
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reimagining drug nanocarriers: clinical realities and smarter strategies for targeted drug delivery
Andrew George
Baker
 *ab and
Ljiljana
Fruk
*a
*ab and
Ljiljana
Fruk
*a
aDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Drive, Cambridge, CB3 0AS, UK. E-mail: agb59@cam.ac.uk
bDepartment of Pharmacology, University of Cambridge, Tennis Court Road, Cambridge CB2 1PD, UK
First published on 25th June 2025
Abstract
Recent advances in nanomedicine have significantly improved the delivery of small molecule therapies and biologics for cancer treatment, gene therapy, and vaccines, leading to better patient outcomes and improved standard of care. Yet, despite the promise of targeted drug delivery, clinical trials of targeted nanoparticle-based systems have frequently underperformed. In this opinion piece, we explore the persistent obstacles facing nanomedicine, analyse the shortcomings of targeted strategies, and propose a path forward. We argue that progress will require a re-examination of nanoparticle pharmacokinetics, optimisation of dosing regimens, and solutions to antigen depletion. These steps are critical for realising the full potential of nanomedicine.
Clinical success of untargeted nanoparticles
In cancer treatment, small-molecule chemotherapeutic agents were designed to target rapidly dividing cells. However, many of these drugs are rapidly cleared from the body, requiring prolonged administration to maintain therapeutic levels. This not only complicates treatment regimens but also increases systemic toxicity. Nanomaterials have significantly improved these outcomes by enabling encapsulation of drugs within carriers that extend circulation time and reduce side effects, thereby maintaining therapeutic concentrations for longer.A prime example is Doxil (Caelyx), a liposomal formulation of doxorubicin, which increases the drug's circulation time by nearly 500-fold.1 Although Doxil did not dramatically improve efficacy and median overall survival compared to free doxorubicin, its substantially reduced cardiotoxicity resulted in improved patient outcomes and benefits in both progression-free and overall survival.1–3 A similar success story is Vyxeos, a liposomal formulation that co-delivers cytarabine and daunorubicin in a specific 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, enabling coordinated pharmacokinetics, prolonging elimination time, and simplifying dosing compared to administering the drugs separately. Unlike Doxil, Vyxeos demonstrated a clear benefit in overall survival over standard therapy.4,5
:1 ratio, enabling coordinated pharmacokinetics, prolonging elimination time, and simplifying dosing compared to administering the drugs separately. Unlike Doxil, Vyxeos demonstrated a clear benefit in overall survival over standard therapy.4,5
Beyond small-molecules, nanoparticles have also transformed the delivery of biologics, particularly RNA-based therapies. For example, Onpattro (patisiran), a lipid nanoparticle-encapsulated siRNA, is used to treat hereditary transthyretin induced amyloidosis (hATTR), a progressive condition characterized by the buildup of abnormal transthyretin protein deposition.6,7 The lipid nanoparticles enable siRNA delivery to the liver while protecting the fragile siRNA payload from degradation by endogenous RNAses.
This early work on siRNA delivery paved the way for mRNA vaccines such as Spikevax (Moderna) and Comirnaty (Pfizer-BioNTech), developed in response to COVID-19.8–10 These vaccines use lipid nanoparticles to deliver mRNA into cells following intramuscular injection, enabling viral protein expression and a robust immune response. However, none of these success stories were targeted therapies in the traditional sense but relied on lipid/liposomal nanoparticles as delivery vehicles to protect and transport the sensitive payloads effectively, prolong circulation and minimise the side effects.
As we enter the era of precision medicine, accelerated by AI-driven diagnostics and the growing use of biologics, targeted drug delivery will play a critical role in cancer therapy and beyond. By directing therapeutics specifically to cancer cells through molecular recognition, or by engineering nanomaterials to respond to features of the tumour microenvironment, targeted delivery offers a path to overcome many of the limitation of conventional therapies, including off-target toxicity and suboptimal efficacy.
