
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into extended coupled polymethines through the investigation of dual UV-to-NIR acidochromic switches based on heptamethine–oxonol dyes†
Benjamin
Mourot
 a,
Valérie
Mazan
b,
Mourad
Elhabiri
b,
Rudraditya
Sarkar
cd,
Denis
Jacquemin
*ce,
Olivier
Siri
a and
Simon
Pascal
*ac
a,
Valérie
Mazan
b,
Mourad
Elhabiri
b,
Rudraditya
Sarkar
cd,
Denis
Jacquemin
*ce,
Olivier
Siri
a and
Simon
Pascal
*ac
aAix Marseille Univ, CNRS UMR 7325, Centre Interdisciplinaire de Nanoscience de Marseille (CINaM), Campus de Luminy, Case 913, Marseille Cedex 09 13288, France. E-mail: simon.pascal@cnrs.fr
bCNRS – Université de Strasbourg – Université de Haute-Alsace, LIMA, CNRS UMR 7042, Equipe Chimie Bioorganique et Médicinale, ECPM, 25 Rue Becquerel, 67200 Strasbourg, France
cUniversité de Nantes, CEISAM UMR 6230, CNRS, Nantes F-44000, France. E-mail: Denis.Jacquemin@univ-nantes.fr
dPresent Address: Institut de Química Computacional i Catàlisi (IQCC), Universitat de Girona, 17003 Girona, Catalonia, Spain
eInstitut Universitaire de France (IUF), Paris F-75005, France
First published on 14th December 2023
Abstract
A series of heptamethine–oxonol dyes featuring different heterocyclic end groups were designed with the aim to explore structure–property relationships in π-extended coupled polymethines. These dyes can be stabilised under three different protonation states, affording dicationic derivatives with an aromatic core, cationic heptamethines, and zwitterionic bis-cyanine forms. The variation of the end groups directly impacts the absorption and emission properties and mostly controls reaching either a colourless neutral dispirocyclic species or near-infrared zwitterions. The acidochromic switching between the three states involves profound electronic rearrangements leading to notable shifts of their optical properties that were investigated using a parallel experiment–theory approach, providing a comprehensive description of these unique systems.
Introduction
Among the common strategies used to prepare compact organic near-infrared (NIR) dyes,1–6 the “cyanine” approach is one of the most powerful. It consists in the delocalization of an even number of π-electrons along a path including an odd number of sp2-hybridized carbon atoms, which is practically achieved by linking two electron-donating (or electron-withdrawing) moieties via a polymethine bridge, allowing the cationic (or anionic) charge to be delocalized between the two terminal units.7–10 When the electronic richness from each of the two terminal groups is adequately balanced, a key electronic structure called the ideal polymethine state (i.e., the cyanine state) is reached, corresponding to the symmetrical delocalization of the charge along the polymethine bridge and resulting in bond length equalization. Such strategy is nearly foolproof to access NIR absorptions and more seldom short-wavelength IR absorbers with high molar extinction coefficients (ε ≥ 104–105 M−1 cm−1),11–15 as illustrated with the omnipresence of NIR polymethines in contemporary applications, e.g., bio-imaging, sensing, and photovoltaics.16–21The coupling principle in polymethines formulates that the connection of at least two polymethine units via one or multiple single bonds generates an interaction reducing the HOMO–LUMO gap and affecting the photophysical properties of the chromophore. As a simple illustration, the coupling of two trimethine units such as the streptocyanine cation Cy3 gives rise to an enhancement of the optical properties going beyond simple additive or stiffening effects (Fig. 1). This principle was theoretically predicted in 1966 by Dähne & Leupold,22 and experimentally confirmed twenty years later, following the oxidation of octamethyl-tetraaminobenzene to the corresponding dication diCy3.23 Such bis-cyanine illustrates the principle since the coupling effect is accompanied by a strong decrease of the energy gap and thus a redshift towards the yellow region, compared to the corresponding trimethine cyanine Cy3 absorbing in the UV region.24 This concept was further corroborated in 2003 following the crystallization of the 2,5-diamino-1,4-benzoquinonediimine cation and dication upon protonation,25,26 and by the more recent isolation of zwitterionic coupled trimethines DABQDI showing far-red absorption albeit with low ε < 8 × 103 M−1 cm−1, due to the cyanine-to-cyanine charge transfer occurring from the anionic subunit towards the cationic one.27–30 Furthermore, strong bathochromic shifts were also found in simple diaminobenzoquinone monoimine derivatives (BQMI), which are highly stable ground-state zwitterions that can be viewed as an assembly of cyanine and oxonol trimethines Cy3 and Ox3.31,32
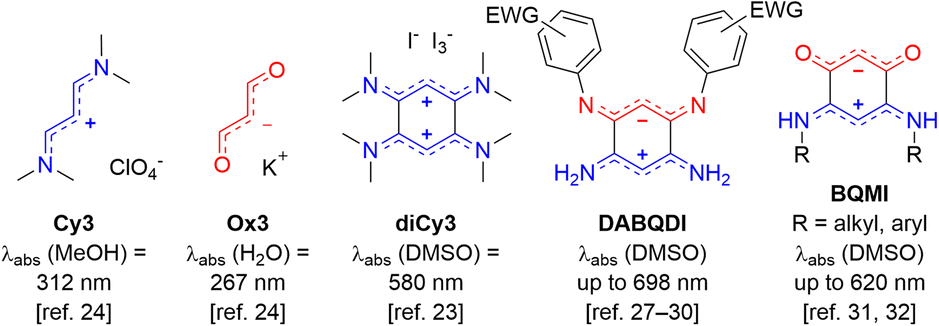 | ||
| Fig. 1 Illustration of the coupling principle with trimethine derivatives (EWG = electron-withdrawing group). | ||
To date, reports of new coupled polymethines (CoPo) remain scarce and usually arise from the serendipitous discoveries of original zwitterions, dianions or dications with moderately intense far-red to NIR absorption, as exemplified by the previously reported oxonol-containing bis-indene,33 the coupled barbiturate-merocyanine,34 the 7-chloropyrido[1,2-a]benzimidazole-8,9-dione core,35 or the recently isolated dicationic pyrazine-linked bis-trimethine absorbing beyond 800 nm.36 These rare examples highlight that the coupling principle appears as an unexplored strategy to design unique NIR dyes, and that structure–property relationships are still to be unravelled to amplify both the absorption as well as the emission properties of the resulting compounds in a way to open up new spectroscopic and applicative horizons.
With these perspectives in mind, we aimed at developing a new generation of π-extended CoPo by replacing one cationic trimethine subunit with a heptamethine one. Rigidified heptamethines such as Cy7 (Fig. 2A) are popular platforms conveniently reaching the NIR spectral range due to their ideal polymethine state that implies particularly high molar extinction coefficients (ε > 2 × 105 M−1 cm−1) and non-negligible emission properties, with however a moderate Stokes shift. Importantly, Shabat and co-workers previously designed the asymmetric enone-coupled heptamethine QCy7 (Fig. 2B) and pointed out an orbital mixing between the two subunits, which was detrimental for the cyanine transition and consequently induced a blueshift of the optical properties while significantly improving the Stokes shift.37,38 It is also relevant to note that close analogues of benzothiazolium- and indolium-containing dyes 3·H+ and 1·H+ (Fig. 2C) were recently synthesized by He, Zeng, and co-workers for selective mitochondria or lysosome staining.39,40 However these two reports did not identify either the zwitterionic species (z-3) or any spirocyclic isomer (e.g.dsp-1) forming at high pH values (vide infra). Yet, a series of such dispiropyran photochromic species dsp-1 were isolated by Metelitsa and co-workers in 2012 and they reported the opening to the monospiropyran under UV irradiation, nevertheless without mentioning any doubly open form.41
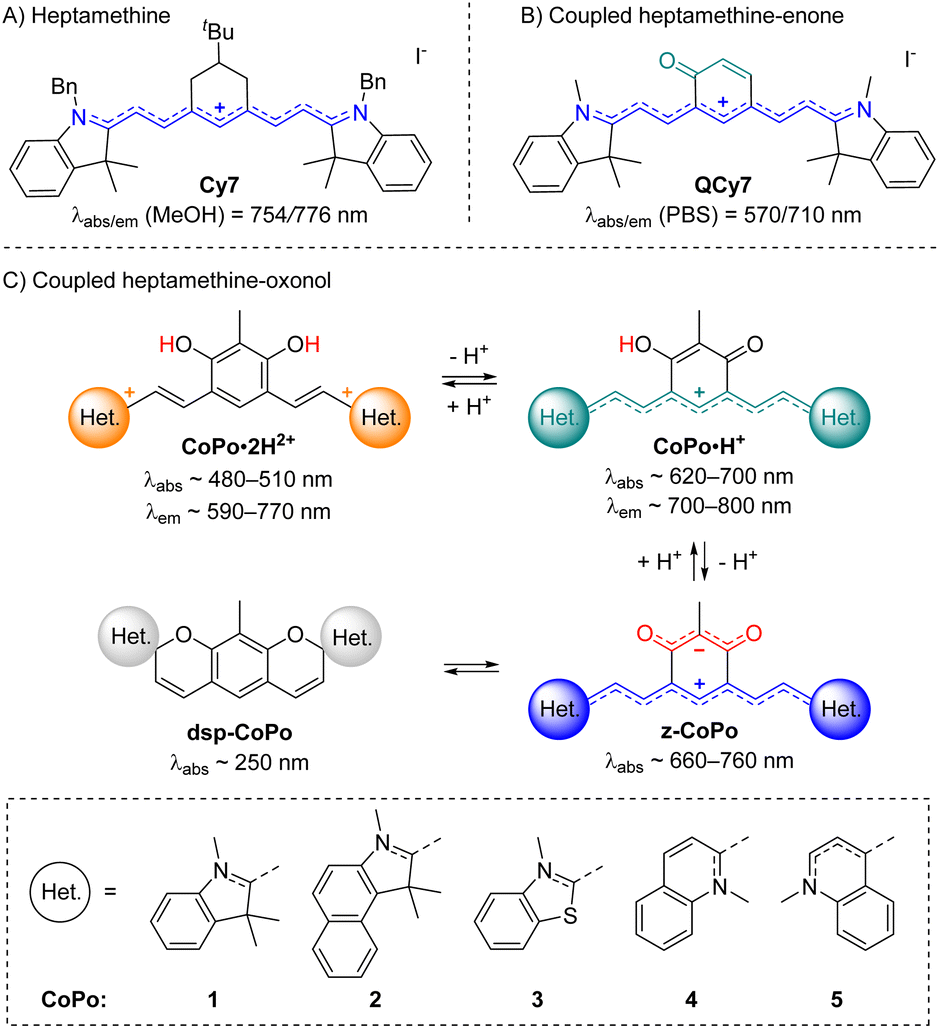 | ||
| Fig. 2 Previously reported rigidified heptamethine (A) and heptamethine–enone (B) dyes, and coupled heptamethine–oxonol dyes investigated in this work with key optical properties in methanol (C). | ||
These fragmented precedents encouraged us to bring to light the missing pieces to assemble the puzzle and provide a detailed overview of coupled heptamethine–oxonol dyes. More importantly, such chromophores could illustrate a new strategy to eventually reach zwitterionic CoPo with molar extinction coefficients potentially ten times higher than for DABQDI or BQMI. Compounds 1–5 isolated and characterized in this work (Fig. 2C) are dual acidochromic switches enabling to form dicationic quadrupoles (CoPo·2H2+), cationic heptamethines with NIR emissions (CoPo·H+), and to reach highly delocalized zwitterions with intense NIR absorption (3–5) or spirocyclic leuco dyes upon two consecutive intramolecular ring closures (1 & 2). Such ring closure is well known in the field of spiropyrans and paved the way for the development of exceptional materials with photochromic and/or acidochromic properties.42–44 However, double-closure has been scarcely reported in the case of molecules that present two spiropyran moieties linked through a π-conjugated moiety.41,45–50 Herein, the variation of the heterocyclic end groups is useful to control the formation of the bis-spiropyran or to favour the zwitterionic species, while also tuning the optical properties in the NIR range. The different forms were explored by means of pH-dependent absorption spectrophotometric titrations and by studying their optical properties, which were supported by a theoretical investigation, providing a deeper insight into the structure–property relationships in coupled polymethines.
