubMed.
- O. Argyros, N. Lougiakis, E. Kouvari, A. Papafotika, C. P. Raptopoulou, V. Psycharis, S. Christoforidis, N. Pouli, P. Marakos and C. Tamvakopoulos, Eur. J. Med. Chem., 2017, 126, 954–968 CrossRef CAS PubMed.
- J.-H. Zhang, C.-D. Fan, Z. Bao-Xiang, D.-S. Shin, W.-L. Dong, Y.-S. Xie and J.-Y. Miao, Bioorg. Med. Chem., 2008, 16(24), 10165–10171 CrossRef CAS PubMed.
- S. Akhlaghi, A. Mostoufi, H. Kalantar and M. Fereidoonnezhad, Med. Chem. Res., 2022, 31, 580–593 CrossRef CAS.
- Zi-Q. Liu, Q. Zhang, Yu-L. Liu, X.-Q. Yu, R.-H. Chui, L.-L. Zhang, B. Zhao and L.-Y. Ma, Bioorg. Med. Chem., 2024, 111, 117847 CrossRef CAS PubMed.
- M. Tan Uygun, K. Amudi, İ. D. Turaçlı and N. Menges, Mol. Diversity, 2022, 26, 113–124 CrossRef PubMed.
- P. Parsonidis, M. Shaik, A. Panagiota Serafeim, I. Vlachou, V. Daikopoulou and I. Papasotiriou, Molecules, 2019, 24(23), 4389 CrossRef CAS PubMed.
- E. S. Tantawy, A. M. Amer, E. K. Mohamed, M. M. Abd Alla and M. S. Nafie, J. Mol. Struct., 2020, 1210, 128013 CrossRef CAS.
- J. Akhtar, A. Ahmed Khan, Z. Ali, R. Haider and M. Shahar Yar, Eur. J. Med. Chem., 2017, 125, 143–189 CrossRef CAS PubMed.
- M. Youssef Jaballah, R. Taha Serya and K. Abouzid, Drug Res., 2017, 67(03), 138–148 CrossRef PubMed.
- K. Fang, X.-H. Zhang, Y.-T. Han, G.-R. Wu, De-S. Cai, N.-N. Xue, W.-B. Guo, Yu-Q. Yang, M. Chen, X.-Y. Zhang, H. Wang, T. Ma and P.-L. Wang, Int. J. Mol. Sci., 2018, 19(10), 2994 CrossRef PubMed.
- A. Alsfouk, Future Med. Chem., 2024, 16(18), 1899–1921 CrossRef PubMed.
- Y.-D. Gong, Mi-S. Dong, S.-B. Lee, N. Kim, Mi-S. Bae and N.-S. Kang, Bioorg. Med. Chem., 2011, 19(18), 5639–5647 CrossRef CAS PubMed.
- K. Fang, X.-H. Zhang, Y.-T. Han, G.-R. Wu, De-S. Cai, N.-N. Xue, W.-B. Guo, Yu-Q. Yang, M. Chen, X.-Y. Zhang, H. Wang, T. Ma, P.-L. Wang and H.-M. Lei, Int. J. Mol. Sci., 2018, 19(10), 2994 CrossRef PubMed.
- F. A. López-Huerta, M. Teresa Ramírez-Apan, C. A. Méndez-Cuesta, A. Nieto-Camacho, S. Hernández-Ortega, E. K. P. Almeida-Aguirre, M. A. Cerbón and G. Delgado, Bioorg. Chem., 2022, 125, 105924 CrossRef PubMed.
- P. Bera, A. Aher, P. Brandao, S. K. Manna, I. Bhattacharyya, G. Mondal, A. Jana and A. S. Pulakesh Bera, New J. Chem., 2021, 45, 11999–12015 RSC.
- X. De Wang, T. Li, Y. Li, W. Hui Yuan and Y. Q. Zhao, Eur. J. Pharmacol., 2020, 881, 173211 CrossRef PubMed.
- S. Rostampour, F. Eslami, E. Babaei, H. Mostafavi and M. Mahdavi, Anti-Cancer Agents Med. Chem., 2024, 24(10), 203–212 CrossRef CAS PubMed.
- J. Hodoň, I. Frydrych, Z. Trhlíková, J. Pokorný, L. Borková, S. Benická, M. Vlk, B. Lišková, A. Kubíčková, M. Medvedíková, M. Pisár, J. Šarek, V. Das, A. Ligasová, K. Koberna, P. Džubák, M. Hajdúch and M. Urban, Eur. J. Med. Chem., 2022, 243, 114777 CrossRef PubMed.
- K. Zurbonsen, A. Michel, P. A. Bonnet, M. N. Mathieu and C. Chevillard, Gen. Pharmacol., 1999, 32(1), 135–141 CrossRef CAS PubMed.
- R. Khanam, R. Kumar, I. Iqbal Hejazi, S. Shahabuddin, R. Meena, V. Jayant, P. Kumar, A. R. Bhat and F. Athar, Apoptosis, 2018, 23, 113–131 CrossRef CAS PubMed.
- S. Behzad, E. Karim, M. Mosaddegh and H. Ali, Iran. J. Pharm. Res., 2016, 15(1), 311–322 CAS.
- R. Prabaharan, R. Ramesh, S. Kamatchi Thangavel and J. G. Małecki, ACS Omega, 2023, 8(13), 12584–12591 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 455–461 Search PubMed.
- S. K. Wahan, S. Sharma and P. A. Chawla, Med. Chem., 2023, 23(19), 700–718 CAS.
- N. W. Hassan, M. N. Saudi, Y. S. Abdel-Ghany, A. Ismail, P. A. Elzahhar, D. Sriram, R. Nassra, M. M. Abdel-Aziz and S. A. El-Hawash, Bioorg. Chem., 2020, 96, 103610 CrossRef PubMed.
- L. Semelkova, K. Konecna, P. Paterova, V. Kubicek, J. Kunes, L. Novakova, J. Marek, L. Naesens, M. Pesko, K. Kralova, M. Dolezal and J. Zitko, Molecules, 2015, 20(5), 8687–8711 CrossRef CAS PubMed.
- S. kumar, P. Dinesha, D. Udayakumar, V. Prakash Shetty and V. Kumar Deekshit, J. Mol. Struct., 2024, 1304, 137657 CrossRef CAS.
- A. A. Siddiki, S. Parmar, H. K. Chaudhari, S. SP Yadav and R. S. Chauhan, ChemistrySelect, 2024, 9(36), e202402487 CrossRef CAS.
- M. M. El-Zahed, M. A. Diab, A. Z. El-Sonbati, S. G. Nozha, H. R. Issa, M. A. El-Mogazy and Sh. M. Morgan, Mater. Sci. Eng., B, 2024, 299, 116998 CrossRef CAS.
- T. Bhat and T. J. Richmond, Discovery Studio Visualizer software, Dassault Systèmes BIOVIA, Version 9, 2021 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Dharavath Ravid and
Ajmeera Rameshe
*a,
Dharavath Ravid and
Ajmeera Rameshe