
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-opening polymerisation of alkyl-substituted ε-caprolactones: kinetic effects of substitution position†
Cinzia
Clamor
a,
James
Beament
b,
Peter M.
Wright
b,
Beatrice N.
Cattoz
b,
Rachel K.
O'Reilly
 *a and
Andrew P.
Dove
*a
*a and
Andrew P.
Dove
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: r.oreilly@bham.ac.uk; a.dove@bham.ac.uk
bInfineum UK Ltd, Milton Hill, Abingdon OX13 6BB, UK
First published on 20th February 2024
Abstract
Ring-opening polymerisation (ROP) of lactones has been proven as a powerful technique to generate polyesters with high levels of control over molar mass and polymer dispersity. However, the introduction of functional groups on the monomer ring structure can dramatically influence the ability of a monomer to undergo ROP. Therefore, understanding the structure–reactivity relationship of functional monomers is essential to gain access to materials with chemical functionality via direct polymerisation. Herein, we report how structural modifications of alkyl-substituted ε-caprolactones affected their reactivity towards the ring-opening of the functional monomer. We observed that the reactivity was strongly influenced by the substituent position, wherein the δ-substituted monomer exhibited the fastest polymerisation kinetics. In contrast, a substituent placement in the ε-position significantly reduced polymerisation time compared to other substituent positions. Moreover, the thermal properties of the resultant functional ε-polycaprolactones were investigated and showed no significant change in the thermal transitions. This demonstrates that functional caprolactone monomers with sterically demanding functional groups can still undergo direct ring-opening polymerisation and that careful positioning of these functional groups enables control of the rate of polymerisation, a crucial parameter to be considered for the design of new prospective functional monomers and their industrial applications.
Introduction
Aliphatic polyesters are an attractive and versatile class of polymeric materials that can undergo degradation by hydrolysis, as well as, in some cases, degrade in the environment.1–3 Their application ranges from commodity thermoplastics to biomaterials for tissue engineering or drug delivery devices.4–6 In particular, polycaprolactones (PCLs) have been the subject of considerable interest owing to their low toxicity, biocompatibility and biodegradability.7–9 Synthetically, PCL can be obtained either by step-growth polycondensation of hydroxycarboxylic acids or ring-opening polymerisation (ROP) of lactones.4 While polycondensation reactions allow the production of a wider range of functional polyesters, they often require long reaction times and high temperatures, resulting in poor control over the molar mass.4 By contrast, ROP enables the generation of polyesters with good control over polymerisation, predictable molar mass and narrow dispersities.10,11 As a consequence, the ROP of lactones constitutes the method of choice for the synthesis of PCL and has been extensively studied using a variety of catalytic systems and initiators.12–15 However, a major limitation towards the development of new materials results from the lack of readily tuneable side chain functionality, which prevents direct functionalisation to be performed on the polymer backbone. Indeed, the presence of functional groups is highly desirable for the design of new potential monomers as it allows important polymer properties such as crystallinity, biodegradation rate or mechanical properties to be tailored.16Over the past two decades several strategies for the synthesis of functional polycaprolactones have been developed. A common approach is to graft functional groups onto preformed polymer chains bearing a reactive pendant group such as azides,17 alkynes18 or epoxides.19 This approach allows the introduction of a variety of functional groups via e.g. click chemistry,20,21 cycloadditions22 or metathesis reactions.23 However, these reactions are often limited by functionality, scalability and contamination by catalysts and/or chemicals used to perform the post-polymerisation functionalisation.4 To overcome these predicaments, the direct polymerisation of functional monomers offers highly effective and efficient synthesis with greater control over monomer purity, polymer composition and the ability to produce block copolymers without the need of a further modification step.24,25 Therefore, the direct ROP of monomers containing functional groups is often more desirable to develop more universal polymerisation strategies for e.g. industrial applications.
The ROP of functional ε-caprolactones has been widely studied using simple functional monomers including lactone substituted with halogenides,26,27 acrylates28 or terminal olefins.29 However, the addition of new functional groups, particularly those in close proximity to the active ring-opening centre, can promote steric hindrance and influence the ring-opening polymerisation kinetics. In order to enhance the understanding of substituent effects on monomer polymerisability, Hillmyer and co-workers systematically studied the polymerisation thermodynamics and kinetics of a series of n-alkyl-substituted δ-valerolactones with a focus on the effect of alkyl substituent sizes and regiochemistry on the polymerisation rate.30 The authors found that the regiochemistry of a methyl substituent influenced the polymerisation rate. Monomers substituted in α- and β-position polymerised at a rate comparable to the unsubstituted monomer, whereas a substituent placed in the δ-position made the polymerisation an order of magnitude slower.30 Considering the importance of exploiting novel functional caprolactones, we envisioned that a comparative study of how sterically demanding alkyl substituents influence the ring-opening kinetics would enhance the understanding of the structure–reactivity relationship, a fundamental parameter to be evaluated prior to polymer applications on a larger scale.