Results and discussion
Synthesis
The core building block 6 depicted in Scheme 1 was synthesized by electrophilic formylation of commercially available 2-methylresorcinol,51 and was then used to carry out Knoevenagel condensations with salts 7–11 prepared by methylation of the corresponding heterocycles (see the ESI†). The condensations of 6 with 2 equivalents of indoleniums 7 or 8 were first carried out in ethanol using piperidine as base, following a recently published protocol reporting the isolation of cationic 1·H+ after acid hydrolysis and recrystallization.40 Surprisingly, in our case the recrystallisation step was unsuccessful and we had to resort to column chromatography on silica gel to isolate the desired compounds 1·H+ and 2·H+ with low yields (ca. 10%). However, when the hydrolysis step was replaced with a basic extraction of the reaction mixture using a saturated aqueous solution of NaHCO3, the neutral dispiropyrans dsp-1 and dsp-2 were isolated with good to excellent yields (>80%), as a result of the intramolecular cyclization of the two oxygen atoms attacking the electrophilic carbons in the alpha position from the nitrogen atoms.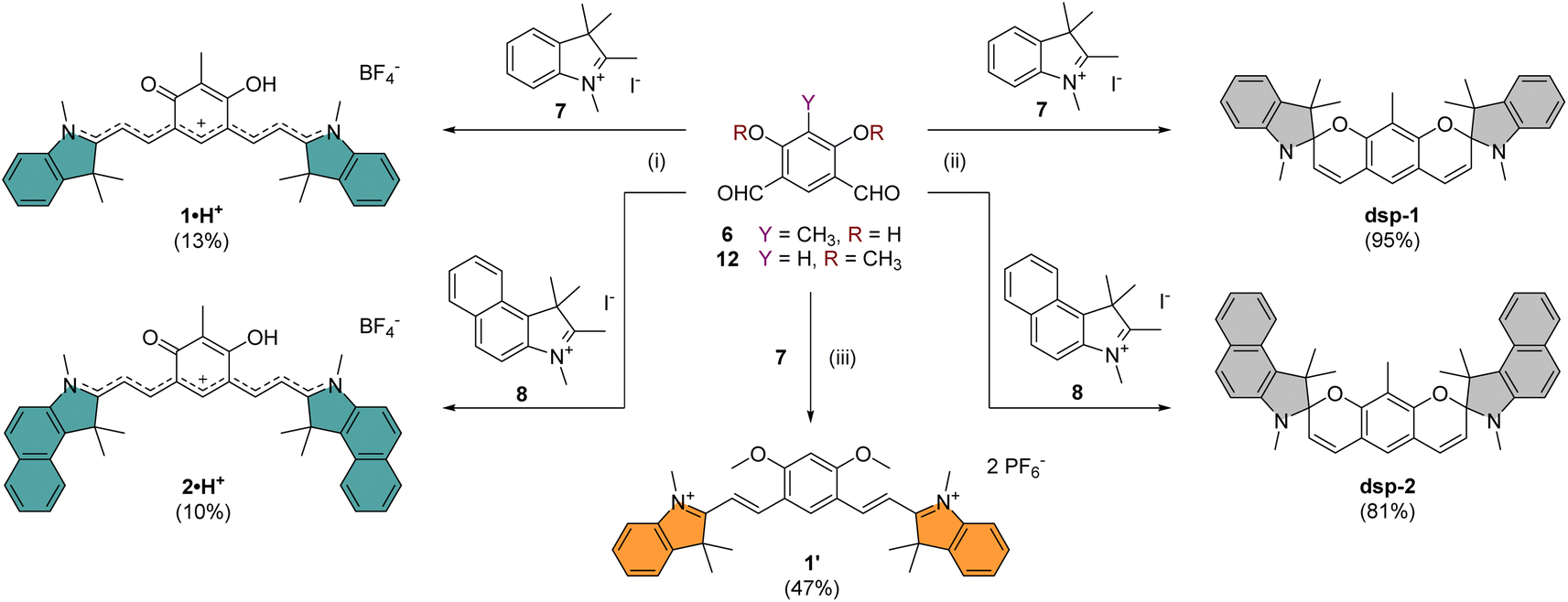 | ||
| Scheme 1 Synthesis of chromophores 1 and 2 under their cationic or neutral forms. Conditions: (i) 6, EtOH, piperidine, 95 °C, 16 h, then aq. HBF4, 25 °C; (ii) 6, EtOH, piperidine, 95 °C, 16 h, then aq. NaHCO3; (iii) 12, Ac2O, AcONa, 100 °C, 16 h, then aq. KPF6. | ||
As already mentioned, the original work describing the preparation of dsp-1 did not indicate the opening of the compound under acidic conditions,41 and the more recent report of a closely related analogue of 1·H+ did not identify either the dicationic or spirocyclic forms of the dye at low and high pH conditions, respectively.40 In our case, the cationic or dispiropyran forms of compounds 1 and 2 were successfully isolated using either acidic or basic treatment of the reaction and both forms could be easily differentiated. Firstly, the appearance of compounds 1 and 2 in solution markedly varies from green for the cationic species to colourless for the less conjugated dispiropyrans. Secondly, 1H NMR analysis unambiguously revealed a trans configuration of the ethylenic protons for the open cationic forms 1·H+ and 2·H+, with proton–proton coupling constants 3J in the 13–15 Hz range, and a cis configuration for the spirocyclic ones, with 3J of ca. 10 Hz. The dicationic species 1·2H2+ and 2·2H2+ were also characterized by NMR spectroscopy following in situ generation from dsp-1 and dsp-2 in deuterated trifluoroacetic acid (see the ESI†). Alternatively, the condensation between 6 and two equivalents of 1,2-dimethylbenzo[cd]indolium tetrafluoroborate was attempted because such heterocyclic end groups generally afford heptamethines absorbing above 1000 nm.52 Unfortunately, the reaction did not yield the expected coupled polymethine or spiropyran derivatives but an inextricable mixture. Eventually, care was taken to synthesize compound 1′ as a reference to identify the optical signature of the dicationic form of CoPo 1 (i.e., 1·2H2+, vide infra). Following the condensation in acetic anhydride between two equivalents of 7 and the isophthalaldehyde derivative 12 (prepared in three steps from 1-bromo-2,4-difluorobenzene, see the ESI†), 1′ was isolated as a red solid in 47% yield.
Compound 3·2H2+ was prepared by Knoevenagel condensation of benzothiazolium salt 9 in ethanol in the presence of a catalytic amount of piperidine (Scheme 2).39 During the course of the reaction, it was noticed that the mono-condensed intermediate precipitates and that the treatment of the reaction with concentrated aqueous tetrafluoroboric acid at room temperature is essential for the reaction to proceed towards the formation of the bis-condensed 3·2H2+ derivative, isolated as a red solid in 65% yield following precipitation. By applying the same synthetic strategy, dications 4·2H2+ and 5·2H2+ were prepared from dialdehyde 6 and quinolinium salts 10 or 11 with 87% and 74% yields, respectively. The HRMS analysis of compounds 3–5·2H2+ dissolved in methanol showed both the dicationic (z = 2) and cationic species (z = 1), highlighting that the compounds were prompt to deprotonate. These results encouraged us to investigate the acid–base features of these new dyes and the influence of pH on their photophysical signatures.
