
Stretching the future: strategies and emerging trends in stretchable organic photovoltaic materials†
Jingyu
Zuo
a,
Dexia
Han
a,
Huifeng
Yao
 b,
Vakhobjon
Kuvondikov
c and
Long
Ye
*ad
b,
Vakhobjon
Kuvondikov
c and
Long
Ye
*ad
aSchool of Materials Science and Engineering, State Key Laboratory of Advanced Materials for Intelligent Sensing, Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Key Laboratory of Organic Integrated Circuits, Ministry of Education, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin University, Tianjin, 300350, China. E-mail: yelong@tju.edu.cn
bSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing, 211189, China
cInstitute of Ion-Plasma and Laser Technologies, Uzbekistan Academy of Sciences, Tashkent 100125, Uzbekistan
dHubei Longzhong Laboratory, Xiangyang 441000, China
First published on 12th May 2025
Abstract
As the demand for wearable electronics continues to rise, the development and fabrication of intrinsically stretchable organic photovoltaics have become a significant area of research within the energy sector. To attain the necessary stretchability, researchers have invested substantial effort in improving the performance of photoactive materials. This feature article offers a concise overview and summarizes key research findings related to the design, optimization, and applications of stretchable photovoltaic materials for highly stretchable organic photovoltaics, particularly highlighting exciting progress since 2022.
Broader contextStretchable photovoltaics, as an emerging solar technology, offer significant advantages over traditional rigid cells, particularly in terms of their ability to meet the demand for thin, lightweight devices and convenience. The increasing global demand for wearable and flexible electronics has spurred significant interest in stretchable organic photovoltaics (s-OPVs) as a promising energy-harvesting solution. These lightweight, mechanically resilient, and solution-processable solar cells have immense potential for self-powered wearable devices, soft robotics, and bio-integrated electronics. However, achieving high efficiency while maintaining mechanical durability remains a major challenge in s-OPV development. Recent research efforts have focused on two key approaches: tailoring the chemical structure of photoactive materials to enhance intrinsic stretchability and optimizing thin-film morphology through advanced processing/casting protocols. Additionally, addressing environmental stability concerns is crucial for real-world applications. This feature article provides a comprehensive overview of recent advancements in stretchable organic photovoltaic materials, particularly highlighting progress since 2022. By examining innovative material design strategies, processing methodologies, and stability improvements, this work aims to guide future research directions toward the commercialization of s-OPVs, ultimately contributing to sustainable and flexible energy solutions. |
1. Introduction
The global demand for reducing carbon emissions, lowering energy costs and decreasing dependence on traditional energy sources is still on the rise. Solar energy, an inexhaustible renewable energy, will continue to be a prominent spot for research in the field of clean energy. Organic photovoltaics (OPVs) exhibit several advantages, including light weight, high transparency, large-area manufacturing, solution processing and mechanical flexibility.1–3 Silicon-based cells inherently suffer from brittleness, which greatly limits their application in flexible and wearable electronics. For developing wearable applications that conform to the human body, the materials used in OPVs must withstand over 40% strain.4 Therefore, there is a critical need to develop organic photovoltaic cells with mechanical flexibility and intrinsic stretchability. Due to its mechanical properties, the organic active layer makes OPVs a highly promising candidate for wearable electronic applications.5The research on the mechanical properties of OPVs can be summarized into three stages: rigid OPVs, flexible OPVs, and stretchable OPVs (hereafter abbreviated as s-OPVs) (Fig. 1a–c).6 Rigid OPVs have minimal stretchability, while flexible OPVs can tolerate some degree of vertical deformation but exhibit low stress resistance under horizontal forces, typically below 10%. This limitation often leads to structural damage under large strains. In contrast, s-OPVs, a recent advancement in the field of deformable devices, can undergo significant deformation from both vertical and horizontal forces without compromising the device structure or photovoltaic performance.6 s-OPVs are able to withstand higher stress and accommodate a wide range of deformations, allowing them to be integrated onto arbitrary surfaces. This enhanced stretchability enables designers to incorporate solar cells into wearable devices more freely, leading to innovative and personalized products. The manufacturing of OPVs has progressed from the initial stage of rigid OPVs to the second stage of flexible OPVs. Researchers are now focusing on improving the mechanical properties of OPVs to ensure their performance meets the demands of portable electronic devices. s-OPVs offer solutions for applications that rigid or flexible OPVs cannot address, opening up new possibilities for innovation in the field. In contrast to flexible OPVs, s-OPVs require the stretchability of all components/layers of the device. The flexibility of OPVs is constrained by the mechanical properties of every layer. The substrate, serving as the foundation, typically requires good mechanical stability and flexibility to support deformation.6 Electron transport layers and the anode/cathode layers are usually located at the bottom or top of the OPVs, and their compatibility with the photoactive layer and other layers must be maximized to prevent cracking or failure during stretching.7,8 The photoactive layer is the most critical component of OPVs, responsible for absorbing photons and converting them into charge carriers. The stretchability of this active layer directly impacts the performance stability and efficiency of the OPVs during deformation.
 | ||
| Fig. 1 Three stages of the research on boosting the mechanical properties of OPVs. (a) Rigid OPVs. (b) Flexible OPVs. (c) Stretchable OPVs (s-OPVs). (d) The development of s-OPVs and several key advances. | ||
For OPVs, two main types of materials are used to fabricate active layers: conjugated polymers and small molecules. Advances in the development of polymer donors (PDs) and small molecule acceptors (SMAs) have led to a significant increase in the efficiency of laboratory-scale single junction OPVs, with efficiencies now exceeding 20%.9,10 However, the state-of-the-art OPVs based on PD:SMA combinations often exhibit poor stretching performance. These materials often possess highly rigid backbones with π–π conjugated structures to achieve tight molecular packing and high crystallinity, which allow electrons to delocalize effectively and help achieve the appropriate energy levels. While this rigidity is crucial for high performance, it restricts the stretchability and deformability of the active layer, limiting its ability to withstand mechanical stress in all directions. SMAs, in particular, are brittle due to their low molecular weights and their lack of long chains that are capable of bridging crystalline domains. When OPVs based on these high-performance active layers are subjected to stress, the material may crack or even break, leading to efficiency declines and, in some cases, complete failure.11–13 Polymer acceptors (PAs) offer significant advantages for s-OPVs compared to SMAs due to their longer polymer chains, which provide greater flexibility and structural integrity.13,14 Consequently, researchers are increasingly focusing on all-polymer solar cells (all-PSCs) that integrate PAs. Despite this, the progress of all-PSCs has been constrained by the limited availability of suitable PAs. Therefore, in addition to striving for high photovoltaic performance, it has become a pressing necessity for researchers to develop OPVs that can endure significant mechanical stresses and adapt to a multitude of deformations. This review consolidates the latest advancements in stretchable organic photovoltaic materials. Before delving into the new progress made from 2022 onward, this feature article first presents a brief overview of earlier studies to establish a foundational understanding of the evolution of the technology. To facilitate a clearer understanding, this paper categorizes the methods of preparing stretchable photovoltaic materials into two primary approaches: (1) designing new photovoltaic materials with high stretchability, which involves tailoring the material's chemical structure for enhanced stretchability and performance, (2) adjusting the thin-film morphology of high-efficiency binary photovoltaic blends through the incorporation of a third component or other functional additives, which aims to improve the material's mechanical properties and overall stretchability. These resulted in a more robust and flexible active layer in s-OPVs. By organizing the review in this manner, the ultimate goal is to offer valuable insights that will inform future research and development efforts in the field of s-OPVs, ultimately driving innovation and facilitating practical applications in stretchable and wearable electronics.
2. Brief overview of the development of s-OPVs
The flexible thin-film transistor15 and the all-polymer field-effect transistor16 are among the earliest forms of flexible devices, which demonstrated the potential of flexible devices. Stretchable electronics is a more recent research area based on flexible electronics (Fig. 1d).In 2011, Bao et al.17 fabricated the first s-OPV with a bucking structure. They deposited the electrode and active layer on the elastomeric substrate which was pre-strained. Upon releasing the pre-strain, the device bulked. The buckling phenomenon provides the devices with stretchability, and the resulting OPVs exhibited elasticity under tensile strain of up to 27%. In 2012, Bauer et al.18 prepared OPVs on polyethylene terephthalate (PET) substrates with a thickness of only 1.4 μm. These devices were then transferred to a pre-stretched elastic support, demonstrating a power conversion efficiency (PCE) of 4.2% and a reversible tensile strain of over 300%. Similarly, methods based on the structural design of OPVs have been developed and applied to induce stretchability in rigid materials, such as spring-like structures,19 textile structures20 and mesh structures.21 However, these methods are relatively complex and costly, compared with designing the structure of the OPVs, and the use of intrinsically stretchable materials enables large-area production at a lower cost, such as solution processing methods and printing methods.22 Notably, the bulking method, which is the most prevalent among these strategies, allows for stretchability in only a predetermined direction, limiting its suitability for wearable devices. As a result, there is a pressing need to develop intrinsically s-OPVs by replacing the rigid layer with stretchable materials.
In 2012, Bao et al.23 initially reported intrinsically s-OPVs based on the blending of two conjugated polymers (poly(3-hexylthiophene) (P3HT) and poly(2,5-bis(2-octyldodecyl)-3,6-di(thiophen-2-yl)diketopyrrolo[3,4-c]pyrrole-1,4-dione-alt-thieno[3,2-b]thiophen) (DPPT-TT)) with [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM), which exhibited reversible stretchability in the resultant OPVs. But the PCE of the OPVs based on P3HT:PC61BM and DPPT-TT:PC61BM was extremely low (<1%). Researchers have since focused on enhancing the stretchability of OPVs without compromising their photovoltaic performance. There is a trade-off between the photovoltaic and mechanical properties of active layer materials. Conjugated materials with high photovoltaic performance typically have fused rings in their molecular structure, which enhances crystallization to improve photovoltaic performance, but this rigid structure makes the active layer brittle. In addition, since the active layers often consist of two or more materials, the degree of phase separation of the different materials also affects the charge transport, and the interface between the different materials imposes a limit on the ductility of the film.
The performance of s-OPVs has been on the rise in recent years. In 2015, Kim et al.14 reported the use of all-polymer photoactive layers for s-OPVs for the first time. Polymer acceptors are able to form entanglements with other polymers within the acceptor domains and at the interface, thereby enhancing the stretchability of films. They prepared OPVs by using poly[4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene-alt-1,3-bis(thiophen-2-yl)5(2-hexyldecyl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione] (PBDTTTPD) as the polymer donor and poly[[N,N′-bis(2-hexyldecyl)-naphthalene-1,4,5,8-bis(dicarboximide)2,6-diyl]-alt-5,5′-thiophene] (P(NDI2HD-T)) as the polymer acceptor, and the result revealed that the elongation at break and the toughness of all-PSCs are over 60 times and 470 times higher, respectively, than those of fullerene PSCs. Another notable example was demonstrated by Yang et al.24 in 2018. They synthesized a highly viscous hydrophobic polymer, poly(dimethylsiloxane-co-methyl phenethylsiloxane) (PDPS), and added it as an additive to the poly(6-fluoro-2,3-bis-(3-octyloxyphenyl)quinoxaline-5,8-dyl-alt-thiophene-2,5-diyl) (TQ-F):poly((N,N′-bis(2-octyldodecyl)-naphthalene-1,4,5,8-bis(dicarboximide)-2,6-diyl)-alt-5,5′-(2,2′-bithiophene)) (P(NDI2OD-T2, known as N2200)) matrix. The device exhibited a PCE of 5.6%, and after 100 bending cycles at a radius of 3 mm, the devices maintained 90% of their initial efficiency. Ye et al.25 introduced a low-cost and rather common thermoplastic elastomer, polystyrene-block-poly(ethylene-ran-butylene)-block-polystyrene (SEBS) as the third component. The crack onset strain (COS) of the blend film gradually increased with the increase of the weight content of SEBS. The photovoltaic performance of the film was slightly improved with a 38% increase in COS at 5% SEBS content, and the photovoltaic performance of the film remained almost unchanged with a 62% increase in COS at 10% SEBS content. This study shows the potential of thermoplastic elastomers to improve OPV stretchability.
Interlayers are essential for efficient s-OPVs as they lower the energy barriers at the active layer/electrode interfaces and adjust the work functions of the electrodes. Chen et al.26 developed a stretchable electron-extraction layer by using poly[(9,9-bis(3′-(N,N-dimethylamino)propyl)-2,7-fluorene)-alt-2,7-(9,9-dioctylfluorene)] (PFN) and nitrile butadiene rubber (NBR) blends that can withstand up to 60% strain. They prepared OPVs that maintained a PCE of 2.82% even after 10% stretching based on this electron-extraction layer. More recently, Chen et al.27 developed a hybrid stretchable electrode with modified PH1000 and silver nanowires (AgNWs). The incorporation of AgNWs leads to the enhancement of charge transport and the modified PH1000 improved the stretchability of the hybrid electrode. The rigid OPV based on this electrode showed a PCE over 17%, while the s-OPV exhibited over 16%.
In addition to the design of the different layers of the OPV, other findings may also contribute to the enhancement of the performance of the s-OPVs. Zhou et al.28 fabricated stretchable all polymer OPVs with an entangled polymer additive. They observed a 4% increase of the power output after stretching to a certain strain, and this feature is of particular significance for s-OPVs to be used in wearable devices. In a more recent study, they achieved a PCE of 15% for s-OPVs, and they also found that the fracture strain values increased with film thickness.
These advancements facilitate the use of wearable electronics and other applications, thereby driving ongoing progress in traditional solar technology.
3. Strategies of designing stretchable organic photovoltaic materials
3.1. Regulation of molecular weight
The effect of molecular weight on the mechanical properties of P3HT has been extensively studied, with significant findings reported as early as 2013.29 It was observed that when P3HT is in the solid state and its molecular weight (Mw) exceeds the critical molecular weight (Mc), the amorphous domains and the degree of chain entanglement increase. This enhanced entanglement enables the material to undergo considerable deformation, which is crucial for stretchable applications. Recent studies have further demonstrated the impact of molecular weight on charge transport and mechanical properties of other photovoltaic polymers. It was concluded that a higher Mw leads to more molecular chain entanglement and higher quality of entanglements.30,31 Entanglement between polymer chains helps to distribute the stress and dissipate the tensile energy, enhancing film ductility.Xu et al.32 synthesized a series of fluorine substituted benzotriazole and bithienyl-benzodithiophene-based copolymers (PBZ-2Si) based on copolymer donor J52, then the low, medium and high molecular weight siloxane-terminated side chain substituted copolymers PBZ-2SiL, PBZ-2SiM and PBZ-2SiH were synthesized. The results showed that the COS of PBZ-2Si:N2200 blends increased upon the molecular weight of PBZ-2Si (Fig. 2a). In particular, the film based on PBZ-2SiH exhibited a significantly improved COS (19.5% to 50.7%) (Fig. 2b). Moreover, after 1000 times stretching cycles at 25% strain, the OPV based on PBZ-2SiM still retained over 75% of the initial PCE, while J52 based OPV only retained 25%. Lee et al.33 reported carboxylate-containing poly(thiophene vinylene) (PTV) with adjustable molecular weight to dissipate tensile stresses (Fig. 2c). Compared to PETTCVT-L:L8-BO, the s-OPV based on PETTCVT-H:L8-BO exhibited a PCE of 10.1%, a PCE80% of 16%, and an increase in COS from 1.3% to 7.1% was observed (Fig. 2d). In addition, the crystallinity of PTV increases gradually with the increase of Mw, resulting in higher hole mobility, lower molecular recombination rate, and higher efficiency of the device.
 | ||
| Fig. 2 (a) The chemical structure of PBZ-2SiL, PBZ-2SiM and PBZ-2SiH. (b) The stress–strain curves of the J52/PBZ-2SiL/PBZ-2SiM/PBZ-2SiH:N2200 blend films under the pseudo free-standing tensile test. (c) The chemical structure of PETTCVT. (d) The stress–strain curves of the PETTCVT-L/PETTCVT-M/PETTCVT-H:L8-BO. (a) and (b) Reproduced with permission from ref. 32. Copyright 2022, Elsevier. (c) and (d) Reproduced with permission from ref. 33. Copyright 2023, Wiley-VC. | ||
3.2. Molecular design
However, the currently reported oligomeric acceptor-based OPVs usually have poor stretchable properties (COS < 5–10%). The rigid structure of oligomer acceptors leads to the discontinuous crystal domains, and cracks are generated at weak interfaces between crystals and spread rapidly when the blend film is subjected to stress.