Herein, we describe the direct preparation of functional lipophilic polyesters via well-controlled ring-opening polymerisation using alkyl-functionalised ε-caprolactone (εCL) monomers. In particular, we sought to investigate the relationship between the substituent position on the εCL ring and the reaction rate, by using four different alkyl-functionalised caprolactone isomers: γC18CL, a mixture of two isomers substituted in β- and δ-position (β/δC18CL isomers), and εC18CL. Moreover, the effect of the alkyl chain position on the thermal properties of the formed polymers was also investigated.
Experimental
Materials
Chemicals and solvents, unless otherwise stated, were purchased from Sigma-Aldrich and Fisher Scientific and used without further purification. Deuterated benzene was ordered from Apollo Scientific, degassed by repeated freeze-pump-thawing and then dried under reflux over sodium and benzophenone before storing it under an inert atmosphere of N2. Deuterated chloroform was ordered from Apollo Scientific, dried over molecular sieves 3 times and deoxygenated with N2 before storing it under an inert atmosphere of N2. Diphenyl phosphate was dried under reduced pressure at 50 °C for 3 days. 4-Methoxybenzyl alcohol was dried over CaH2 for 24 h and distilled under reduced pressure before storing it under an inert atmosphere of N2. Amberlyst A21 free base was washed repeatedly with methanol and dried under reduced pressure prior to use. The alkyl-substituted monomers, γC18CL, β/δC18CL isomers and εC18CL were received from Infineum UK Ltd (Schemes S1–S3†), dissolved in dry CH2Cl2 and dried over CaH2 for 24 h (and this was repeated 1× with fresh CaH2). Excess CaH2 was removed by cannula filtration and CH2Cl2 was distilled off under reduced pressure before storing the monomers under an inert atmosphere of N2.General considerations
All polymerisations were performed under an inert nitrogen atmosphere in a glovebox unless otherwise stated. All other chemical manipulations were performed using standard Schlenk-line techniques. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX-300, DPX-400 or HD500 spectrometer. Chemical shifts are reported in ppm. Solvent residual signals were used as reference (CDCl3, 1H: δ = 7.26 ppm for CHCl3, 13C: δ = 77.16 ppm for CDCl3; C6D6, 1H: δ = 7.16 ppm for C6D5H and 13C: δ = 128.06 ppm for C6D6). The signals are characterised as s = singlet, d = doublet, t = triplet, q = quartet and m = multiplet. The coupling constant J is recorded in Hertz (Hz). Size exclusion chromatography (SEC) measurements were performed on an Agilent 1260 Infinity II Multi-Detector GPC/SEC System fitted with RI, ultraviolet (UV, λ = 309 nm), and viscometer detectors. The polymers were eluted through an Agilent guard column (PLGel 5 μM, 50 × 7.5 mm) and two Agilent mixed-C columns (PLGel 5 μM, 300 × 7.5 mm) using CHCl3 (buffered with 0.5% v/v NEt3) as the mobile phase (flow rate = 1 mL min−1, 40 °C). Number average molar mass (Mn), weight average molar mass (Mw) and dispersities (ĐM = Mw/Mn) were determined using Agilent GPC/SEC software (vA.02.01) against a 15-point calibration curve (Mp = 162–3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 187000 g mol−1) based on poly(styrene) standards (Easivial PS-M/H, Agilent). Number average molar mass (Mn), weight average molar mass (Mw) are given in g mol−1. The thermal characteristics of the polymers were determined using differential scanning calorimetry (DSC) on a STARe DSC3 system from Mettler Toledo and analysed in 40 μL aluminium pans from -100–200 °C at a heating rate of 10 °C min−1 for two heating/cooling cycles or 1 °C min−1 for one heating/cooling cycle. TGA data was obtained using a Q50 Thermogravimetric Analyzer (TA instruments). Thermograms were recorded under an N2 atmosphere using a heating rate of 10 °C min−1, from 25–500 °C, with an average sample weight of ca. 10 mg. Decomposition temperatures were reported as the 5% weight loss temperature (Td 5%).
