DOI:
10.1039/D5CE00071H
(Paper)
CrystEngComm, 2025,
27, 2070-2085
Exploring intermolecular interactions and energetics in crystalline substituted thieno[2,3-d]pyrimidines†
Received
18th January 2025
, Accepted 25th February 2025
First published on 25th February 2025
Abstract
This study explores the synthesis and crystallographic characterization of eight thieno[2,3-d]pyrimidine derivatives. Keeping in mind their practical importance in different biological applications, the understanding of the crystal structure in terms of the existence of various intermolecular interactions is of relevance. The presence of strong N–H⋯N dimers in the crystal structures of five compounds, followed by N–H⋯O and C–H⋯O interactions in the remaining three compounds, due to the presence of the amide and the sulphonamide substituents, is ascertained from crystal packing analysis. In addition, several weak intermolecular interactions, including C–H⋯π/N/S, S⋯C (σ-hole/π-hole), and S⋯S contacts, contribute towards their additional stability in the crystal. To understand this, the nature and energetics associated with these intermolecular interactions were characterized via Crystal Explorer21.5. Further, the electrostatic complementarity amongst the robust N–H⋯N dimers are evidenced via noticing the large negative and positive molecular surface electrostatic potential (MESP) values across the nitrogen and the hydrogen atoms, respectively. Moreover, carbon-bonding was explicitly described for S⋯C interactions. This implied the participation of the nucleophilic sulphur with the electrophilic carbon atom (either mediated through a σ-hole or a π-hole on the carbon atom). Besides this, the change in the directionality of these interactions, from the linear to the orthogonal orientation for tetrel interactions, was observed and justified through an analysis of the MESP, the deformation density plots and the QTAIM analysis.
1. Introduction
The importance of heterocyclic compounds has now been well-recognized in the field of drug discovery and development.1,2 However, the importance of molecular recognition in the design and discovery of fused heterocycles with improved biological activity is indispensable. In this regard, understanding the role of intermolecular interactions in crystalline fused heterocycles, on account of their varying biological applications via slight chemical modifications, is of interest.3,4 Moreover, the recognition of strong hydrogen bonding motifs among molecules in the crystalline lattice in structurally related compounds is of profound interest.5
Thieno[2,3-d]pyrimidines (TPs) are molecular scaffolds containing a thiophene ring fused with a pyrimidine moiety. Amongst these, pyrimidines are heterocyclic aromatic compounds with two nitrogen atoms at the 1 and 3 positions of a six-membered ring.6 Pyrimidines are considered as essential building blocks with widespread therapeutic applications. They are found to be the most ubiquitous member in uracil and thymine constituents of ribonucleic acid (RNA) and deoxyribonucleic acid (DNA), and are also present in many natural products such as vitamin B1 (thiamine) and in synthetic compounds like barbituric acid and veranal. Likewise, thiophenes also exhibit biological activities.7 Thiophene is a five-membered heteroaromatic compound with a sulphur atom at the 1-position.6–8 The electron pair on sulphur is significantly delocalized into the aromatic ring and thus is extremely reactive towards the synthesis of thieno[2,3-d]pyrimidine fused heterocycles (Fig. S1†). Therefore, the derivatives of thieno[2,3-d]pyrimidines display a broad spectrum of biological activities2 as antibacterial, antifungal, anti-inflammatory,9 antiviral, or anticancer agents.10–17 Moreover, their pharmacological properties can be utilized for drug discovery and studies related to therapeutics.18–22 These properties, however, inspired us to analyse their crystal packing features.23
Based on the knowledge of crystal engineering,24–26 understanding the crystalline geometry and different intermolecular interactions that govern the crystal packing becomes essential. This allows us to recognize the hierarchy of intermolecular interactions that are present in the crystal. To this extent, this justifies the probable approach of molecular pairs that form strong hydrogen bonds/dimers. So, the overall crystalline arrangement of molecules is primarily controlled by interactions between the strong donor and the strong acceptor, along with some weak intermolecular interactions of the type C–H⋯π/N/S, S⋯S, S⋯C (σ-hole/π-hole), etc. In addition to this, the energetic contributions from different intermolecular interactions toward the stabilization of crystal packing allows detailed comprehension of molecules that stack in the crystalline lattice.27,28
The purpose of the present work is to explore the effect of the structural modifications by varying functional group substituents, which could optimize the biological activity.29 This also has an impact on the crystal structure and hence requires the study of intermolecular interactions like hydrogen bonding, tetrel bonding, and chalcogen bonding that govern the overall packing of molecules in the crystalline assembly of thieno[2,3-d]pyrimidine derivatives30–33 (Fig. 1). In addition to this, the energetic contributions of these interactions towards crystal formation are also addressed. Further, the study provides an in-depth analysis of the transition in S⋯C tetrel interactions, from the σ-hole (in 2) to the π-hole (in 10 & 11) centered towards the nucleophilic sulphur atom. Moreover, type I chalcogen interactions were observed in 3. These results were then corroborated from molecular electrostatic surface potentials, deformation density plots, and quantitative inputs from the QTAIM approach. A significant distinction between the natures of interactions across the different sets of compounds was determined and thoroughly discussed. Consequently, elucidating these interactions helps in structure–property analysis,32 which can be leveraged for drug molecule design. Moreover, the presence of multiple molecules in the asymmetric unit (high Z′)34 is also of interest and have been reported as well.
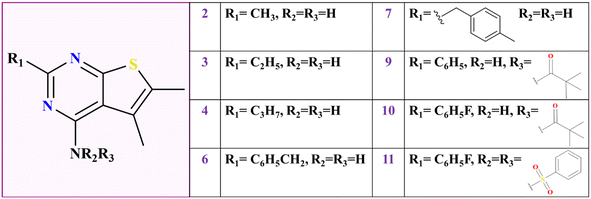 |
| | Fig. 1 Chemical structure of all eight thieno[2,3-d]pyrimidine derivatives. | |
2. Experimental
2.1 Chemistry and materials
Compounds 2, 3, 4, 6, 7, 9, 10, and 11 were synthesized following the steps shown in Scheme 1. All chemicals were obtained from Sigma Company and were used without purification. The progress of the chemical reactions was followed using thin layer chromatography (TLC), and visualized under UV light (TLC silica gel 60 F254, 25 aluminium sheets 60 F254 20 × 20 CCM Gel de silica 60 F254 UV indicators). Melting points (uncorrected) were measured with Stuart's scientific melting point apparatus (SMP30).
 |
| | Scheme 1 Synthesis of new 2,4-disubstituted-5,6-dimethylthieno[2,3-d]pyrimidine derivatives. | |
2.1.1 General procedure for the preparation of 5,6-dimethyl-2-substituted thieno[2,3-d]pyrimidin-4-amines (2, 3, 4, 5, 6, 7 and 8).