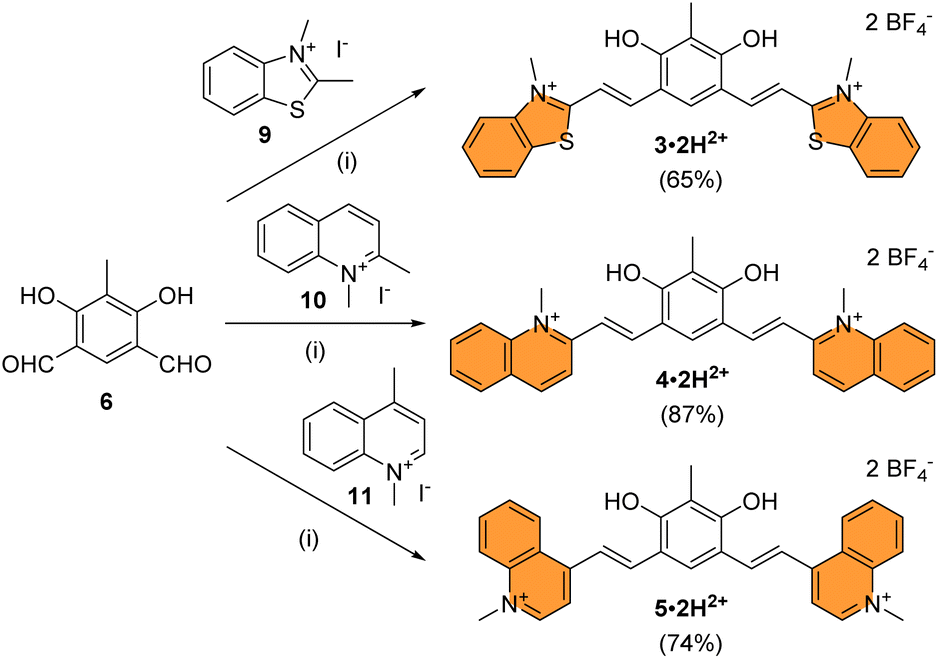 | ||
| Scheme 2 Synthesis of chromophores 3–5 under their dicationic forms. Conditions: (i) EtOH, piperidine, 95 °C, 16 h, then aq. HBF4, 25 °C, 5 min to 5 h. | ||
Photophysical properties
To initiate the investigation of the optical properties, the electronic absorption of all the dicationic derivatives was recorded in methanol solutions in the presence of 0.5 vol% trifluoroacetic acid (TFA) to ensure the protonation of the central resorcinol core. The spectra displayed in Fig. 3 and the data summarized in Table 1 show that all the dications absorb in the 350–600 nm range, which corresponds to the spectral range of the reference dye 1′ (λmax = 461 nm) and is also consistent with the dicationic precursor of QCy7 reported by Shabat and co-workers (λmax∼450 nm).38 Within the series of dications, the indole-containing dyes 1·2H2+ and 2·2H2+ show the most redshifted and intense bands, with maxima at 498 (ε498 = 4.6 × 104 M−1 cm−1) and 513 nm (ε513 = 4.8 × 104 M−1 cm−1), respectively. When comparing the electronic absorption spectra of 1′ and 1·2H2+, rather unexpected bathochromic and hyperchromic shifts appear for the latter dication. This effect can be attributed to the structural differences pointed out by theoretical calculations, implying a slightly lower aromatic character of the central ring in 1·2H2+ (see the theoretical section below).| Experimental | Theoretical | |||||||
|---|---|---|---|---|---|---|---|---|
| Species | Conditions | λ expabs [nm] (ε [104 M−1cm−1]) | λ em [nm] | Stokes shift [cm−1] | Φ [%] | τ [ns] | Species | λ theovert–abs [nm] (f)b |
| a Methanol or mixture of methanol + 0.5% (vol.) TFA or DBU. b Theoretical vertical absorption maximum determined in methanol (f: corresponding oscillator strength, see the ESI for details). c Underestimated fluorescence quantum yield due to detection limit at 850 nm. d The errors on the λ, the ε values and the quantum yields are estimated to be 1 nm, 10% and 10%, respectively. sh: shoulder. | ||||||||
| 1′ | 1′ in MeOH | 461 (2.6) | 585 | 4600 | 4 | <0.2 | 1′ | 408 (3.01) |
| 438 (sh, 2.54) | 387 (0.39) | |||||||
| 1·2H2+ | dsp-1 in MeOH/TFAa | 498 (4.55) | 746 | 6700 | 4 | 0.5 | 1·2H2+ | 430 (1.88) |
| 411 (3.57) | 384 (0.41) | |||||||
| 1·H+ | 1·H+ in MeOH | 640 (7.95) | 694 | 1200 | 27 | 1.1 | 1·H+ | 601 (1.81) |
| 429 (1.38) | 422 (0.58) | |||||||
| sp-1 | 1·H+ in MeOH/DBUa | 501 (4.27) | 549 | 1700 | 0.1 | <0.2 | sp-1 | 507 (0.88) |
| 421 (2.52) | 411 (0.06) | |||||||
| dsp-1 | dsp-1 in MeOH | 340 (0.58) | — | — | — | — | dsp-1 | 310 (0.07) |
| + z-1 | ||||||||
| 290 (sh, 1.84) | 298 (0.06) | |||||||
| 251 (4.72) | 279 (0.15) | |||||||
| + 578 (0.29) | — | — | — | — | z-1 | 698 (0.03) | ||
| 550 (0.28) | 636 (0.73) | |||||||
| 444 (0.18) | ||||||||
| 2·2H2+ | dsp-2 in MeOH/TFAa | 513 (4.82) | 767 | 6500 | 2c | 0.5 | 2·2H2+ | 457 (1.05) |
| 450 (3.45) | 417 (0.32) | |||||||
| 2·H+ | 2·H+ in MeOH | 670 (9.44) | 723 | 1100 | 13 | 0.6 | 2·H+ | 640 (0.54) |
| 443 (2.33) | 442 (1.85) | |||||||
| sp-2 | 2·H+ in MeOH/DBUa | 521 (5.69) | 560 | 1300 | 0.1 | <0.2 | sp-2 | 522 (1.05) |
| 441 (2.67) | 414 (0.05) | |||||||
| 372 (0.98) | 399 (0.66) | |||||||
| dsp-2 | dsp-2 in MeOH | 360 (sh, 0.72) | — | — | — | — | dsp-2 | 337 (0.02) |
| + z-2 | ||||||||
| 341 (1.15) | 336 (0.07) | |||||||
| 292 (2.46) | 312 (0.04) | |||||||
| 247 (10.11) | 301 (0.15) | |||||||
| + 700 (sh, 0.27) | — | — | — | — | z-2 | 699 (0.03) | ||
| 604 (0.65) | 661 (0.86) | |||||||
| 459 (0.34) | ||||||||
| 3·2H2+ | 3·2H2+ in MeOH/TFAa | 477 (3.77) | 769 | 8000 | 4c | 0.5 | 3·2H2+ | 410 (1.74) |
| 415 (3.09) | 374 (0.48) | |||||||
| 3·H+ | 3·2H2+ in MeOH | 615 (5.62) | 695 | 1900 | 8 | 1.7 | 3·H+ | 603 (1.83) |
| 469 (3.74) | 428 (0.54) | |||||||
| z-3 | 3·2H2+ in MeOH/DBUa | 656 (7.06) | — | — | — | — | z-3 | 672 (0.03) |
| 463 (3.90) | 646 (0.87) | |||||||
| 4·2H2+ | 4·2H2+ in MeOH/TFAa | 473 (3.45) | 594 | 4300 | 2 | 0.4 | 4·2H2+ | 404 (1.81) |
| 430 (3.08) | 376 (0.30) | |||||||
| 4·H+ | 4·2H2+ in MeOH | 626 (4.70) | 731 | 2300 | 2c | <0.2 | 4·H+ | 617 (1.96) |
| 498 (2.48) | 464 (0.13) | |||||||
| z-4 | 4·2H2+ in MeOH/DBUa | 690 (7.09) | — | — | — | — | z-4 | 665 (0.03) |
| 508 (2.35) | 682 (2.60) | |||||||
| 5·2H2+ | 5·2H2+ in MeOH/TFAa | 490 (1.94) | 642 | 4800 | 2 | <0.2 | 5·2H2+ | 429 (1.45) |
| 455 (1.89) | 399 (0.27) | |||||||
| 5·H+ | 5·2H2+ in MeOH | 702 (4.48) | 794 | 1700 | 4c | <0.2 | 5·H+ | 691 (1.80) |
| 521 (1.99) | 500 (0.35) | |||||||
| z-5 | 5·2H2+ in MeOH/DBUa | 757 (4.05) | — | — | — | — | z-5 | 755 (2.74) |
| 557 (1.73) | 716 (0.05) | |||||||
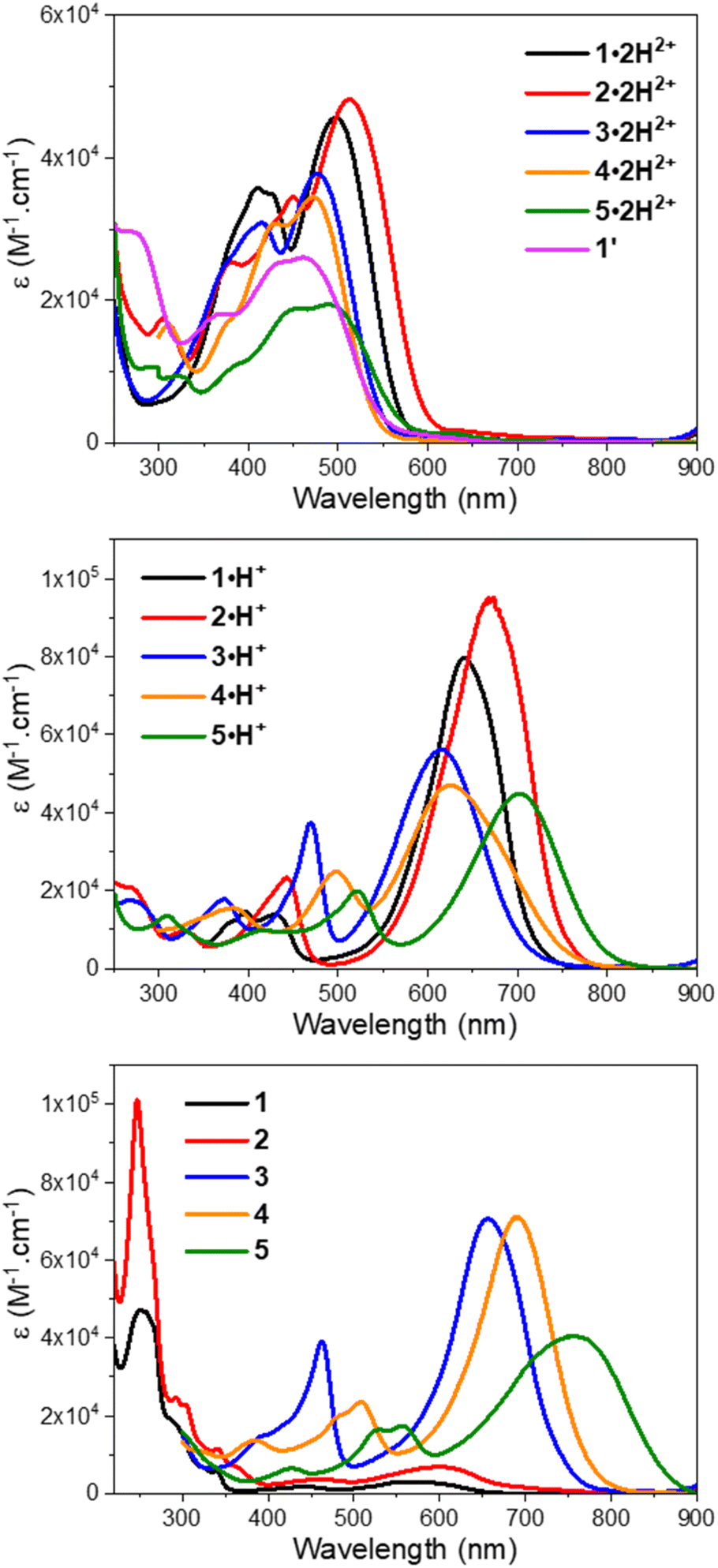 | ||
| Fig. 3 UV-vis-NIR electronic absorption spectra of compounds 1–5 in methanol with TFA (top), methanol (middle), and methanol with DBU (bottom). | ||
Interestingly, when a dicationic species is dissolved in solution in the absence of TFA, the molecule undergoes spontaneous deprotonation to the monocationic form, characterized by a green coloration (see the solvatochromism of dications in Fig. S71 of the ESI†). This proved particularly convenient for recording the absorption of the monocationic species, all of which exhibit, compared to their dicationic counterpart, strong bathochromic (∼140–150 nm for 1–4 and ∼210 nm for 5) and hyperchromic shifts of both their low energy transition, that reaches the red region (600–700 nm), and their less intense second transition that appears in the 400–500 nm spectral range (Fig. 3 and Table 1). As discussed below, this strong redshift is reproduced by theory and originates from a rather subtle change of the nature of the excited state. While the thiazole 3·H+ and quinoline 4·H+ derivatives have their maxima centred around 620 nm, the indole 1·H+ and benzo[e]indole 2·H+ derivatives peak at 640 and 670 nm, respectively, with ε ≥ 8 × 104 M−1 cm−1, nearly twice the ε of the other cations of the series. The lepidine-containing dye 5·H+ features a low energy-transition peaking at 702 nm, which is expectedly redshifted due to a more extended delocalization pathway than in the 2-quinoline derivative 4·H+.9,53 We underline that the low energy transitions of these cationic derivatives remain blueshifted compared to the absorptions of previously reported Cy7 with λmax = 754 nm (Fig. 2A), and other related rigidified chloro-heptamethines featuring various heterocyclic end groups that show absorption maxima in the 790–860 nm spectral range (see Table S1, ESI†).54–57 This difference can be mainly attributed to the electronic contribution of the keto–enol subunit to the low energy transition, decreasing the cyanine character of the cationic subunit, as illustrated below in the theoretical section.