The clinical reality of targeted nanoparticles
In recent years, biologically targeted therapies made a significant step towards precision cancer treatment. Strategies such as immune checkpoint inhibitors, antibody drug conjugates (ADCs), bispecific T cell engagers, and chimeric antigen receptor (CAR)-T cells all rely on specific cell surface targets and high-affinity binding to a defined molecular epitope.11–14 While these approaches have markedly improved outcomes for many patients, they still face significant challenges, including limited delivery efficiency, antigen depletion, off-target leakage of cytotoxic payloads in ADCs, and severe immune responses such as cytokine storms in CAR-T therapies.12,13,15,16Despite the clinical success of biologically targeted therapies, nanoparticle-based targeted delivery systems have yet to achieve comparable breakthroughs. Many formulations have failed to meet key clinical endpoints, impeding regulatory approval.
For instance, MM-302 from Merrimack, a HER2-targeted antibody–liposomal doxorubicin, showed promising Phase 1 results.17 However, in a Phase II trial, it failed to significantly improve progression-free survival (PFS) compared to standard chemotherapy combined with trastuzumab.18
Another example is BIND-014, developed by Bind Therapeutics, a docetaxel-loaded poly-lactic acid (PLA) nanoparticle decorated with the small-molecule targeting ligand ACUPA, which binds to prostate-specific antigen (PSA). This system initially demonstrated a favourable safety profile and a distinct pharmacokinetic profile in Phase I trials.19 Although some patients experienced reductions in PSA levels and circulating tumour cells, Phase II results showed no meaningful improvements in survival or off-target effects compared to free docetaxel.20,21
Similarly, targeting Cornell Dots (C’ Dots), sub 8 nm silica nanoparticles with customizable surface developed by Elucida Oncology did not progress into the Phase II trials. First generation of C Dots were functionalised 124I for PET imaging and cyclo-(Arg-Gly-Asp-Tyr) (cRGDY) peptide for integrin targeting.22 These targeted PET-contrast agents demonstrated excellent renal clearance and tissue distribution patterns correlated with integrin expression. Building on this, a second-generation C’ Dots formulation, ELU001, was engineered to deliver a topoisomerase-1 inhibitor exatecan through cathepsin-B cleavable linker. While Phase I trials showed a favourable safety profile and early signs of efficacy,23 subsequent trials were halted due to lack of funding.24
Why targeting strategies often fail?
Despite decades of research, no active targeting strategy using nanoparticles, whether through peptides, antibodies, or small molecule ligands, has achieved clinical success (Fig. 1). Recent meta-analysis of over 200 preclinical studies in murine models showed no significant improvement in therapeutic delivery with targeted nanoparticles compared to untargeted counterparts.25 These findings suggest that the addition of targeting ligands does not enhance tumour accumulation or therapeutic efficacy in vivo (Fig. 1). | ||
| Fig. 1 Targeting strategies used in clinical trials that have failed, such as peptides, antibodies and small molecules. As well as those that have led to clinical approval such as unmodified, charged, and apolipoprotein E (ApoE) acquired in the body. | ||
Nevertheless, the academic focus remains heavily skewed toward developing increasingly complex nanoparticle systems, often incorporating multiple targeting ligands and antibodies. These designs significantly raise production complexity and cost, yet offer minimal, if any, benefit in terms of clinical translation.26 This is in stark contrast to biologically targeted therapies such as checkpoint inhibitors or ADCs, where the targeting directly correlates with therapeutic success.
One fundamental distinction may lie in the pharmacokinetics (PK) of the most common targeting nanoparticles: nanoparticle-antibody conjugates. While antibodies alone typically exhibit long circulation times, ranging from days to weeks,27,28 conjugating them to nanoparticles often reduces circulation to 20–40 hours.22,29 A shortened circulation time likely reflects modified elimination routes, and significantly reduces the likelihood of effective tumour targeting thus compromising the therapeutic impact.