Compound 1 (1.83 g, 0.012 mol) was dissolved in dioxane (12.2 mL, 0.12 mol), and then the corresponding nitrile (0.12 mol) and HCl (12 M, 13.1 mL, 0.12 mol) were added. The reaction mixture was stirred under reflux for 4–6 hours, keeping the temperature between 85 and 90 °C. Afterward, the mixture was cooled to 25 °C and neutralized dropwise with 6 M NaOH while keeping it on ice. The resulting product was filtered, dried, and recrystallized using chloroform and methanol, or chloroform and dimethylformamide (DMF), to yield pure compounds 2, 3, 4, 5, 6, 7, and 8. The molecular formula, molecular weight, percentage yield, melting points, and Rf values for all the eight synthesized compounds (2–4, 6, 7, 9–11) are presented in Table S1.†
2.1.2 General procedure for the preparation of N-(5,6-dimethyl-2-phenylthieno[2,3-d]pyrimidin-4-yl)pivalamide (9) and N-(2-(4-fluorophenyl)-5,6-dimethylthieno[2,3-d]pyrimidin-4-yl)pivalamide (10).
Compounds 5/8 (0.51/0.55 g, 0.002 mol) were dissolved in pyridine (6.00 mL, 0.07 mol), and the mixture was stirred for 2–3 hours before adding trimethylacetyl chloride (1.2 mL, 0.01 mol). The reaction mixture was then stirred at room temperature for 10 hours. After 10 hours, the mixture was maintained at 25 °C and neutralized dropwise with 6 M HCl while on ice. The reaction mixture was then diluted with water and extracted with ethyl acetate. The ethyl acetate layer was collected, dried, and recrystallized using chloroform and dimethylformamide (DMF) to yield the final compounds 9 and 10.
2.1.3 General procedure for the preparation of N-(2-(4-fluorophenyl)-5,6-dimethylthieno[2,3-d]pyrimidin-4-yl)benzenesulfonamide (11).
Compound 8 (0.55 g, 0.002 mol) was dissolved in pyridine (6.00 mL, 0.07 mol), and the mixture was stirred for 2–3 hours before adding benzene sulfonyl chloride (1.28 mL, 0.01 mol). The reaction was then stirred at room temperature for 48 hours. After 48 hours, the mixture was maintained at 25 °C and neutralized dropwise with 6 M HCl while on ice. The reaction mixture was then diluted with water and extracted with ethyl acetate. The ethyl acetate layer was collected, dried, and recrystallized using chloroform and dimethylformamide (DMF) to yield the final compound 11.
2.2 Characterization via nuclear magnetic resonance (NMR) spectroscopy
1H and 13C NMR spectra were recorded for all the eight compounds with CDCl3 and DMSO-d6 as solvents. The NMR spectra were recorded on a Bruker AVANCE-III 500 MHz NMR spectrometer. A detailed analysis is given in Fig. S2–S9.†
2.3 Liquid chromatography mass spectrometry (LCMS)
These experiments were conducted using a Bruker microTOF QII low resolution mass spectrometer. A good correlation amongst the calculated and experimentally observed mass was observed for all the compounds (Fig. S10†).
2.4 Crystallization
All the bulk synthesized compounds were crystallized by a slow evaporation method in a library of solvents and kept at low (4 °C) and room temperatures (22–24 °C). The solvents used here for crystallization are of HPLC grade. The resulting crystals of different morphologies (plate, needle, and block) so obtained were then analysed via diffraction studies using X-ray.
2.5 Single crystal X-ray diffraction (SCXRD)
The obtained crystals of all eight compounds were examined under an optical microscope and the X-ray diffraction data were collected on a Bruker D8 Venture and D8 Quest with MoKα radiation (λ = 0.71073 Å). The collected data were then reduced and analysed to obtain the initial structural model. The absorption correction was done using SADABS. Followed by this, the crystal structure refinement was performed in Olex2, by the full matrix least squares method using SHELXL. The software Mercury 4.2.0 was used for molecular ORTEPs to investigate the crystal packing involving sulphur centered interactions. The crystallographic information files (CIFs) obtained are employed in analysing the non-covalent interactions, particularly hydrogen bonds and sulphur centered interactions. All the interactions discussed has a maximum contact distance of ≤ (∑vdWradii + 0.2) Å.
2.6 Structural overlay
The molecular conformational analysis was performed via structural overlay using Mercury 4.2.0 software. The changes in the conformations of the conformers present in the asymmetric unit were studied.
2.7 Hirshfeld surface analysis and fingerprint plots
The space occupied by the molecule in a crystal is mapped via Hirshfeld surface analysis, by partitioning the crystal electron density into molecular fragments. For all the eight molecular structures, the variation in the molecular electrostatic surface potentials (MESPs) was mapped over the Hirshfeld surface over a range of −0.02 au to +0.02 au. The red and blue in these surfaces indicate the corresponding negative and positive electrostatic potentials. The distance from the Hirshfeld surface to the nearest atomic nucleus inside (di) and outside (do) the surface is measured. This gives the contribution of various intermolecular interactions experienced by the molecule in the form of 2D fingerprint plots. The fingerprint plots for all the differently substituted thienopyrimidines have also been discussed.
2.8 Energy frameworks and lattice energy calculations
The theoretical calculations were carried out by CrystalExplorer21.5 using the in-built TONTO program for generating a wavefunction using the B3LYP/6-31G(d,p) basis set. Further, the energy framework was constructed, to identify the chemical environment of molecules to thereby gain insight into the nature of intermolecular interactions that govern the crystal packing. For this, molecules within the radius of 3.8 Å were modelled with the corresponding basis set. However, all the intermolecular interactions that are electrostatic and dispersion driven are justified by understanding the energy decomposition representation. The strength of each interaction is visualized via the difference in magnitude of the cylindrical radius, in energy frameworks. Further, the lattice energy for all the molecules were computed by plotting the cluster with a radius of 20 Å.
2.9 3D deformation density
Crystal Explorer21.5 helps to map the 3D deformation density at the crystal geometry for a molecule, with an electron density isosurface of 0.008 e au−3. Here, the charge depleted and charge concentrated regions are shown in red and blue lobes.
2.10 Computations based on the quantum theory of atoms in molecules (QTAIM) approach
All the molecular motifs involving tetrel interactions were selected and analysed using the QTAIM approach.35 The electronic features involved in chalcogen and tetrel interactions were computed using the M062X/6311G++(d,p) level of theory via the AIMALL programme suite.36 The presence of bond critical points and a bond path validates the existence of bonding interactions (of non-covalent type) amongst the interacting atoms.37 Further, the topological parameters derived from the topological analysis of the electron density distribution for various dimeric motifs were reported and thoroughly discussed.