Lee et al.34 developed a star-shaped trimeric acceptor (TYT-S) based on Y-SMA, and the s-OPVs obtained by utilizing the TYT-S achieved a PCE of 14.4%. In addition, the prepared OPVs exhibit high mechanical stability, with a PCE of 80% of the initial value when subjected to 31% strain. These star-shaped molecular structures prevent the formation of rigid crystalline domains while increasing the extent of amorphous regions, which improves the stretchability of the resulting films.
The synthesis of dimer acceptors can be achieved through three distinct methodologies: end-to-end connection, face-to-face connection, and back-to-back connection. Among these, the end-to-end connection, due to its linear structure akin to polymerized small molecule acceptors (PSMAs), is frequently employed. The spacers currently in use are conjugated rigid fused rings,35 and flexible spacers.36 The introduction of these spacers has been observed to increase the conformational uncertainty and reduce the persistence length of polymers, suppressing the crystallinity of the active layer material and preventing the crystallization domains from aggregation.
Based on 2,2′-((2Z,2′Z)-((3,9-bis(2-butyloctyl)-12,13-bis(2-octyldodecyl)-12,13-dihydro-[1,2,5]thiadiazolo[3,4-]thieno[2′′,3′′:4′,5′]thieno[2′,3′:4,5]pyrrolo[3,2-]thieno[2′,3′:4,5] thieno[3,2-b]indole-2,10-diyl)bis(methanylyli-dene))bis(5,6-dichloro-3-oxo-2,3-dihydro-1H-indene-2,1-diyli-dene)) dimalononitrile (MY-BO), Yin et al.37 synthesized the conjugated dimer acceptor DY-TVCl and the non-conjugated dimer acceptor DY-3T. The COS of the MY-BO based film was 4.98%, while the COS of PM6:DY-TVCl and PM6:DY-3T reached 8.26% and 10.31%, respectively. Ding et al.38 utilized thiophene–alkane–thiophene (TAT) as a conjugate-break linker, and synthesized dimer acceptors through halogen atom substitution and modulation of the linking site (Fig. 3a). In contrast to the end-to-end conformation, the above dimeric acceptors were developed in center-to-center conformation in this paper.39 The COS of the PM6:Y6 film is 8.50%, while the PM6:FDY-m-TAT film has a COS of 18.23% (Fig. 3b). The larger molecular size of FDY-m-TAT is favorable for the formation of a more robust blend film. In addition, the FDY-m-TAT molecules are not only entangled with themselves, but also form an entangled structure with PM6, whereas Y6 is only entangled between the molecules, which makes the former favorable for the improvement of tensile properties of the blend film. Lee et al.40 developed a novel series of dimeric SMAs, designated as DYBT-CX (where X = 0, 4, and 8) (Fig. 3a), by incorporating various flexible units. Among the OPVs fabricated with these materials, the PBQx-TF:DYBT-C4 blend exhibited the highest COS of 32%, while the PBQx-TF:MYT-based devices achieved only 10% (Fig. 3c). Furthermore, the is-OPVs utilizing PBQx-TF:DYBT-C4 demonstrated excellent mechanical stability, retaining 80% of the initial PCE under a 36% strain. Although the DYBT-C8 has the longer FS units than DYBT-C4, the blends based on DYBT-C8 showed domain size crossing multiple length scales (63 and 18 nm), while the PBQX-TF:DYBT-C4 showed a small and uniform values of 21 nm. This leads to the different mechanical performances. Liu et al.41 designed a series of dimer acceptors 2BOHD-TCXT (X = 4 or 6); these dimer acceptors with flexible segments can form more entanglements with donors, and PM6:2BOHD-TC4T exhibited a COS value of 9.69%, which is almost twice as much as PM6:BOHD (COS = 4.81%). According to the study by Zhang et al.,42 they introduced a flexible alkyl linker between the precursor CH8-T, synthesizing two dimeric acceptors CH8-6 and CH8-7 (Fig. 3a). The COS of the PM6:CH8-6, PM6:CH8-7, and PM6:CH8-T blend films was 40.2%, 41.9%, and 28.3%, respectively (Fig. 3d). Song et al.43 designed and synthesized the dimer acceptor (DOY-TVT) (Fig. 3a) and doped DOY-TVT into the D18:N3 system. The addition of 15% DOY-TVT to the D18:N3 binary blended films resulted in a nearly 50% increase in COS (from 7.5 ± 0.6% to over 11%) and a 23.2% and 25.8% decrease in modulus of elasticity and stiffness (Fig. 3e). The flexible OSCs based on the oligomers exhibited excellent photovoltaic performance, and reached the PCE of 18.06%. The ternary flexible devices retained 97% of the initial PCE after 3000 bending cycles, while the binary device maintained only 88.4%. Qi et al.44 introduced an alkyl linker into the Y6-derivative T9TBO, synthesizing a new dimer acceptor dT9TBO, and the mechanical properties of the ternary OPV based on PM6:Y6:dT9TBO were improved due to the incorporation of flexible alkyl linker. You et al.45 developed a series of back-to-back connected dimer acceptors (2Qx-TT,2Qx-C3, and 2Qx-C6) (Fig. 3a). When incorporated into the PM6:BTP-eC9 system, these dimers have been shown to optimize the film-forming kinetics and improve the PCE of the devices. The OPVs based on PM6:BTP-eC9 exhibited a PCE of 17.63%, and the addition of the dimer acceptors improved the PCE to over 18%. Furthermore, the incorporation of dimers with flexible linkage resulted in an enhanced robustness of the active layer, and a significant increase in the COS value of PM6:BTP-eC9:2Qx-C3 to 15.0% (Fig. 3f). Ye et al.46 developed and incorporated a series of ductile oligomeric acceptors (DOY-C2, DOY-C4, and TOY-C4) (Fig. 3a) into the D18:N3 system. A substantial enhancement in all COS values of the ternary films was observed, with a notable increase from 7.8% to approximately 12% (Fig. 3g). The D18:N3:DOY-C4 based flexible OPVs exhibited a PCE of 17.80%, while that for the binary flexible OPVs was 16.85%.
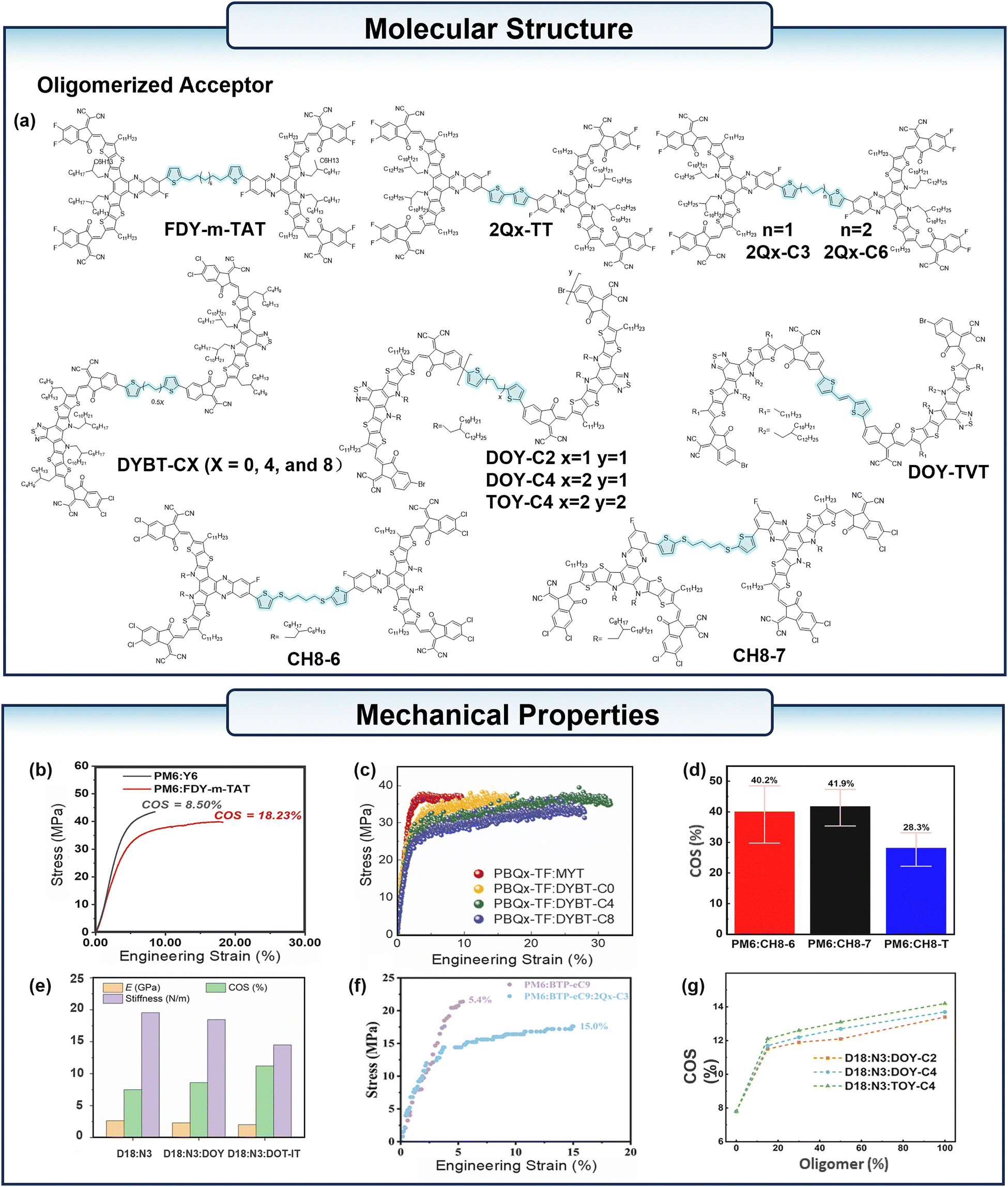 | ||
| Fig. 3 (a) The chemical structure of the oligomerized acceptors mentioned in the context. (b) The stress–strain curves of the PM6:Y6 and PM6:FDY-m-TAT blend films. (c) The stress–strain curves of the PBQx-TF:MYT, PBQx-TF:DYBT-CX (X = 0, 4, and 8) blend films. (d) Histograms of COS of PM6:CH8-6, PM6:CH8-7, and PM6:CH8-T blend films. (e) Comparison of COS, elastic modulus, and stiffness between D18:N3-based binary and ternary films. (f) The stress–strain curves of PM6:BTP-eC9 and PM6:BTP-eC9:2Qx-C3 blend films. (g) The stress–strain curves of D18:N3:DOY-C2, D18:N3:DOY-C4, and D18:N3:TOY-C4 blend films. (b) Reproduced with permission from ref. 38. Copyright 2024, Wiley-VCH. (c) Reproduced with permission from ref. 40. Copyright 2024, Elsevier. (d) Reproduced with permission from ref. 42. Copyright 2024, Royal Society of Chemistry. (e) Reproduced with permission from ref. 43. Copyright 2023, Wiley-VCH. (f) Reproduced with permission from ref. 45. Copyright 2024, Wiley-VCH. (g) Reproduced with permission from ref. 46. Copyright 2023, Wiley-VCH. | ||
Lee et al.47 linked two Y-based SMA units with polydimethylsiloxane (PDMS), resulting in the synthesis of an elastomer-incorporated dimer acceptor, referred to as DYPDMS (Fig. 4b). Additionally, they integrated PDMS into PM6 (Fig. 4a), thereby developing a block copolymer PM6-b-PDMS. By combining these two materials, the resulting is-OPV with a PDMS-incorporated active layer achieved a power conversion efficiency (PCE) of 12.7%. More importantly, this is-OPVs system demonstrated a notable enhancement in power output under strain (Fig. 4e). This study confirms the feasibility of incorporating elastomers into SMA to enhance the mechanical performance of the blended film.