Moreover, in the context of ADCs, it is well established that the site of antibody–drug conjugation can dramatically alter pharmacokinetics and biodistribution.30,31 However, this level of control is rarely applied to nanoparticle-targeted formulations. Common conjugation strategies, such as amide coupling, result in random attachment through lysine residues, leading to heterogenous distribution of antibody's anchoring sites. Thus, more refined, site-specific conjugation approaches, and subsequent studies mimicking those used in advanced biologics are needed.
Perhaps most critically, nanoparticle targeting may unintentionally trigger antigen depletion, a phenomenon already observed in therapies like ADCs and CAR-T cells.12,32 Recent studies in nanoparticle-mediated protein degradation suggest that nanoparticles can accelerate antigen loss compared to free antibodies.33–35 If this holds true, nanoparticle formulations could be actively diminishing their own targets, limiting clinical efficacy and long-term therapeutic outcomes. This mechanism warrants deeper investigation, as it may represent a key obstacle to successful clinical translation.
The power of passive targeting: when nanoparticles work without targeting ligands
Despite the underwhelming clinical performance of actively targeted nanoparticles, there are cases where nanoparticles achieve organ- or cell-specific delivery through a non-active, endogenous mechanism.One of the most prominent examples is the natural tropism of lipid nanoparticles, which consistently deliver their cargo to the liver and the liver cells.36,37 This targeting is primarily mediated by the spontaneous adsorption of apolipoprotein E (ApoE) onto the nanoparticle surface, forming a protein corona. ApoE then facilitates receptor-mediated uptake in the liver by binding to LDL or APoE receptors.7 Despite this success, attempts to redirect lipid nanoparticles to other organs have proven challenging. To date, the overwhelming majority of nanoparticle-based therapies in clinical development still default to hepatic delivery.36
COVID-19 vaccines presented a compelling case for immune targeting, with some reports attributing dendritic cell uptake to a combination of nanoparticle size, surface charge, and the presence of ionizable lipids.8 However, we must be careful not to overstate this point. These nanoparticles are delivered intramuscularly, not intravenously, which bypasses many of the physiological barriers that hinder systemic targeting. In this context, the interaction with dendritic cells might be more a function of anatomical proximity and formulation chemistry than true targeting.
Future directions
Most nanoparticles that have made it to the clinic are chemically simple and biologically passive. They are not functionalised with targeting ligands or engineered for complex biodistribution. Instead, they succeed by leveraging material properties and administration routes that align with biological reality, without overly complex designs.So, where do we go from here? It is time to rethink our focus on active targeting. Instead of chasing ever-more elaborate conjugation strategies, we need to be honest about the likelihood of clinical success and acknowledge the shortfalls of current strategies. First, we must improve our understanding of nanoparticle pharmacokinetics and their biological interactions (Fig. 2). Insights from ADCs have shown that the addition of chemotherapeutic payloads, can dramatically alter circulation times. Critically, and in contrast to free antibodies, nanoparticle-antibody conjugates often exhibit significantly shorter half-lives than expected. Investigating how these changes occur, both with or without antibody attachment, could uncover critical insights and lead to improved therapeutic efficacy.