3. Results and discussion
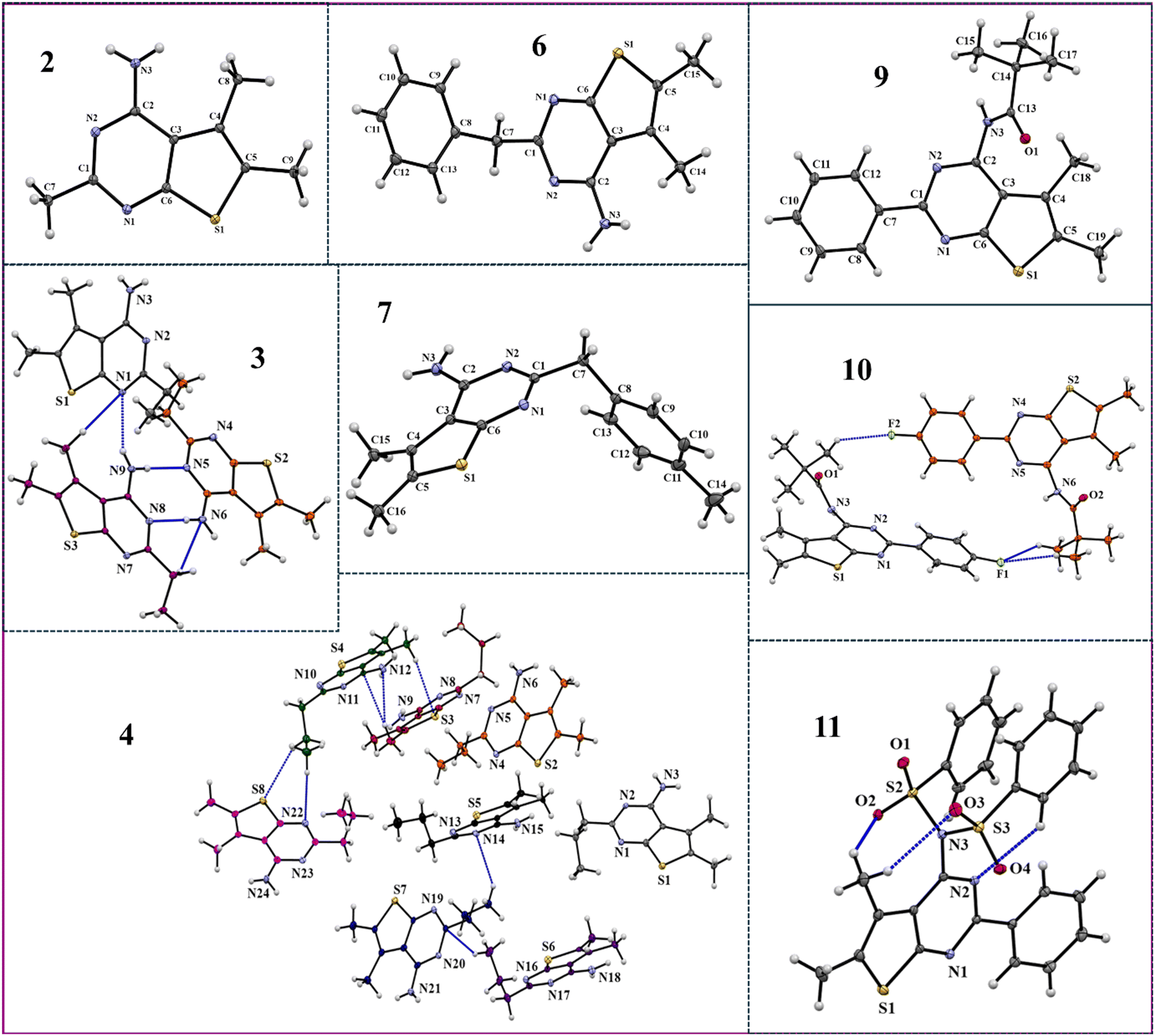
All eight synthesised compounds were crystallized and characterized via SCXRD. The crystals so obtained after crystallization from different solvents were screened for polymorphism (Table S2†). ORTEPs for all eight compounds are displayed in Fig. 2, while the crystallographic data for each compound are provided in Table S3.†
 |
| | Fig. 2
ORTEPs with 50% ellipsoidal probability. Dotted blue lines depict intermolecular interactions among molecules in the asymmetric unit. | |
The packing diagram illustrates the formation of strong and directional N–H⋯N hydrogen bonds along with several weak dispersive C–H⋯π/N/S, S⋯C (σ-hole/ π-hole), and S⋯S interactions. Further, the details of these intermolecular interactions are listed in Table S4.† Moreover, the energetic contributions from different intermolecular interactions in different molecular pairs are presented in Table S5.† In addition to this, the molecular surface electrostatic potential, fingerprint plots and energy frameworks illustrated in this study offer an in-depth analysis of the role of these interactions in constructing the molecular crystalline assembly.
3.1 Compound 2
The molecule crystallizes in a centrosymmetric monoclinic P21/c space group with Z = 4. The crystalline arrangement of compound 2 reflects the very short and highly directional centrosymmetric (2.00 Å, 178°) N3–H3A⋯N2 hydrogen bond dimer with a total IE of −62.1 kJ mol−1, and forms a sheet like structure when viewed down the b-axis (Fig. 3(a)). Here, the electrostatic component plays a prominent role in the overall stabilization of this motif (80%) and can be attributed to the presence of strong donor–acceptor moieties (Fig. 3(c)). The dimers are extended along the b-direction via C8–H8A⋯S1 and C8–H8C⋯π(C6) and along the a-direction via C9–H9A⋯N2 interactions, thus resulting in a brick layer type of stacking when viewed along the ab-plane (Fig. 3(b)). Dispersion majorly accounts for the overall stabilization involving C–H⋯π/S/N interactions. Among these, the C–H⋯π interactions have a greater total IE of −50.2 kJ mol−1 (Fig. 3(d)). Additionally, these molecular layers are linked along the c-axis through bifurcated N–H⋯N, C–H⋯N (involving H3B and H8A with N1) and C7–H7A⋯N3 hydrogen bonds, utilizing the c-glide (x, 1/2 − y, −1/2 + z), resulting in a herringbone pattern. These electrostatically stabilize the molecular pairs with an interaction energy of −26.2 kJ mol−1 (Fig. 3(c)).
 |
| | Fig. 3 (a) The packing of molecules through N–H⋯N and C–H⋯N interactions; (b) the brick layer type of stacking through N–H⋯N and C–H⋯N/S/π interactions (viewed along the ab-plane) of compound 2; (c) and (d) energy framework diagram depicting the total interaction energy values in kJ mol−1. | |
3.2 Compound 3
The molecule crystallizes in the centrosymmetric monoclinic P21/c space group with Z = 12. The three molecules in the asymmetric unit are depicted in grey (A), orange (B), and purple (C) colors and were held via N–H⋯N and C–H⋯N interactions (Fig. 4). Two centrosymmetric dimers are generated: (i) within the asymmetric unit between B and C (via N6–H6A⋯N8 and N9–H9A⋯N5 interactions) and (ii) between A (via the N3–H3A⋯N2 interaction) type molecules. The bifurcated C/N–H⋯N hydrogen bond within the unit cell has a lower total IE of −38.8 kJ mol−1, amongst the molecular pairs due to the reduced directionality of the bonds. The bifurcated C–H⋯N and N–H⋯N interactions (involving H29A and H9B with N1) connect A to C within the unit cell (Fig. 4(a)). The asymmetric unit is further extended along the a-axis through bifurcated hydrogen bonds involving H3B and H9E with acceptor N7. Further, these molecules extend down the b-axis via C–H⋯N hydrogen bonds (H28C with N3 and H28B with N4). Furthermore, the crystal structure is stabilized via C28–H28B⋯N4 and C–H⋯π interactions (Fig. 4(b)). The net energetic contributions involved in the crystalline packing are shown in Fig. 4(c) and (d).