 | ||
| Fig. 4 (a) The chemical structure of the conventional PM6 and PM6-b-PDMS. (b) The chemical structure of the conventional DYBT and DYPDMS. (c) The stress–strain curves of the PM6:DYBT and PM6-b-PDMS:DYBT blend films. (d) The stress–strain curves of the PM6-b-PDMS:acceptors (DYBT, DYBT:PDMS, and DYBT:DYPDMS). (e) Strain-induced power output changed in the resulting devices: normalized power output vs. engineering strain curves. Reproduced with permission from ref. 47. Copyright 2025, Royal Society of Chemistry. | ||
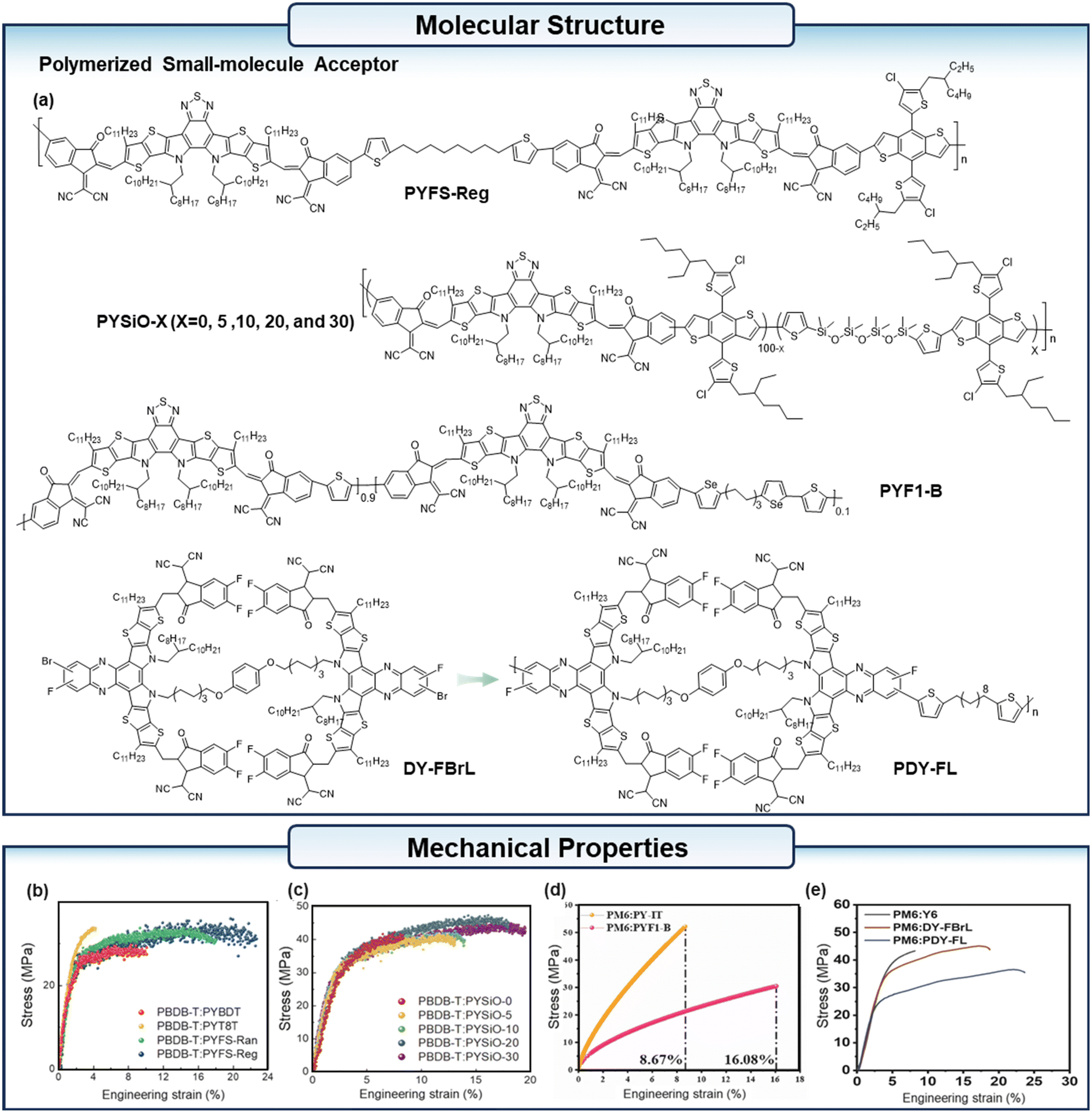 | ||
| Fig. 5 (a) The chemical structure of the polymerized small-molecular acceptors mentioned in the context. (b) The stress–strain curves of the PBDB-T:PYBDT, PBDB-T:PYT8T, PBDB-T:PYFS-Ran, and PBDB-T:PYFS-Reg blend films. (c) The stress–strain curves of the PBDB-T:PYSiO-X (X = 0, 5, 10, 20, and 30) blend films. (d) The stress–strain curves of the PM6:PY-IT and PM6:PYF1-B blend films. (e) The stress–strain curves of the PM6:Y6, PM6:DY-FBrL and PM6:PDY-FBr blend films. (b) Reproduced with permission from ref. 48. Copyright 2022, Royal Society of Chemistry. (c) Reproduced with permission from ref. 49. Copyright 2022, Royal Society of Chemistry. (d) Reproduced with permission from ref. 50. Copyright 2024, Elsevier. (e) Reproduced with permission from ref. 51. Copyright 2025, Wiley-VCH. | ||
In another study, Lee et al.49 synthesized a new series of PSMAs, designated as PYSiO-X (where X = 0, 5, 10, 20, and 30) (Fig. 5a), by incorporating SiO-functionalized flexible spacer (SiO-FS) units into the backbone of the PSMA. The results revealed that the inclusion of 10 mol% SiO-FS helps to enhance the performance of the all-polymer solar cells (PSCs), achieving a COS of 15.2% (Fig. 5c). In contrast, the all-PSCs fabricated with the PSMA lacking SiO-FS exhibited a substantially lower COS of 9.6%. Hu et al.50 synthesized four distinct acceptors—PY-IT, PY-IF1, PYF1-A, and PYF1-B, by integrating both rigid-bridge and flexible-bridge strategies (Fig. 5a). The introduction of flexible-bridges, which partially replace the thiophene rigid-bridges, significantly enhances the performance of the resulting material. Notably, the PM6:PYF1-B blend demonstrates a COS of 16.08%, a substantial improvement compared to the 8.67% achieved by the PM6:PY-IT blend (Fig. 5d). Ding et al.51 synthesized a dimer acceptor DY-FBrL first, with the reaction sites of DY-FBrL, they designed a new polymerized acceptor PDY-FL (Fig. 5a). With the donor PM6, the active layer based on the DY-FBrL showed a COS of 18.54%, and the blend film based on PDY-FL reached a COS of 23.45% (Fig. 5e).
Lee et al.54 synthesized a PD of poly[didecyl5-(4,8-bis(5-(2-ethylhexyl)-4-fluorothiophen-2-yl)-6-methylbenzo [1,2-b:4,5-b′]dithiophen-2-yl)-5′′-methyl-[2,2′:5′,2′′-terthiohene]-3,3′′-dicarboxylate] (PBET-TF) (Fig. 6a). Then they paired PBET-TF with MYBO, and the blends showed a significantly improved COS of 31%. (Fig. 6b). After 30% strain, the PBET-TF:MYBO blend films did not show any crack, while the PBDB-TF-MYBO blend films showed a sharp crack after 3% strain. It is important to note that PBET-TF:MYBO-based s-OPVs demonstrated a power output enhancement of over 5% at 40% strain. In contrast, PBDB-TF:MYBO-based is-OPVs maintained only 13% of the initial power output at 20% strain.
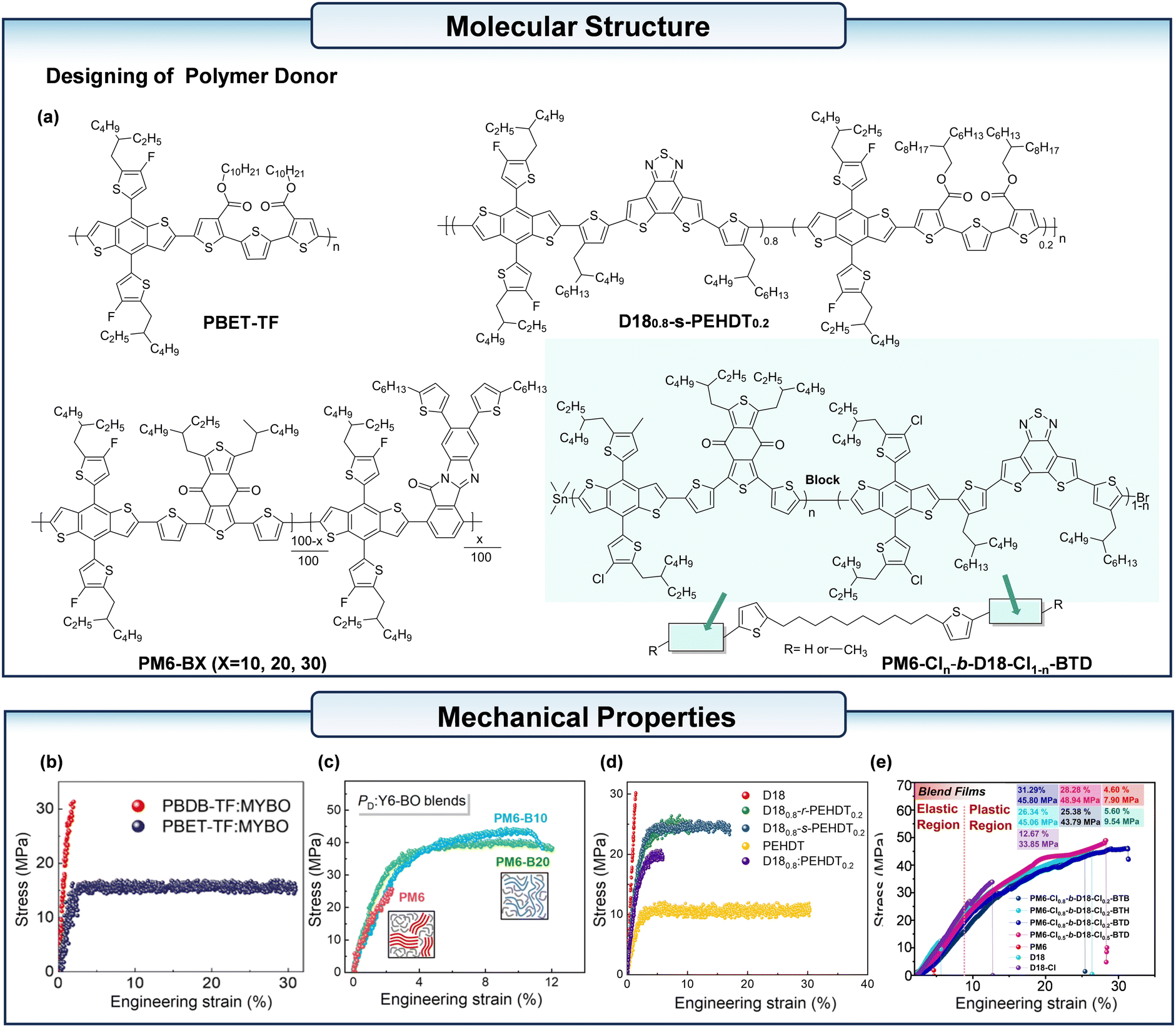 | ||
| Fig. 6 (a) The chemical structure of the polymer acceptors mentioned in the context including the incorporation of conjugation-break spacers, and block copolymerization. (b) The stress–strain curves of the PBDB-FT:MYBO and PBDB-TF:MYBO blend films. (c) The stress–strain curves of the PM6:Y6-BO, PM6-B10:Y6-BO, and PM6-B20:Y6-BO films. (d) The stress–strain curves of the PD: L8-BO blend films (PD: D18, D180.8-r-PEHDT0.2, D180.8-s-PEHDT0.2, PEHDT, and D180.8-PEHDT0.2). (e) The stress–strain curves of the Fs-MCD:BTP-eC9 blend films. (b) Reproduced with permission from ref. 54. Copyright 2024, Elsevier. (c) Reproduced with permission from ref. 55. Copyright 2023, Royal Society of Chemistry. (d) Reproduced with permission from ref. 56. Copyright 2023, Elsevier. (e) Reproduced with permission from ref. 57. Copyright 2024, Wiley-VCH. | ||
To reduce the stiffness of the polymer backbone and enhance the film's ductility, incorporating bulky electroactive units into the polymer chains is also a feasible approach. Kim et al.55 synthesized a series of terpolymers (PM6-BX, X = 10–30) (Fig. 6a) based on PM6 by adding 7,8-bis(5-hexylthiophen-2-yl)-11H-benzo[4,5]imidazo[2,1-a]isoindol-11-one (BID). The mechanical properties of the PM6-B10:L8-BO blend films were much greater than those of PM6:L8-BO (COSavg = 11.4% vis COSavg = 2.0%; toughness = 4.1 MJ m−3 vis toughness = 0.3 MJ m−3) (Fig. 6c). The stretchable device based on the PM6-B10:L8-BO blend film with TPU substrate cracked only when subjected to 30% stress, while the stretchable device based on the PM6:L8-BO blend film cracked at only 10% stress.
The incorporation of multi-component copolymerized donors (MCDs) into the active layer has been demonstrated to enhance the stretchability of blends. This is achieved by reducing the excessive rigidity of the polymer donor backbone and the formation of large crystalline domains, combining the benefits of the mechanical and electrical properties of the two blocks that are phase separated at the nanoscale. Lee et al.56 synthesized the block copolymer D180.8-s-PEHDT0.2 with rigid D18 and soft PEHDT (Fig. 6a), a polymer donor composed of alternating soft and hard segments by sequential block copolymerization. This allowed the OPV to balance stretchability with PCE. The stretchable device based on D180.8-s-PEHDT0.2 achieved an efficiency of 14.3% and preserved 80% of the initial PCE at 31% strain. The COS value of D180.8-s-PEHDT0.2:L8-BO reached to 17.2%, while the D18:L8-BO showed only 1.5% (Fig. 6d). In another study, Lin et al.57 developed a series of flexible linker-sequential block MCDs, they incorporated flexible functional groups into the conjugated polymer backbone, by incorporating 1,10-bis(5-(trimethylstannyl)thiophen-2-yl)decane (BTD) as the flexible linker, and the pure film of PM6-Cl0.8-b-D18-Cl0.2-BTD (Fig. 6a) achieved a COS of 49.88%. This Fs-MCDs:BTP-eC9 based blend exhibited a COS value of 31.29% (Fig. 6e). Furthermore, the flexible OPVs reached a PCE of 16.63%. Lee et al.58 developed a series of Mw-controlled PD-b-elastomer block copolymers (D18x-b-PDMS, x = L, M, and H). The D18:L8-BO:PDMS ternary blend films physically mixed with PDMS had significantly lower mechanical robustness (toughness = 0.1 MJ m−3) than the D18:L8-BO blend. In contrast, the active layer based on D18x-b-PDMS exhibits excellent mechanical robustness with a toughness of 1.8–2.6 MJ m−3, which is 4–5 times higher than the reference D18:L8-BO active layer (0.5 MJ m−3). The s-OPVs based on D18:L8-BO and D18H-b-PDMS:L8-BO exhibited PCE values of 12.4% and 11.9%, respectively. However, the strain at PCE80 was 8% for the D18:L8-BO s-OPVs and 16% for the D18H-b-PDMS:L8-BO s-OPVs.
Hydrogen bonding (H-bonding) is stronger than other interaction forces, and this strong intermolecular interaction with a bonding energy of 10–40 kJ mol−1 improves the stretchability of polymer films.59–61 A novel polymer donor, PhAmX (where X = 3, 5 and 10) (Fig. 7a) was synthesized based on a flexible spacer with the ability to form hydrogen bonds by Lee and his coworkers.59 As the value of X increased, the COS value of the PhAmX:Y7 hybrid film increased from 10.4% (PhAm3:Y7) to 22.6% (PhAm10:Y7) (Fig. 7b). The toughness increased from 0.30 MJ m−3 for PM6:Y7 to 8.42 MJ m−3 for PhAm10:Y7. Furthermore, the s-OPVs based on the PhAm5:Y7 system reached a PCE of 12.7%, with a strain of 32% at 80% PCE, which represents a significantly higher level of stretchability than the brittle PM6:Y7 based system (PCE80% = 15%). The improved ductility of the active layer was attributed to the reversible hydrogen bonding between the PhAmX molecular chains. In order to illustrate the effect of hydrogen bonding on the mechanical properties of the films, a comparison was made between the properties of the blend films obtained from aliphatic flexible spacer-containing units and those from flexible spacer units with the ability to form hydrogen bonds. It was found that the stretchability of the PhAm10-based blend film (COS = 23%) was significantly improved compared to the blend film obtained by incorporating normal flexible spacers (PM6-C10, COS = 9%). Wan et al.60 synthesized a conjugated polymer acceptor, N2200-ThyDap (Fig. 7d), which has greatly enhanced intermolecular interactions through the formation of hydrogen bonds between thymine (Thy) and diaminopyrazine (Dap) on N2200-ThyDap. In addition, the results revealed that the hydrogen bonding in N2200-ThyDap helps to connect the isolated domains between the SMAs, allowing the amorphous behavior to be enhanced and contributing to the improvement of the ternary OPV stretchability. The blend film based on PM6:Y6:N2200-ThyDap achieves a COS value of 4.8%, while the blend film based on PM6:Y6:N2200 has a COS value of only 2.1% (Fig. 7e). Wan et al.62 synthesized a series of derivative polymers of PM7 (PM7-Thy5, PM7-Thy10 and PM7-Thy20) (Fig. 7g) by introducing difluoro quinoxaline with a thymine (Thy) end group in the side chain. The stretchability of the blend films was enhanced due to the ability of the introduced Q-Thy units to form hydrogen bonding between the chains. The COS of the PM7:L8-BO based film is only 2.6%, whereas the COS of the PM7-Thy10:L8-BO based film is greater than 13% (Fig. 7h). And the PM7-Thy10-based s-OPVs showed higher PCE than the PM7-based s-OPV, with a strain at PCE80% of 43.1%. In the study by Wang et al.,63 PM6 and PY-IT were selected as the basis polymer, and amide units with alkyl segments and hydrogen bonds were introduced into the basis polymer. The introduction of the amide units resulted in a decrease in the crystallinity of the polymer due to its flexible segments and led to an improvement in the intermolecular force because of the strong hydrogen bonding energy, promoting the stretchability of the blend films.