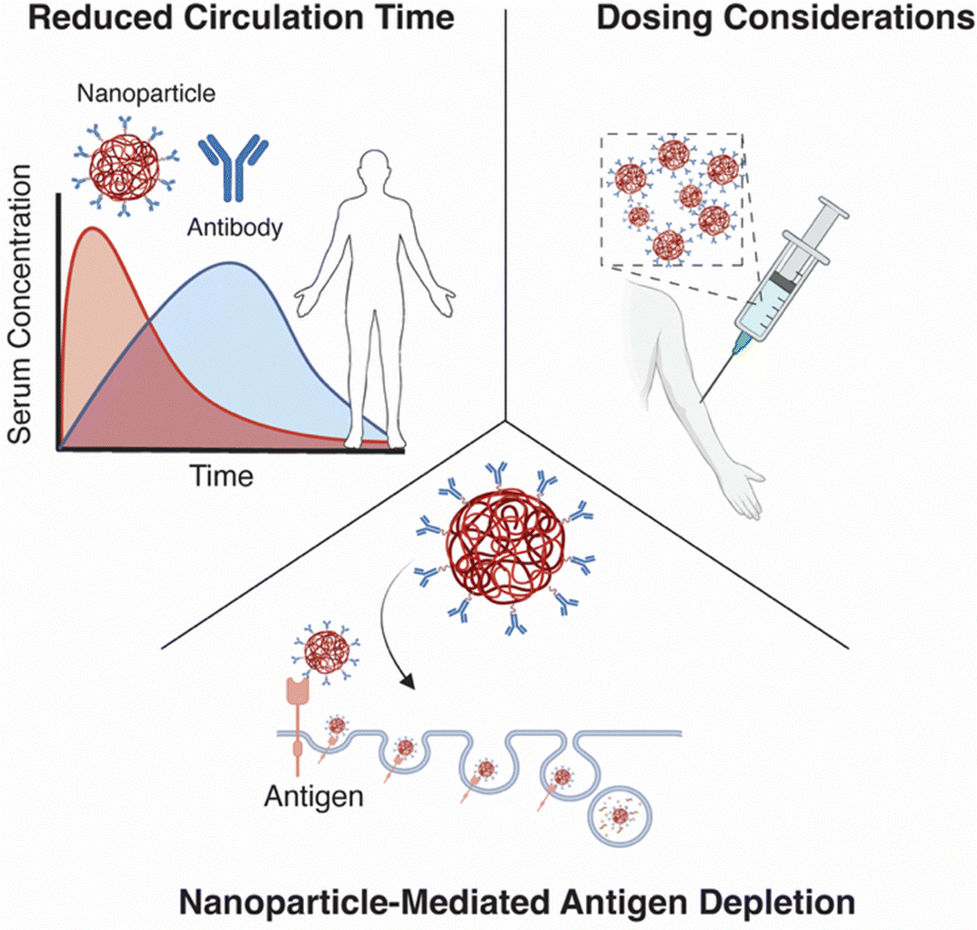 | ||
| Fig. 2 The major challenges of nanoparticles entering the clinic. Changes in biodistribution as well as reduced elimination times of nanoparticle antibody formulations versus antibodies alone. The calculation of dose and the decision of which dose to use. Finally, the mechanism of nanoparticle mediated targeted protein degradation, and resultant antigen depletion. | ||
Secondly, nanoparticle dosing and data reporting, particularly around dosage, remains inadequate. While this issue has been acknowledged in the field, we still lack a standardised unit analogous to the “mole” in molecular chemistry to quantify number of nanoparticles delivered. A study from Warren Chan's group highlights the critical importance of this: they found that not only the nanoparticles type, but the absolute number of nanoparticle administered significantly influences in vivo efficacy.38 Using untargeted Doxil formulations, they demonstrated that co-injection of both therapeutic and ‘dummy’ (empty) liposomes increased the delivery to tumours. They hypothesised that the dummy particles saturated the liver's clearance mechanisms, allowing more therapeutic nanoparticles to bypass filtration and accumulate at tumour site. This points to a threshold phenomenon; a minimal particle counts necessary to overcome hepatic clearance and enable meaningful tumour targeting, which they defined as 1 trillion particles.
Finally, we need to consider antigen depletion. A major reason for the clinical failure of targeted nanoparticles may lie in a well-documented phenomenon from the biologic's world: each dose depletes the very antigen it seeks to target. This has been observed in case of monoclonal antibodies, ADCs and CAR-T therapies, yet remains underexplored in the context of nanoparticle delivery. And yet, evidence from the emerging field of nanoparticle-mediated protein degradation suggests this mechanism is not only possible, it is occurring.34
If nanoparticles are indeed accelerating antigen clearance, then we must ask: how do cells respond? Do they upregulate alternate targets? Is there a time-dependant recovery of the original antigen? If so, timing and dosing intervals may need to be optimised for targeted nanoparticles to take into account the target availability. Furthermore, in the context of targeted drug delivery this would mean that the nanoparticle is not just acting as a passive carrier but actively altering the protein landscape of the cell it targets.