 |
| | Fig. 4 (a) Herringbone arrangement formed via centrosymmetric N–H⋯N and C–H⋯N interactions in compound 3; (b) the overall packing of the molecule through C–H⋯π and C–H⋯N interactions; (c) and (d) energy frameworks depicting the total interaction energy from the N–H⋯N dimer along the ac-plane and C–H⋯π/N interactions in the bc-plane. | |
3.3 Compound 4
The crystal structure of compound 4 contains eight molecules in the asymmetric unit. The high value of Z′ may result in the molecule to crystallize in the low symmetry centrosymmetric P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group.13 The molecules in the asymmetric unit are linked through highly directional N–H⋯N and C–H⋯N/S/π interactions. These N–H⋯N dimeric interactions among different molecular pairs extended across the ab-plane are presented in the crystal packing (Fig. 5). Additionally, numerous other interactions are present in the crystal structure that gives stability to the overall crystal packing.
space group.13 The molecules in the asymmetric unit are linked through highly directional N–H⋯N and C–H⋯N/S/π interactions. These N–H⋯N dimeric interactions among different molecular pairs extended across the ab-plane are presented in the crystal packing (Fig. 5). Additionally, numerous other interactions are present in the crystal structure that gives stability to the overall crystal packing.
 |
| | Fig. 5 Crystal packing depicts the crystalline arrangement of eight molecules through N–H⋯N and C–H⋯N interactions in compound 4. | |
3.4 Compound 6
The molecule crystallizes in the triclinic P space group with one molecule in the asymmetric unit (Fig. 6). The molecule forms a centrosymmetric dimer via N3–H3A⋯N2 and C13–H13A⋯N3 interactions (Fig. 6(a)). The extremely short and directional N–H⋯N hydrogen bond (1.99 Å, 171°) was observed in the crystal packing. The dimers were then extended along the body diagonal of the unit cell utilizing C10–H10⋯N1 interactions with a total IE of −23.6 kJ mol−1 (Fig. 6(c)). C–H is a moderate hydrogen bond donor which leads to higher contribution from dispersion (73%). Further, the crystal packing also involves the formation of dimeric motifs through C–H⋯N/π interactions (involving H14A with N1 and C6), which are further extended down the a-axis via C11–H11⋯N1 generating a herringbone pattern (Fig. 6(b)). It is noted that the stability of the molecular pairs interacting through C–H⋯N/π interactions in the crystalline lattice was due to the higher percentage (74%) of the dispersion energy contributions (Fig. 6(d)).
 |
| | Fig. 6 (a) The packing of the molecules via N–H⋯N and C–H⋯N interactions (viewed down the ab-plane) in compound 6; (b) the herringbone structure generated through C–H⋯N and C–H⋯π interactions; (c) and (d) energy framework diagrams presenting the total interaction energy component. | |
3.5 Compound 7
The molecule crystallizes in the monoclinic P21/c space group with Z = 4. The N3–H3A⋯N2 interaction generates short (2.12 Å) and highly directional (173°) centrosymmetric dimers (Fig. 7(b)). The resulting dimer then propagates via the c-glide plane through bifurcated C/N–H⋯N (involving H15A and H3B with the acceptor N1) and C14–H14A⋯N3 interactions forming a herringbone pattern. Here, the C–H⋯N interaction is dispersive dominant. Further, these molecules in the crystalline arrangement are extended along the crystallographic b-axis via C12–H12⋯S1 and C7–H7A⋯π(C13) interactions (Fig. 7(a) and (c)). Further, the weak dispersive C–H⋯π (C9–H9⋯π(C9) and C16–H16B⋯π(C13)) interactions propagate the dimers along the crystallographic a-axis (Fig. 7(a) and (c)). The net energetic contributions involved among interacting components are presented in Fig. 7(d)–(f), based on the crystal packing analysis.
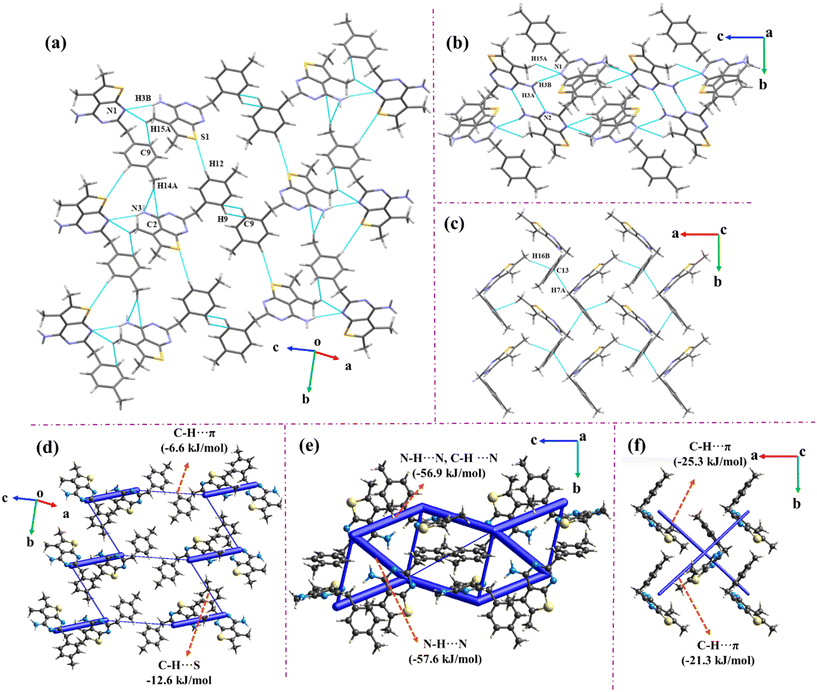 |
| | Fig. 7 (a) Crystal packing down the b-axis involving C–H⋯S/N/π interactions in compound 7; (b) herringbone pattern generated through dimeric N–H⋯N and C–H⋯N interactions in the bc-plane; (c) crystal packing through C–H⋯π along the a-axis; (d)–(f) energy framework diagrams depicting the total energy component. | |
3.6 Compound 9
The molecule crystallizes in a centrosymmetric monoclinic P21/c space group with Z′ = 1. The trifurcated N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–H⋯OC hydrogen bonds (involving H3, H16C and H15A with O1) coupled with C18–H18C⋯N2 interactions arrange the molecules in a chain like fashion along the c-axis, with the utilization of the c-glide perpendicular to the crystallographic b-axis (Fig. 8(a)). These interactions form the most stable molecular pair in the entire crystal lattice with a total IE of −75.5 kJ mol−1 (Fig. 8(b)). Further, the C–H⋯π interactions namely C19–H19C⋯π(C8) and C19–H19C⋯π(C7) extend the chains along the ab-plane forming the three-dimensional lattice. These chains are extended in such a way that the orientations of molecules are orthogonal to one another in the adjacent chains.