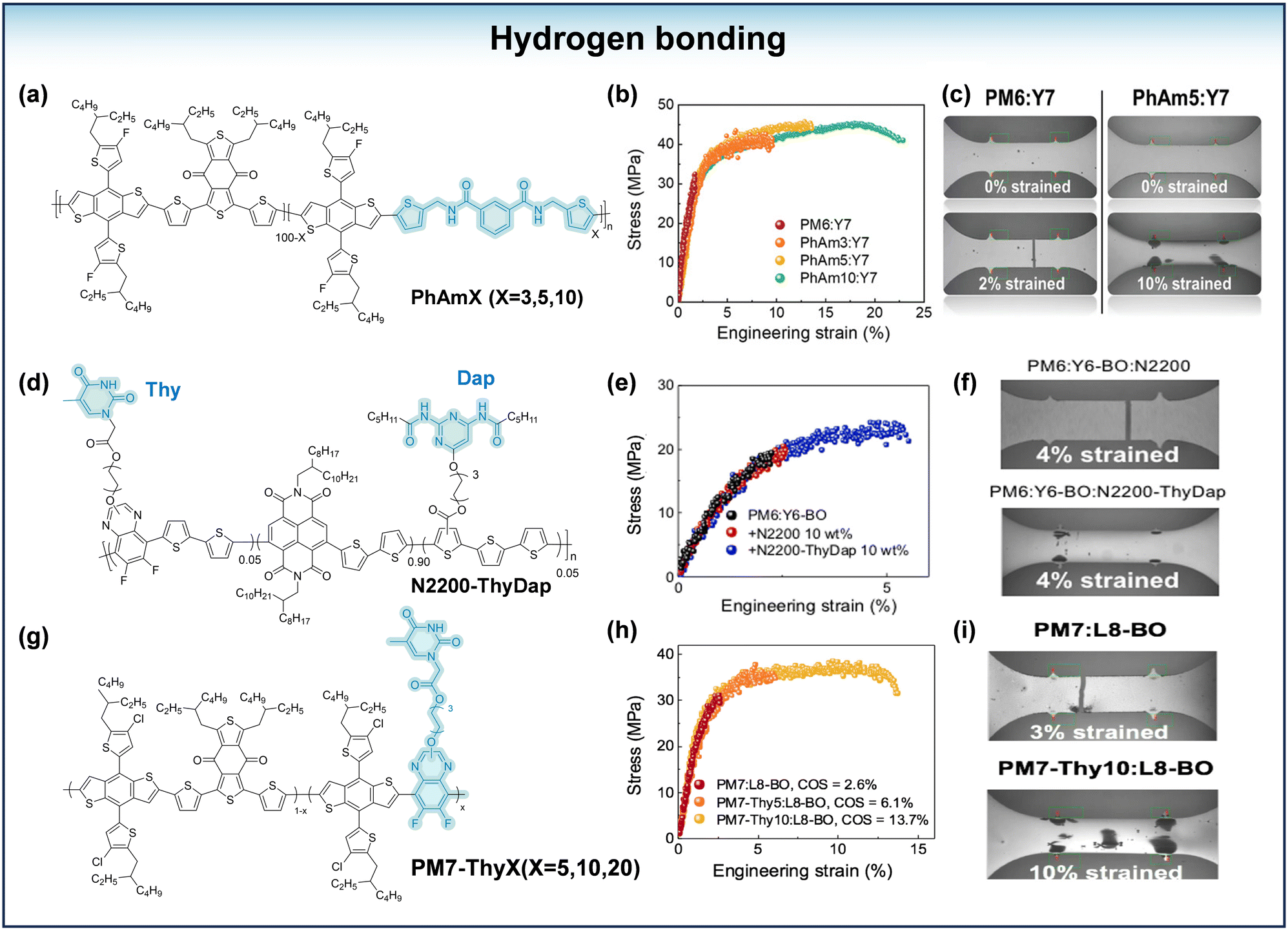 | ||
| Fig. 7 (a) The chemical structure of PhAmX (X = 3,5,10). (b) The stress–strain curves of the blend films of PM6:Y7, PhAm3:Y7, PhAm5:Y7 and PhAm10:Y7. (c) Images of blend films during the tensile test. (d) The chemical structure of N2200-ThyDap. (e) The stress–strain curves of the blend films of PM6:Y6-BO, PM6:Y6-BO:N2200 and PM6:Y6-BO:N2200-ThyDap (wt% value of PA indicates the weight of PA compared to (PA + SMA) weight). (f) Images of blend films during the tensile test. (g) The chemical structure of PM7-ThyX (X = 5, 10, 20). (h) The stress–strain curves of the blend films of PM7:L8-BO, PM7-Thy5:L8BO, and PM7-Thy10:L8-BO. (i) Images of blend films during the tensile test. (b) and (c) Reproduced with permission from ref. 59. Copyright 2022, Wiley-VCH. (e) and (f) Reproduced with permission from ref. 60. Copyright 2023, American Chemical Society. (h) and (i) Reproduced with permission from ref. 62. Copyright 2023, American Chemical Society. | ||
 | ||
| Fig. 8 (a) The chemical structure of PM6-HD and the stress–strain curves of the blend films with different contents of PM6-HD. (b) The chemical structure of PY-EH and PY-SiO and the stress–strain curves of the blend films based on PY-EH and PY-SiO. (c) The chemical structure of BTP-Si4 and the stress–strain curves of the blend films of PNTB6-Cl:Y6 and PNTB6-Cl:BTP-Si4. (d) The chemical structure of BTP-C3, BTP-EH and BTP-HD and the stress–strain curves of the blend films of D18:BTP-C3, D18:BTP-EH and D18:BTP-HD. (a) Reproduced with permission from ref. 66. Copyright 2024, Wiley-VCH. (b) Reproduced with permission from ref. 67. Copyright 2024, American Chemical Society. (c) Reproduced with permission from ref. 68. Copyright 2025, Science. (d) Reproduced with permission from ref. 69. Copyright 2025, Royal Society of Chemistry. | ||
On the other hand, rational design of side chains can improve the mechanical properties of conjugated polymers. In 2014, Lipomi et al.70 demonstrated that appropriate alkyl chain lengths are beneficial in improving both photovoltaic and mechanical performance. They discovered that for the same number of carbon atoms, branched chains have a greater positive impact than side chains. And compared with normal terminal alkane side chains, terminal siloxane conjugated polymers have larger free volume and longer interchain distance of the polymer backbone, which may produce lower film modulus and better film ductility. He et al.67 synthesized a polymer acceptor PY-SiO with siloxane-terminated side chains and compared its properties with those of PY-EH, which has ethylhexyl-terminated side chains (Fig. 8b). The results demonstrated that the incorporation of siloxane side chains facilitated the aggregation of PY-SiO molecules and led to the promotion of phase separation between the donor and acceptor, thereby enhancing the efficiency of the resulting OPV (10.85% to 12.04%). The siloxane side chains resulted in larger molecular chain spacing, larger chain free volume, and easier chain rotation and reconfiguration among the backbones of the polymerized acceptors, which led to an increase in the COS of the film to 18.32% (Fig. 8b), in comparison to 11.15% for the PY-EH-based film. In a study by Wang et al.,32 the siloxane-functionalized polymer PBZ-2SiM:N2200 blend film exhibited a COS value of 38.4% and an E-modulus value of 0.30 GPa, which were significantly higher than those of the J52:N2200 system (COS = 19.5%, E = 0.42 GPa).
In a more recent study by Wang et al.,68 they reported an organosilane-functionalized SMA BTP-Si4 (Fig. 8c) and blended BTP-Si4 with PNTB6-Cl. It is established that SMAs usually suppress ductility, but the addition of BTP-Si4 has been shown to enhance this property. The dichroic ratio (DR) of the PNTB6-Cl:BTP-Si4 blend films exhibited a consistent increase up to a maximum strain of 100% (Fig. 8c), indicative of remarkable strain tolerance. The s-OPVs on the basis of PNTB6-Cl:BTP-Si4 exhibited a PCE of 14.6%. In addition, only a mild decrease in PCE (approximately 25%) was experienced after 1000 cycles under 30% strain. They also synthesized a series of BTP-Si4-like SMA (BTP-Si4, BTP-Si6, and BTP-Si8), which were different in the length preceding the branching point. The design of the branch of BTP-Si4-like SMAs promoted the miscibility with the donor polymer and increased the free volume in blend films, thus led to an improvement of PCE and mechanical performance. Zhang et al.69 synthesized three SMAs: BTP-C3, BTP-EH and BTP-HD (Fig. 8d), and these molecules share an identical dithienothiophen[3,2-b]-pyrrolobenzothiadiazole core but they vary in terms of branching position on the pyrrole rings and length of the branching alkyl chains. The blend films based on BTP-C3, BTP-EH and BTP-HD showed a COS of 19.6%, 25.8% and 32.5%, respectively (Fig. 8d). The BTP-EH exhibited the best miscibility with D18, and the s-OPVs based on BTP-EH achieved a PCE of 15.6%. After 1000 cycles, the BTP-EH and BTP-HD based s-OPVs exhibited 30% and 25% decrease in PCE while the BTP-C3 based s-OPVs decreased to 70% of the initial value after only 100 cycles.
3.3. Incorporation of a third component
Unlike the complicated chemical synthesis, the ternary blends can be prepared by adding another donor or acceptor component to the binary blends, and the introduction of the third component can play a facilitating role in various aspects such as light absorption and charge transport, which can improve the stretchability of the film more simply, and this method has been recognized as one of the most effective methods to improve the properties of the blend films, as shown in Fig. 9. | ||
| Fig. 9 The chemical structure of the donors and acceptors mentioned in the secondary donor/acceptor strategy. | ||
Ma et al.71 improved the ductility of the blend film by introducing PBDB-TF, a polymer donor with high Mw into the PBQx-TF:PY-IT system. The high Mw PBDB-TF effectively dissipates stress, improves the stretchability of blend films, and even after 2000 continuous bending cycles, the device can still maintain 91% of its initial values. Li et al.72 introduced a conjugated polymer PTQ10 as the second donor to the PM6:N3 blend. When 20 wt% of the third component PTQ10 was added, the PCE and COS were the best among the tested OPVs. The introduction of PTQ10 lead to the π–π stacking and molecular arrangement being more ordered, which results in the improvement of the stretchability of film. The modulus of elasticity decreased monotonically with the increasing PTQ10 content. In another example, Li et al.66 synthesized a PM6 derivative with a long alkyl chain, PM6-HD, which was incorporated into binary blending systems PM6:BTP-eC9 and PM6:N2200. The addition of PM6-HD resulted in a notable enhancement in the PCE80% strain, with increases observed in both the PM6:BTP-eC9 (from 11% to 21%) and PM6:N2200 (from 31% to 50%) systems. s-OPVs based on PM6:PM6-HD:BTP-eC9 blends demonstrated remarkable resilience, retaining 52% of the initial PCE after 500 cycles. In comparison, OPVs based on PM6:PM6-HD:N2200 blends exhibited superior durability, with more than 80% retention of PCE after 1000 cycles of stretching. Li et al.3 introduced PM6-OD, a PM6-like donor polymer containing long branched alkyl chains, into PM6:PY-IT blends to build intrinsically stretchable active layers of all-polymer OPVs. The incorporation of the third component PM6-HD has been shown to result in a consistent and incremental rise in COS. When the donor in the active layer is neat PM6-OD, the COS exhibited an increase of more than fourfold in comparison with the reference value. The underlying causes of these observations are likely attributable to the formation of chain entanglements in extended chains of high molecular weight, in addition to the modification of side chains.
Liu et al.73 constructed a ternary system by adding a polymerized fullerene material, PPCBMB, to the PM6:PYIT system. The results demonstrated that an increase in the concentration of PPCBMC in the blends led to a rise in both the COS (9.6 ± 1.2% to 13.1 ± 1.3%) and strain-at-break (6.6% to 7.4%), thus enhancing the ductility of the film. The smaller domain size in the optimal ternary blend contributes to improved ductility by minimizing the number of sites where cracks can initiate. Kim et al.74 developed novel electroactive compatibilizers P1 and P2 as the third component of the active layer of PBQx-TF:PYIT for all-polymer solar cells, whereas P2-based OPVs exhibited a PCE of 13.7%, with a PCE80% enhancement of 35%, whereas the efficiency of the stretchable device for the binary system of PBQx-TF:PYIT without the use of compatibilizers is only 12.1%, with a PCE80% was 27%. In addition, the P2-based PBQx-TF:P2:PYIT ternary hybrid films exhibited enhanced mechanical durability with Gc and COS values of 2.6 J m−2 and 20.4%, respectively, compared to the binary hybrid films (Gc = 1.1 J m−2 and COS = 16.5%). It was found that the addition of these compatibilizers strengthened the donor–acceptor interface and the ternary blends were more cohesive, which prevented the film from being damaged by external stresses.
Wang et al.75 introduced N2200 into the PM6:Y6 active layer blends. N2200 act as tie molecules to improve the interfacial interaction between PM6 and Y6, leading to the enhancement of stretchability. The ductility of the film increases with N200 content, the E of films show a decrease, with E values of 0.84, 0.79, and 0.60 GPa for 10%, 20%, and 30% N2200 incorporation, respectively. Zhou et al.28 employed a strategy of entangled polymer additives to optimize the morphology of the film. By incorporating conjugated polymer PNDI with a molecular weight exceeding its critical entanglement molecular weight (Mc) as a polymer additive, it was found that P180k (PNDI with a Mw of 180 kg mol−1) exhibited the best miscibility with the host photovoltaic material. Its entangled structure effectively hindered the propagation of cracks in the film, resulting in COS exceeding 50% for the ternary blend. When the s-OPV based on PM6:PY-IT:P180k was subjected to 30% strain and stretched 600 times repeatedly, its efficiency remained above 70% of the initial PCE. Xian et al.76 incorporated a thiophene-dicarboxylate spacer tethered molecule TDY-α as a function aid into the PM6:eC9 system. With the addition of the thick bulk-heterojunction strategy, the stretchability of the blend film has been greatly improved. Furthermore, they discovered that the COS of TDY-α toughened blend films increased with the thickness of films. The COS for the 70 nm thick blend film is the smallest (6.1%), while the 100 nm thick film has a COS of 8.6%. In contrast, the film with 300 nm thickness shows a much higher COS of 15.6%. When the active layer thickness exceeded 300 nm, thick film s-OPVs maintained more than 80% of their initial efficiency after 1000 stretch–release cycles at 15% strain. In contrast, thin-film devices show significant damage after repeated mechanical stress.