In the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), the dicationic compounds 3–5·2H2+ are instantaneously deprotonated to generate the corresponding zwitterionic species. The UV-vis-NIR absorption spectra of these CoPo derivatives reveal redshifted absorptions compared to the monocationic species, with maxima centered at 656, 690, and 757 nm for z-3, z-4, and z-5, respectively, and molar extinction coefficients in the order of 4–7 × 104 M−1 cm−1 (Fig. 3 and Table 1). These values are among the most intense reached for zwitterionic coupled polymethines, with a nearly ten-fold increase in ε compared to the best DABQDI and BQMI dyes (Fig. 1).30,32 Such intense, narrow and redshifted absorption bands strongly support the view that the cationic heptamethine subunit is in its ideal polymethine state and has a rather weak coupling with the anionic oxonol subunit (see also the electron density difference plots below). The experimental absorption data of these zwitterionic species were well correlated with the theoretical predictions, further confirming the attribution of the dicationic, cationic and zwitterionic forms of dyes 3–5 (Table 1 and Fig. 4).
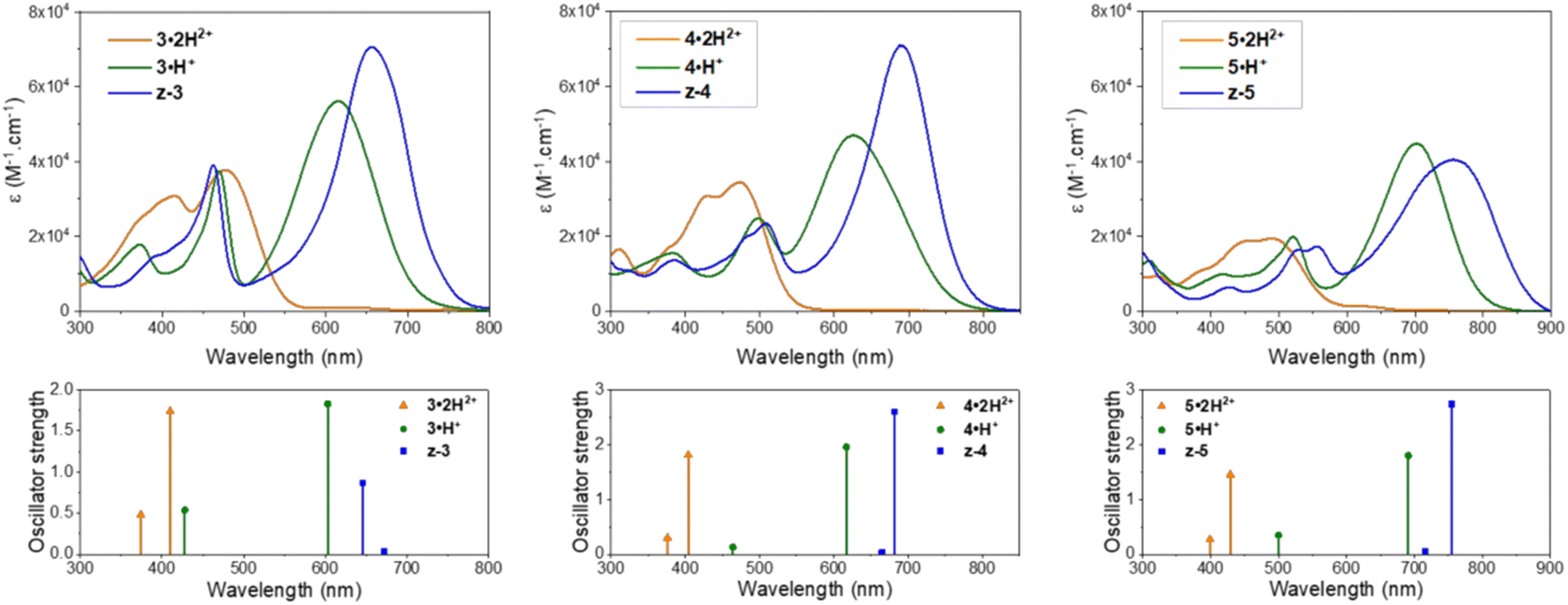 | ||
| Fig. 4 Experimental UV-vis-NIR electronic absorption spectra (top) and calculated lowest two vertical transitions (bottom) for the different protonation states and forms adopted by compounds 3–5 in acidic, neutral or basic methanolic solutions (see Table 1 for conditions). | ||
Remarkably, when the cationic dyes 1·H+ or 2·H+ are dissolved in the presence of DBU, the zwitterionic or leuco dispiropyran forms are not reached but instead, a red-coloured species is formed. With the help of theoretical calculations, the absorption bands peaking at 501 or 521 nm were attributed to the monospiropyrans sp-1 and sp-2, respectively (Scheme 3 and Fig. 5). In contrast, the leuco species dsp-1 and dsp-2 are characterized by the absence of coloration when dissolved in solution. This translates in no marked transition in the visible, while a strong absorption band remains in the UV domain below 300 nm for both compounds. The visible bleaching is complete in dichloromethane (Fig. S74, ESI†), suggesting the presence of only fully closed species, whereas a weak band is found at ca. 600 nm in polar protic solvents like methanol, which may presumably result from the opening of a small portion of dsp-1 and dsp-2 affording the mixed open–closed forms sp-1 and sp-2 and the completely open zwitterions z-1 and z-2, according to theoretical predictions (Fig. 5 and Table 1). The proportion of open zwitterionic species is even more pronounced in a methanol–water mixture used for pH titration (vide infra). It is important to note that, for 1, the bands at ca. 540–630 nm were originally attributed to the monospirocyclic species sp-1 formed under UV photoirradiation in acetonitrile;41 however, the identification of both sp-1 and z-1 in the present study, supported by theoretical calculations, indicates that the zwitterion was also formed.
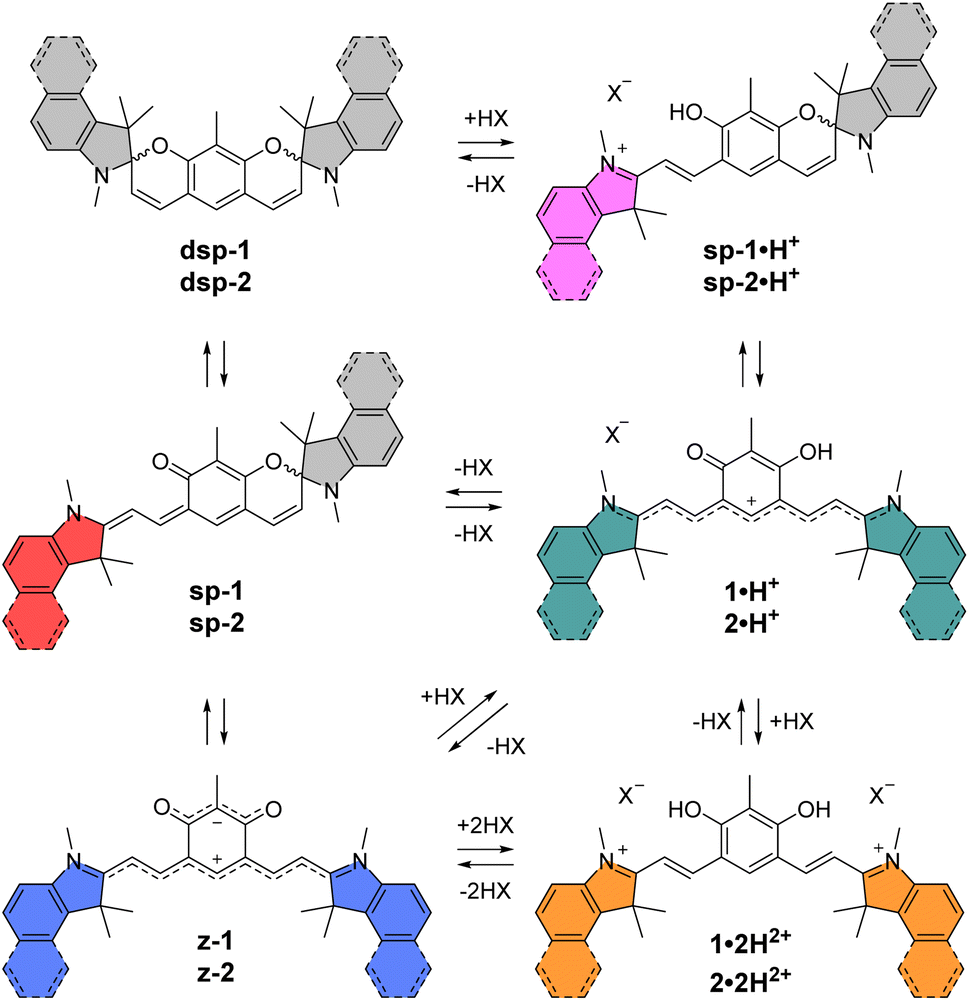 | ||
| Scheme 3 Different forms and protonation states identified for indole-based compound 1 and benzo[e]indole-based compound 2. | ||
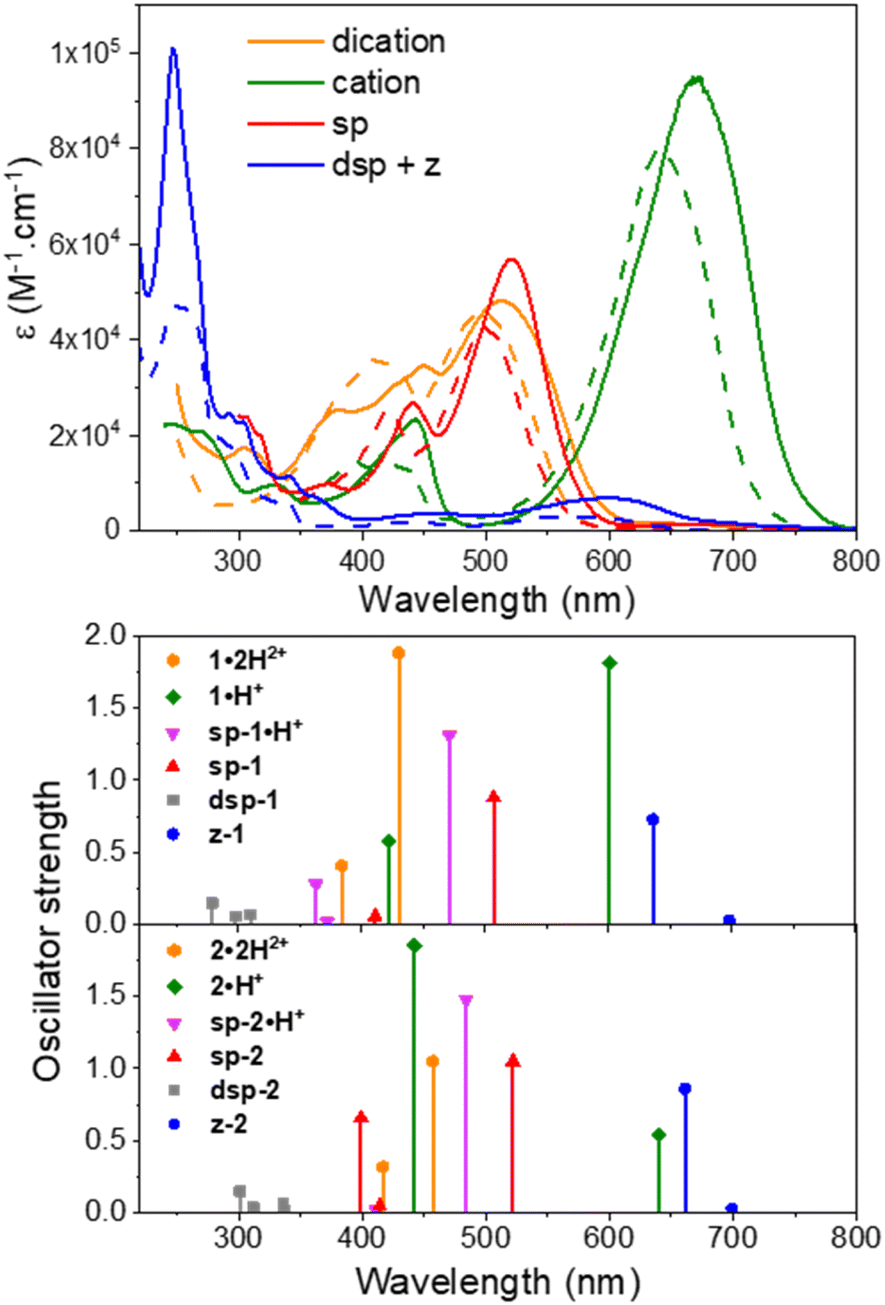 | ||
| Fig. 5 Experimental UV-vis-NIR electronic absorption spectra (top) and calculated vertical transitions (bottom) for the different protonation states and forms adopted by compounds 1 (dashed lines) and 2 (plain lines) in acidic, neutral or basic methanolic solutions (see Table 1 for conditions). | ||
Fluorescence was recorded for the dicationic and cationic species of 1–5 in MeOH, showing emission in the red to NIR spectral ranges (Table 1, Fig. 6 and S75–S78, ESI†). While the dications 1·2H2+, 2·2H2+ and 3·2H2+ display fluorescence beyond 750 nm characterized by particularly large Stokes shifts in the 6500–8000 cm−1 range (i.e. Δλ = 248–292 nm), the emissions of 4·2H2+ and 5·2H2+ are blueshifted in the orange-red window with smaller Stokes shifts of 4300–4800 cm−1 (i.e. Δλ = 121–152 nm). Such important Stokes shifts presumably evidence a strong stabilization at the excited state. Furthermore, all the monocationic species display emission maxima in the 700–800 nm range, characterized by smaller Stokes shifts in the 1100–2300 cm−1 range (Δλ ca. 50–100 nm), values closer to those of typical chloro-heptamethine cyanine emitters that usually feature partially overlapping absorption and emission bands, with Stokes shifts in the order of 400 cm−1 (Δλ ∼20 nm).58
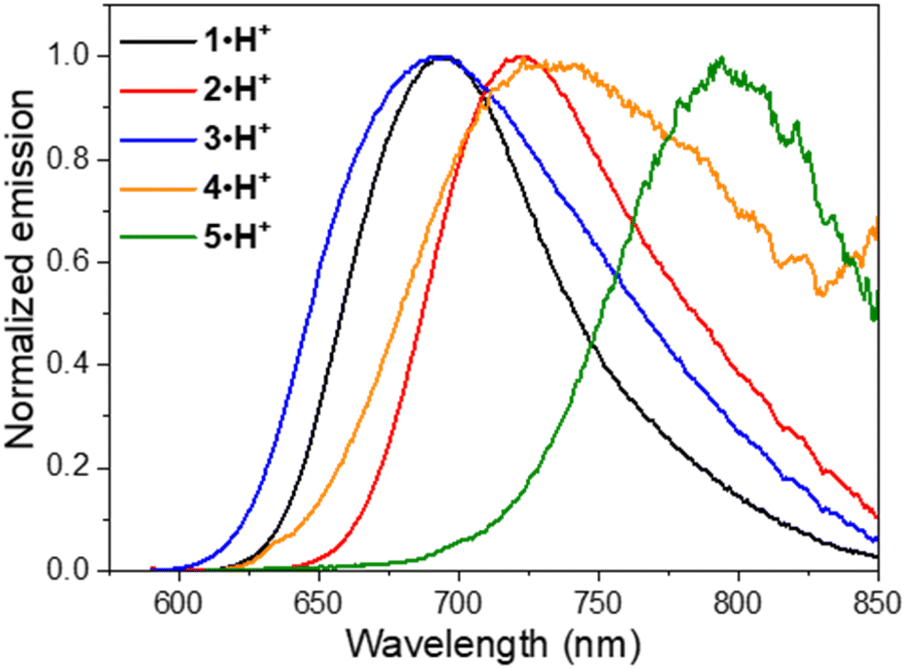 | ||
| Fig. 6 Normalized fluorescence emission spectra of the cationic species of 1–5 in methanol. | ||