C and C–H⋯OC hydrogen bonds (involving H3, H16C and H15A with O1) coupled with C18–H18C⋯N2 interactions arrange the molecules in a chain like fashion along the c-axis, with the utilization of the c-glide perpendicular to the crystallographic b-axis (Fig. 8(a)). These interactions form the most stable molecular pair in the entire crystal lattice with a total IE of −75.5 kJ mol−1 (Fig. 8(b)). Further, the C–H⋯π interactions namely C19–H19C⋯π(C8) and C19–H19C⋯π(C7) extend the chains along the ab-plane forming the three-dimensional lattice. These chains are extended in such a way that the orientations of molecules are orthogonal to one another in the adjacent chains.
 |
| | Fig. 8 (a) Infinite chains formed through N–H⋯O, C–H⋯O, and C–H⋯N hydrogen bonds in compound 9. (b) Energy framework diagram depicting C–H⋯O and C–H⋯N interactions and their total energetic contributions. | |
3.7 Compound 10
Compound 10 crystallizes in the non-centrosymmetric orthorhombic Pca21 space group with Z′ = 2. The molecules are connected via C–H⋯F interactions (involving H17A with F2 and H35C and H36A with F1) in the asymmetric unit and further extended along the crystallographic c-direction via C31–H31⋯π(C12) and C12–H12⋯π(C31) (Fig. 9(c)). This results in a zigzag sheet like structure along the bc-plane. The C–H⋯F interactions in the asymmetric unit are stabilized with an energy value of −16.9 kJ mol−1 and have a significant contribution from dispersion (84%) (Fig. 9(f)). The grey-colored molecule is extended along the crystallographic a-axis through C16–H16A⋯N2, C17–H17C⋯O1 and N3–H3⋯O1 interactions (Fig. 9(b)). The molecular pair thus generated is highly stable with total IE = −81 kJ mol−1 (Fig. 9(e)). It is noted that the C–H⋯N and C–H⋯O interactions overpower the N–H⋯O interaction, thereby leading to the considerable value for the dispersion component. Additionally, the bifurcated acceptor in C–H⋯F (involving H36B and H36A with F1), C36–H36C⋯N5, and C9–H9⋯O2 interactions, and the bifurcated donor in C–H⋯π interactions (involving C10 and C11 with H38A) extend the zigzag sheets in three dimensions utilizing the 21-screw symmetry element (Fig. 9(a)).
 |
| | Fig. 9 (a) Crystal packing via C–H⋯N/O/F/π interactions; (b) packing of the molecule along the b-axis through C–H⋯N/O and N–H⋯O interactions in compound 10; (c) zigzag sheets formed by C–H⋯F/π interactions along the bc-plane; (d)–(f) energy framework diagrams depicting the total interaction energy values of major interactions. | |
3.8 Compound 11
Compound 11 crystallizes in the monoclinic P21/c space group with Z′ = 1. The ORTEP presents intramolecular C–H⋯N and C–H⋯O interactions in the molecule.
The molecule forms centrosymmetric dimers through C12–H12⋯O2 interactions. The interaction is significantly important with a total pairwise IE of −71.2 kJ mol−1. The dimers are oriented in two ways such that one of the orientations is perpendicular to the other. These are extended in an alternate fashion along the c-axis through C23–H23⋯O2 utilizing the c-glide symmetry (Fig. 10(a)). Along the b-axis, the molecules are extended through C16–H16⋯O4 in addition to C15–H15⋯O1 and C14–H14⋯O3 interactions that utilize the 21-screw axis. The C16–H16⋯O4 interaction is stabilized with a total IE of −35.1 kJ mol−1. Further, C10–H10⋯O4, C16–H16⋯O4, C9–H9⋯N1, C9–H9⋯π(C12), C9–H9⋯π(C7), and C9–H9⋯π(C1) interactions are responsible for the propagation of molecules diagonal to the crystallographic bc-plane (Fig. 10(b)). The net energetic contributions from all the interactions are shown in Fig. 10(c) and (d).
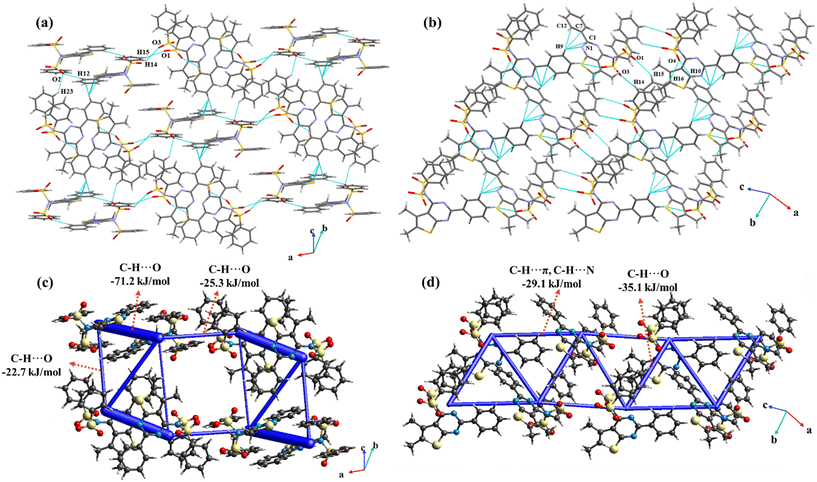 |
| | Fig. 10 (a) Crystal packing of compound 11 through C–H⋯O interactions; (b) crystal packing of compound 11 through C–H⋯O and C9–H9⋯π/N interactions; (c) and (d) energy framework diagrams representing the total interaction energy in kJ mol−1. | |
4. Structural overlay
Any change in molecular conformations influences the nature of intermolecular interactions, which, depending on their strength, determines the preferred molecular orientation in the crystalline lattice. To understand this, multiple conformers within the asymmetric unit were structurally constrained across thiophene and pyrimidine rings, followed by structural overlay analysis. Notably, the flexibiltity was observed in the ethyl, propyl and amide side chains of compounds 3, 4 and 10, which can be attributed to variations in the torsion angles across the C–C bond, as indicated in red arrows (Fig. 11). This corroborated to the formation of strong dimeric N–H⋯N interactions of a similar type in compounds 3 and 4. The root mean square deviation (RMSD) values obtained from the structural overlay for compounds 3, 4 and 10 were found to be 0.046, 0.063 and 0.005, respectively.