The fabrication of ternary OPVs by adding polymer acceptors with larger molecular weight is just as good an approach. Song et al.77 introduced the polymer PY-IT into the PM6:BTP-eC9 system to form a chain-entangled structure to produce a high-performance, intrinsically heat-resistant s-OPV. In this work, the ternary system of PM6:BTP-eC9:PY-IT OPV exhibited a PCE of 15.30%. In addition, the similar backbone structure of PY-IT and BTP-eC9 makes PY-IT more compatible with the donor material leading to the presence of a large number of connecting molecules and the formation of entangled chain networks, and the COS of the ternary blend films with 10% and 20% PY-IT were increased by 17.14% and 27.14%, respectively. The efficiency of the best ternary device was retained by 88% after 200 cycles at 8% tensile strain, which was 23.8% higher than that of the binary device, and it indicated that the entangled structure could dissipate the stresses well and avoid the device from degrading or damaging under stretching. Peng et al.78 added a highly ductile conjugated polymer acceptor P(NDI2OD-T2) into the system of PM6:N3. They found that adding 10 wt% of P(NDI2OD-T2) can enhance the photovoltaic performance by increasing the π–π stacking order and surface accumulation, optimizing domain size, and increasing the planarity of acceptor domains. The long P(NDI2OD-T2) chains formed linker molecules and entangled structures, which effectively connected the isolated regions in the active layer and provided a channel for the transport of carriers, and this entangled structure also facilitates the mechanical properties of the blend film and enhances the stretchability of the OPVs.
The second donor is typically a polymer with high molecular weight, which enhances the mechanical properties of blend films through intermolecular interactions and chain entanglements. The complementary absorption spectra of different donors broaden the spectral response range, thereby elevating the PCE. However, precise tuning of donor energy levels via copolymerization or side-chain modification is essential to minimize inter-donor disparities. Furthermore, solvent compatibility must be evaluated to prevent phase separation during blending. In high-efficiency active layer systems, acceptors are often rigid molecules that compromise film stretchability. Additionally, energy level mismatches between acceptors may induce charge recombination at interfaces, further limiting device stability. To reconcile photovoltaic performance with mechanical robustness, the development of high molecular weight acceptors and non-fullerene acceptors must be considered. These strategies remain constrained by challenges such as complex synthesis ways and limited scalability. When evaluating the two strategies for optimizing s-OPVs, while both offer benefits, the second donor strategy exhibits a more pronounced synergy in simultaneously improving mechanical properties and PCEs. This is attributed to its capacity to enhance both light-harvesting and mechanical resilience through polymer design.
Elastomers currently employed in s-OPVs typically exhibit low modulus and high tensile strength as their defining characteristics. These materials, for instance SEBS, achieve such mechanical performance through their structures, where soft segments composed of flexible chains mediate energy dissipation via molecular chain sliding under strain. Meanwhile, rigid segments (e.g., crystalline domains or aromatic units) act as crosslinking points, preventing the fracture of films by stabilizing the polymer network. This synergistic interplay enables precise tuning of the blend film's mechanical properties. Additionally, elastomers containing hydrogen-bonding groups (such as PVA) endow the blended film with self-healing capabilities. Such dynamic bonds reorganize under mechanical deformation, suppressing crack propagation and enhancing device durability under strain. Beyond mechanical considerations, economic feasibility can also be prioritized in photovoltaic applications. The selection of elastomers should balance their cost-effectiveness and availability with performance requirements.
To optimize elastomer efficacy, the optimal concentration within the active layer must be explored. The percolation threshold—the critical composition ratio at which a minority polymer forms a network within the matrix represents a pivotal parameter in blend design. Below this threshold, isolated elastomer domains fail to establish effective stress-dissipating pathways; above it, excessive elastomer loading may disrupt the conjugated polymer domain organization, compromising charge transport.82 By adjusting the elastomer-to-conjugated polymer ratio, an interconnected network can be achieved. The conjugated polymers form ordered π-stacked domains for efficient charge transport, while the elastomer phase establishes a network that redistributes strain energy. This dual functionality ensures that the active layer retains high photovoltaic performance even under extreme mechanical deformation.
Li et al.83 demonstrated the potential of elastomeric diluents for a wide range of applications in s-OPVs by using a commercially available polymer elastomer as a diluent for PM6:PYFT-o (Fig. 10a). At a fraction of 41% elastomer in the active layer, the fracture strain of the blend film was >50% and PCE >10%, and at up to 90% elastomer mass fraction, the fracture strain of the blend film was >1000% and elastic recovery (ER) >90% (Fig. 10b). Lee et al.84 developed an active layer based on the co-continuous structure of the conjugated polymer D18 and the thermoplastic elastomer SEBS. It was found that when the ratio of D18 to SEBS is 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 w/w, D18 and SEBS can form an optimal co-continuous structure, which provides a channel for the dissipation of stress. The s-OPV based on D180.4:SEBS0.6:L8-BO retained 86% of the initial PCE at 50% strain and 90% PCE retention after 200 cycles of cyclic stretching at 15% strain. Tang et al.85 successfully induced the formation of a high-density fibrous network with low crystallinity in the bottom D18 layer. Additionally, they effectively inhibited the generation of a large phase separation between Y6 and SEBS in the top layer by employing a sequential deposition technique, wherein the bottom D18 layer and the top Y6:SEBS layer were deposited with the addition of the additive p-xylene in the primary solvent. Furthermore, the solid additive 1,3-dibromo-5-chlorobenzene was employed to facilitate the formation of more ordered crystalline domains of Y6 within the SEBS matrix. This resulted in the effective dissipation of stress on the active layer, and the s-OPV based on this active layer exhibited up to 26.38% COS with a 16.54% PCE. Zhang et al.86 incorporated SEBS into small-molecule donor B1 and acceptor BTP-eC9-4F to fabricate s-OPVs. The crystalline quality of the blend film can be enhanced by SEBS through its ability to prolong the duration of both nucleation and rearrangement during the process. This leads to a reduction in recombination losses and an improvement in PCE. Furthermore, during the stretching process, the SEBS segments within the films absorb and release mechanical stresses, enhancing tensile ability. The blend film with 10% SEBS achieved a COS of 5.28%. After 10% stretching, the stretchable devices based on B1:BTP-ec9-4F containing 0%, 1% and 10% SEBS demonstrated a maintenance of 24.18%, 34.06%, and 62.36% of their initial PCEs, respectively. These findings outlined a straightforward strategy for achieving s-OPVs using all small molecules. Zheng et al.87 fabricated intrinsically s-OPVs with top-illuminated structures by adding 5–10% styrene–ethylene–propylene–styrene triblock copolymer (SEPS) to the PM6:L8-BO:BTP-eC9 active layer system, achieving an efficiency of 15.71% and a PCE retention rate of more than 80% after 200 stretching cycles at 10% strain. Yang et al.88 added ethylene-vinyl acetate (EVA) (Fig. 10a) into the PTzBI-oF:PYIT all-polymer blends. The addition of EVA proves that the formation of dual-network helps dissipate applied strains in the ternary system. Intrinsically stretchable devices based on active layers of PTzBI-oF:PYIT and PTzBI-oF:PYIT:15% EVA were fabricated to verify the effectiveness of EVA. The initial PCE of the PTzBI-oF:PYIT:15% EVA device was 10.73%, and its PCE retained 8.74% at 30% strain. In contrast, the EVA-free devices exhibited a similar PCE of 10.81%, and the PCE quickly dropped to 7.33% when only 20% strain was applied (Fig. 10c). Li et al.89 enhanced the mechanical properties of PM6:PBQx-TF:PY-IT blends by adding polyurethane (PU) (Fig. 10a). The COS value increased with the content of PU (Fig. 10d). The incorporation of thermoplastic elastomer led to the PCE retention of 85.75% and 96.06% in 25 cm2 flexible and super-flexible devices with 1% PU addition. In the study by Kang et al.,90 isophorone diisocyanate (IPDI) was used as the linking unit in the design and synthesis of polyurethane insulation K-BA20 (or 50), which contains PDMS and 2D aryl block 9,9-diphenyl-9H-fluorene (DPF). The incorporation of 15 wt% K-BA20 into the PM6:Y6 blend film resulted in a 3.2-fold enhancement in the elongation at break, from 5.81% to 18.46% (Fig. 10e). And the presence of intermolecular hydrogen bonds has been demonstrated to result in the formation of strong interlayer adhesion, thereby ensuring the stability of the photovoltaic devices under stress conditions.
:60 w/w, D18 and SEBS can form an optimal co-continuous structure, which provides a channel for the dissipation of stress. The s-OPV based on D180.4:SEBS0.6:L8-BO retained 86% of the initial PCE at 50% strain and 90% PCE retention after 200 cycles of cyclic stretching at 15% strain. Tang et al.85 successfully induced the formation of a high-density fibrous network with low crystallinity in the bottom D18 layer. Additionally, they effectively inhibited the generation of a large phase separation between Y6 and SEBS in the top layer by employing a sequential deposition technique, wherein the bottom D18 layer and the top Y6:SEBS layer were deposited with the addition of the additive p-xylene in the primary solvent. Furthermore, the solid additive 1,3-dibromo-5-chlorobenzene was employed to facilitate the formation of more ordered crystalline domains of Y6 within the SEBS matrix. This resulted in the effective dissipation of stress on the active layer, and the s-OPV based on this active layer exhibited up to 26.38% COS with a 16.54% PCE. Zhang et al.86 incorporated SEBS into small-molecule donor B1 and acceptor BTP-eC9-4F to fabricate s-OPVs. The crystalline quality of the blend film can be enhanced by SEBS through its ability to prolong the duration of both nucleation and rearrangement during the process. This leads to a reduction in recombination losses and an improvement in PCE. Furthermore, during the stretching process, the SEBS segments within the films absorb and release mechanical stresses, enhancing tensile ability. The blend film with 10% SEBS achieved a COS of 5.28%. After 10% stretching, the stretchable devices based on B1:BTP-ec9-4F containing 0%, 1% and 10% SEBS demonstrated a maintenance of 24.18%, 34.06%, and 62.36% of their initial PCEs, respectively. These findings outlined a straightforward strategy for achieving s-OPVs using all small molecules. Zheng et al.87 fabricated intrinsically s-OPVs with top-illuminated structures by adding 5–10% styrene–ethylene–propylene–styrene triblock copolymer (SEPS) to the PM6:L8-BO:BTP-eC9 active layer system, achieving an efficiency of 15.71% and a PCE retention rate of more than 80% after 200 stretching cycles at 10% strain. Yang et al.88 added ethylene-vinyl acetate (EVA) (Fig. 10a) into the PTzBI-oF:PYIT all-polymer blends. The addition of EVA proves that the formation of dual-network helps dissipate applied strains in the ternary system. Intrinsically stretchable devices based on active layers of PTzBI-oF:PYIT and PTzBI-oF:PYIT:15% EVA were fabricated to verify the effectiveness of EVA. The initial PCE of the PTzBI-oF:PYIT:15% EVA device was 10.73%, and its PCE retained 8.74% at 30% strain. In contrast, the EVA-free devices exhibited a similar PCE of 10.81%, and the PCE quickly dropped to 7.33% when only 20% strain was applied (Fig. 10c). Li et al.89 enhanced the mechanical properties of PM6:PBQx-TF:PY-IT blends by adding polyurethane (PU) (Fig. 10a). The COS value increased with the content of PU (Fig. 10d). The incorporation of thermoplastic elastomer led to the PCE retention of 85.75% and 96.06% in 25 cm2 flexible and super-flexible devices with 1% PU addition. In the study by Kang et al.,90 isophorone diisocyanate (IPDI) was used as the linking unit in the design and synthesis of polyurethane insulation K-BA20 (or 50), which contains PDMS and 2D aryl block 9,9-diphenyl-9H-fluorene (DPF). The incorporation of 15 wt% K-BA20 into the PM6:Y6 blend film resulted in a 3.2-fold enhancement in the elongation at break, from 5.81% to 18.46% (Fig. 10e). And the presence of intermolecular hydrogen bonds has been demonstrated to result in the formation of strong interlayer adhesion, thereby ensuring the stability of the photovoltaic devices under stress conditions.
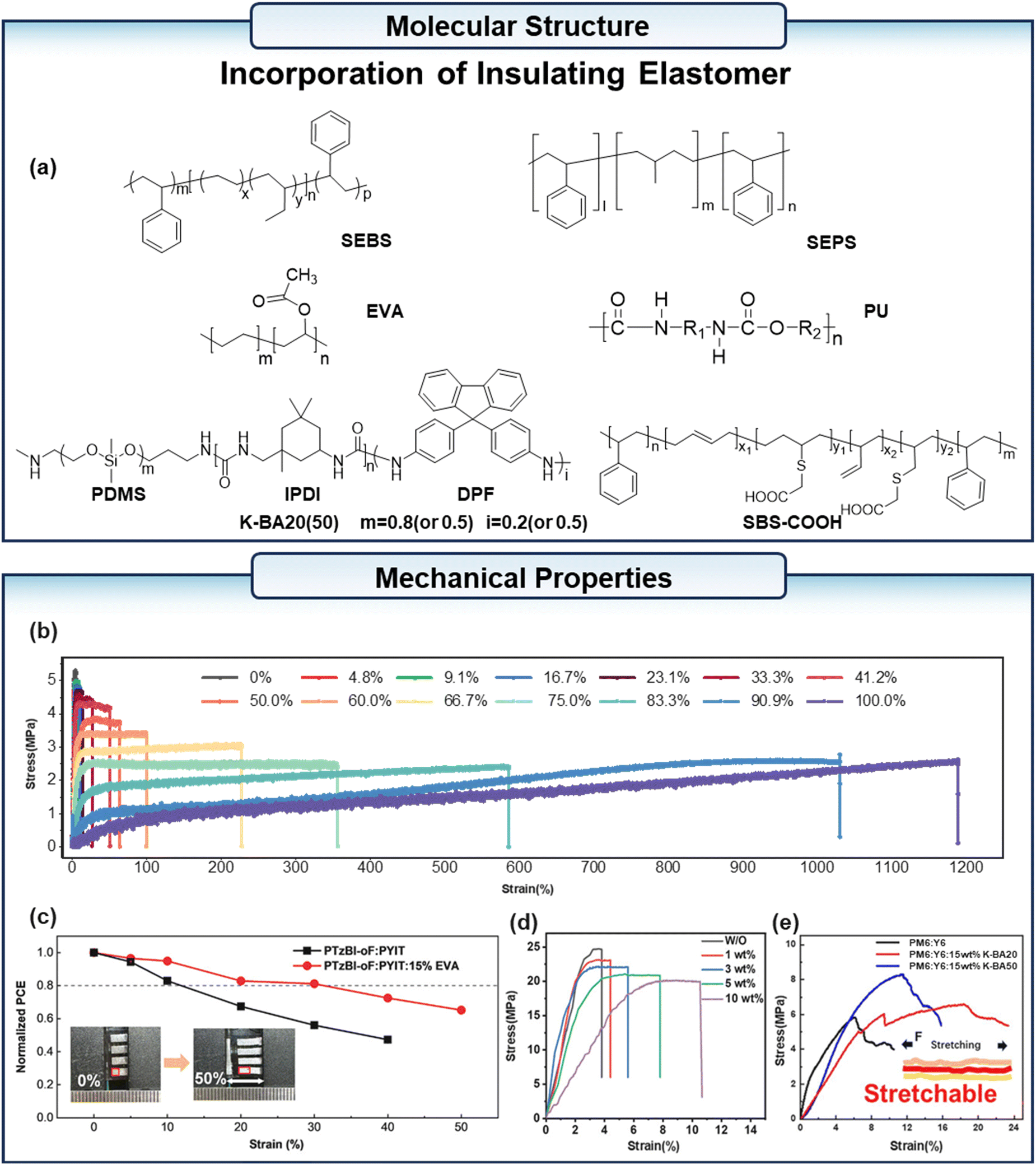 | ||
| Fig. 10 (a) The chemical structure of the elastomers used as the third component in the active layer. (b) The stress–strain curves of dilute-absorber blend films with different weight fractions of elastomer. (c) Normalized PCE as a function of the strain for devices with and without 15% EVA. (d) Stress–strain curves of the blend films with different contents of PU. (e) The stress–strain curves of PM6:Y6 and PM6:Y6 with 15 wt% of K-BA20 and K-BA50. (b) Reproduced with permission from ref. 83. Copyright 2023, Wiley-VCH. (c) Reproduced with permission from ref. 88. Copyright 2024, Wiley-VCH. (d) Reproduced with permission from ref. 89. Copyright 2024, Wiley-VCH. (e) Reproduced with permission from ref. 90. Copyright 2023, Elsevier. | ||
The elevated COS of these insulating polymers is attributable to their chemical structure. However, the disparity in surface energy between conjugated and insulating polymers inevitably results in the segregation of disparate domains, thereby influencing the performance of OPVs. To improve the miscibility between conjugated polymer and insulating polymer, Zhang et al.91 introduced varied carboxyl groups into the side chains of SBS, synthesizing SBS-COOH (Fig. 10a) with differing surface energy. The resultant films (PM6:SBS-COOH) exhibited significantly enhanced mechanical stretchability, attributable to both enhanced miscibility and hydrogen bonding interactions between PM6 and SBS-COOH. Following the incorporation of SBS-COOH, the PM6: 20% SBS-COOH blend demonstrated a COS of 21.48%, which is significantly higher than that of the pure PM6 film (8.32%) and the PM6:20% SBS blend (5.93%).