187000 g mol−1) based on poly(styrene) standards (Easivial PS-M/H, Agilent). Number average molar mass (Mn), weight average molar mass (Mw) are given in g mol−1. The thermal characteristics of the polymers were determined using differential scanning calorimetry (DSC) on a STARe DSC3 system from Mettler Toledo and analysed in 40 μL aluminium pans from -100–200 °C at a heating rate of 10 °C min−1 for two heating/cooling cycles or 1 °C min−1 for one heating/cooling cycle. TGA data was obtained using a Q50 Thermogravimetric Analyzer (TA instruments). Thermograms were recorded under an N2 atmosphere using a heating rate of 10 °C min−1, from 25–500 °C, with an average sample weight of ca. 10 mg. Decomposition temperatures were reported as the 5% weight loss temperature (Td 5%).
Standard procedure for DPP catalysed polymerisations
Using standard glovebox techniques, a stock solution was prepared containing 4-methoxybenzyl alcohol initiator (55.0 mg, 0.40 mmol) and dry benzene-d6 (500 μL). The stock solution (50 μL) was added to the appropriate monomer (2 mmol) and catalyst (50.0 mg, 0.20 mmol) in dry benzene-d6 (1.95 mL) to form a 1 M solution. The solution was then transferred into a vial. Aliquots were taken at allotted time points and quenched by the addition of Amberlyst® A21 free base. After determining the polymer conversion by 1H NMR spectroscopy, Amberlyst® was removed via filtration through a pipette plugged with cotton wool and the polymer was precipitated into cold MeOH, cooled using liquid nitrogen. Polymers were dried under vacuum.
13C NMR (101 MHz, CDCl3, 299 K, ppm): δ = 173.9 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 62.8 (CH2O), 61.0 (CH2), 34.4 (CH), 33.3 (CH2), 32.3 (CH2), 32.08 (CH2), 31.7 (CH2), 30.2 (CH2), 29.9 (CH2), 29.9 (CH2), 29.8 (CH2), 29.5 (CH2), 28.8 (CH2), 26.6 (CH2), 22.8 (CH2), 14.3 (CH3).
O), 62.8 (CH2O), 61.0 (CH2), 34.4 (CH), 33.3 (CH2), 32.3 (CH2), 32.08 (CH2), 31.7 (CH2), 30.2 (CH2), 29.9 (CH2), 29.9 (CH2), 29.8 (CH2), 29.5 (CH2), 28.8 (CH2), 26.6 (CH2), 22.8 (CH2), 14.3 (CH3).
SEC (CHCl3): Mn = 11350, Mw = 12700, ĐM = 1.12.
13C NMR (101 MHz, CDCl3, 299 K, ppm): δ = 173.6 (CO), 66.9 (CH2O), 36.9 (CH), 34.5 (CH2), 32.0 (CH2), 31.1 (CH2), 30.9 (CH2), 30.0 (CH2), 29.8 (CH2), 29.7 (CH2), 29.7 (CH2), 29.4 (CH2), 26.7 (CH2), 26.6 (CH2), 22.7 (CH2), 22.2 (CH2), 14.2 (CH3).
SEC (CHCl3): Mn = 6350, Mw = 6900, ĐM = 1.09.
O), 74.0 (CHO), 50.9 (CH2), 34.5 (CH2), 34.0 (CH2), 33.9 (CH2), 32.0 (CH2), 29.8 (CH2), 29.7 (CH2), 29.7 (CH2), 29.7 (CH2), 29.6 (CH2), 29.6 (CH2), 29.4 (CH2), 25.4 (CH2), 25.0 (CH2), 25.0 (CH2), 22.7 (CH2), 14.1 (CH3).
SEC (CHCl3): Mn = 10100, Mw = 12500, ĐM = 1.23.
Results and discussion
Ring-opening polymerisation of n-alkyl-substituted caprolactones
The alkyl-substituted monomers, γC18CL, εC18CL and the mixture of the β- and δC18CL isomers, were each polymerised under identical conditions using diphenyl phosphate (DPP) (10 mol%) as the catalyst and 4-methoxybenzyl alcohol as the initiator ([M]0/[I]0 = 50) (Scheme 1). DPP enables the ROP of lactones with high control over molar mass regardless of the monomer structure and was therefore chosen as the catalyst for this study.30,31 | ||
| Scheme 1 General scheme for the ring-opening polymerisation of n-alkyl-substituted ε-caprolactone monomers. | ||
Each polymerisation was monitored independently by 1H NMR spectroscopy following the disappearance of CH2OCO monomer resonance (δ = 3.7 ppm, Fig. 1) and the appearance of the CH2OCO polymer resonance (δ = 4.2 ppm, Fig. 1); as shown for γC18CL in Fig. 1. For this particular monomer, a decrease in the monomer concentration was observed, reaching a monomer conversion of 90% within 2.5 h. In order to study the polymerisation control, aliquots were taken from the individual polymerisations, quenched by the addition of a solid-support base (Amberlyst®) and further analysed by size exclusion chromatography (SEC) and 1H NMR spectroscopy.