 |
| | Fig. 11 Superposition of different conformers crystallized in the asymmetric unit of compounds (a) 3; (b) 4; (c) 10. Red curved arrows indicate the flexible torsions. | |
5. Hirshfeld surface analysis and fingerprint plots
The molecular electrostatic potential plotted over the Hirshfeld surface from Crystal Explorer21.5 depicts the different regions of positive (blue) and negative (red) potentials. The negative potential regions around the pyrimidine nitrogens interact with the positive potential across the amine group to preferably form the strong dimeric N–H⋯N hydrogen bonding interactions in compounds 2, 3, 4, 6, and 7. The molecular electrostatic surface potentials (MESPs) plotted for all the eight molecules are shown in Fig. 12. The MESP plot of compound 2 reflects that the nitrogen atom (N2) exhibits a highly negative ESP value of −250.5 kJ mol−1 owing to its electron rich nature, while the hydrogen atom in the amine group (H3A) shows a positive ESP of +296.41 kJ mol−1 indicating electron deficiency. The significant electrostatic complementarity between them contributes to the formation of a strong N–H⋯N hydrogen bond. Further, the N–H⋯N dimer generated in compound 3 among B and C molecules within the unit cell displays a high electrostatic contribution of 78%. This could be attributed to the high negative ESP values on N8 and N5 (−250.2 kJ mol−1 and −243.6 kJ mol−1) and positive ESP values on H6A and H9A (361.2 kJ mol−1 and 312.4 kJ mol−1). Also, the electrostatic complementarity existing among A, B, and C molecules in the asymmetric unit is shown. In compound 6, the interactions effectively account for 66% of electrostatic contribution with the electron rich nitrogen atom (ESP value of −114.9 kJ mol−1) forming a strong hydrogen bond with electron deficient amine hydrogen, which has a positive ESP value of +129.2 kJ mol−1. Likewise, compound 7 contributes 71% electrostatic contribution with an energy value of −57.6 kJ mol−1. The MESP plot also suggests that N2 is highly electron rich due to the negative ESP value of −277.3 kJ mol−1, and H3A is electron deficient with a positive ESP value of +218.7 kJ mol−1. In addition to this, the presence of strong donor N3–H3B (+209.2 kJ mol−1) and acceptor N1 (−197.2 kJ mol−1) moieties results in the formation of a strong bond H-bond.
 |
| | Fig. 12 MESP values (in kJ mol−1) plotted over the Hirshfeld surface for compounds (a) 2; (b) 3; (c) 6; (d) 7; (e) 9; (f) 10; (g) 11. | |
In contrast, substituting the amine group with an amide in compounds 9 and 10 results in N–H⋯O hydrogen bonds instead of N–H⋯N interactions. The CO group of compound 9 exhibits a negative ESP value of −181 kJ mol−1, while the hydrogen atoms on the amide nitrogen and methyl groups show positive ESP values of +194 kJ mol−1 and +98.4 kJ mol−1, respectively. In comparison to compound 9, the presence of fluorine in compound 10 gives rise to low lattice energy (Table S6†). As fluorine being highly electronegative and a poor H-bond acceptor or electron donor, this donation is not feasible. In addition to this, the low ESP values for the fluorine (−72.7 kJ mol−1 and −66.1 kJ mol−1) and hydrogen atoms (+76.1 kJ mol−1 and +67.4 kJ mol−1) lead to the highly dispersive nature of the bond. In compound 11, the sulphonamide group contributes significantly to the dispersion part of the total energy. This observation is consistent with the ESP values obtained from the MESP plot where O2 shows a negative ESP value of −106 kJ mol−1 and H12 exhibits a positive ESP value of 78.5 kJ mol−1.
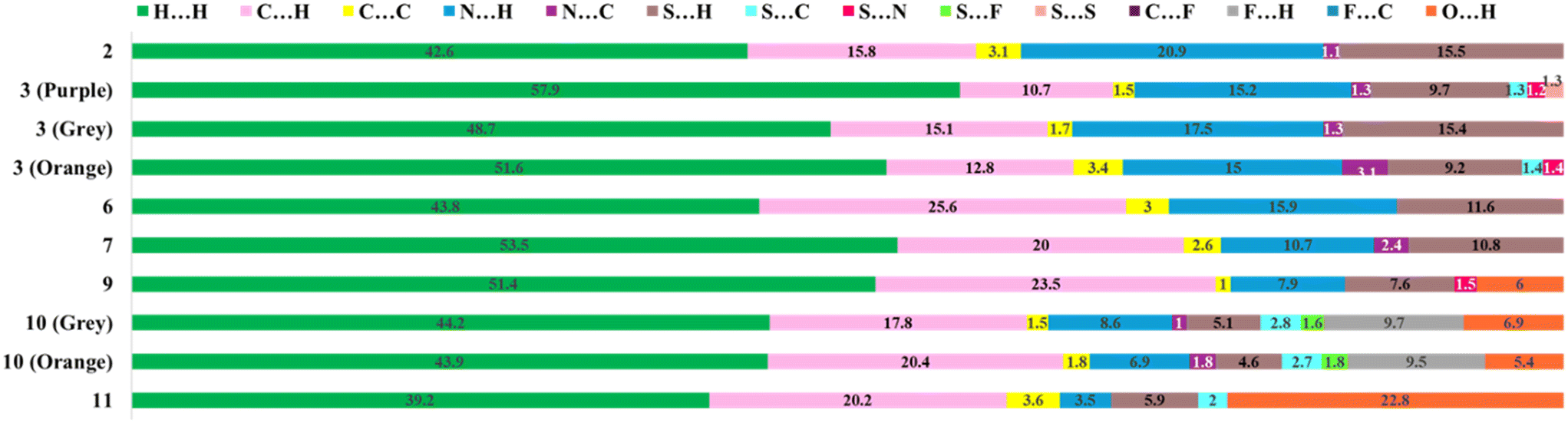
The 2D fingerprint plots depict the overall existing atomic contacts and their relative contributions in terms of percentage (Fig. S11†). This plot displays that the major surface coverage involves H⋯H contacts in all the molecules (>40%). Fig. 13 highlights the significant contributions from sulphur-related contacts, including S⋯H/C/S/N/F contacts. On increasing the substitution, the extent of reciprocal H⋯S contacts was minimal. Interestingly, for all five molecules, these plots displayed sharp spikes corresponding to reciprocal H⋯N contacts, which are indicative of their high directionality, whereas in compounds 9, 10, and 11, the sharp spikes represent reciprocal H⋯O contacts.
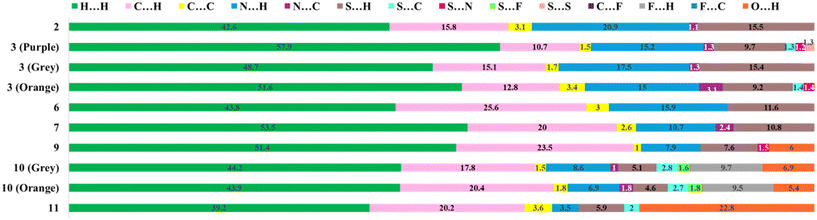 |
| | Fig. 13 The relative percentage contribution of various contacts based on 2D fingerprint plots present in all eight compounds. | |
6. QTAIM analysis
This analysis was performed to obtain quantitative insights into the electronic features of all sulphur bonded motifs. This revealed gradual variations in the directionality of S⋯C interactions, suggesting the potential occurrence of σ-hole and π-hole centered tetrel interactions. Among the non-covalent interactions, carbon bonding or tetrel bonding refers to the interaction where an electrophilic carbon atom forms a non-covalent bond with a nucleophilic centre.38,39 Additionally, the presence of methyl groups at adjacent and vicinal positions augmented the nucleophilicity of the sulphur atom and evidenced that its lone pairs were more pronounced for these interactions. Besides, the results from MESP indicate the presence of a σ-hole along the extension of the methyl group in 2, with the MESP value being 62.5 kJ mol−1, highlighting the electrophilic nature of the carbon atom. Furthermore, the deformation density plot depicts the S⋯C (σ-hole) interaction, where the blue charge concentrated (CC) region on the sulphur interacts with the red charge depleted (CD) region of the carbon atom, with a linear directionality of 174° (Fig. 14). A detailed evaluation of the distance and directionality for all S⋯C interactions has also been discussed for all cases. In contrast to this, 10 and 11 exhibit S⋯C (π-hole) mediated interactions (Fig. 15 and 16). Notably, this transition from linear (σ-hole) to orthogonal (π-hole) tetrel interactions is typically driven by an increase in the steric bulk of the substituents present across the pyrimidine ring. To elucidate this further, the topological parameters obtained from the QTAIM approach confirm the chemical bonding between the sulphur and the tetrel atoms and hence unequivocally establishes their electronic features as well (Table S7†). This includes the values of electron density (ρ), Laplacian of the density (∇2ρ) and the ratio of the local potential energy density to the kinetic energy density. The estimation of the relative strength of carbon bonding was extensively determined by the value of |V|/G. Interestingly, in all the cases, the kinetic energy density (G) at the BCP is more dominant than the potential energy density (V), and the Laplacian of the density is positive (∇2ρ > 0; charge depletion); this characterises the closed-shell nature of these interactions (|V|/G < 1). Moreover, the presence of bond critical points and bond paths originating from the sulphur to carbon atoms explicitly demonstrates the significant S⋯C interactions in compounds 2, 10, and 11.