 | ||
| Fig. 11 (a) The chemical structure of the solvent additive CN, the stress–strain curves of blend films with different content of CN, and the schematic morphology of blend films. (b) The chemical structure of a photo-crosslinkable BAC and the stress–strain curves of blend films with different content of BAC and different cross-linking time, and the schematic morphology of blend films with appropriate crosslinking density. (c) The chemical structure of PM6-b-PYIT and the schematic morphology of blend films. (a) Reproduced with permission from ref. 92. Copyright Wiley-VCH, 2022. (b) Reproduced with permission from ref. 93. Copyright Wiley-VCH, 2022. (c) Reproduced with permission from ref. 94. Copyright Wiley-VCH, 2024. | ||
Adding crosslinking agents to limit the rearrangement of polymers or small molecules in the blend film is one of the ways to improve the stretchability of OPVs. Using a photo-crosslinkable small molecule 2,6-bis(4-azidobenzylidene) cyclohexanone (BAC) (Fig. 11b) as the crosslinking agent, Wang et al.93 controlled the crosslink density by controlling the concentration of crosslinker added and the crosslinking time. resulting in an increase in the elongation of the PM6:Y6 blend film from 4.5% to 18%, and they found that with a cross-linking time of 90 seconds, the stretchability of the films reached 20%. The decrease in elongation at 120 seconds may be due to the long crosslinking time and the high crosslinking density that stiffens the polymer network structure and greatly reduces the ability of the polymer chains to move under external strains. The s-OPV prepared on this basis retained 64% of the initial PCE after 1000 tensile cycles and the strain at 80% of PCE was 20%. Tseng et al.94 synthesized a D–A block copolymer (PM6-b-PYIT) (Fig. 11c). This copolymer worked as a compatibilizer in the PM6:L8-BO based active layer, and the high molecular structure similarity between the PM6-b-PYIT and the host materials improved the miscibility between the donor and acceptor in the BHJ, leading to more ordered morphology of the active layer, and when the blends were stretched by 5%, the film with 0.5 wt% PM6-b-PYIT showed smaller and less cracking area than the control film. Wang et al.95 synthesized a new block copolymer additive PTB7-b-PNDI. The incorporation of PTB7-b-PNDI into the PM6:Y7 active layer connecting both small-molecule-rich and polymer-rich domains and reducing the pores between molecular chains in blend films, acts to increase the PCE and improve mechanical properties.
4. Conclusions and perspectives
The development of stretchable solar cells remains an ongoing challenge, particularly in meeting the demanding requirements of wearable electronics. Achieving reliable performance in these applications requires photovoltaic materials that combine excellent tensile properties with highly efficient energy conversion. However, designing new materials that maintain mechanical flexibility without compromising optical or electronic performance presents significant hurdles. This review provides a concise summary of recent advancements in stretchable organic photovoltaic materials since 2022, highlighting key strategies, material innovations, and performance improvements aimed at advancing the practical implementation of s-OPVs. The main performance parameters of the stretchable and flexible OPV devices mentioned above are summarized in Tables 1 and 2, respectively. The main ways to improve the film stretchability of active layer materials are mainly categorized into the following: (1) molecular design of stretchable donor and acceptor materials via structural modification. These methods commence with the most fundamental molecular structure, and the experimental process is more complex and costly. In order to achieve the greatest possible elasticity from a given material without compromising charge transport, it is essential to ensure that key material properties are carefully balanced. It is now established that the molecular weight, the regioregularity of the molecules, the side chain and the rigidity of the backbone all exert a significant influence on the ductility of the material and the electronic properties of the films. (2) Addition of a third component and additive: the incorporation of a third component and additives, such as an insulating polymer rubber with ductility, a secondary donor or acceptor, and an electroactive compatibility agent, can serve to enhance the mechanical properties of the blend film. Further developments will be placed on self-healing, easily crosslinkable and stimuli-responsive/smart organic photovoltaic materials with high intrinsic stretchability.| Active layer | Initial PCE (%) | Strain at PCE80% (%) | COS (%) | Applied strain (%) | PCE retention (Cycles) | Ref. |
|---|---|---|---|---|---|---|
| J52:N2200 | 4.30 | <20 | 19.5 | 25 | 25% (1000) | 32 |
| PBZ-2SiM:N2200 | 6.00 | 50 | 38.4 | 25 | 75% (1000) | 32 |
| PETTCVT-L:L8-BO | 6.27 | 7 | 1.3 | — | — | 33 |
| PETTCVT-M:L8-BO | 8.04 | 11 | 3.7 | — | — | 33 |
| PETTCVT-H:L8-BO | 10.1 | 16 | 7.1 | — | — | 33 |
| D18:MYT | 12.19 | 8 | 1.3 | 10 | 3% (120) | 34 |
| D18:TYT-L | 13.10 | 16 | 6.4 | 10 | 37% (120) | 34 |
| D18:TYT-S | 14.37 | 31 | 21.6 | 10 | 79% (120) | 34 |
| PM6:Y6 | 12.80 | ∼10 | 8.5 | — | — | 38 |
| PM6:FDY-m-TAT | 14.29 | ∼20 | 18.2 | — | — | 38 |
| PBQx-TF:MYT | 12.14 | 15 | 10.3 | 20 | 9% (120) | 40 |
| PBQx-TF:DYBT-C0 | 13.19 | 21 | 17.3 | 20 | 27% (120) | 40 |
| PBQx-TF:DYBT-C4 | 14.25 | 35 | 31.8 | 20 | 70% (120) | 40 |
| PBQx-TF:DYBT-C8 | 12.55 | 30 | 28.0 | 20 | 75% (120) | 40 |
| PM6-b-PDMS:DYBT;DYPDMS | 12.74 | 41 | 31.9 | 10 | 80% (100) | 47 |
| PBDB-T:PYDBT | 8.54 | 18.0 | 11.7 | — | — | 48 |
| PBDB-T:PYFS-Ran | 8.17 | 32.1 | 18.1 | — | — | 48 |
| PBDB-T:PYFS-Reg | 10.64 | 36.7 | 22.4 | — | — | 48 |
| PM6:Y6 | 12.79 | 11 | 8.17 | — | — | 51 |
| PM6:DY-FBrL | 14.31 | 23 | 18.54 | — | — | 51 |
| PM6:PDY-FL | 11.61 | 31 | 23.45 | — | — | 51 |
| PBDB-TF:MYBO | 12.89 | 11.7 | 2 | 10 | <25% (100) | 54 |
| PBET-TF:MYBO | 12.93 | 50.7 | 31 | 10 | >75% (150) | 54 |
| D18:L8-BO | 12.77 | 7 | 1.5 | 10 | 8% (100) | 56 |
| D180.8-r-PEHDT0.2:L8-BO | 10.97 | 18 | 10.4 | 10 | 54% (100) | 56 |
| D180.8-s-PEHDT0.2:L8-BO | 14.31 | 31 | 17.2 | 10 | 75% | 56 |
| D180.8:PEHDT0.2:L8-BO | 11.71 | 12 | 5.6 | 10 | 31% (100) | 56 |
| D18:L8-BO | 12.4 | 8 | 1.9 | — | — | 58 |
| D18H-b-PDMS:L8-BO | 11.9 | 16 | 6.6 | — | — | 58 |
| PM6:Y7 | 11.05 | 15 | 1.8 | 15 | 41% (120) | 59 |
| PhAm5:Y7 | 12.73 | 31.6 | 13.8 | 15 | 86% (120) | 59 |
| PM7:L8-BO | 11.28 | 16.5 | 2.6 | — | — | 62 |
| PM7-Thy10:LB-BO | 13.69 | 43.1 | 13.7 | — | — | 62 |
| PM6:BTP-eC9 | 8.20 | 11 | 4.4 | 30 | 34% (100) | 66 |
| PM6:PM6-HD:BTP-eC9 | 7.12 | 21 | 12.1 | 30 | 52% (500) | 66 |
| PM6:N2200 | 3.21 | 31 | — | 30 | 53% (1000) | 66 |
| PM6:PM6-HD:N2200 | 2.28 | 50 | — | 30 | 80% (1000) | 66 |
| PBDB-T:PY-EH | 8.2 | <10 | 11.15 | — | — | 67 |
| PBDB-T:PY-SiO | 9.8 | ∼15 | 18.32 | — | — | 67 |
| PNTB6-Cl:BTP-Si4 | 14.1 | ∼80 | — | 30 | 75% (1000) | 68 |
| D18:BTP-C3 | 14.9 | ∼20 | 19.6 | 10 | 70% (100) | 69 |
| D18:BTP-EH | 15.6 | ∼30 | 25.8 | 10 | 70% (1000) | 69 |
| D18:BTP-HD | 13.7 | ∼30 | 32.5 | 10 | 75% (1000) | 69 |
| PBQx-TF:PYIT | 12.14 | 27 | 19.45 | 15 | 72% (400) | 74 |
| PBQx-TF:P2:PYI | 13.71 | 35 | 20.42 | 15 | 90% (400) | 74 |
| PM6:N2200:Y6 | 13.3 | 20 | — | 15 | 67% (1000) | 75 |
| PM6:PY-IT:P180k | 12.35 | 51.2 | 15.0 | 50 | >70% | 28 |
| PM6:eC9:TDY-α | 10.16 | — | 17.7 | 15 | 83% (1000) | 76 |
| PM6:BTP-eC9:PY-IT | 15.30 | — | 8.2 | 5 | 88% (200) | 77 |
| PM6:PYFT-o:SEBS | 10.16 | — | — | 30 | ∼80% (500) | 83 |
| D180.4:SEBS0.6/L8-BO | 8.51 | ∼50 | 126 | 15 | 90% (200) | 84 |
| D18:Y6:SEBS | 16.54 | — | 26.38 | — | — | 85 |
| PM6:L8-BO:BTP-eC9:SEPS | 15.71 | — | 14.0 | 10 | 71.2% (200) | 87 |
| PTzBI-oF:PYIT:EVA | 10.73 | 30 | 17.23 | — | — | 88 |
| PM6:Y6:BAC | 13.4 | 20 | — | 20 | 64% (1000) | 93 |
| Active layer | Initial PCE (%) | COS (%) | Bending radius (mm) | PCE retention (Cycles) | Ref. |
|---|---|---|---|---|---|
| PM6:CH8-6 | 16.2 | 40.2 | 5 | >96% (1200) | 42 |
| PM6:CH8-7 | 15.8 | 41.9 | 5 | >96% (1200) | 42 |
| PM6:CH8-T | 15.9 | 28.3 | 5 | ∼92% (600) | 42 |
| D18:N3 | 17.35 | 7.5 | 2 | 88.4% (3000) | 43 |
| D18:N3:DOY-TVT | 18.06 | 11.2 | 2 | 97% (3000) | 43 |
| PM6:BTP-eC9 | 14.60 | 5.4 | 3 | 77% (1000) | 45 |
| PM6:BTP-eC9:2Qx-C3 | 16.09 | 15.0 | 3 | 84.9% (1000) | 45 |
| D18:N3 | 17.06 | 7.8 | 2 | 89% (2000) | 46 |
| D18:N3:DOY-C2 | 17.23 | 11.5 | 2 | 96% (2000) | 46 |
| D18:N3:DOY-C4 | 17.91 | 11.7 | 2 | 98% (2000) | 46 |
| D18:N3:TOY-C4 | 16.41 | 12.1 | 2 | 98% (2000) | 46 |
| PM6:MY-BO | 13.52 | 4.98 | 5 | 57% (1500) | 37 |
| PM6:DY-TVCl | 15.23 | 8.26 | 5 | 79% (1500) | 37 |
| PM6:DY-3T | 14.34 | 10.31 | 5 | 90% (1500) | 37 |
| PM6:BOHD | 11.06 | 4.81 | 3 | ∼65% (800) | 41 |
| PM6:2BOHD-T | 12.45 | 6.23 | 3 | ∼65% (800) | 41 |
| PM6:2BOHD-TC4T | 12.77 | 9.69 | 3 | 84.2% (800) | 41 |
| PM6:2BOHD-TC6T | 10.94 | 10.51 | 3 | 88.1% (800) | 41 |
| PM6:Y6 |
13.83 | — | 7.35 | ∼80% (1500) | 44 |
| PM6:Y6:dT9TBO |
14.86 | — | 7.35 | ∼95% (1500) | 44 |
| PM6:PY-IT | 12.29 | 8.67 | — | 89% (1000) | 50 |
| PM6:PYF1-B | 13.42 | 16.08 | — | 92% (1000) | 50 |
| PM6:PY-IT | 12.86 | 9.67 | 4 | 82.46% (2000) | 53 |
| PM6-A:PYTCl-A | 13.81 | 20.10 | 4 | 91.34% (2000) | 53 |
| PM6-Cl0.8-b-D18-Cl0.2-BTD:BTP-eC9 | 16.63 | 31.29 | 8 | 95% (2000) | 57 |
| PBQx-TF:PBDB-TF:PY-IT | 16.5 | 6.1 | 8 | 91% (2000) | 71 |
| PM6:PBQx-TF:PY-IT:PU | 19.40 | — | — | 96.98% (2000) | 89 |
| PM6:Y6:K-BA20 | 11.70 | — | 6 | 37% (600) | 90 |
| PM6:Y6:K-BA50 | 11.31 | — | 6 | 53% (600) | 90 |
| B1:BTP-eC9-4F:SEBS | 8.57 | 5.28 | — | 61.7% (100) | 86 |
| PM6:L8-BO:SBS-COOH | 15.43 | — | 1 | 88.9% (40000) |
91 |
Despite the rapid development of s-OPVs, their efficiency remains inferior to that of their rigid and flexible counterparts within the same system. There is a clear need for further research and innovation in the field of stretchable photoactive materials to fully realize the advancement of wearable devices.