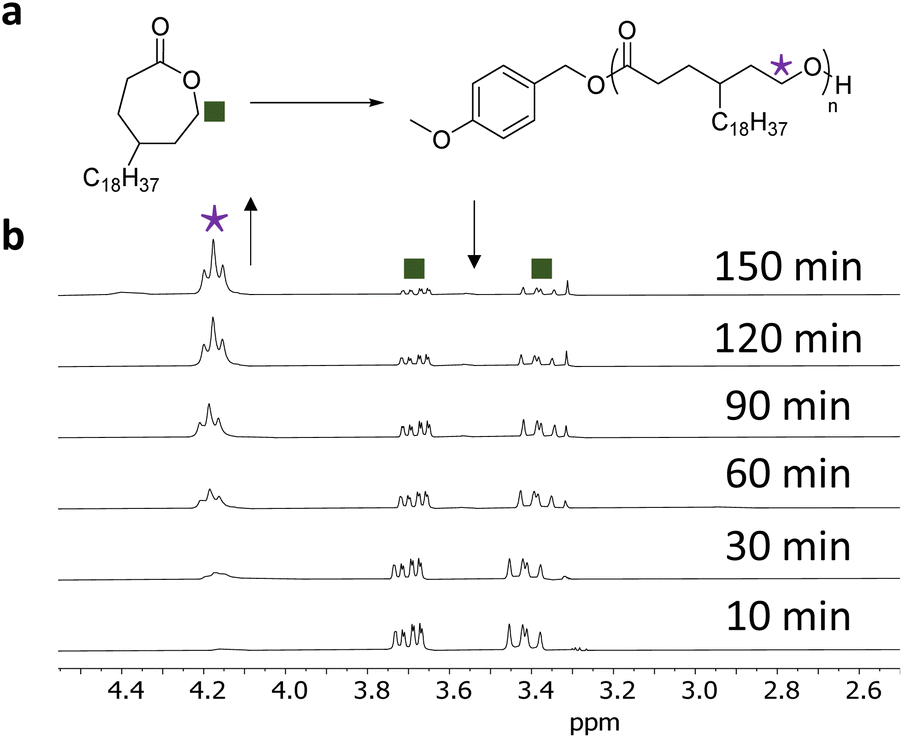 | ||
| Fig. 1 (a) ROP of γC18CL using DPP (10 mol%), 4-methoxybenzyl alcohol ([M]0/[I]0 = 50), benzene-d6. (b) Stacked 1H NMR spectra (benzene-d6, 300 MHz) during ROP of γC18CL showing the CH2OCO monomer resonance between δ = 3.37–3.70 ppm and CH2OCO polymer resonance at δ = 4.2 ppm. | ||
All studied polymerisations demonstrated a linear increase of molar mass with conversion (Fig. 2), indicating a well-controlled polymerisation. This could be further confirmed by 1H NMR spectroscopy of the precipitated polymers, which revealed the characteristic peaks of the PCL backbone with the pendant alkyl moieties and well-defined end-group signals (Fig. S4–S6†). These were used to confirm a degree of polymerisation (DP) close to the targeted DP. SEC analysis further confirmed the well-controlled nature of the polymerisations with dispersities between 1.09 and 1.23. (Table 1 and Fig. S7–S9†) It should be noted that εC18PCL showed a small shoulder at higher molar mass, which can be explained by the presence of transesterification side reactions occurring at high monomer conversions (Fig. S9†). Overall, these results confirm that ROP is a method that enables the synthesis of a range of functional polycaprolactones while maintaining control over polymerisation parameters such as molar mass and dispersity.
 | ||
| Fig. 2 Number-average molar mass (Mn) plotted against monomer conversion for the ROP of γC18CL, εC18CL and β/δC18CL isomers using DPP (10 mol%) and 4-methoxybenzyl alcohol as initiator ([M]0/[I]0 = 50). Number-average molar mass (Mn) determined by SEC analysis in CHCl3 (0.5% NEt3), calibrated against polystyrene standards. | ||
| Monomer | Reaction Time (h) | Monomer Conversion (%)b | M n (kg mol−1)c | M n (kg mol−1)d | Đ M |
|---|---|---|---|---|---|
|
a DPP (10 mol%) and 4-methoxybenzyl alcohol as initiator ([M]0/[I]0 = 50) at 25 °C in benzene-d6.
b Determined by integrating end-groups using 1H NMR spectroscopy (benzene-d6, 298 K).