Overall, the dications exhibit weak fluorescence quantum yields between 2 and 4% and short lifetimes < 0.5 ns (Fig. S76 and S77, ESI†). These low values are expected in view of the strong redshifted emission and the usual dominant non-radiative decay pathways when the gap is so small. In that vein, significantly higher quantum yields are measured in methanol for the cations 1·H+, 2·H+ and 3·H+ with 27%, 13% and 8%, respectively, and lifetimes between 0.7 and 1.7 ns, because the emissions are taking place at ca. 700 nm. These values are comparable to the performances of the analogous rigidified chloro-heptamethines (Table S1, ESI†)56,57 and higher than the QCy7 derivatives.38 Nevertheless, dyes 4·H+ and 5·H+, which feature the most redshifted emissions, show markedly lower quantum yields estimated at ca. 2–4%, more in accordance with the energy gap law.59
Spectrophotometric titrations versus pH
In order to characterise and quantify more precisely the different protonated species of the CoPo derivatives, absorption spectrophotometric titrations coupled with potentiometry were carried out on compounds 1–3, and compound 1′ that was used as reference. These studies were carried out in a mixture of 80% methanol and 20% water by weight, conditions in which the compounds retain good solubility. As an example, the absorption versus pH titration curve of compound 1 is shown in Fig. 7, while the others for compounds 2 and 3 are available in the ESI (Fig. S83–85).† This first titration, carried out from basic to acidic conditions, clearly shows spectral variations (Fig. 7A) in agreement with the data recorded in pure methanol in the presence or absence of TFA or DBU (Fig. 5). Statistical processing of the spectroscopic and potentiometric data revealed the presence of three protonated species whose electronic spectra could be accurately determined (Fig. 7B). The deprotonated species 1 is characterised by a weak absorption band centred in the visible at 581 nm (ε581 = 0.85 × 104 M−1 cm−1) and accompanied by a strong absorption in the UV (below 300 nm). According to theoretical calculations and experimental data in methanol (Fig. 5 and Table 1), this hints at a mixture of zwitterionic z-1 and dispiropyran dsp-1 species coexisting in equilibrium under neutral to basic conditions with dsp-1 likely predominant. Upon a first protonation (pKa2 = 5.98 ± 0.02), the low-energy absorption experienced significant hyperchromic and bathochromic shifts (λmax = 635 nm, ε635 = 3.38 × 104 M−1 cm−1). This is in excellent agreement with the formation of a cationic heptamethine dye 1·H+ (Scheme 3 and Fig. 5), most likely in equilibrium with the protonated monospiropyran species sp-1·H+ as indicated by the broad absorption at ∼500 nm partially masked by the sharp and intense transitions of the 1·H+ species at 471 nm (ε471 = 3.95 × 104 M−1 cm−1). A second protonation reaction then takes place under more acidic conditions (pKa1 = 3.49 ± 0.02) and affords the predominant dicationic species with significant hyperchromic shifts of the main absorption peaks (λmax = 495 nm, ε495 = 3.98 × 104 M−1 cm−1 and λmax = 409 nm, ε409 = 3.44 × 104 M−1 cm−1). The spectral properties of this diprotonated species 1·2H2+ are again in excellent agreement with both the data measured in methanol with TFA (Table 1) and the theoretical predictions (Fig. 5).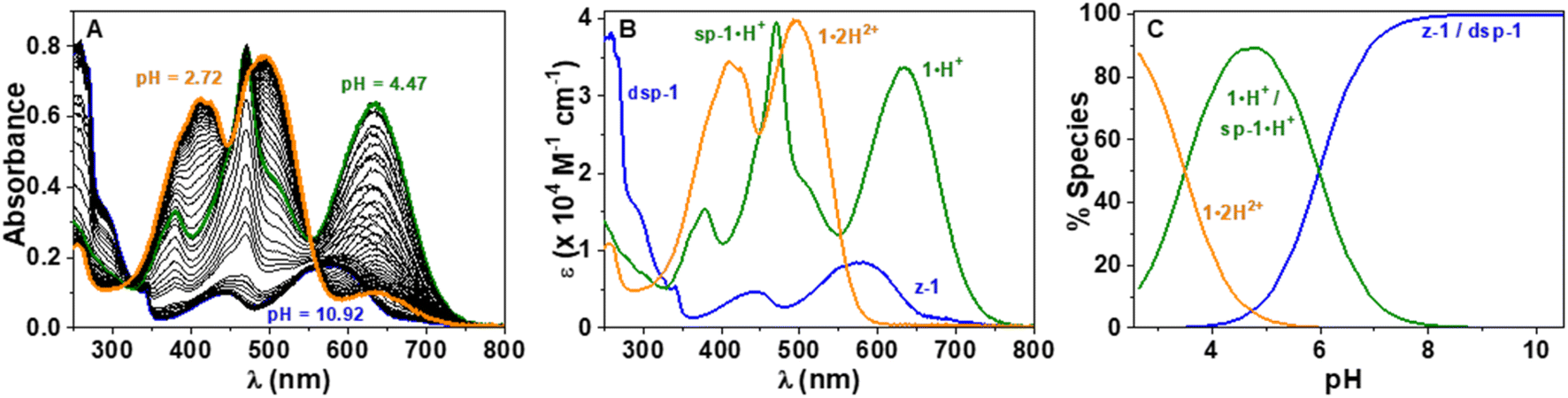 | ||
Fig. 7 (A) UV-vis absorption spectrophotometric properties versus pH titration of 1 in CH3OH/H2O (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 by weight) from basic to acidic pH values; (B) electronic absorption spectra of 1 (z-1 and dsp-1) and its protonated species 1·H+ (1·H+ and sp-1·H+) and 1·2H2+; (C) distribution diagrams of the protonated species of 1. I = 0.1 M NBu4ClO4; T = 25.0 ± 0.1 °C; [1] = 2.13 × 10−5 M. The absorption spectra are not corrected for the dilution effects. The data have been processed from pH 10.92 to 2.72 with pKa1 = 3.49 ± 0.02 and pKa2 = 5.98 ± 0.02. :20 by weight) from basic to acidic pH values; (B) electronic absorption spectra of 1 (z-1 and dsp-1) and its protonated species 1·H+ (1·H+ and sp-1·H+) and 1·2H2+; (C) distribution diagrams of the protonated species of 1. I = 0.1 M NBu4ClO4; T = 25.0 ± 0.1 °C; [1] = 2.13 × 10−5 M. The absorption spectra are not corrected for the dilution effects. The data have been processed from pH 10.92 to 2.72 with pKa1 = 3.49 ± 0.02 and pKa2 = 5.98 ± 0.02. | ||
These spectrophotometric and protonation properties were further confirmed by an absorption–pH titration carried out in reverse mode from acidic to basic pH (Fig. S80, ESI†), demonstrating the complete reversibility of the system and the occurrence of the very same forms of deprotonated (z-1 and dsp-1) and monocationic (1·H+ and sp-1·H+) species. The data obtained with CoPo 1 are in excellent agreement with the o-hydroxymerocyanine dye described in the literature,60 for which a merocyanine–spirocyanine protonation equilibrium was highlighted with a calculated pKa of 2.9 and spectral changes in line with those observed for 1. Finally, the species formed in solution with CoPo 1 are stable over time under acid or basic conditions in the dark (Fig. S81, ESI†) and slightly affected by daylight exposure under basic conditions (Fig. S82, ESI†).