(1) A comprehensive understanding of the alterations in film morphology of the active layer during stretching and releasing is essential to elucidate the underlying mechanism behind the changes in the mechanical properties of the active material and to provide insights for the design of new stretchable photoactive materials. In situ stretching analysis of thin films is a crucial method that can help us to observe nanoscale morphological transition points during film stretching. For example, in situ grazing-incidence X-ray scattering96 and in situ wide/small angle X-ray scattering78 characterizations can achieve this purpose. Notably, a very recent study97 developed a temperature-controlled, rotatable in situ stretching sample stage device capable of performing in situ GIWAXS tests under variable temperature stretching conditions, which provides a new method for characterizing the molecular packing in the films. Moving forward, the development of novel in situ experimental set-ups and AI-driven data analysis methods should be continued in order to further deepen the understanding of the behavior of polymer films under tensile conditions, thus contributing to the improvement of film properties.
(2) Beyond ensuring the stability of mechanical properties, maintaining the operational stability of s-OPVs is equally crucial for their long-term reliability and performance.98,99 External factors such as oxygen and humidity pose significant threats to device efficiency and lifespan by accelerating material degradation.100 Consequently, effective environmental protection is a key priority in s-OPV development. To mitigate these risks, the development of effective encapsulation materials is essential. These materials must form an effective barrier to prevent moisture and oxygen infiltration, safeguarding the delicate active layers within the device. Additionally, achieving high optical transparency in these encapsulation materials is critical to ensure minimal interference with light absorption and energy conversion. Balancing these protective properties with optical performance is vital for enhancing the durability and efficiency of s-OPVs in real-world applications, particularly in wearable electronics and other flexible technologies.
(3) Mechanical properties of polymer blend films are often characterized by their COS values. Beyond this point, cracks begin to appear in the film, resulting in a degradation of the film properties. However, COS does not adequately represent the alterations in the film during elastic deformation. Furthermore, it has been demonstrated that, upon the surpassing of the yield point, the material will undergo irreversible deformation, which will have a significant effect on the film properties. Consequently, in addition to augmenting the value of COS, it is imperative to investigate methodologies for extending the range of elastic deformation and quantifying the fracture/adhesion energy of the film. Characterisation of the mechanical properties of free-standing films is also crucial.
(4) In addition to the design of the active layer, optimizing the fabrication process of s-OPVs is crucial for streamlining the preparation process while enhancing the performance.101–103 This is a highly significant step towards advancing the commercialization of s-OPV. Solution-based continuous deposition has proven effective for laboratory-scale OPV production; however, it is more complex for large-scale fabrication. This approach leverages molecular self-assembly to construct active and transport layers. While self-assembling deposition offers notable advantages, such as improved interface quality and enhanced cell performance, it requires precise molecular design to ensure effective self-organization. When successful, this method not only simplifies the fabrication process but also achieves an improvement in the performance of the cell, making it a promising avenue for scalable s-OPV production.
Author contributions
J. Z. conducted the literature review, performed data analysis, prepared figures, and wrote the initial draft of the manuscript. D. H., H. Y., and V. K. contributed to scientific discussions and assisted in manuscript editing. L. Y. supervised the overall research project and provided critical revisions to the manuscript. All authors reviewed and approved the final version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We are grateful for the financial support from the Science Fund for Distinguished Young Scholars of Tianjin Municipality (No. 23JCJQJC00240), the National Natural Science Foundation of China (No. 52121002), the start-up grant of Peiyang Scholar program from Tianjin University, and the Fundamental Research Funds for the Central Universities, and the Opening Project of Hubei Longzhong Laboratory (2022KF-01). We also appreciate the Agency of Innovative Development of the Republic of Uzbekistan (Project FL-9024093524) for supporting this work.References
- W. Zhang, C. Sun, I. Angunawela, L. Meng, S. Qin, L. Zhou, S. Li, H. Zhuo, G. Yang, Z. G. Zhang, H. Ade and Y. Li, Adv. Mater., 2022, 34, e2108749 CrossRef PubMed.
- Y. Cui, Y. Xu, H. Yao, P. Bi, L. Hong, J. Zhang, Y. Zu, T. Zhang, J. Qin, J. Ren, Z. Chen, C. He, X. Hao, Z. Wei and J. Hou, Adv. Mater., 2021, 33, e2102420 CrossRef PubMed.
- M. F. Li, K. H. Xian, W. C. Zhao, D. Sheng, C. H. Liu, X. Li, W. W. Li and L. Ye, Chem. Eng. J., 2023, 476, 146723 CrossRef CAS.
- M. Amjadi, Y. J. Yoon and I. Park, Nanotechnology, 2015, 26, 375501 CrossRef PubMed.
- S. Li, J. Liu, V. Kuvondikov, J. Yan and L. Ye, Matter, 2025, 8, 102062.
- J. S. Park, G. U. Kim, S. Lee, J. W. Lee, S. Li, J. Y. Lee and B. J. Kim, Adv. Mater., 2022, 34, e2201623 CrossRef PubMed.
- D. Koo, S. Jung, J. Seo, G. Jeong, Y. Choi, J. Lee, S. M. Lee, Y. Cho, M. Jeong, J. Lee, J. Oh, C. Yang and H. Park, Joule, 2020, 4, 1021–1034 CrossRef CAS.
- C. Xie, Y. Liu, W. Wei and Y. Zhou, Adv. Funct. Mater., 2022, 33, 2210675 CrossRef.
- K. Chong, X. Xu, H. Meng, J. Xue, L. Yu, W. Ma and Q. Peng, Adv. Mater., 2022, 34, e2109516 CrossRef PubMed.
- K. Xian, K. Zhou, M. Li, J. Liu, Y. Zhang, T. Zhang, Y. Cui, W. Zhao, C. Yang, J. Hou, Y. Geng and L. Ye, Chin. J. Chem., 2023, 41, 159–166 CrossRef CAS.
- K. K. Zhou, K. H. Xian, Q. C. Qi, M. Y. Gao, Z. X. Peng, J. W. Liu, Y. Liu, S. M. Li, Y. D. Zhang, Y. H. Geng and L. Ye, Adv. Funct. Mater., 2022, 32, 2201781 CrossRef CAS.
- J. W. Lee, C. Sun, B. S. Ma, H. J. Kim, C. Wang, J. M. Ryu, C. Lim, T. S. Kim, Y. H. Kim, S. K. Kwon and B. J. Kim, Adv. Energy Mater., 2020, 11, 2003367 CrossRef.
- Q. P. Fan, W. Y. Su, S. S. Chen, W. Kim, X. B. Chen, B. Lee, T. Liu, U. A. Méndez-Romero, R. J. Ma, T. Yang, W. L. Zhuang, Y. Li, Y. W. Li, T. S. Kim, L. T. Hou, C. Yang, H. Yan, D. H. Yu and E. G. Wang, Joule, 2020, 4, 658–672 CrossRef CAS.
- T. Kim, J. H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T. S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS PubMed.
- T. P. Brody, IEEE Trans. Electron Devices, 1984, 31, 1614–1628 Search PubMed.
- F. Garnier, R. Hajlaoui, A. Yassar and P. Srivastava, Science, 1994, 265, 1684–1686 CrossRef CAS PubMed.
- D. J. Lipomi, B. C. Tee, M. Vosgueritchian and Z. Bao, Adv. Mater., 2011, 23, 1771–1775 CrossRef CAS PubMed.
- M. Kaltenbrunner, M. S. White, E. D. Glowacki, T. Sekitani, T. Someya, N. S. Sariciftci and S. Bauer, Nat. Commun., 2012, 3, 770 CrossRef PubMed.
- Z. Yang, J. Deng, X. Sun, H. Li and H. Peng, Adv. Mater., 2014, 26(2643–2647), 2613 Search PubMed.
- Y. Ding, M. A. Invernale and G. A. Sotzing, ACS Appl. Mater. Interfaces, 2010, 2, 1588–1593 CrossRef CAS PubMed.
- J. A. Fan, W. H. Yeo, Y. Su, Y. Hattori, W. Lee, S. Y. Jung, Y. Zhang, Z. Liu, H. Cheng, L. Falgout, M. Bajema, T. Coleman, D. Gregoire, R. J. Larsen, Y. Huang and J. A. Rogers, Nat. Commun., 2014, 5, 3266 CrossRef PubMed.
- E. Dauzon, X. Sallenave, C. Plesse, F. Goubard, A. Amassian and T. D. Anthopoulos, Adv. Mater., 2021, 33, e2101469 CrossRef PubMed.
- D. J. Lipomi, H. Chong, M. Vosgueritchian, J. G. Mei and Z. A. Bao, Sol. Energy Mater. Sol. Cells, 2012, 107, 355–365 CrossRef CAS.
- S. Chen, S. Jung, H. J. Cho, N. H. Kim, S. Jung, J. Xu, J. Oh, Y. Cho, H. Kim, B. Lee, Y. An, C. Zhang, M. Xiao, H. Ki, Z. G. Zhang, J. Y. Kim, Y. Li, H. Park and C. Yang, Angew. Chem., Int. Ed., 2018, 57, 13277–13282 CrossRef CAS PubMed.
- Z. Peng, K. Xian, Y. Cui, Q. Qi, J. Liu, Y. Xu, Y. Chai, C. Yang, J. Hou, Y. Geng and L. Ye, Adv. Mater., 2021, 33, 2106732 CrossRef CAS PubMed.
- Y. T. Hsieh, J. Y. Chen, S. Fukuta, P. C. Lin, T. Higashihara, C. C. Chueh and W. C. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 21712–21720 CrossRef CAS PubMed.
- X. Wu, X. Zheng, T. Chen, S. Zhang, Y. Zhou, M. Wang, T. Chen, Y. Wang, Z. Bi, W. Fu, M. Du, W. Ma, L. Zuo and H. Chen, Adv. Mater., 2024, 36, e2406879 CrossRef PubMed.
- K. K. Zhou, D. X. Han, K. H. Xian, S. M. Li, M. Y. Gao, K. Zhang, B. Zhao, X. Li, Y. Chen, Y. H. Geng and L. Ye, Energy Environ. Sci., 2024, 17, 5950–5961 RSC.
- F. P. V. Koch, J. Rivnay, S. Foster, C. Müller, J. M. Downing, E. Buchaca-Domingo, P. Westacott, L. Y. Yu, M. J. Yuan, M. Baklar, Z. P. Fei, C. Luscombe, M. A. McLachlan, M. Heeney, G. Rumbles, C. Silva, A. Salleo, J. Nelson, P. Smith and N. Stingelin, Prog. Polym. Sci., 2013, 38, 1978–1989 CrossRef CAS.
- H. Kang, M. A. Uddin, C. Lee, K. H. Kim, T. L. Nguyen, W. Lee, Y. Li, C. Wang, H. Y. Woo and B. J. Kim, J. Am. Chem. Soc., 2015, 137, 2359–2365 CrossRef CAS PubMed.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- M. C. Xu, D. Zhang, Z. Y. Wang, Z. T. Liu, X. Gao, J. Y. He, Y. R. Gao, Z. L. Li and M. Shao, Chem. Eng. J., 2022, 440, 2201589 Search PubMed.
- J. W. Lee, T. N. L. Phan, E. S. Oh, H. G. Lee, T. S. Kim and B. J. Kim, Adv. Funct. Mater., 2023, 33, 2305851 CrossRef CAS.
- J. W. Lee, C. Sun, J. Lee, D. J. Kim, W. J. Kang, S. Lee, D. Kim, J. Park, T. N. L. Phan, Z. Tan, F. S. Kim, J. Y. Lee, X. Bao, T. S. Kim, Y. H. Kim and B. J. Kim, Adv. Energy Mater., 2024, 14, 2303872 CrossRef CAS.
- W. Liu, J. Yuan, C. Zhu, Q. Wei, S. Liang, H. Zhang, G. Zheng, Y. Hu, L. Meng, F. Gao, Y. Li and Y. Zou, Sci. China: Chem., 2022, 65, 1374–1382 CrossRef CAS.
- J. W. Lee, C. Sun, C. Lee, Z. P. Tan, T. N. L. Phan, H. Jeon, D. Jeong, S. K. Kwon, Y. H. Kim and B. J. Kim, ACS Energy Lett., 2023, 8, 1344–1353 CrossRef CAS.
- H. Yin, G. Xie, T. Wu, S. Liu, D. Chen and Y. Chen, Macromol. Rapid Commun., 2024, e2400433 Search PubMed.
- Y. Ding, W. A. Memon, D. Zhang, Y. Zhu, S. Xiong, Z. Wang, J. Liu, H. Li, H. Lai, M. Shao and F. He, Angew. Chem., Int. Ed., 2024, 63, e202403139 CrossRef CAS PubMed.
- H. B. Chen, Z. Zhang, P. R. Wang, Y. X. Zhang, K. Q. Ma, Y. Lin, T. A. Duan, T. F. He, Z. F. Ma, G. K. Long, C. X. Li, B. Kan, Z. Y. Yao, X. J. Wan and Y. S. Chen, Energy Environ. Sci., 2023, 16, 1773–1782 RSC.
- J. W. Lee, C. Sun, S. Lee, D. J. Kim, E. S. Oh, T. N. L. Phan, T. H. Q. Nguyen, S. Seo, Z. Tan, M. J. Lee, J. Y. Lee, X. C. Bao, T. S. Kim, C. Lee, Y. H. Kim and B. J. Kim, Nano Energy, 2024, 125, 109541 CrossRef CAS.
- J. Liu, W. Zhou, J. Deng, X. Geng, S. Y. Jeong, Y. Cui, H. Y. Woo, F. Wu, F. Liu and L. Chen, Nano Energy, 2024, 121, 109218 CrossRef CAS.
- Z. Zhang, S. Yuan, T. Chen, J. Wang, Y.-Q.-Q. Yi, B. Zhao, M. Li, Z. Yao, C. Li, X. Wan, G. Long, B. Kan and Y. Chen, Energy Environ. Sci., 2024, 17, 5719–5729 RSC.
- W. Song, Q. Ye, S. Yang, L. Xie, Y. Meng, Z. Chen, Q. Gu, D. Yang, J. Shi and Z. Ge, Angew. Chem., Int. Ed., 2023, 62, e202310034 CrossRef CAS PubMed.
- F. Qi, Y. Li, R. Zhang, F. R. Lin, K. Liu, Q. Fan and A. K. Jen, Angew. Chem., Int. Ed., 2023, 62, e202303066 CrossRef CAS PubMed.