c Determined by 1H NMR spectroscopy (CDCl3, 298 K) analysis by integrating end-group signal and CH2OCO polymer resonance (δ = 4.2 ppm).
d Determined by SEC analysis in CHCl3 (0.5% NEt3) as eluent and calibrated against polystyrene standards.
|
|||||
| γC18CL | 3.4 | 86 | 17.5 | 11.4 | 1.12 |
| β/δC18CL (1:1 ratio) |
4 | 78 | 17.1 | 6.4 | 1.09 |
| εC18CL | 207 | 82 | 17.7 | 10.1 | 1.23 |
Kinetic resolution of the β- and δC18CL isomers
Notably, while the increase of Mn with conversion showed a linear behaviour in all cases, the kinetic plots showed first order behaviour only for the pure monomers γC18CL and εC18CL. Not surprisingly, the mixture of the β- and δC18CL isomers displayed a non-linear relationship of the semilogarithmic plot, giving first indication that both monomers polymerise at different rates (Fig. 3b). 1H NMR spectroscopic analysis in benzene-d6 did not show any resolution between the CH2OCO polymer resonance (δ = 4.2 ppm) of the β- or δ-substituted C18PCL. Therefore, to assess the progression of the polymerisation of the two isomers, individual kinetic points were taken at allotted times, precipitated into methanol and re-dissolved in chloroform-d. The ratio of the β- to δ-isomers was evaluated by comparing the CH2OCO integrals (δ = 4.0 and 3.9 ppm, respectively), which showed distinct splitting patterns (Tables 2 and S1†). The CH2OCO resonance splitting into a doublet can be assigned to δC18PCL, whereas the triplet corresponds to the βC18PCL isomer (Fig. 3a). The linearity of the resolved semi-logarithmic plot indicates first order kinetics in all cases with the δ-substituted C18PCL displaying the fastest polymerisation rate (Fig. 3c). Using this kinetic resolution experiment allows the comparison of the polymerisation rate of all 4 alkyl-substituted monomers at fixed catalyst and initiator concentration (Fig. 4). Not surprisingly, the rate of polymerisation is strongly dependent on the position of the substituent. While δ- and γ-substituted C18εCL displayed the fastest kinetics, with monomer conversion of around 85% reached within 3 h, the β-position seemed to have a greater impact on the ROP kinetics, possibly owing to an increased steric hindrance. In contrast to these findings, when the C18-substituent was placed in the ε-position, a substantially reduced rate of polymerisation was observed such that a monomer conversion of only 80% could be obtained within 9 days. We propose that the significantly slower kinetics are the result of high levels of steric hindrance around the carbonyl functionality (kinetics: δC18 > γC18 > βC18 ≫ εC18). These results are similar to those observed by Hillmyer and coworkers, where a series of alkyl-substituted δ-valerolactones were investigated. Among the methyl-substituted lactones, the γ-substituted derivative exhibited the fastest polymerisation rate, whereas the δ-substituted δ-valerolactone was reported to have the slowest polymerisation rate, likely as the result of the relatively low reactivity of the propagating secondary alcohol.30 Nevertheless, the sluggish polymerisation rate of εC18CL could be further improved by increasing the temperature from 25 to 50 °C or by changing the catalyst from DPP to Mg(BHT)2(THF)2 (Fig. S10†).