While CoPo 2 behaves similarly to 1 with comparable spectral features and protonation constants (Fig. S83 and S84, ESI†), the benzothiazole analogue 3 showed markedly different properties. Insertion of the sulphur atom at the end groups hinders the formation of spiropyran derivatives, as shown by the absence of strong absorption in the UV region and the high absorptivity of the low-energy absorption band (Fig. 8) for any of the protonated species (z-3: λmax = 625 nm, ε625 = 7.45 × 104 M−1 cm−1; 3·H+: λmax = 608 and 469 nm, ε608 = 6.05 × 104 M−1 cm−1 and ε469 = 3.79 × 104 M−1 cm−1; 3·2H2+: λmax = 475 and 413 nm, ε475 = 3.86 × 104 M−1 cm−1 and ε413 = 3.25 × 104 M−1 cm−1). In addition, both pKa values leading to the mono- and dicationic species were found to be increased by more than 1.5 units, as compared to 1. The potentiometric data obtained for CoPo 1–3 are consistent with systems having two identical but non-independent protonation sites due to a π-conjugated system allowing delocalisation throughout the whole system. Indeed, the differences in pKa (ΔpKa = 2.49, 2.53 and 2.84 for 1, 2 and 3, respectively) are much greater than would be expected for two identical and independent sites (ΔpKa = 0.6).61
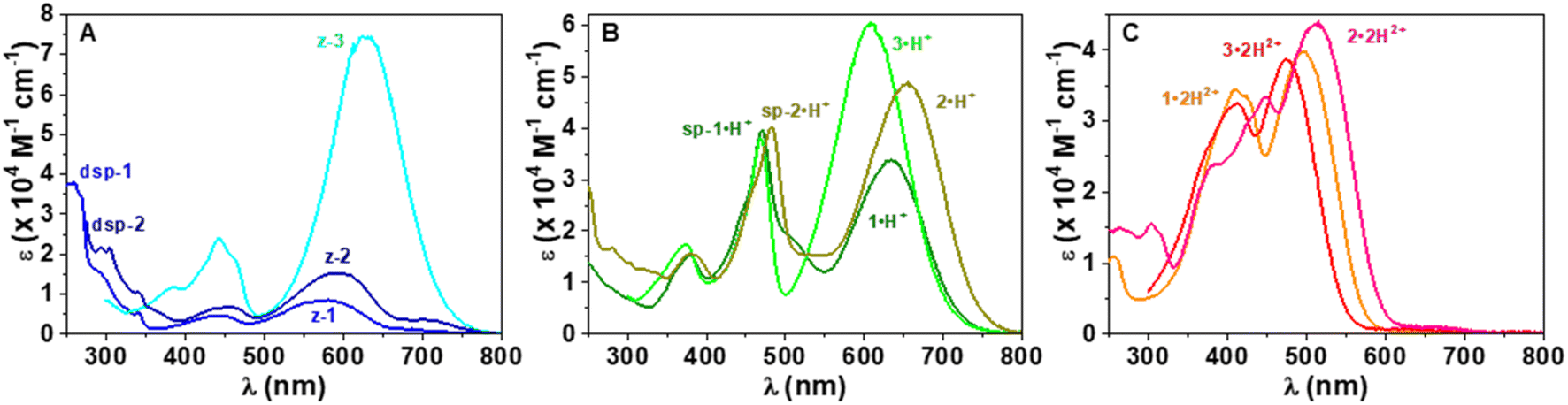 | ||
| Fig. 8 (A) Comparison of the UV-vis electronic absorption spectra of fully deprotonated 1, 2 and 3 showing the predominant formation of the zwitterionic z-3 species with 3 and a mixture of two species in equilibrium with CoPo 1 and 2 (the zwitterionic z-1 or z-2 and the dispiropyran dsp-1 or dsp-2). (B) Comparison of the UV-visible electronic absorption spectra of monoprotonated 1·H+, 2·H+ and 3·H+ showing the predominant formation of the heptamethine species 3·H+ with 3 and a mixture of two species in equilibrium with CoPo 1 and 2 (1·H+ or 2·H+ and the protonated monospiropyrans sp-1·H+ or sp-2·H+). (C) Comparison of the UV-visible electronic absorption spectra of deprotonated species 1·2H2+, 2·2H2+ and 3·2H2+. Solvent: CH3OH/H2O (80:20 by weight); I = 0.1 M NBu4ClO4; T = 25.0 ± 0.1 °C. | ||
In contrast to CoPo derivatives 1 and 2, further deprotonation reactions were observed under basic conditions with 3 (Fig. S85B, ESI†). The increase towards higher pH values leads to a strong hypochromic shift of the characteristic band of the zwitterionic species at 627 nm as well as a significant and concomitant broadening (Fig. S85, ESI†). This is most likely due to the predominance of the zwitterionic species z-3 (Fig. 8A) that can be prone to hydroxylation reactions of the end groups leading to a reduction in the dye's conjugated π-network and fading of the visible transitions, as reported for o-hydroxymerocyanine (pKa = 10.7).60 This was further confirmed using the reference compound 1′ (Scheme 1) for which two reversible equilibria and two basic protonation constants (pKa1 = 7.8 ± 0.3 and pKa2 = 10.29 ± 0.06) were determined (Fig. S86, ESI†).
Thin film acidochromic switching
To assess the acidochromic properties at the solid state, a transparent thin film of dsp-1 was prepared by spin-coating and exposed to hydrochloric acid vapours, triggering an immediate change in the state of the substrate from colourless to orange (Fig. 9), characterized by a broad absorption band centred at 494 nm corresponding to the dication 1·2H2+. In the presence of ammonia vapours, the substrate colour changes to green within a few seconds and the electronic absorption measurements revealed two main transitions at 482 and 664 nm, which were attributed to the formation of the cationic species sp-1·H+ and 1·H+, respectively (Scheme 3). The reversibility of the acidochromic switching was monitored at 660 nm and highlighted the good fatigue resistance of the system over six acid–base cycles.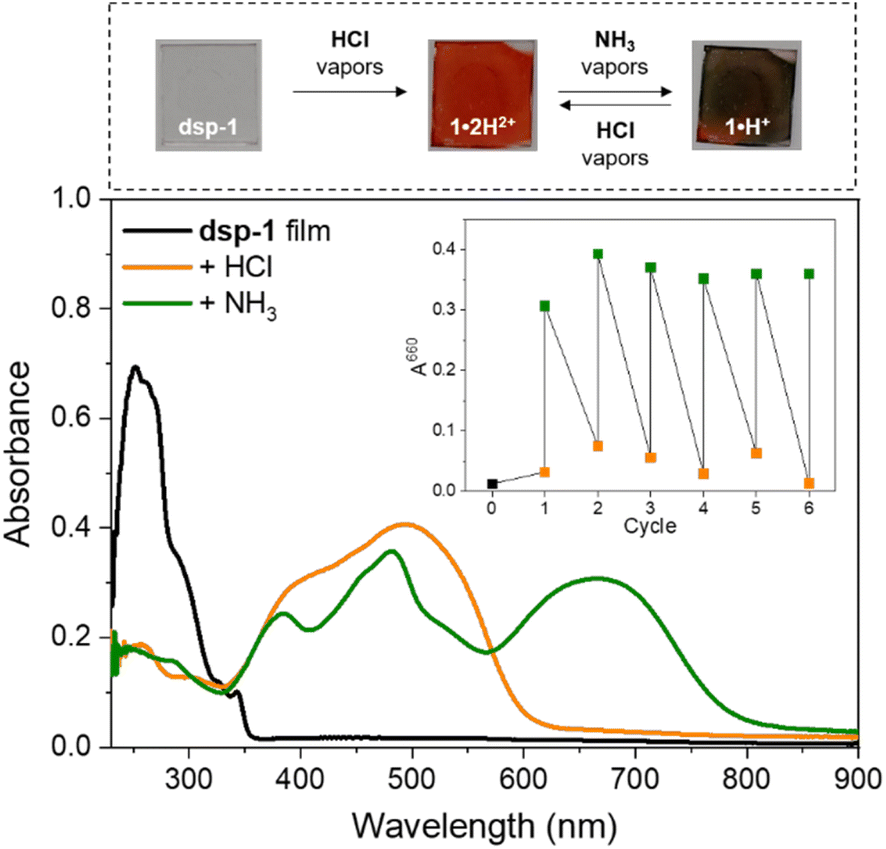 | ||
| Fig. 9 Top: glass substrate spin-coated with dsp-1 and exposed to ammonia and then hydrochloric acid vapours. Bottom: electronic absorption spectra of the thin film before and after exposure to vapours. Inset: reversibility of pH-switching monitored at 660 nm. | ||
Computational study
We have used first-principles calculations to model the structures and optical signatures of all compounds, using a method detailed in the ESI.† Whilst we stick to the vertical approximation for obvious computational reasons, all calculations of transition energies are determined on the basis of second-order coupled-cluster (CC2) calculations. As can be seen from the discussion above, and in particular the data listed in Table 1, the selected approach provides data consistent with the main experimental trends, with errors typical of the selected level of theory, and we focus here on providing additional insights into the nature of the electronic (ground and excited) states.In Fig. S87 in the ESI,† we compare the structure of the s-cis and s-trans forms of various forms of 1, and it can be seen that the energy difference between the two is negligible (less than 1 kcal.mol−1, smaller than the typical DFT error bar). However, as compared in Table S2 in the ESI,† both structures show very similar optical signatures but for a slight redshift of the longest wavelength band in 1·2H2, 1·H+, and z-1, not affecting the trends. For obvious computational reasons, we therefore performed our analysis for the s-trans conformers only.