 |
| | Fig. 14 (a) Distance and directionality of S⋯C (σ hole) tetrel interactions in compound 2; (b) MESP mapped over the Hirshfeld surface (values in kJ mol−1); (c) deformation density plot depicting the CC and CD regions; (d) QTAIM analysis validates the existence of bond critical points (highlighted in blue circles) in dimeric motifs. | |
 |
| | Fig. 15 Distance and directionality of S⋯C (π-hole) tetrel interactions in compound 10 (a) among grey molecules and (e) among orange molecules; (b) and (f) MESP mapped over the Hirshfeld surface (values in kJ mol−1); (c) and (g) deformation density plots depicting the CC and CD regions; (d) and (h) QTAIM analysis validates the existence of bond critical points (highlighted in blue circles) in dimeric motifs. | |
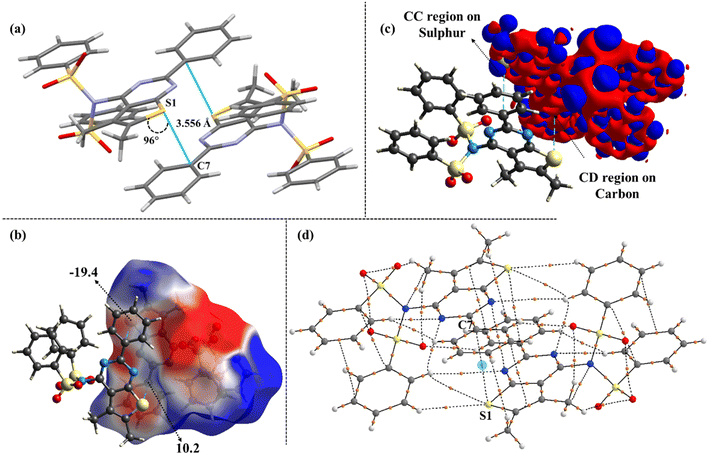 |
| | Fig. 16 (a) Distance and directionality of S⋯C (π-hole) tetrel interactions in compound 11; (b) MESP mapped over the Hirshfeld surface (values in kJ mol−1); (c) deformation density plot depicting the CC and CD regions; (d) QTAIM analysis validates the existence of bond critical points (highlighted in blue circles) in dimeric motifs. | |
In addition to this, the presence of type I S⋯S chalcogen interactions in compound 3 is of relevance. Fig. 17 depicts the deformation density plot and bond critical points, establishing the existence of these interactions. In contrast to the above, the other compounds predominantly exhibited C–H⋯S interactions as shown in Fig. S12.†
 |
| | Fig. 17 (a) Distance and directionality of S⋯S chalcogen interactions in compound 3; (b) MESP mapped over the Hirshfeld surface (values in kJ mol−1); (c) deformation density plot depicting the CC and CD regions; (d) QTAIM analysis validates the existence of bond critical points (highlighted in blue circles) in dimeric motifs. | |
7. Conclusions
A total of eight molecules comprising a thiophene ring fused with the pyrimidine ring, forming substituted thieno[2,3-d]pyrimidines with various substituents, were synthesized and structurally characterized. Among these, five compounds (2, 3, 4, 6, & 7) prominently exhibited strong dimeric N–H⋯N interactions. Meanwhile, in the remaining compounds (9, 10 & 11), the amine was replaced with amide and sulphonamide groups, thus favouring the formation of N–H⋯O and C–H⋯O interactions, respectively. The net energetic contributions of these molecules were quantitatively discussed in terms of electrostatics and dispersion. Additionally, the relative percentage contribution of individual intermolecular contacts was evaluated, and detailed fingerprint plots were addressed. The strong dimeric N–H⋯N interactions were evidenced by large negative and positive MESP values across nitrogen and hydrogen atoms, respectively. Moreover, the specific interactions involving the sulphur atom were investigated and the presence of both tetrel and chalcogen interactions was observed. The MESP mapped over the Hirshfeld surface illustrates the S⋯C tetrel interaction for compounds 2, 10 and 11. Further, the deformation density plots explore the directional S⋯C interactions in compound 2, wherein the charge concentrated (CC) region on sulphur interacts with the charge depleted (CD) region of carbon, within a suitable distance, forming tetrel bonds. Meanwhile, in the case of compounds 10 and 11, the directionality of the S⋯C interactions is changed due to the relatively large size of the substituents. This resulted in the nucleophilic interaction of sulphur with a carbon atom (π-hole) in compounds 10 and 11, in comparison to the directional interaction (σ-hole) with the electrophilic carbon atom in compound 2. Furthermore, a type I chalcogen interaction was observed in compound 3. The rational contribution of all these interactions was then justified by QTAIM analysis.
Data availability
The data supporting this article have been included as part of the ESI.† The crystallographic data for all the reported structures have been deposited in CSD and are available at https://www.ccdc.cam.ac.uk. The CCDC numbers are 2402902–2402905, 2402899, 2402900, 2402896 and 2402893.
Author contributions
Dr. Pran Kishore Deb: conceptualization, design, synthesis, crystallization, synthetic procedure writing, supervision, and validation; Anila M. Menon: conceptualization, manuscript writing, SCXRD experimentation, crystallographic data analysis and QTAIM analysis; P. S. R. Fathima Nida: crystallization, Hirshfeld surface analysis, and energy frameworks; Ipsha Shruti: SCXRD experimentation and data analysis; Sara Nidal: synthesis and purification; Dr. Katharigatta N. Venugopala: supervision and validation; Prof. Deepak Chopra: conceptualization, supervision and validation.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Author PKD thanks BIT Mesra, Ranchi, for providing the research fund under the Seed Money Scheme (Grant Number BIT/DRIE/SMS/2024-25/1877) and the Department of Pharmaceutical Sciences and Technology for providing the necessary research facilities. AM, FN, and DC thank IISER Bhopal for its research facilities and infrastructure. AM also thanks UGC for its senior research fellowship. We thank Amen Anees, a BS-MS student at IISERB for the dedicated efforts in designing the cover page.