 | ||
| Fig. 3 (a) 1H NMR spectrum (CDCl3, 400 MHz) of β and δC18PCL. * = MeOH. (b) Semi-logarithmic kinetic plot time for the ROP of β/δC18CL isomers. (c) Resolved semi-logarithmic kinetic plot for the ROP of β/δC18CL isomers. [M]0 = 0.5 M (calculated by NMR spectroscopy based on a starting ratio of β isomer:δ isomer = 1:1). | ||
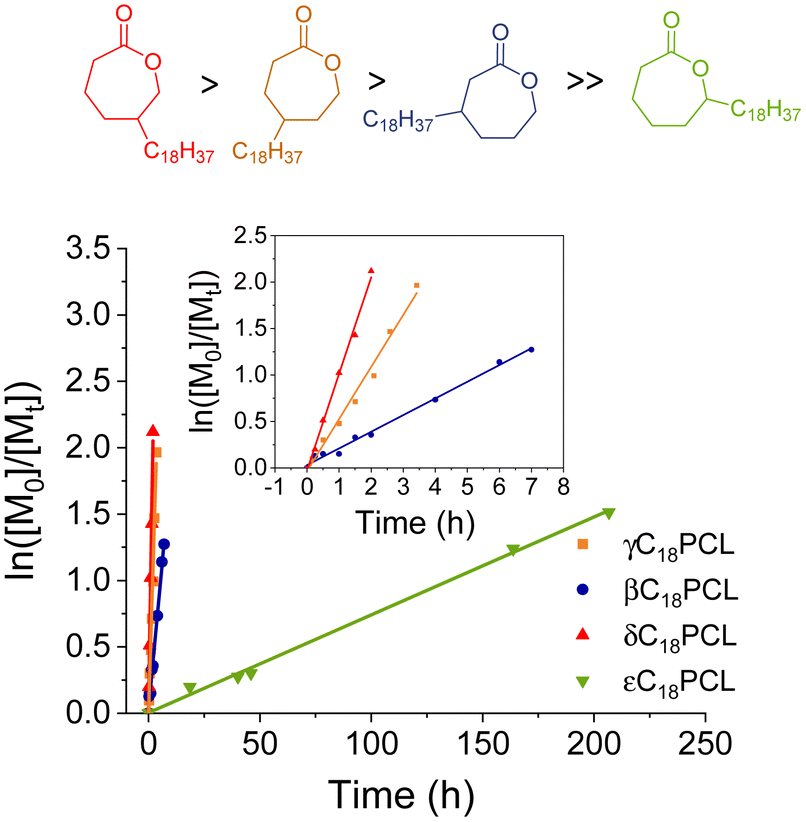 | ||
| Fig. 4 Semi-logarithmic kinetic plot for ROP of εC18CL, γC18CL and β/δC18CL isomers using DPP (10 mol%) and 4-methoxybenzyl alcohol as initiator ([M]0/[I]0 = 50). ln([M]0/[M]t) was determined by 1H NMR spectroscopy. | ||
| Time (min) | Total conversionb (%) | β-C18PCL isomer:δ-C18PCL isomerc |
Resolved conversion (β isomer)d (%) | Resolved conversion (δ isomer)d (%) |
|---|---|---|---|---|
|
a ROP of β/δ(C18)CL isomers using DPP and 4-methoxybenzyl alcohol as initiator ([M]0/[I]0 = 50). [M] = 1 M.
b Determined by 1H NMR spectroscopy in benzene-d6.
c Determined by 1H NMR spectroscopy in CDCl3 by integration of CH2OCO integrals at δ = 4.0 and 3.9 ppm.
d Calculated from isomer ratios obtained by 1H NMR spectroscopy based on a total monomer concentration of 1 M and a starting ratio of β isomer:δ isomer = 1:1.
|
||||
| 15 | 15 | 1:1.6 |
12 | 18 |
| 30 | 27 | 1:3 |
14 | 40 |
| 60 | 39 | 1:4.5 |
14 | 64 |
| 90 | 53 | 1:2.7 |
28 | 76 |
| 120 | 60 | 1:2.9 |
30 | 88 |
| 240 | 78 | 1:1.9 |
52 | — |
| 360 | 85 | 1:1.2 |
68 | — |
| 420 | 92 | 1:1.1 |
72 | — |
Thermal properties of the C18-substituted PCLs
In order to probe how the substituent position affects the thermal properties of the obtained polymers, differential scanning calorimetry (DSC) analysis was performed on γC18PCL εC18PCL and β/δC18PCL isomers with a degree of polymerisation (DP) of 50 ([M]0/[I]0 = 50) (Fig. 5 and S11†). All three polymers exhibited a sharp first-order melting and crystallisation transition (Tm and Tc). The glass transition temperature (Tg) could not be observed, possibly owing to it being masked by the intensity of the Tm and Tc transition. The observed crystallinity is likely the result of a cooperative organisation of the pendant C18 alkyl chains, which has been previously reported to enhance the overall order in the polymers.32,33 However, when comparing the three different constitutional isomers only slight differences in the Tm and Tc could be observed with γC18PCL displaying the highest Tm of 49 °C while β/δC18PCL isomers displayed the lowest Tm of 42 °C (Table 3). The lower Tm of the β/δC18PCL isomers is likely the result of the presence of two substituent sides in the polymers, which thereby disrupt the packing of the C18-alkyl chains in the polymer. Thermogravimetric analysis (TGA) revealed a weight loss of 5% at 280 °C for γC18PCL and at around 260 °C for both εC18PCL and β/δC18PCL isomers, although εC18PCL isomers showed a much sharper weight loss compared β/δC18PCL (Fig. 5a). Nevertheless, all three polymers exhibit thermal stability below 250 °C, making them a suitable material for a range of industrial applications. Overall, it could be demonstrated that the position of the alkyl chain on the PCL backbone has only minor effects on the thermal properties of the resultant polymers. This demonstrates that the discrete substituents play a major role in polymerisation kinetics, but only have minor effect on the resulting thermal properties.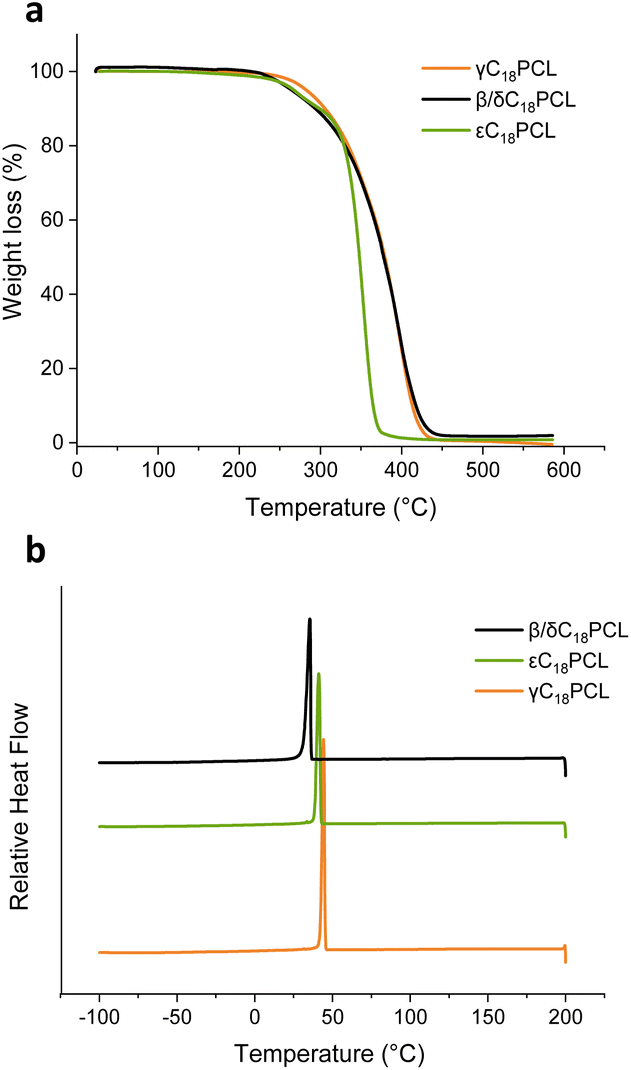 | ||