By addressing these issues, improving pharmacokinetic understanding, standardising dosage metrics, and investigating the consequences of antigen depletion, we may finally move past conventional thinking about targeting and toward a more evidence-based framework. It is entirely possible that the future of nanoparticle delivery does not lie in complex ligand engineering, but in smarter, biology-aware design choices. Perhaps, active targeting, as we currently define it, is not the path forward at all.
Author contributions
A. G. B. and L. F. conceptualization, writing the first draft, and writing – review & editing.Conflicts of interest
A. G. B. and L. F. are co-founders of Senesys Bio, a company improving the formulation of senolytics.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
We would like to acknowledge all members of the Fruk lab past and present who have contributed to conversations of nanoparticle targeting over the past years. All figures were created on BioRender.com as well as Adobe Illustrator. We would also like to acknowledge EPSRC funding EP/W035049/1 and EP/S009000/1 supporting our work on targeting nanocarriers.References
- A. Gabizon, R. Catane, B. Uziely, B. Kaufman, T. Safra, R. Cohen, F. Martin, A. Huang and Y. Barenholz, Cancer Res., 1994, 54, 987–992 CAS.
- E. A. Forssen and Z. A. Tökès, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 1873–1877 CrossRef CAS PubMed.
- B. Uziely, S. Jeffers, R. Isacson, K. Kutsch, D. Wei-Tsao, Z. Yehoshua, E. Libson, F. M. Muggia and A. Gabizon, J. Clin. Oncol., 1995, 13, 1777–1785 CrossRef CAS PubMed.
- J. E. Lancet, G. L. Uy, J. E. Cortes, L. F. Newell, T. L. Lin, E. K. Ritchie, R. K. Stuart, S. A. Strickland, D. Hogge, S. R. Solomon, R. M. Stone, D. L. Bixby, J. E. Kolitz, G. J. Schiller, M. J. Wieduwilt, D. H. Ryan, A. Hoering, K. Banerjee, M. Chiarella, A. C. Louie and B. C. Medeiros, J. Clin. Oncol., 2018, 36, 2684–2692 CrossRef CAS PubMed.
- J. E. Lancet, G. L. Uy, L. F. Newell, T. L. Lin, E. K. Ritchie, R. K. Stuart, S. A. Strickland, D. Hogge, S. R. Solomon, D. L. Bixby, J. E. Kolitz, G. J. Schiller, M. J. Wieduwilt, D. H. Ryan, S. Faderl and J. E. Cortes, Lancet Haematol., 2021, 8, e481–e491 CrossRef CAS PubMed.
- D. Adams, A. Gonzalez-Duarte, W. D. O’Riordan, C.-C. Yang, M. Ueda, A. V. Kristen, I. Tournev, H. H. Schmidt, T. Coelho, J. L. Berk, K.-P. Lin, G. Vita, S. Attarian, V. Planté-Bordeneuve, M. M. Mezei, J. M. Campistol, J. Buades, T. H. Brannagan, B. J. Kim, J. Oh, Y. Parman, Y. Sekijima, P. N. Hawkins, S. D. Solomon, M. Polydefkis, P. J. Dyck, P. J. Gandhi, S. Goyal, J. Chen, A. L. Strahs, S. V. Nochur, M. T. Sweetser, P. P. Garg, A. K. Vaishnaw, J. A. Gollob and O. B. Suhr, N. Engl. J. Med., 2018, 379, 11–21 CrossRef CAS PubMed.
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. J. Hope, T. D. Madden, B. L. Mui, S. C. Semple, Y. K. Tam, M. Ciufolini, D. Witzigmann, J. A. Kulkarni, R. van der Meel and P. R. Cullis, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed.
- N. Chaudhary, D. Weissman and K. A. Whitehead, Nat. Rev. Drug Discovery, 2021, 20, 817–838 CrossRef CAS PubMed.