References
- P. Lagardère, C. Fersing, N. Masurier and V. Lisowski, Pharmaceuticals, 2022, 15, 35 CrossRef PubMed.
- M. Abdel-Megid, K. M. Elmahdy, A. M. Elkazak, M. H. Seada and O. F. Mohamed, J. Pharm. Appl. Chem., 2016, 2, 103–127 CrossRef.
- J. A. Cisneros, M. J. Robertson, B. Q. Mercado and W. L. Jorgensen, ACS Med. Chem. Lett., 2017, 8, 124–127 CrossRef CAS PubMed.
- L. Zhang, H. Zhu, A. Mathiowetz and H. Gao, Bioorg. Med. Chem., 2011, 19, 5763–5770 CrossRef CAS PubMed.
- G. A. Jeffrey, Crystallogr. Rev., 2003, 9, 135–176 CrossRef CAS.
-
J. A. Joule and K. Mills, Heterocyclic Chemistry, Black Science, Oxford, U.K., 4th edn, 2000, ch. 25 Search PubMed.
- K. V. Rao, C. Balakumar, B. L. Narayana, P. K. Deb, R. Kaur and R. R. Akkinepally, Tetrahedron Lett., 2013, 54, 1274–1278 CrossRef.
- V. R. Kaki, R. R. Akkinepalli, P. K. Deb and M. R. Pichika, Synth. Commun., 2015, 45, 119–126 CrossRef CAS.
- L. T. Mun, J. K. K. Hing, P. K. Deb, K. N. Venugopala, R. P. Mailavaram, A. Y. Binsaleh and S. A. R. Shilbayeh, Indian J. Pharm. Educ. Res., 2024, 58, s515–s520 CrossRef.
- R. Shah and P. K. Verma, Chem. Cent. J., 2018, 12, 1–22 CrossRef PubMed.
- S. Malasala, A. Polomoni, M. N. Ahmad, M. Shukla, G. Kaul, A. Dasgupta, S. Chopra and S. Nanduri, J. Mol. Struct., 2021, 1234, 130168 CrossRef CAS.
- M. T. M. Sayed, R. A. Hassan, P. A. Halim and A. K. El-Ansary, Med. Chem. Res., 2023, 32, 659–681 CrossRef CAS.
- V. P. Litvinov, Adv. Heterocycl. Chem., 2006, 92, 83–143 CrossRef CAS.
- Y. A. Ibrahim, A. H. M. Elwahy and A. M. Kadry, Adv. Heterocycl. Chem., 1996, 65, 235–281 CrossRef CAS.
- M. Raghu Prasad and P. K. Deb, Chem. Pharm. Bull., 2007, 55, 776–779 CrossRef PubMed.
- M. Raghu Prasad, J. Prashanth, K. Shilpa and P. K. Deb, Chem. Pharm. Bull., 2007, 55, 557–560 CrossRef PubMed.
- M. Raghu Prasad, S. Razia and P. K. Deb, J. Chem. Res., 2007, 3, 133–135 CrossRef.
- M. Raghu Prasad, A. Raghuram Rao, P. Shanthan Rao, K. S. Rajan, S. Meena and K. Madhavi, Eur. J. Med. Chem., 2008, 43, 614–620 CrossRef PubMed.
- E. M. H. Ali, M. S. Abdel-Maksoud and C. H. Oh, Bioorg. Med. Chem., 2019, 27, 1159–1194 CrossRef CAS PubMed.
- K. Miwa, T. Hitaka, T. Imada, S. Sasaki, M. Yoshimatsu, M. Kusaka, A. Tanaka, D. Nakata, S. Furuya, S. Endo, K. Hamamura and T. Kitazaki, J. Med. Chem., 2011, 54, 4998–5012 CrossRef CAS PubMed.
- P. K. Deb, R. Mailavaram, B. Chandrasekaran, V. R. Kaki, R. Kaur, S. Kachler, K. N. Klotz and R. R. Akkinepally, Chem. Biol. Drug Des., 2018, 91, 962–969 CrossRef CAS PubMed.
- R. Kaur, P. K. Deb, B. L. Narayana, K. V. Rao, C. Balakumar, V. Rajkumar and A. R. Rao, J. Chem. Sci., 2011, 123, 69–73 CrossRef CAS.
- A. Hossan, M. Alsahag, A. Alisaac, M. A. Bamaga, A. I. Alalawy and N. M. El-Metwaly, J. Taibah Univ. Sci., 2023, 17, 2164993 CrossRef.
- P. Munshi and T. N. G. Row, J. Phys. Chem. A, 2005, 109, 659–672 CrossRef CAS PubMed.
- P. Munshi, K. N. Venugopala, B. S. Jayashree and T. N. Guru Row, Cryst. Growth Des., 2004, 4, 1105–1107 CrossRef CAS.
- P. Panini, K. N. Venugopala, B. Odhav and D. Chopra, Proc. Natl. Acad. Sci., India, Sect. A, 2014, 84, 281–295 CrossRef CAS.
- R. Shukla, T. P. Mohan, B. Vishalakshi and D. Chopra, CrystEngComm, 2014, 16, 1702–1713 RSC.
- I. Khan, P. Panini, S. U. D. Khan, U. A. Rana, H. Andleeb, D. Chopra, S. Hameed and J. Simpson, Cryst. Growth Des., 2016, 16, 1371–1386 CrossRef CAS.
- V. Sharma, N. Chitranshi and A. K. Agarwal, Int. J. Med. Chem., 2014, 2014, 1–31 Search PubMed.
-
M. P. Callahan, Encyclopedia of Astrobiology, Springer, New York, 2011 Search PubMed.
- R. Guha, Methods Mol. Biol., 2013, 993, 81–94 CrossRef CAS PubMed.
- M. K. Tripathi and V. Ramanathan, J. Phys. Chem. A, 2023, 127, 2265–2273 CrossRef CAS PubMed.
- K. Mandal, A. Hasija, R. Shukla, V. R. Hathwar and D. Chopra, Phys. Chem. Chem. Phys., 2023, 25, 19427–19434 RSC.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895–2933 CrossRef CAS PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
-
T. A. Keith, AIMAll (Version 19.10.12) TK Gristmill Software, Overland Park KS, USA, 2019 Search PubMed.
- J. Pilmé, E. Renault, F. Bassal, M. Amaouch, G. Montavon and N. Galland, J. Chem. Theory Comput., 2014, 10, 4830–4841 CrossRef PubMed.
- S. P. Thomas, M. S. Pavan and T. N. Guru Row, Chem. Commun., 2014, 50, 49–51 RSC.
- T. N. Guru Row and R. Parthasarathy, J. Am. Chem. Soc., 1981, 103, 477–479 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Anila M.
Menon‡
*a,
Anila M.
Menon‡