| Fig. 5 (a) Thermogravimetric analysis (TGA) of γC18PCL, εC18PCL and β/δC18PCL isomers. Analytical data of the analysed polymers is summarised in Table 1. (b) Differential scanning calorimetry (DSC) for 2nd cooling cycle (200 to −100 °C, 10 °C min−1) showing crystallisation and melting temperature of the polymers. | ||
| Polymer |
T
ma (°C) |
T
c
a (°C) |
ΔHmb (J g−1) |
T
dc 5% (°C) |
|---|---|---|---|---|
| a T m and Tc were determined by differential scanning calorimetry and were obtained from the 2nd heating cycle and cooling cycle, respectively. b Total enthalpy of melting (ΔHm) was calculated by integration of the first-order endothermic transition. c Determined by TGA at 5% weight loss. | ||||
| γC18PCL | 49 | 44 | −60.2 | 280 |
| β/δC18PCL isomers | 42 | 35 | −35.8 | 262 |
| εC18PCL | 47 | 41 | −32.6 | 265 |
Conclusions
We report the well-controlled ring-opening polymerisation of 4 alkyl-substituted ε-caprolactone monomers. All 4 monomers could be polymerised using the commercially available organocatalyst DPP to obtain lipophilic polyesters. Although the synthesis of all functional polyesters could be achieved with good control over molar mass and dispersity, the polymerisation rate was strongly dependent on the substituent placement. While the substituents in γ- and δ-position had only a minor effect on the overall polymerisation time, with 85% conversion reached in 3 h, the substituent placed in β-position increased polymerisation time to 7 h owing to increasing steric hindrance around the active carbonyl centre. Using identical polymerisation conditions, the ε-caprolactone monomer with a εC18-substituent further increased the polymerisation time drastically to 9 days. However, analysis of the thermal properties by DSC and TGA revealed that the substituent position in the resulting functional polyester has only minor impact on properties such as crystallisation and degradation temperature. Overall, this shows that by changing the position of the C18-alkyl substituent the relative polymerisation rates can be significantly changed without impacting the thermal properties of the functional polyesters. This highlights how simple substituent placement can be utilised to favour the kinetics of different monomer systems. These fundamental studies are essential to understand synthetic limitations for the design and development of new functional monomers with tailored property profiles and can be utilised to establish guidelines for the design of future materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Infineum UK Ltd for funding and delivery of the monomers.References
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- H. R. Kricheldorf, Chemosphere, 2001, 43, 49–54 CrossRef CAS PubMed.