In Table 2, we provide selected structural data for various forms of 1, together with the NICS of the central six-membered ring. In both 1′ and 1·2H2+, one notices quite short bonds between the top and bottom moieties, accompanied with sizeable bond length alternation (BLA) and NICS values compatible with an aromatic character for the central six-membered ring. We note nevertheless that there are small differences between 1′ and 1·2H2+, the former being slightly closer to the pure aromatic character for the phenyl ring with a single/double bond alternation in line with the one of polyenes. These slight variations provide a conceivable explanation regarding the more intense and redshifted absorption of 1·2H2+ compared to 1′ (Fig. 3). In 1·H+, significant changes are found with elongation of the C4–C1′ and C6–C3′ bonds, and the upper section of the compound acquires a clear alternating character, consistent with the presence of a hydroxyl and a keto group, whereas the lower section shows a significantly smaller BLA. In other words, 1·H+ presents a cyanine-like heptamethine subunit. These changes are enhanced in z-1, in which aromaticity is essentially lost and two single-like bonds connect the oxonol and heptamethine units, both having vanishing BLA.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| C4–C1′ | C6–C3′ | BLA Oa–Ob | BLA C1–C5 | BLA C9–C5 | NICS(0) | NICS(1zz) | |
| 1′ | 1.413 | 1.414 | 0.009 | 0.067 | 0.068 | −7.16 | −7.89 |
| 1·2H2+ | 1.420 | 1.420 | 0.000 | 0.062 | 0.061 | −6.48 | −7.20 |
| 1·H+ | 1.475 | 1.448 | 0.104 | 0.035 | 0.022 | −1.09 | −3.65 |
| z-1 | 1.496 | 1.494 | 0.000 | 0.008 | 0.008 | +1.82 | −1.87 |
Let us now discuss the nature of the excited states starting with 1·2H2+ for which the lowest transition is strongly dipole-allowed. The electron density different (EDD) plot displayed in Fig. 10 unsurprisingly shows a highly delocalized π–π* transition along the long sp2-C atom pathway, with the double bonds of the central unit acting as mild donor groups (mostly in blue). The topology of the excited state of 1′ is very similar. In the 1·H+ form, the topology of the excited state is interestingly not strongly modified, yet one notes a clear asymmetric character, since the keto oxygen atom is a quite potent donor group enhancing the charge-transfer (CT) character of the transition, which, together with the equalization of bond lengths (see above), can account for the observed redshift in going from 1·2H2+ to 1·H+. The situation changes more drastically in z-1 in which the lowest excited state, presenting a very small oscillator strength (Table 1), corresponds to a clear CT from the anionic oxonol to the cationic heptamethine, whereas the second excited state presents the typical ideal cyanine topology (Fig. 10). We note in the measured spectrum of Fig. 8 the presence of a very weak redshifted band at ca. 700 nm, followed by a more intense absorption at ca. 580 nm, which is consistent with the present computational analysis. In dsp-1 the two first transitions appearing in the UV (Table 1), as expected, correspond to π–π* excitations localized on the central core. In the mixed open–closed species sp-1·H+, the emergence of a long-delocalized path allows for a significant redshift with an excited-state topology quite alike the one of “half” of 1·2H2+ (Fig. 10). Conversely, the topology of sp-1 also shows strong similarities to the ones of 1·2H2+ and 1·H+. Table S3 and Fig. S88–S91 in the ESI† respectively provide extra structural data and EDD plots for the other compounds.
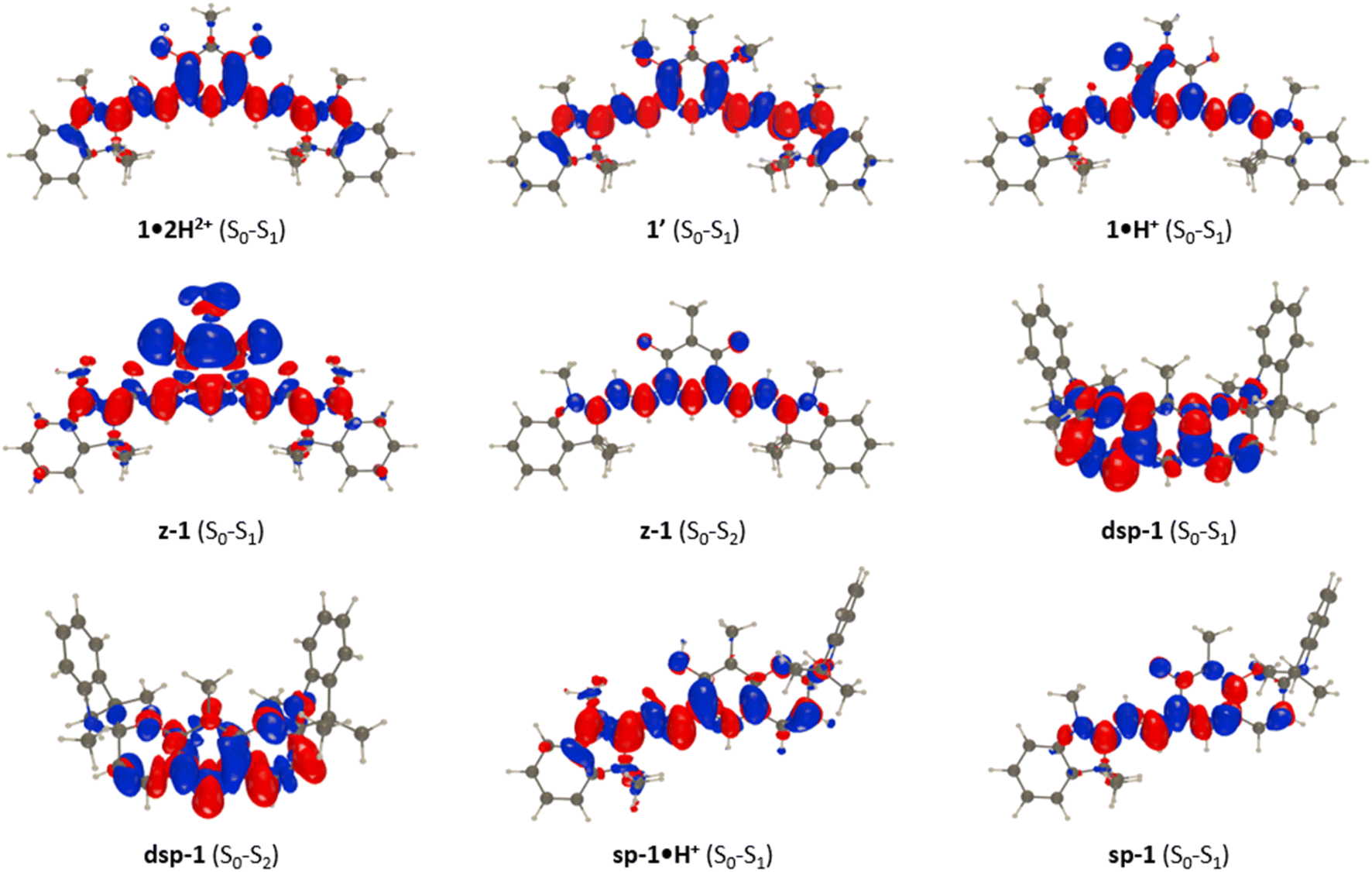 | ||
| Fig. 10 EDD plots corresponding to key transitions in various forms of 1. The blue and red lobes indicate the decrease and increase of electron density upon photon absorption, respectively. | ||
Conclusions
This work reported the design and characterization of π-extended coupled polymethines featuring various heterocyclic end groups that affect both their absorption and emission properties, while also controlling the formation of colourless dispirocyclic species (1 and 2) or red-NIR-absorbing zwitterionic bis-cyanines (3–5) in basic conditions. These extended heptamethine–oxonol dyes allow reaching absorptions at much longer wavelengths in their cationic and zwitterionic forms, compared to the previously designed benzoquinoneimine-based coupled trimethines, with up to ten times higher molar absorptivities in the visible and NIR regions. Importantly, this work bridges the gap between the previous reports on coupled heptamethine–oxonol dyes and related spirocyclic species by fully rationalizing their multiple acidochromic switching properties spanning from the UV to the NIR. Additionally, the joined experimental–theoretical approach pointed out the influence of the oxonol subunit that promotes an intramolecular CT towards the heptamethine subunit, at the origin of the broadened absorption bands, high Stokes shifts and overall the blueshifted optical properties of these coupled derivatives compared to classical rigidified heptamethine cyanines. These established structure–property relationships in extended CoPo lay the foundation for their use in the many domains where polymethines shine.Data availability
Most of the data are already available in the ESI.† If any data is needed, it is available on reasonable request to the authors.Author contributions
BM synthesized and characterized the compounds. VM and ME carried out the spectrophotometric and potentiometric titrations, and analysed the corresponding data. RS and DJ performed and analysed the quantum chemical calculations. DJ designed the theoretical protocol. SP and OS conceived the idea. SP designed the experiments and supervised the project. All authors critically reviewed the manuscript draft and approved the final version for submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Agence Nationale de la Recherche, in the frame of the SOCOOL project (ANR-20-CE07-0024). R. S. and D. J. are indebted to the Région des Pays de la Loire for financial support in the framework of the Opt-Basis PSR project. The authors thank the CNRS for continuous support, the Spectropole (Fédération Sciences Chimiques Marseille) for HRMS analyses, and the CCIPL/GliCid computational mesocenter for generous allocation of computational resources.References
- J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197–1226 CrossRef CAS.
- G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006–1029 CrossRef CAS PubMed.
- A. Barbieri, E. Bandini, F. Monti, V. K. Praveen and N. Armaroli, Top. Curr. Chem., 2016, 374, 47 CrossRef PubMed.