- L. R. Baden, H. M. El Sahly, B. Essink, K. Kotloff, S. Frey, R. Novak, D. Diemert, S. A. Spector, N. Rouphael, C. B. Creech, J. McGettigan, S. Khetan, N. Segall, J. Solis, A. Brosz, C. Fierro, H. Schwartz, K. Neuzil, L. Corey, P. Gilbert, H. Janes, D. Follmann, M. Marovich, J. Mascola, L. Polakowski, J. Ledgerwood, B. S. Graham, H. Bennett, R. Pajon, C. Knightly, B. Leav, W. Deng, H. Zhou, S. Han, M. Ivarsson, J. Miller and T. Zaks, N. Engl. J. Med., 2021, 384, 403–416 CrossRef CAS PubMed.
- F. P. Polack, S. J. Thomas, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, J. L. Perez, G. Pérez Marc, E. D. Moreira, C. Zerbini, R. Bailey, K. A. Swanson, S. Roychoudhury, K. Koury, P. Li, W. V. Kalina, D. Cooper, R. W. Frenck, L. L. Hammitt, Ö. Türeci, H. Nell, A. Schaefer, S. Ünal, D. B. Tresnan, S. Mather, P. R. Dormitzer, U. Şahin, K. U. Jansen and W. C. Gruber, N. Engl. J. Med., 2020, 383, 2603–2615 CrossRef CAS PubMed.
- C. Dumontet, J. M. Reichert, P. D. Senter, J. M. Lambert and A. Beck, Nat. Rev. Drug Discovery, 2023, 22, 641–661 CrossRef CAS PubMed.
- M.-E. Goebeler and R. C. Bargou, Nat. Rev. Clin. Oncol., 2020, 17, 418–434 CrossRef PubMed.
- R. C. Sterner and R. M. Sterner, Blood Cancer J., 2021, 11, 69 CrossRef PubMed.
- J. A. Wells and K. Kumru, Nat. Rev. Drug Discovery, 2024, 23, 126–140 CrossRef PubMed.
- R. Chen, J. Hou, E. Newman, Y. Kim, C. Donohue, X. Liu, S. H. Thomas, S. J. Forman and S. E. Kane, Mol. Cancer Ther., 2015, 14, 1376–1384 CrossRef CAS PubMed.
- F. Loganzo, M. Sung and H.-P. Gerber, Mol. Cancer Ther., 2016, 15, 2825–2834 CrossRef CAS PubMed.
- P. Munster, I. E. Krop, P. LoRusso, C. Ma, B. A. Siegel, A. F. Shields, I. Molnár, T. J. Wickham, J. Reynolds, K. Campbell, B. S. Hendriks, B. S. Adiwijaya, E. Geretti, V. Moyo and K. D. Miller, Br. J. Cancer, 2018, 119, 1086–1093 CrossRef CAS PubMed.
- K. Miller, J. Cortes, S. A. Hurvitz, I. E. Krop, D. Tripathy, S. Verma, K. Riahi, J. G. Reynolds, T. J. Wickham, I. Molnar and D. A. Yardley, BMC Cancer, 2016, 16, 352 CrossRef PubMed.
- D. D. Von Hoff, M. M. Mita, R. K. Ramanathan, G. J. Weiss, A. C. Mita, P. M. LoRusso, H. A. Burris, L. L. Hart, S. C. Low, D. M. Parsons, S. E. Zale, J. M. Summa, H. Youssoufian and J. C. Sachdev, Clin. Cancer Res., 2016, 22, 3157–3163 CrossRef CAS PubMed.
- K. A. Autio, R. Dreicer, J. Anderson, J. A. Garcia, A. Alva, L. L. Hart, M. I. Milowsky, E. M. Posadas, C. J. Ryan, R. P. Graf, R. Dittamore, N. A. Schreiber, J. M. Summa, H. Youssoufian, M. J. Morris and H. I. Scher, JAMA Oncol., 2018, 4, 1344 CrossRef PubMed.
- K. A. Autio, J. A. Garcia, A. S. Alva, L. L. Hart, M. I. Milowsky, E. M. Posadas, C. J. Ryan, J. M. Summa, H. Youssoufian, H. I. Scher and R. Dreicer, J. Clin. Oncol., 2016, 34, 233 Search PubMed.