- A. G. A. Coombes, S. C. Rizzi, M. Williamson, J. E. Barralet, S. Downes and W. A. Wallace, Biomaterials, 2004, 25, 315–325 CrossRef CAS PubMed.
- C. Jérôme and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef PubMed.
- O. Coulembier, P. Degée, J. L. Hedrick and P. Dubois, Prog. Polym. Sci., 2006, 31, 723–747 CrossRef CAS.
- T. K. Dash and V. B. Konkimalla, J. Controlled Release, 2012, 158, 15–33 CrossRef CAS PubMed.
- R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed.
- P. Joshi and G. Madras, Polym. Degrad. Stab., 2008, 93, 1901–1908 CrossRef CAS.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS PubMed.
- E. Martin, P. Dubois and R. Jérôme, Macromolecules, 2003, 36, 7094–7099 CrossRef CAS.
- A. Kowalski, J. Libiszowski, T. Biela, M. Cypryk, A. Duda and S. Penczek, Macromolecules, 2005, 38, 8170–8176 CrossRef CAS.
- M. Trollsås, V. Y. Lee, D. Mecerreyes, P. Löwenhielm, M. Möller, R. D. Miller and J. L. Hedrick, Macromolecules, 2000, 33, 4619–4627 CrossRef.
- L. Ciani, S. Bortolussi, I. Postuma, L. Cansolino, C. Ferrari, L. Panza, S. Altieri and S. Ristori, Int. J. Pharm., 2013, 458, 340–346 CrossRef CAS PubMed.
- X. Lou, C. Detrembleur, P. Lecomte and R. Jérôme, Macromolecules, 2001, 34, 5806–5811 CrossRef CAS.
- D. Tian, P. Dubois, C. Grandfils and R. Jérôme, Macromolecules, 1997, 30, 406–409 CrossRef CAS.
- J. Xu, F. Prifti and J. Song, Macromolecules, 2011, 44, 2660–2667 CrossRef CAS PubMed.
- S. Tempelaar, I. A. Barker, V. X. Truong, D. J. Hall, L. Mespouille, P. Dubois and A. P. Dove, Polym. Chem., 2013, 4, 174–183 RSC.
- R. Riva, S. Schmeits, C. Jérôme, R. Jérôme and P. Lecomte, Macromolecules, 2007, 40, 796–803 CrossRef CAS.
- R. H. Grubbs, Tetrahedron, 2004, 60, 7117–7140 CrossRef CAS.
- H. Seyednejad, A. H. Ghassemi, C. F. van Nostrum, T. Vermonden and W. E. Hennink, J. Controlled Release, 2011, 152, 168–176 CrossRef CAS PubMed.
- J. Hao, P. Kos, K. Zhou, J. B. Miller, L. Xue, Y. Yan, H. Xiong, S. Elkassih and D. J. Siegwart, J. Am. Chem. Soc., 2015, 137, 9206–9209 CrossRef CAS PubMed.
- S. Gautier, V. D'Aloia, O. Halleux, M. Mazza, P. Lecomte and R. Jérôme, J. Biomater. Sci., Polym. Ed., 2003, 14, 63–85 CrossRef CAS PubMed.
- M. Su, H. Huang, X. Ma, Q. Wang and Z. Su, Macromol. Rapid Commun., 2013, 34, 1067–1071 CrossRef CAS PubMed.
- D. Mecerreyes, J. Humes, R. D. Miller, J. L. Hedrick, C. Detrembleur, P. Lecomte, R. Jérôme and J. San Roman, Macromol. Rapid Commun., 2000, 21, 779–784 CrossRef CAS.
- A. Chen, J. Xu, W. Chiang and C. L. L. Chai, Tetrahedron, 2010, 66, 1489–1495 CrossRef CAS.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2016, 49, 2419–2428 CrossRef CAS.
- P. V. Persson, J. Schröder, K. Wickholm, E. Hedenström and T. Iversen, Macromolecules, 2004, 37, 5889–5893 CrossRef CAS.
- S. Mete, K. G. Goswami, E. Ksendzov, S. V. Kostjuk and P. De, Polym. Chem., 2019, 10, 6588–6599 RSC.
- S. Zhou, Y. Zhao, Y. Cai, Y. Zhou, D. Wang, C. C. Han and D. Xu, Polymer, 2004, 45, 6261–6268 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01380d |
| This journal is © The Royal Society of Chemistry 2024 |