- R. S. Rao, Suman and S. P. Singh, Chem. – Eur. J., 2020, 26, 16582–16593 CrossRef CAS PubMed.
- L. Li, X. Dong, J. Li and J. Wei, Dyes Pigm., 2020, 183, 108756 CrossRef CAS.
- S. Pascal, S. David, C. Andraud and O. Maury, Chem. Soc. Rev., 2021, 50, 6613–6658 RSC.
- A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973–2012 CrossRef CAS PubMed.
- M. Panigrahi, S. Dash, S. Patel and B. K. Mishra, Tetrahedron, 2012, 68, 781–805 CrossRef CAS.
- J. L. Bricks, A. D. Kachkovskii, Y. L. Slominskii, A. O. Gerasov and S. V. Popov, Dyes Pigm., 2015, 121, 238–255 CrossRef CAS.
- H. Mustroph, Dyes Pigm., 2022, 208, 110783 CrossRef CAS.
- S. Barlow, J.-L. Bredas, Y. A. Getmanenko, R. L. Gieseking, J. M. Hales, H. Kim, S. R. Marder, J. W. Perry, C. Risko and Y. Zhang, Mater. Horiz., 2014, 1, 577–581 RSC.
- E. D. Cosco, J. R. Caram, O. T. Bruns, D. Franke, R. A. Day, E. P. Farr, M. G. Bawendi and E. M. Sletten, Angew. Chem., Int. Ed., 2017, 56, 13126–13129 CrossRef CAS PubMed.
- L. Štacková, P. Štacko and P. Klán, J. Am. Chem. Soc., 2019, 141, 7155–7162 CrossRef PubMed.
- B. Li, M. Zhao, L. Feng, C. Dou, S. Ding, G. Zhou, L. Lu, H. Zhang, F. Chen, X. Li, G. Li, S. Zhao, C. Jiang, Y. Wang, D. Zhao, Y. Cheng and F. Zhang, Nat. Commun., 2020, 11, 3102 CrossRef CAS PubMed.
- E. D. Cosco, B. A. Arús, A. L. Spearman, T. L. Atallah, I. Lim, O. S. Leland, J. R. Caram, T. S. Bischof, O. T. Bruns and E. M. Sletten, J. Am. Chem. Soc., 2021, 143, 6836–6846 CrossRef CAS PubMed.
- L. Feng, W. Chen, X. Ma, S. H. Liu and J. Yin, Org. Biomol. Chem., 2020, 18, 9385–9397 RSC.
- N. G. Medeiros, C. A. Braga, V. S. Câmara, R. C. Duarte and F. S. Rodembusch, Asian J. Org. Chem., 2022, 11, e202200095 CrossRef CAS.
- X. Zhao, F. Zhang and Z. Lei, Chem. Sci., 2022, 13, 11280–11293 RSC.
- W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed.
- H. Janeková, M. Russo and P. Štacko, Chimia, 2022, 76, 763 CrossRef PubMed.
- D. Saccone, S. Galliano, N. Barbero, P. Quagliotto, G. Viscardi and C. Barolo, Eur. J. Org Chem., 2016, 2016, 2244–2259 CrossRef CAS.
- S. Dähne and D. Leupold, Angew Chem. Int. Ed. Engl., 1966, 5, 984–993 CrossRef.
- K. Elbl, C. Krieger and H. A. Staab, Angew Chem. Int. Ed. Engl., 1986, 25, 1023–1024 CrossRef.
- S. S. Malhotra and M. C. Whiting, J. Chem. Soc., 1960, 3812–3822 RSC.
- O. Siri, P. Braunstein, M.-M. Rohmer, M. Bénard and R. Welter, J. Am. Chem. Soc., 2003, 125, 13793–13803 CrossRef CAS PubMed.
- S. Pascal and O. Siri, Coord. Chem. Rev., 2017, 350, 178–195 CrossRef CAS.
- S. Pascal, L. Lavaud, C. Azarias, G. Canard, M. Giorgi, D. Jacquemin and O. Siri, Mater. Chem. Front., 2018, 2, 1618–1625 RSC.
- S. Pascal, L. Lavaud, C. Azarias, A. Varlot, G. Canard, M. Giorgi, D. Jacquemin and O. Siri, J. Org. Chem., 2019, 84, 1387–1397 CrossRef CAS PubMed.
- J.-F. Longevial, Z. Chen, S. Pascal, G. Canard, D. Jacquemin and O. Siri, Chem. Commun., 2021, 57, 548–551 RSC.
- T. Horáčková, M. H. E. Bousquet, A. Morice, U. Triballier, G. Canard, P. Lhoták, D. Jacquemin, S. Pascal and O. Siri, Dyes Pigm., 2022, 206, 110681 CrossRef.
- O. Siri and P. Braunstein, Chem. Commun., 2002, 208–209 RSC.
- A. T. Ruiz, M. H. E. Bousquet, S. Pascal, G. Canard, V. Mazan, M. Elhabiri, D. Jacquemin and O. Siri, Org. Lett., 2020, 22, 7997–8001 CrossRef CAS PubMed.
- Y. Zheng, M.-S. Miao, Y. Zhang, T.-Q. Nguyen and F. Wudl, J. Am. Chem. Soc., 2014, 136, 11614–11617 CrossRef CAS PubMed.
- Y. Nagao, T. Sakai, K. Kozawa and T. Urano, Dyes Pigm., 2007, 73, 344–352 CrossRef CAS.
- A. Gaile, S. Belyakov, B. Turovska and N. Batenko, J. Org. Chem., 2022, 87, 2345–2355 CrossRef CAS PubMed.
- T. Munteanu, V. Mazan, M. Elhabiri, C. Benbouziyane, G. Canard, D. Jacquemin, O. Siri and S. Pascal, Org. Lett., 2023, 25, 3886–3891 CrossRef CAS PubMed.
- N. Karton-Lifshin, E. Segal, L. Omer, M. Portnoy, R. Satchi-Fainaro and D. Shabat, J. Am. Chem. Soc., 2011, 133, 10960–10965 CrossRef CAS PubMed.
- N. Karton-Lifshin, L. Albertazzi, M. Bendikov, P. S. Baran and D. Shabat, J. Am. Chem. Soc., 2012, 134, 20412–20420 CrossRef CAS PubMed.
- X. Yin, Y. Cai, S. Cai, X. Jiao, C. Liu, S. He and X. Zeng, RSC Adv., 2020, 10, 30825–30831 RSC.
- Y. Cai, C. Liu, Z. Lei, Z. Wang, Y. Bian, S. He and X. Zeng, Spectrochim. Acta, Part A, 2022, 265, 120404 CrossRef CAS PubMed.
- N. A. Voloshin, E. V. Solov’eva, S. O. Bezugliy, A. V. Metelitsa and V. I. Minkin, Chem. Heterocycl. Compd., 2012, 48, 1361–1370 CrossRef CAS.
- B. S. Lukyanov and M. B. Lukyanova, Chem. Heterocycl. Compd., 2005, 41, 281–311 CrossRef CAS.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- Sh. A. Samsoniya, M. V. Trapaidze, N. N. Nikoleishvili, K. G. Japaridze, J. P. Maisuradze and U. Kazmaier, Chem. Heterocycl. Compd., 2010, 46, 1016–1019 CrossRef CAS.
- L. Kortekaas, O. Ivashenko, J. T. van Herpt and W. R. Browne, J. Am. Chem. Soc., 2016, 138, 1301–1312 CrossRef CAS PubMed.
- C. Peng, W. Yuyang, Z. Yu-Mo and Z. S. Xiao-An, Acta Chim. Sin., 2016, 74, 669–675 CrossRef.
- Z. Wu, K. Pan, S. Mo, B. Wang, X. Zhao and M. Yin, ACS Appl. Mater. Interfaces, 2018, 10, 30879–30886 CrossRef CAS PubMed.
- I. V. Ozhogin, E. L. Mukhanov, A. V. Chernyshev, A. D. Pugachev, B. S. Lukyanov and A. V. Metelitsa, J. Mol. Struct., 2020, 1221, 128808 CrossRef CAS.
- L. Kortekaas, J. D. Steen, D. R. Duijnstee, D. Jacquemin and W. R. Browne, J. Phys. Chem. A, 2020, 124, 6458–6467 CrossRef CAS PubMed.
- S. H. M. Mehr, H. Depmeier, K. Fukuyama, M. Maghami and M. J. MacLachlan, Org. Biomol. Chem., 2017, 15, 581–583 RSC.
- C. Elian, B. Mourot, C. Benbouziyane, J.-P. Malval, S. Lajnef, F. Peyrot, F. Massuyeau, O. Siri, D. Jacquemin, S. Pascal and D.-L. Versace, Angew. Chem., Int. Ed., 2023, 62, e202305963 CrossRef CAS PubMed.
- K. Ilina and M. Henary, Chem. – Eur. J., 2021, 27, 4230–4248 CrossRef CAS PubMed.
- Z. Li, S. Mukhopadhyay, S.-H. Jang, J.-L. Brédas and A. K.-Y. Jen, J. Am. Chem. Soc., 2015, 137, 11920–11923 CrossRef CAS PubMed.
- I. Davydenko, S. Barlow, R. Sharma, S. Benis, J. Simon, T. G. Allen, M. W. Cooper, V. Khrustalev, E. V. Jucov, R. Castañeda, C. Ordonez, Z. Li, S.-H. Chi, S.-H. Jang, T. C. Parker, T. V. Timofeeva, J. W. Perry, A. K. Y. Jen, D. J. Hagan, E. W. Van Stryland and S. R. Marder, J. Am. Chem. Soc., 2016, 138, 10112–10115 CrossRef CAS PubMed.
- S. Pascal, S.-H. Chi, J. W. Perry, C. Andraud and O. Maury, ChemPhysChem, 2020, 21, 2536–2542 CrossRef CAS PubMed.
- A. Kurutos, Y. Shindo, Y. Hiruta, K. Oka and D. Citterio, Dyes Pigm., 2020, 181, 108611 CrossRef CAS.
- S. Pascal, S.-H. Chi, A. Grichine, V. Martel-Frachet, J. W. Perry, O. Maury and C. Andraud, Dyes Pigm., 2022, 203, 110369 CrossRef CAS.
- H. C. Friedman, E. D. Cosco, T. L. Atallah, S. Jia, E. M. Sletten and J. R. Caram, Chem, 2021, 7, 3359–3376 CAS.
- Y. Yue, F. Huo, S. Lee, C. Yin, J. Yoon, J. Chao, Y. Zhang and F. Cheng, Chem. – Eur. J., 2016, 22, 1239–1243 CrossRef CAS PubMed.
- B. Perlmutter-Hayman, Acc. Chem. Res., 1986, 19, 90–96 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed descriptions for all experimental protocols and computational methods used. NMR, HRMS, FTIR, photophysical and theoretical data are also provided. See DOI: https://doi.org/10.1039/d3sc06126d |
| This journal is © The Royal Society of Chemistry 2024 |