- S. You, Y. Zhang, B. Huang, S. Y. Jeong, X. Shuai, S. Huang, H. Y. Woo, F. Wu and L. Chen, Adv. Funct. Mater., 2024, 35, 2414803 CrossRef.
- Q. Ye, Z. Chen, D. Yang, W. Song, J. Zhu, S. Yang, J. Ge, F. Chen and Z. Ge, Adv. Mater., 2023, 35, e2305562 CrossRef PubMed.
- J. W. Lee, T. H. Q. Nguyen, W. J. Kang, S. Seo, S. Lee, S. Lee, J. Choi, J. Park, J. Y. Lee, T. S. Kim and B. J. Kim, Energy Environ. Sci., 2025, 18, 3325–3340 RSC.
- J. W. Lee, C. Sun, S. W. Lee, G. U. Kim, S. Li, C. Wang, T. S. Kim, Y. H. Kim and B. J. Kim, Energy Environ. Sci., 2022, 15, 4672–4685 RSC.
- J. W. Lee, S. W. Lee, J. Kim, Y. H. Ha, C. Sun, T. N. L. Phan, S. Lee, C. Wang, T. S. Kim, Y. H. Kim and B. J. Kim, J. Mater. Chem. A, 2022, 10, 20312–20322 RSC.
- H. P. Hu, W. Zhou, J. B. Liu, J. P. Xie, S. Y. You, S. Y. Jeong, H. Y. Woo, F. Y. Wu and L. Chen, Chem. Eng. J., 2024, 491, 152009 CrossRef CAS.
- Y. Ding, S. Xiong, W. A. Memon, D. Zhang, Z. Wang, M. Li, Z. Deng, H. Li, M. Shao and F. He, Angew. Chem., Int. Ed., 2025, e202421430 CAS.
- X. N. Luo, G. Freychet, Z. Q. Gan, K. An, H. J. Du, C. Wang, N. Li, W. K. Zhong and L. Ying, Macromolecules, 2023, 56, 8928–8938 CrossRef CAS.
- J. Liu, J. Deng, Y. Zhu, X. Geng, L. Zhang, S. Y. Jeong, D. Zhou, H. Y. Woo, D. Chen, F. Wu and L. Chen, Adv. Mater., 2023, 35, e2208008 CrossRef PubMed.
- J.-W. Lee, E. S. Oh, S. Lee, T. N.-L. Phan, T.-S. Kim, J.-Y. Lee, J. R. Reynolds and B. J. Kim, Joule, 2025, 9, 101792 CrossRef CAS.
- J. Kim, G. U. Kim, D. J. Kim, S. Lee, D. Jeong, S. Seo, S. J. Ko, S. C. Yoon, T. S. Kim and B. J. Kim, J. Mater. Chem. A, 2023, 11, 4808–4817 RSC.
- J. W. Lee, H. G. Lee, E. S. Oh, S. W. Lee, T. N. Phan, S. Li, T. S. Kim and B. J. Kim, Joule, 2024, 8, 204–223 CrossRef CAS.
- C. Lin, R. Peng, W. Song, J. Gao, T. Feng, Y. Bai, Q. Liu, M. Yang, J. Zhang and Z. Ge, Angew. Chem., Int. Ed., 2025, 64, e202420121 CrossRef CAS PubMed.
- H. G. Lee, J. W. Lee, E. S. Oh, M. J. Lee, T. S. Kim, C. Lee and B. J. Kim, J. Mater. Chem. A, 2024, 12, 19039–19051 RSC.
- J. W. Lee, S. Seo, S. W. Lee, G. U. Kim, S. Han, T. N. Phan, S. Lee, S. Li, T. S. Kim, J. Y. Lee and B. J. Kim, Adv. Mater., 2022, 34, e2207544 CrossRef PubMed.
- Q. P. Wan, H. Jeon, S. Seo, E. S. Oh, J. W. Lee, C. Wang, T. S. Kim, B. J. Kim and B. C. Thompson, Chem. Mater., 2023, 35, 10476–10486 CrossRef CAS.
- H. Ke, M. Gao, S. Li, Q. Qi, W. Zhao, X. Li, S. Li, V. Kuvondikov, P. Lv, Q. Wei and L. Ye, Sol. RRL, 2023, 7, 2300109 CrossRef CAS.
- Q. Wan, S. Seo, S. W. Lee, J. Lee, H. Jeon, T. S. Kim, B. J. Kim and B. C. Thompson, J. Am. Chem. Soc., 2023, 145, 11914–11920 CrossRef CAS PubMed.
- H. Wang, H. Kou, J. Pan, X. Wang, D. Liu and R. Yang, Small, 2025, e2412767 CrossRef PubMed.
- J. G. Mei and Z. N. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- D. Rodriquez, J. G. Kohl, P. Morel, K. Burrows, G. Favaro, S. E. Root, J. Ramirez, M. A. Alkhadra, C. W. Carpenter, Z. Fei, P. Boufflet, M. Heeney and D. J. Lipomi, ACS Macro Lett., 2018, 7, 1003–1009 CrossRef CAS PubMed.
- X. Li, H. Z. Ke, S. S. Li, M. Y. Gao, S. M. Li, J. F. Yu, H. J. Xie, K. K. Zhou, K. Zhang and L. Ye, Adv. Funct. Mater., 2024, 34, 2400702 CrossRef CAS.
- J. He, D. Zhang, J. Liu, L. Yang, Y. Gao and M. Shao, ACS Appl. Mater. Interfaces, 2024, 16, 22294–22302 CrossRef CAS PubMed.
- Z. Wang, D. Zhang, L. Yang, O. Allam, Y. Gao, Y. Su, M. Xu, S. Mo, Q. Wu, Z. Wang, J. Liu, J. He, R. Li, X. Jia, Z. Li, L. Yang, M. D. Weber, Y. Yu, X. Zhang, T. J. Marks, N. Stingelin, J. Kacher, S. S. Jang, A. Facchetti and M. Shao, Science, 2025, 387, 381–387 CrossRef CAS PubMed.
- D. Zhang, J. F. Liu, X. Gao, Z. Wang, J. Y. He, Z. Y. Wang, L. Yang, Y. R. Gao and M. Shao, Energy Environ. Sci., 2025, 18, 2342–2352 RSC.
- S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Macromolecules, 2014, 47, 1981–1992 CrossRef CAS.
- L. Ma, Y. Cui, J. Zhang, K. Xian, Z. Chen, K. Zhou, T. Zhang, W. Wang, H. Yao, S. Zhang, X. Hao, L. Ye and J. Hou, Adv. Mater., 2023, 35, e2208926 CrossRef PubMed.
- M. F. Li, Z. X. Peng, K. H. Xian, W. C. Zhao, C. X. Liu, Y. Chen, C. H. Cui and L. Ye, J. Mater. Chem. A, 2023, 11, 5606–5614 RSC.
- T. Liu, K. Zhou, R. Ma, L. Zhang, C. Huang, Z. Luo, H. Zhu, S. Yao, C. Yang, B. Zou and L. Ye, Aggregate, 2023, 4, e308 CrossRef CAS.
- G. U. Kim, C. Choi, D. Jeong, D. J. Kim, T. N. L. Phan, S. Song, J. Park, T. S. Kim, Y. H. Kim and B. J. Kim, Adv. Energy Mater., 2023, 13, 2302125 CrossRef CAS.
- Z. Y. Wang, D. Zhang, L. Yang, M. C. Xu, J. F. Liu, Z. Wang, Y. R. Gao, L. Y. Zhang, L. B. Niu and M. Shao, ACS Mater. Lett., 2024, 6, 4710–4718 CrossRef CAS.
- K. H. Xian, K. Zhang, T. Zhang, K. K. Zhou, Z. G. Zhang, J. H. Hou, H. L. Zhang, Y. H. Geng and L. Ye, Energy Environ. Sci., 2025, 18, 2570–2583 RSC.
- W. Song, K. Yu, J. Ge, L. Xie, R. Zhou, R. Peng, X. Zhang, M. Yang, Z. Wei and Z. Ge, Matter, 2022, 5, 1877–1889 CrossRef CAS.
- Z. Peng, K. Xian, J. Liu, Y. Zhang, X. Sun, W. Zhao, Y. Deng, X. Li, C. Yang, F. Bian, Y. Geng and L. Ye, Adv. Mater., 2023, 35, 2207884 CrossRef CAS PubMed.
- Y. Bai, S. Li, Q. Wang, Q. Chen, Z. Zhang, S. Meng, Y. Zang, H. Fu, L. Xue, L. Ye and Z. Zhang, Natl. Sci. Rev., 2025, 12, nwaf019 CrossRef PubMed.
- N. Balar, J. J. Rech, R. Henry, L. Ye, H. Ade, W. You and B. T. O’Connor, Chem. Mater., 2019, 31, 5124–5132 CrossRef CAS.
- A. A. Shafe, H. M. Schrickx, K. Ding, H. Ade and B. T. O’Connor, ACS Energy Lett., 2023, 8, 3720–3726 CrossRef CAS.
- S. A. Mollinger, B. A. Krajina, R. Noriega, A. Salleo and A. J. Spakowitz, ACS Macro Lett., 2015, 4, 708–712 CrossRef CAS PubMed.
- S. Li, M. Gao, K. Zhou, X. Li, K. Xian, W. Zhao, Y. Chen, C. He and L. Ye, Adv. Mater., 2024, 36, 2307278 CrossRef CAS PubMed.
- J. W. Lee, T. H. Q. Nguyen, E. S. Oh, S. Lee, J. Choi, H. S. Kwon, C. Wang, S. Lee, J. Y. Lee, T. S. Kim and B. J. Kim, Adv. Energy Mater., 2024, 14, 2401191 CrossRef CAS.
- W. B. Tang, Z. C. Ding, Y. L. Su, Q. Weng, Y. Zhang, R. P. Li, W. L. Huang, Z. C. Wang, Y. Wu, Y. C. Han, K. Zhao, Z. Yang, X. C. Wang and S. Z. Liu, Adv. Funct. Mater., 2024, 34, 2312289 CrossRef CAS.
- C. Y. Zhang, Y. Q. Liu, H. X. Li, X. Y. Cui, Z. D. Wei, Y. H. Liu, M. H. Li, A. D. Zhang, P. Cheng and Z. S. Bo, Chin. J. Polym. Sci., 2025, 43, 271–277 CrossRef CAS.
- X. Zheng, X. Wu, Q. Wu, Y. Han, G. Ding, Y. Wang, Y. Kong, T. Chen, M. Wang, Y. Zhang, J. Xue, W. Fu, Q. Luo, C. Ma, W. Ma, L. Zuo, M. Shi and H. Chen, Adv. Mater., 2024, 36, e2307280 CrossRef PubMed.
- W. Y. Yang, X. N. Luo, M. K. Li, C. Q. Shi, Z. Y. Wang, Z. Y. Yang, J. M. Wu, X. W. Zhang, W. B. Huang, D. G. Ma, C. Wang, W. K. Zhong and L. Ying, Adv. Energy Mater., 2024, 15, 2403259 CrossRef.
- H. Li, J. Le, H. Tan, L. Hu, X. Li, K. Zhang, S. Zeng, Q. Liu, M. Zhang, L. Shi, Z. Cai, S. Liu, H. Li, L. Ye, X. Hu and Y. Chen, Adv. Mater., 2025, 37, e2411989 CrossRef PubMed.
- X. Kang, Y. Bao, T. Y. Feng, Y. W. Wu, Y. W. Zhang, Y. Zhao, C. M. Yang, M. L. Sun and X. C. Bao, Chem. Eng. J., 2023, 476, 146828 CrossRef CAS.
- J. Zhang, Q. Chen, M. Li, G. Zhang, Z. Zhang, X. Deng, J. Xue, C. Zhao, C. Xiao, W. Ma and W. Li, Adv. Mater., 2024, 36, e2312805 CrossRef PubMed.
- Z. C. Ding, Y. Zhang, Y. L. Su, Y. Wu, Y. C. Han, K. Zhao and S. Z. Liu, Energy Environ. Mater., 2023, 6, e12421 CrossRef CAS.
- Z. Wang, D. Zhang, M. Xu, J. Liu, J. He, L. Yang, Z. Li, Y. Gao and M. Shao, Small, 2022, 18, e2201589 CrossRef PubMed.
- Y. C. Tseng, Q. P. Fan, C. Y. Tsai, J. F. Chang, M. H. Yu, H. Z. Tseng, H. L. Yip, F. R. Lin, A. K. Y. Jen and C. C. Chueh, Adv. Funct. Mater., 2024, 34, 2408993 CrossRef CAS.
- M. S. Wang, K. Zheng and X. M. Cai, Opt. Mater., 2025, 159, 116639 CrossRef CAS.
- M. Y. Aliouat, S. Escoubas, M. C. Benoudia, D. Ksenzov, D. Duché, E. Bènevent, C. Videlot-Ackermann, J. Ackermann, O. Thomas and S. Grigorian, J. Appl. Phys., 2020, 127, 045108 CrossRef CAS.
- Y. Chen, S. M. Li, Z. B. Shen, C. L. Sun, J. T. Feng and L. Ye, Sci. China Mater., 2024, 67, 3917–3924 CrossRef CAS.
- Y. Bae, D. Kim, S. Li, Y. Choi, S. Son, T. Park and L. Ye, Prog. Polym. Sci., 2024, 159, 101899 CrossRef CAS.
- Z. Peng, K. Xian, J. Liu, Y. Zhang, X. Sun, W. Zhao, Y. Deng, X. Li, C. Yang, F. Bian, Y. Geng and L. Ye, Adv. Mater., 2023, 35, 2207884 CrossRef CAS PubMed.
- K. Xian, R. Ma, K. Zhou, J. Liu, M. Gao, W. Zhao, M. Li, Y. Geng and L. Ye, Aggregate, 2024, 5, e466 CrossRef CAS.
- L. Pei, D. Han, Y. Wang, M. Gao, J. Wu, C. Sun, M. Yu, Y. Wang, X. Li, H. Ke and L. Ye, Adv. Funct. Mater., 2025 DOI:10.1002/adfm.202425892.
- K. Zhou, W. Zhai, V. Kuvondikov, K. Dai and L. Ye, Nexus, 2025 DOI:10.1016/j.ynexs.2025.100072.
- R. Ma, Z. Luo, Y. Zhang, L. Zhan, T. Jia, P. Cheng, C. Yan, Q. Fan, S. Liu, L. Ye, G. Zhang, X. Xu, W. Gao, Y. Wu, J. Wu, Y. Li, Y. Liu, F. Liu, J. Song, H. Chen, W. Chen, X. Zhang, Y. Liu, J. Yuan, Q. Liu, Z. Kan, H. Yin, X. Li, Y. Ma, D. Deng, L. Zhu, Y. Huo, B. Fan, H. Fu, X. Liao, H. Hu, C. Li, R. Yu, H. Hu, Z. Yao, Y. Cai, D. Qian, Y. Cui, H. Yao, B. Xu, B. Kan, K. Gao, C. Duan, X. Hu and H. Sun, Sci. China Mater., 2025 DOI:10.1007/s40843-025-3366-9.
Footnote |
| † This work is dedicated to the 130th Anniversary of Tianjin University and the Special Collection celebrating this milestone. |
| This journal is © The Royal Society of Chemistry 2025 |