- E. Phillips, O. Penate-Medina, P. B. Zanzonico, R. D. Carvajal, P. Mohan, Y. Ye, J. Humm, M. Gönen, H. Kalaigian, H. Schöder, H. W. Strauss, S. M. Larson, U. Wiesner and M. S. Bradbury, Sci. Transl. Med., 2014, 6, 260ra149 Search PubMed.
- W. W. Ma, D. Orr, C. A. Perez, Y. R. Murciano-Goroff, E. P. Hamilton, Y. Zhao, C. Anders, G. P. Adams, C. Reddick, H. Wroe McClintock, E. Bayever and A. W. Tolcher, Ann. Oncol., 2023, 34, S482–S483 CrossRef.
- W. W. Ma, A. W. Tolcher, C. A. Perez, A. E. Wahner Hendrickson, D. W. Orr, Y. R. Murciano-Goroff, C. K. Anders, E. P. Hamilton, E. M. Swisher, C. A. Mathews, B. Bashir, L. A. Rojas-Espaillat, G. Colon-Otero, G. P. Adams, C. W. Reddick and E. Bayever, J. Clin. Oncol., 2024, 42, TPS3173 CrossRef.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338(6109), 903–910 CrossRef CAS PubMed.
- A. Paci, A. Desnoyer, J. Delahousse, L. Blondel, C. Maritaz, N. Chaput, O. Mir and S. Broutin, Eur. J. Cancer, 2020, 128, 107–118 CrossRef CAS PubMed.
- A. Deslandes, MAbs, 2014, 6, 859–870 CrossRef PubMed.
- H. Lee, A. F. Shields, B. A. Siegel, K. D. Miller, I. Krop, C. X. Ma, P. M. LoRusso, P. N. Munster, K. Campbell, D. F. Gaddy, S. C. Leonard, E. Geretti, S. J. Blocker, D. B. Kirpotin, V. Moyo, T. J. Wickham and B. S. Hendriks, Clin. Cancer Res., 2017, 23, 4190–4202 CrossRef CAS PubMed.
- P. Strop, S.-H. Liu, M. Dorywalska, K. Delaria, R. G. Dushin, T.-T. Tran, W.-H. Ho, S. Farias, M. G. Casas, Y. Abdiche, D. Zhou, R. Chandrasekaran, C. Samain, C. Loo, A. Rossi, M. Rickert, S. Krimm, T. Wong, S. M. Chin, J. Yu, J. Dilley, J. Chaparro-Riggers, G. F. Filzen, C. J. O’Donnell, F. Wang, J. S. Myers, J. Pons, D. L. Shelton and A. Rajpal, Chem. Biol., 2013, 20, 161–167 CrossRef CAS PubMed.
- G. Ahn, S. M. Banik, C. L. Miller, N. M. Riley, J. R. Cochran and C. R. Bertozzi, Nat. Chem. Biol., 2021, 17, 937–946 CrossRef CAS PubMed.
- H. Lin, X. Yang, S. Ye, L. Huang and W. Mu, Biomed. Pharmacother., 2024, 178, 117252 CrossRef CAS PubMed.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- A. G. Baker, A. P. T. Ho, L. S. Itzhaki and L. Fruk, Angew. Chem., Int. Ed., 2025, e202503958 Search PubMed.
- Y. Liu, R. Liu, J. Dong, X. Xia, H. Yang, S. Wei, L. Fan, M. Fang, Y. Zou, M. Zheng, K. W. Leong and B. Shi, Nat. Nanotechnol., 2025, 20, 296–302 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2021, 6, e10246 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143 CrossRef PubMed.
- B. Ouyang, W. Poon, Y.-N. Zhang, Z. P. Lin, B. R. Kingston, A. J. Tavares, Y. Zhang, J. Chen, M. S. Valic, A. M. Syed, P. MacMillan, J. Couture-Senécal, G. Zheng and W. C. W. Chan, Nat. Mater., 2020, 19, 1362–1371 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |