
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A combined computational and experimental study of metathesis and nucleophile-mediated exchange mechanisms in boronic ester-containing vitrimers†
Jacopo
Teotonico
a,
Daniele
Mantione
 ab,
Laura
Ballester-Bayarri
a,
Marta
Ximenis
a,
Haritz
Sardon
ac,
Nicholas
Ballard
*ab and
Fernando
Ruipérez
*ad
ab,
Laura
Ballester-Bayarri
a,
Marta
Ximenis
a,
Haritz
Sardon
ac,
Nicholas
Ballard
*ab and
Fernando
Ruipérez
*ad
aPolymat, University of the Basque Country UPV/EHU, Joxe Mari Korta zentroa, Tolosa Hiribidea 72, 20018, Donostia-San Sebastian, Spain. E-mail: nicholas.ballard@polymat.eu; fernando.ruiperez@ehu.eus
bIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
cDepartment of Polymers and Advanced Materials: Physics, Chemistry and Technology, Faculty of Chemistry, University of the Basque Country UPV/EHU, Donostia-San Sebastián 20018, Spain
dPhysical Chemistry Department, Faculty of Pharmacy, University of the Basque Country UPV/EHU, 01006 Vitoria-Gasteiz, Spain
First published on 18th December 2023
Abstract
The rheological properties of vitrimer materials are largely controlled by the kinetics of covalent bond exchange and therefore a mechanistic understanding of bond exchange can provide insights into how the performance of vitrimers can be improved. In this work, density functional theory (DFT) calculations are combined with experimental measurements in order to understand the nature of the exchange reaction between boronic esters. Quantum chemical calculations show that both a metathesis and a nucleophile-mediated pathway are possible, although a significant increase in the rate of exchange in the presence of a nucleophile is observed. The DFT studies are supported by kinetic measurements of the exchange of small molecule dioxaborinanes in the presence and absence of nucleophile. Finally, the accelerating effect of nucleophiles is demonstrated in vitrimeric materials synthesized using a dioxaborinane-based crosslinker and a hydroxyl functional monomer as internal catalyst to produce vitrimers with significantly reduced relaxation times.
1 Introduction
Polymeric materials are ubiquitous in daily life, but there is growing concern over their end-of-life disposal.1 This is a particular issue for thermoset materials, which are characterized by a permanently cross-linked network of polymer chains that, once synthesized, cannot be remolded.2 A possible solution to this issue is the insertion of dynamic covalent bonds into the network. This potentially allows the material to flow in response to a specific stimulus, thus facilitating reprocessing and recycling.3–5 These types of dynamic polymers are known as covalent adaptable networks (or CANs) and can be divided into two main categories: dissociative CANs and associative CANs.6 The difference between these two is based on their exchange mechanism: in the former, a new bond is formed only after another one is broken, while in associative CANs bond formation and bond cleavage occur simultaneously.7Over the past decade, research into associative CANs in particular has grown rapidly following the discovery of a new family of dynamic polymers, termed vitrimers.8–11 These materials are unique in that they are covalently crosslinked, yet can be thermally reprocessed through a liquid-like state without a reduction in their network density. The rheological properties of these materials at elevated temperatures are largely determined by the kinetics of bond exchange.12–16 As a result, the rheological response, which is critical when considering potential applications, can be controlled through the chemistry involved in the network. In this context, the ability to predict and modify the behavior of vitrimeric materials on-demand requires a mechanistic understanding of the dynamic exchange process.
Although there are multiple different exchange reactions that have been shown to lead to CANs,17–21 boronic esters have received a significant amount of attention due to their high thermal stability, good tolerance to oxygen, easily tunable dynamic nature in the absence of a catalyst and compatibility with many functional groups.22 Furthermore, the preparation of boronic esters from boronic acid and diols is straightforward: the process is itself an equilibrium, and the forward reaction is fast and particularly favorable when the boronic ester product is insoluble in the reaction solvent.23 There are three principle dynamic exchange mechanisms characteristic of these compounds that can be used in CANs: (A) hydrolysis and reesterification, (B) transesterification with a diol and (C) metathesis with another boronic ester (see Fig. 1).
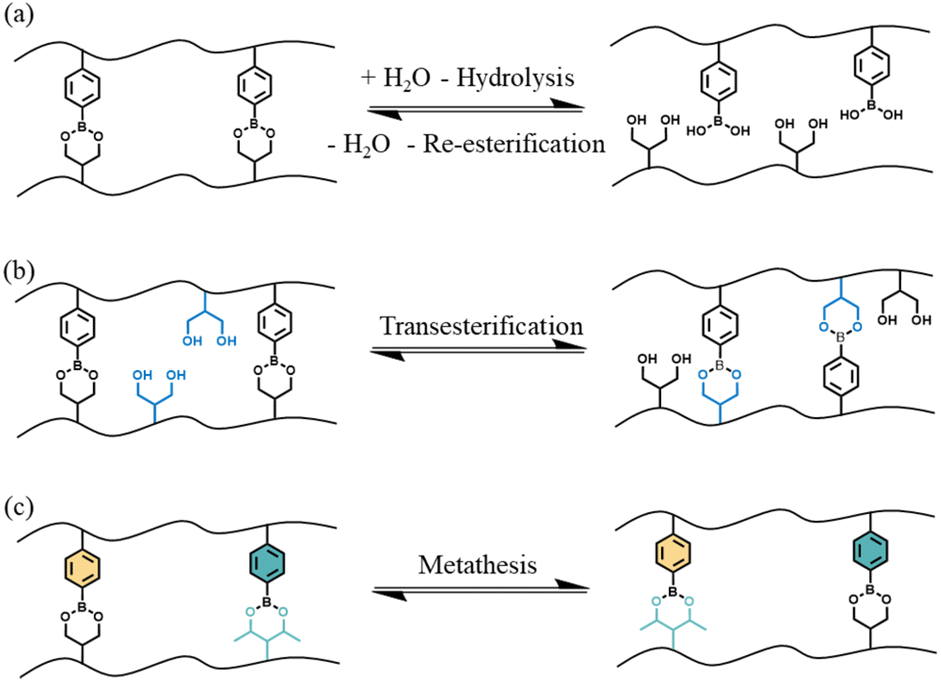 | ||
| Fig. 1 Three possible exchange mechanisms of boronic esters: (a) hydrolysis/re-esterification, (b) transesterification and (c) metathesis. Black lines represent polymer chains of the crosslinked network. | ||
Exploiting the first two reaction mechanisms, boron-containing polymers have been widely used in pH-responsive materials that could be used in smart drug delivery, self-healing or shape memory hydrogels.24–29 More recently, metathesis-like reactions of boronic esters have been also used for the preparation of different dynamic networks, ranging from robust, self-healable elastomers to vitrimers.30–33
With regards to the metathesis reaction and its use in vitrimeric systems, the mechanistic process is not well described and there are relatively few reports in the literature. Röttger et al.34 reported the synthesis of a vitrimer material where the dynamic exchange was proposed to occur through the metathesis of dioxaborolanes (5-membered ring boronic esters) without the addition of any catalyst at temperatures as low as 60 °C. The authors hypothesized that the mechanism of the reaction could occur through a direct process or through successive transesterification of dioxaborolanes with undetectable traces of diols. Winne et al.,35 in their review on dynamic covalent chemistry, suggested that the metathesis reaction is likely a multistep process, initiated by the formation of a zwitterionic adduct. However, the existence of a true metathesis reaction has yet to be confirmed.
More recently, Brunet et al.36 reported the dynamic exchange in styrenic based polymers containing pinacol boronate ester groups. Through experimental analyses and DFT calculations, they concluded that the most probable mechanism involved a nucleophilic ring opening step to accelerate the exchange process and that metathesis was unlikely. In support of the ring-opening mechanism, Yang et al.37 have shown that the presence of free alcohol groups increases the rate of exchange of six-membered phenyl boronic esters. The two potential mechanisms for exchange are summarized in Scheme 1.
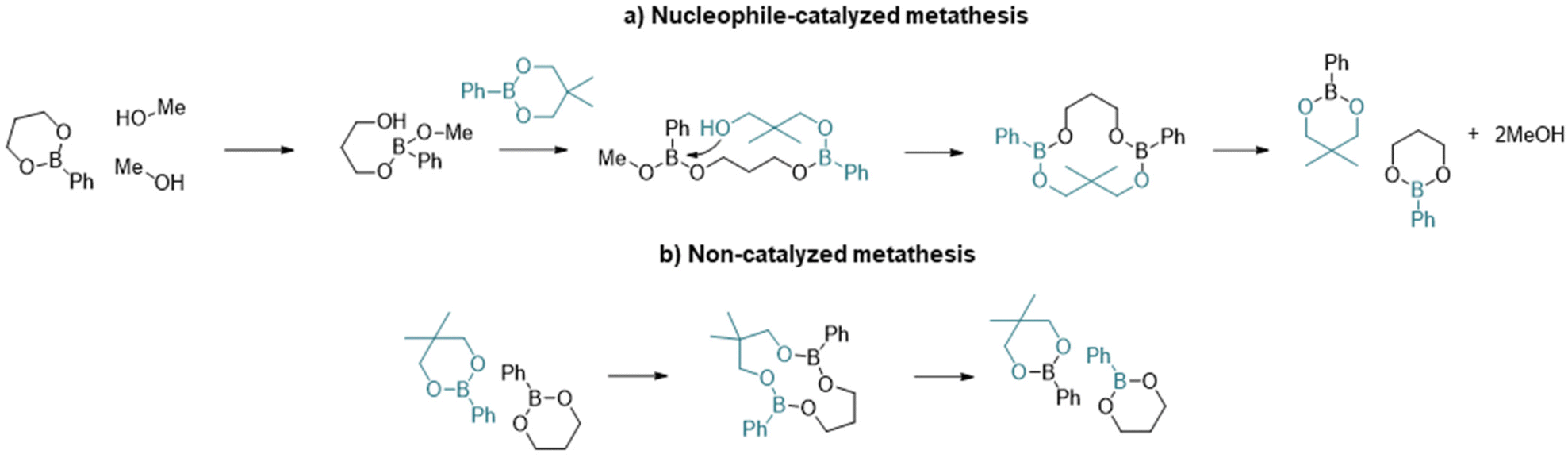 | ||
| Scheme 1 Illustration of the main steps of the two metathesis mechanisms of dioxaboranines: (a) nucleophile-mediated metathesis and (b) direct metathesis. | ||
Although different mechanistic processes for the exchange reaction between boronic esters have been proposed, a full mechanistic study is still missing. Because of this uncertainty and since the correct understanding of the mechanism of exchange is fundamental to tune the kinetics of dynamic bonds, in this work we combine quantum chemical calculations and experimental model studies to elucidate the mechanism of metathesis of boronates, with and without a nucleophilic mediator. Due to the enhanced stability in the presence of water we focus on exchange of dioxaborinanes (6-membered ring boronic esters). First, a comprehensive computational study is reported to outline the potential mechanistic pathways in the exchange of boronic esters. We subsequently describe a comparative kinetic study of the two pathways and discuss the potential influence of hydrolysis on the reaction. Finally, we demonstrate the use of this knowledge to accelerate stress relaxation in materials containing dioxaborinanes through the use of nucleophile-containing comonomer.
2 Experimental section
Materials
4-Methyl phenyl boronic acid, phenyl boronic acid, 4-vinyl phenyl boronic acid, 2,2-dimethyl-1,3-propanediol and 1,3-propanediol were purchased from TCI and used without further purification. Butyl acrylate, 2-hydroxyethyl methacrylate, triethylamine, methacroyl chloride, trimethylol propane, sodium sulfate and the solvents dry THF and CH2Cl2, were purchased from Sigma Aldrich and used without further purification. Deuterated solvents were purchased from Sigma Aldrich and were dried over molecular sieves.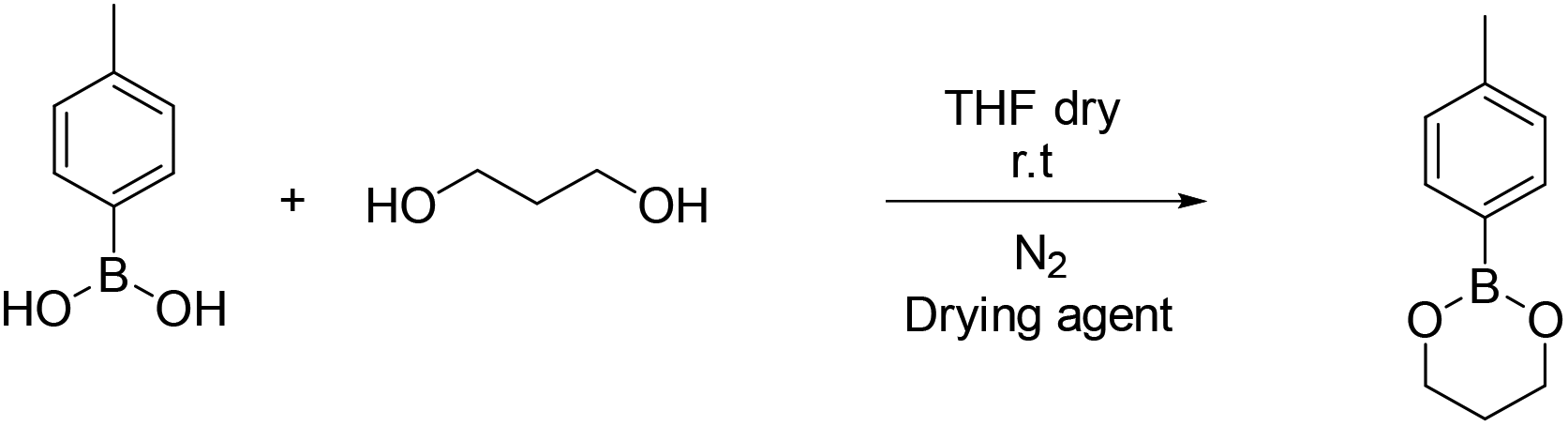 | ||
| Scheme 2 Synthesis of 2-(4-methylphenyl)-1,3,2-dioxaborinane (B1). | ||
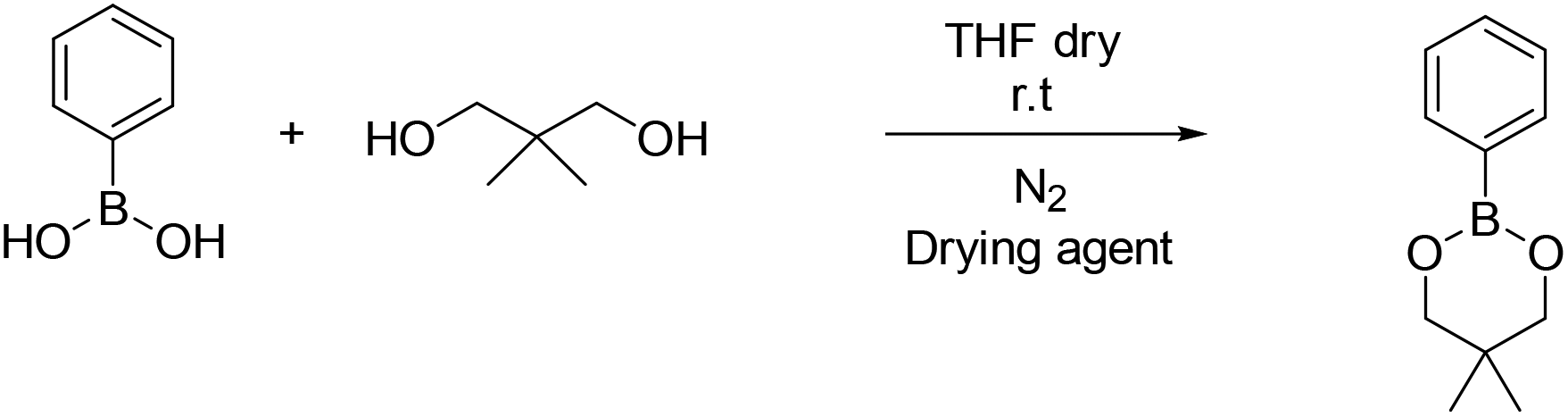 | ||
| Scheme 3 Synthesis of 5,5-dimethyl-2-phenyl-1,3,2-dioxaborinane (B2). | ||
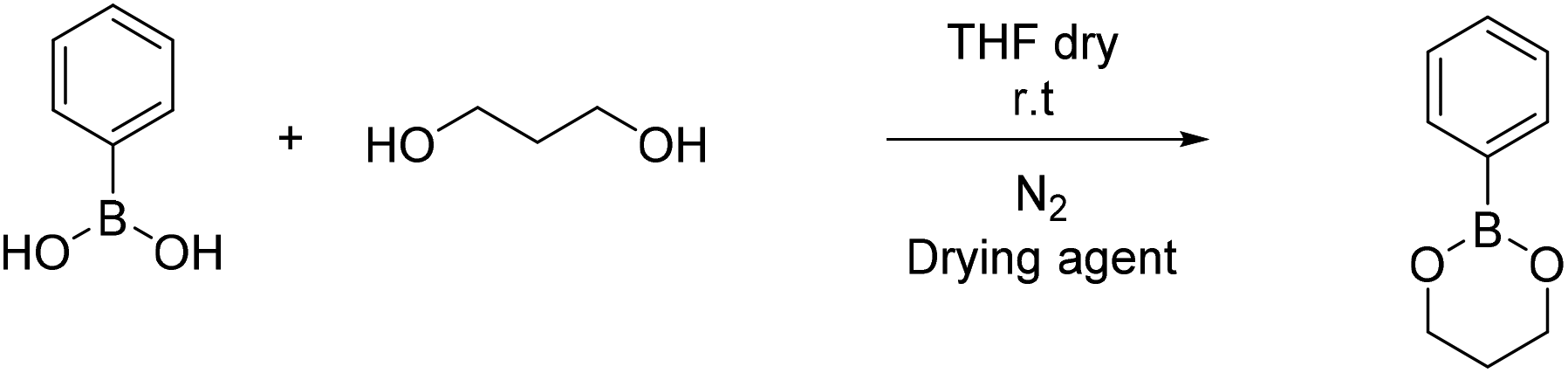 | ||
| Scheme 4 Synthesis of 2-phenyl-1,3,2-dioxaborinane (B3). | ||
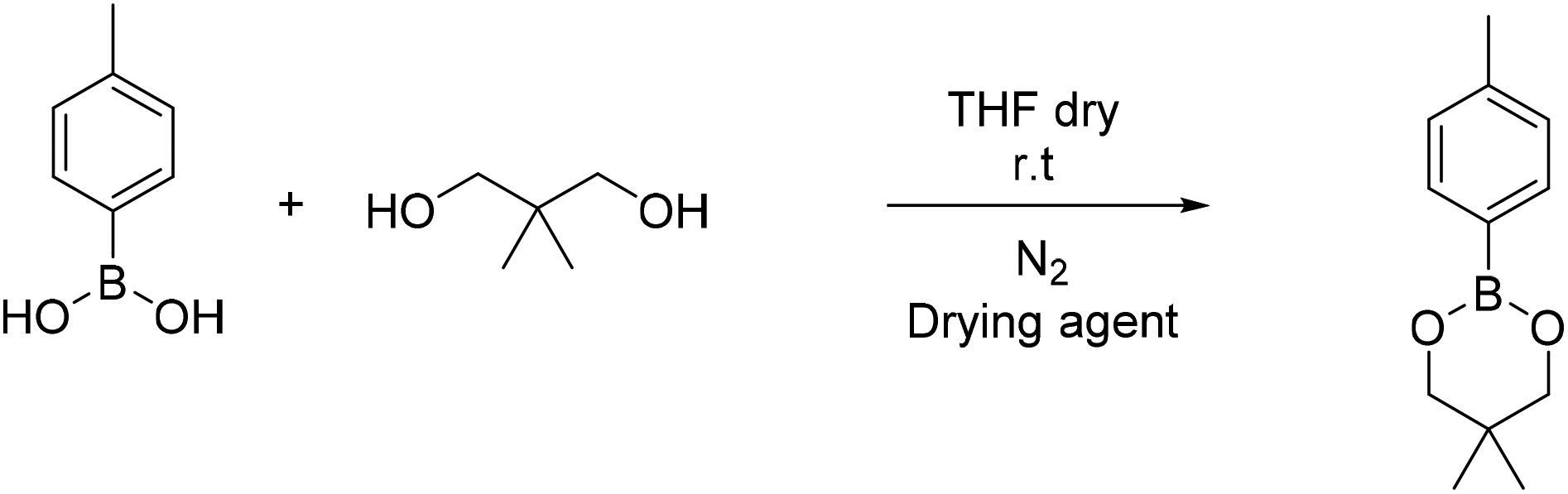 | ||
| Scheme 5 Synthesis of 5,5-dimethyl-2-(4-methylphenyl)-1,3,2-dioxaborinane (B4). | ||
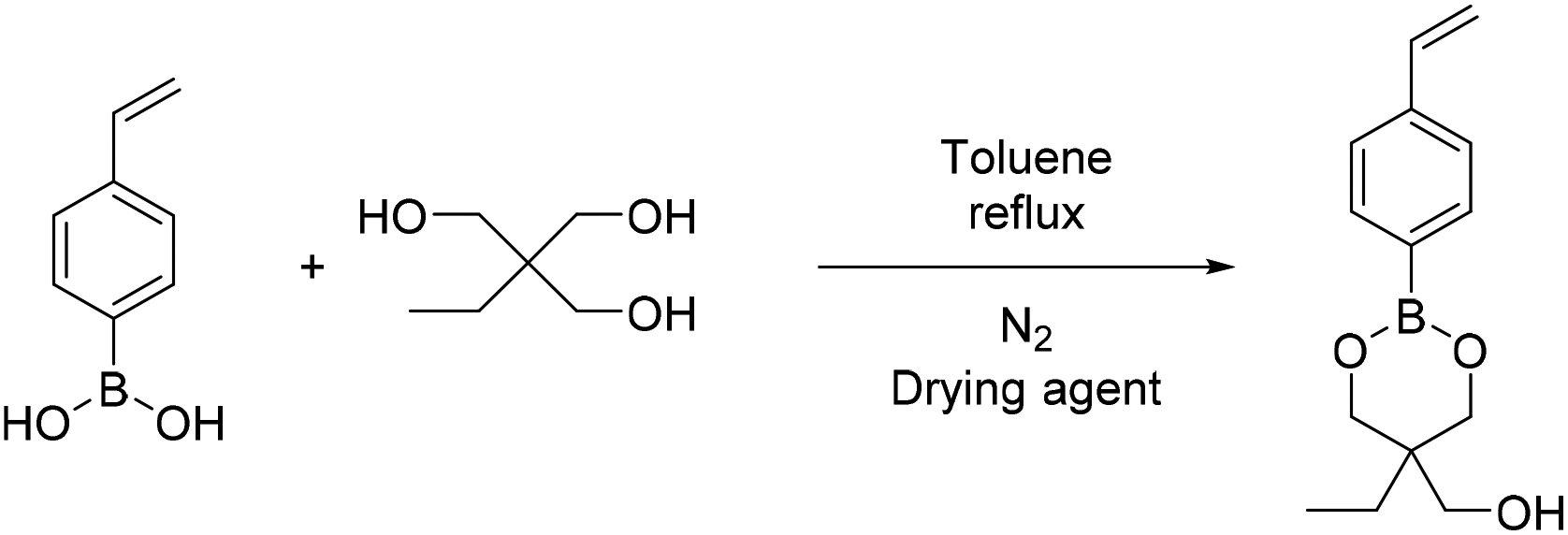 | ||
| Scheme 6 Synthesis of (5-ethyl-2-(4-vinylphenyl)-1,3,2-dioxaborinan-5-yl)methanol (B5). | ||
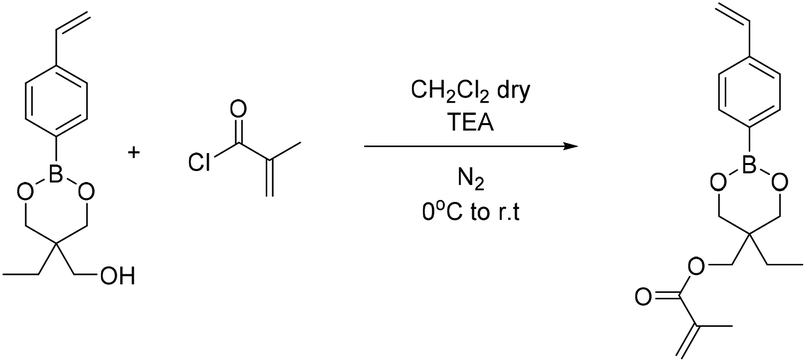 | ||
| Scheme 7 Synthesis of (5-ethyl-2-(4-vinylphenyl)-1,3,2-dioxaborinan-5-yl)methyl methacrylate (B6). | ||
 | ||
| Scheme 8 Exchange reaction between B2 and B1. | ||
 | ||
| Scheme 9 Preparation of crosslinked networks. | ||
Fourier transform-infrared (FT-IR) spectra were recorded using a Nicolet 6700 FTIR with an ATR accessory (MKII Golden Gate) from SPECAC with a diamond crystal. The measurements were recorded using 16 scans and a resolution of 8 cm−1.
Thermogravimetric analysis (TGA) was performed in a TGA 8000 from PerkinElmer under a nitrogen atmosphere from 30 to 900 °C at a heating rate of 10 °C min−1.
Differential Scanning Calorimetry was performed on a TA Instruments DSC 250 calibrated with indium, and the curves were analyzed using the Trios software. The sample was first equilibrated at −85 °C followed by a 1-minute isothermal step. A heating ramp of 20 °C min−1 was then performed until 100 °C. The sample was then kept at 100 °C for 1 minute before being cooled down with a cooling rate of 50 °C min−1 until −85 °C. A second heating ramp was then performed with the same parameters as the first one. Tgs were measured as the inflexion point of the first DSC scan.
Dynamic Mechanical Thermal Analysis (DMTA) was performed in compression mode using a Triton 2000 DMA (Triton Technology). The experiments were carried out using circular samples with a diameter of 8 mm at a frequency of 1 Hz. The chosen temperature range was from −80 °C to 150 °C at a heating rate of 4 °C min−1.
Samples were reprocessed in Collin P200E compression molding machine under the following conditions: (1) preheating the mould for 5 min at 150 °C; (2) heating the sample at 150 °C for 10 min at 300 bar of pressure and finally (3) cooling the sample.
Rheological experiments were performed in ARES-G2 rheometer from TA Instruments. Frequency sweeps were performed in shear geometry with 8 mm diameter plates under linear viscoelastic conditions, in which the storage modulus and the loss modulus of the samples were determined. Confirmation that the measurements were recorded within the linear viscoelastic regime was obtained through strain sweeps at a frequency of 1 Hz. Stress relaxation measurements were performed to obtain the relaxation modulus (G(t)) in shear using plates with 8 mm of diameter under linear viscoelastic conditions. The samples had a thickness between 0.6 mm and 1 mm.
To measure the degree of swelling a piece (7 mm in diameter and 1 mm in thickness) of the polymerized material was immersed in 2 mL of dry CH2Cl2 overnight at room temperature. The swollen sample was weighed and the swelling ratio was obtained following the equation written below:
| Swelling ratio = (Ms − Md)/Md |
3 Results and discussion
Density functional theory (DFT) results
 | ||
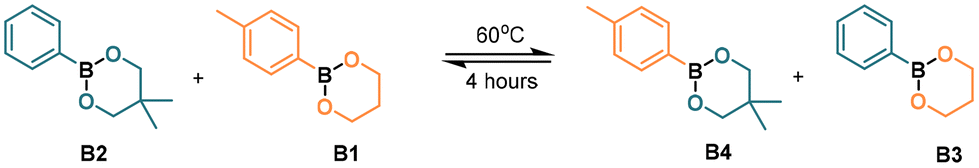
| Fig. 2 Potential energy surface of the MeOH-mediated exchange of dioxaborinanes. Gibbs free energies (ΔG) in kcal mol−1. Red line indicates the r.d.s. of the reaction with the value in kcal mol−1. The molecular structures corresponding to the intermediates and transition states are depicted in the ESI.† | ||
The first step of the mechanism as determined by DFT is the nucleophilic attack of one MeOH molecule to the boron atom (TS1, 15.74 kcal mol−1). Next, a hydrogen transfer from the methanol oxygen to one of the dioxaborinane oxygens takes place, assisted by a second MeOH molecule (TS2, 20.96 kcal mol−1), to generate a zwitterionic species that undergoes a ring-opening process to yield an acyclic intermediate with a free hydroxyl moiety (INT2, 14.74 kcal mol−1). This free hydroxyl is also a nucleophile and may attack a second dioxaborinane molecule (TS3, 26.97 kcal mol−1). Next, a hydrogen transfer between oxygens assisted by a MeOH molecule takes place, through a transition state similar to TS2 (TS4, 30.83 kcal mol−1). Again, the generated zwitterionic species is not stable and ring opens, leading to an intermediate with a free hydroxyl moiety (INT4, 25.12 kcal mol−1). This new free hydroxyl may undergo an intramolecular nucleophilic attack to the boron atom of the first boronic ester (TS5, 34.27 kcal mol−1), leading to a zwitterionic cyclic dimer of dioxaborinanes (INT5, 31.08 kcal mol−1). Next, a hydrogen transfer between the positively charged oxygen to the oxygen of the pendant methoxy group in the first dioxaborinane takes place (TS6, 37.99 kcal mol−1) and the generated methanol group leaves (TS7, 22.67 kcal mol−1), producing a neutral cyclic dimer (INT7, 20.69 kcal mol−1) and recovering the methanol. The last step involves an intramolecular metathesis of the cyclic dimer, where two oxygen atoms simultaneously attack the boron atoms to give the two new dioxaborinanes. This is the rate determining step, with an energy barrier of 17.68 kcal mol−1.
 | ||
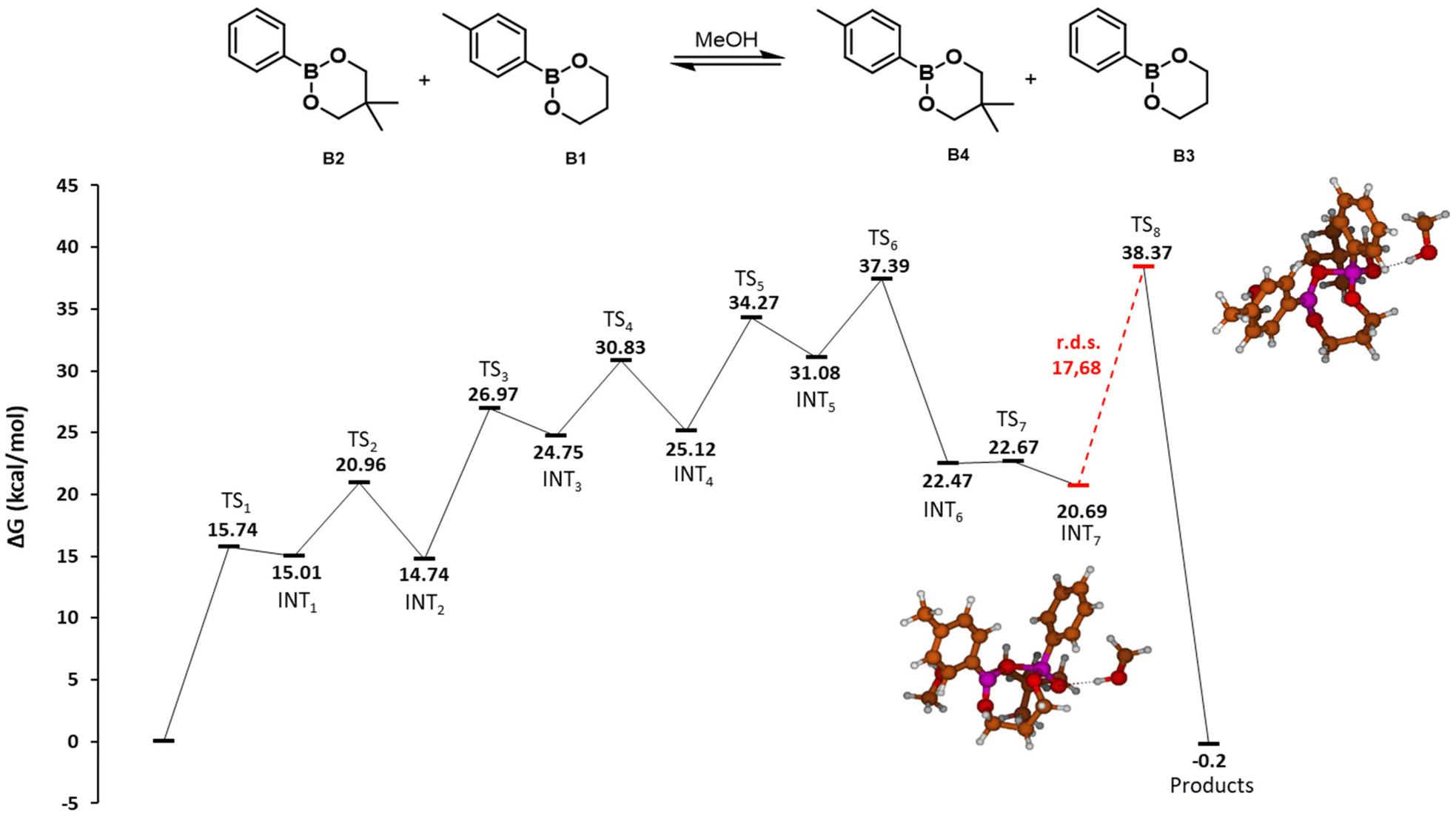
| Fig. 3 Potential energy surface of the direct metathesis reaction of dioxaborinanes. Gibbs free energies (ΔG) in kcal mol−1. Red line indicates the rate determining step with the value in kcal mol−1 The molecular structures of the intermediates and transition states are depicted in the ESI.† | ||
When two dioxaborinanes approach, the oxygens may undergo a nucleophilic attack to the boron atoms in a [2 + 2] cycloaddition reaction (TS1, 29.15 kcal mol−1). This leads to a four-membered cyclic structure that opens to form a macrocycle dimer of the two initial boronic esters (INT1, 17.58 kcal mol−1). This is the rate determining step of the reaction, with an energy barrier of 29.15 kcal mol−1. Next, an intramolecular metathesis may take place (TS2, 32.58 kcal mol−1) to generate the two new dioxoborinane molecules.
The two exchange mechanisms shown above highlight that the exchange reaction can take place through different pathways, with significant differences in the expected energy barriers. Although the proposed MeOH-mediated exchange reaction is more complex, involving several steps, the energy barrier of the rate determining step is just 17.68 kcal mol−1. In contrast, for the direct metathesis mechanism, the reaction takes places in just two steps, but the rate determining step involves a significantly higher barrier (29.15 kcal mol−1). Thus, it is expected that the presence of MeOH (or other nucleophile) in the reaction mixture would facilitate the exchange of boronic esters.
Model studies
In order to determine whether the effects predicted by DFT lead to differences in the exchange dynamics experimentally, a number of exchange experiments were performed using small molecules as model compounds. As previous reports have proposed that adventitious nucleophiles may play an important role even where no nucleophile is specifically added,39 we first explored the stability of the dioxaborinanes used in this work by addition of water and MeOH to B4 at 60 °C for 4 hours (Scheme 10). In order to avoid solubility issues in the case of using water these tests were performed in DMSO. The aromatic peaks at 7.60 ppm for the boronic ester and 7.70 ppm for the hydrolyzed product were integrated to determine the relative percentages of the two components in the mixture. The NMR spectra are shown in Fig. S18–S30 for reference.†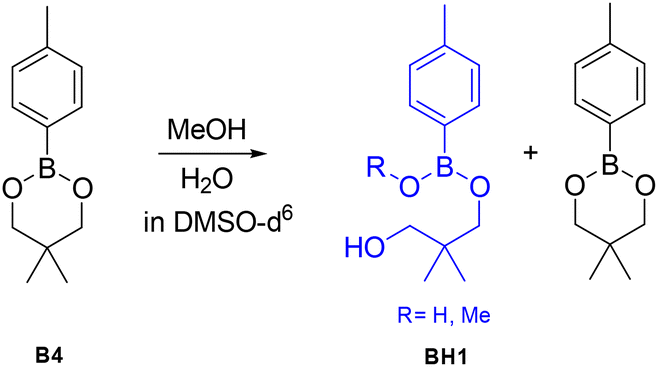 | ||
| Scheme 10 Hydrolysis or MeOH-nucleophilic attack to dioxaborinane B4 to give product BH1. | ||
Dioxaborinane B4 showed very high stability towards MeOH: adding 0.1 and 0.2 molar equivalents to the boronic ester in deuterated DMSO, after 4 hours at 60 °C, resulted in the alcoholysis of just 1% of the boronic ester into product BH1 (see Fig. S19 and S20† for the corresponding NMR spectra). Adding 0.5 eq. or 1 eq. has a similar outcome to the one aforementioned, with only 3% BH1 formed after 4 hours (see Fig. S21 and S22 for the corresponding NMR spectra†). Note that even at higher amounts of MeOH in the system, the extent of hydrolysed material is relatively small (i.e. 8% with 5 molar equivalents of MeOH) (see Fig. S24 for the corresponding NMR spectra†).
Since one of the possible exchange mechanisms of boronic ester is the hydrolysis to boronic acid and the subsequent re-esterification to the initial product (mechanism 1 in Fig. 1), we also tested the effect of water as a nucleophile. Again, here, B4 showed high stability towards hydrolysis. Note that the stability of the six-membered dioxaborinanes used here is significantly higher than the dioxaborolanes that have been used in many previous studies where complete hydrolysis occurs even with relatively small amounts of water.45 Comparing with MeOH, water seems to result in more hydrolysis: when 0.1 eq. and 0.2 eq. were added, a total of, respectively 2% and 3% of the boronic ester was hydrolysed to BH1. However, unlike the case of MeOH, in the case of water as a nucleophile, at higher molar equivalents the amount of hydrolyzed material increased substantially (i.e. 28% with 5 molar equivalents of MeOH).
Following these stability experiments, a series of kinetic measurements were performed in bulk and solution with the addition of MeOH and H2O in different molar percentages at 60 °C in CDCl3. The total concentration of dioxaborinanes in solution experiments was 0.5 mol L−1 in all cases. At regular intervals, a small volume of the reaction mixture was taken out to perform 1H NMR measurements in CDCl3. The signals of the characteristic methylenes (–OCH2−) at 3.80 ppm for B2 and 3.82 ppm for B4 were integrated to determine the relative percentages of the four components in the reaction mixture. The corresponding NMR spectra are shown in the ESI (Fig. S9–S17†).
Fig. 4 shows the evolution of the reaction mixture with time for the various reactions. When B2+B1 were left in solution without any catalyst at 60 °C, the exchange was slow and after 4 hours the ratio between the starting reagent B2 and the metathesis product B4 was just 9/1. In contrast, when the two dioxaborinanes were reacted in bulk with the same conditions after 4 hours the ratio between B2 and B4 was close to equilibrium, being 6.1/3.9. The relatively fast exchange in bulk is simply the result of higher concentrations of reactants but does highlight that exchange occurs even in the absence of intentionally added mediator, thus supporting the idea of a metathesis reaction.
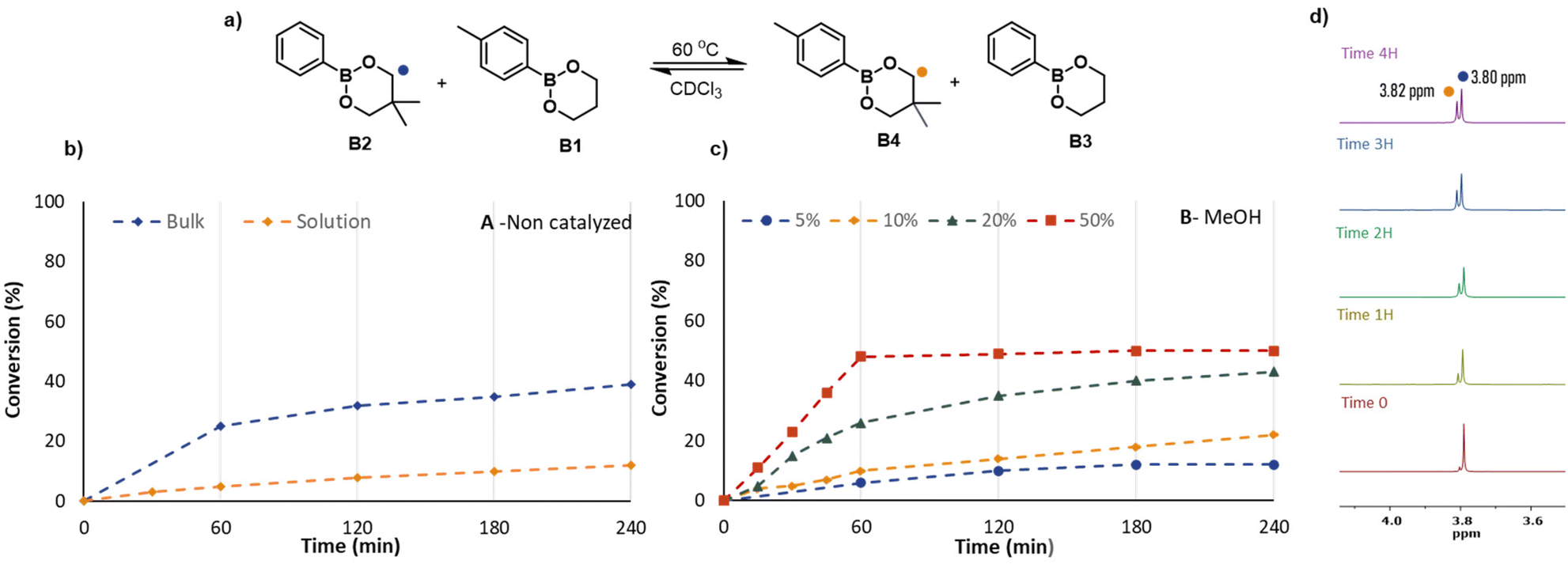 | ||
| Fig. 4 (a) Experimental kinetics of the reaction between dioxaborinane B2 and B1 at 60 °C for 4 hours. (b) Kinetics of B2+B1 without catalyst in solution (CDCl3) and in bulk. (c) Kinetics of B2+B1 in solution with 5, 10, 20 and 50 mol% of MeOH with respect to the dioxaborinanes. (d) Time-dependent 1H-NMR spectra of the bulk kinetic reaction of the two dioxaborinanes. | ||
For the reactions in solution with different amounts of MeOH as a nucleophilic catalyst, higher concentrations of MeOH clearly resulted in faster exchange dynamics. As graph B from Fig. 4 shows, when 5% or 10% of MeOH was added, the rate of exchange did not differ substantially from the uncatalyzed reaction. However, when the concentration of MeOH was increased, i.e., for 20% and 50% molar MeOH, the increase in rate was significant. This behaviour can be explained by the presence of MeOH, which in higher quantity, facilitates the opening of the dioxaborinane cycle. To quantify this effect, the effective rate coefficient for the exchange reaction was determined from the kinetic plots by assuming an identical rate coefficient for the forward and backward reaction. As shown in Fig. S31† although the effective rate coefficient increases with the concentration of MeOH, as the MeOH concentration is reduced it decreases to a non-zero value which suggests that there is a significant contribution from the direct metathesis reaction pathway. In addition to the use of MeOH we also tested the use of water as a nucleophile by addition of 50% water with respect to dioxaborinanes, leading to a water-saturated reaction mixture (see Fig. S32†). Similar to the case of methanol the rate was substantially faster than kinetic experiments without any added nucleophile.
The combination of the kinetic results and DFT results reported above allow us to draw some conclusions on the exchange pathways in the case of boronic esters. Based on the negligible alcoholysis reported in Table 1, even for relatively high alcohol concentration, and the relatively high rate of exchange at zero/low alcohol concentrations shown in Fig. 4, it appears that the direct metathesis pathway detected in the DFT calculations can be important. In the presence of MeOH the strong increase in rate suggests that the alternative, nucleophile-mediated pathway becomes more significant. However, it is important to note that due to the very different hydrolytic stability of dioxaborolanes and dioxaborinanes, these observations made for dioxaborinanes may differ when changing the boronic ester structure. Thus, in the case of dioxaborolanes the presence of small amounts of water can result in a significant amounts of hydrolysis and would therefore promote the ring-opening mechanisms proposed by Raynaud et al.36
| MeOH/H2O eq. | % Alcoholysis | % Hydrolisis |
|---|---|---|
| 0.10 | ≤1% | 2% |
| 0.20 | 1% | 3% |
| 0.50 | 3% | 6% |
| 1 | 3% | 7% |
| 2.5 | 3% | 17% |
| 5 | 3% | 28% |
Preparation and characterization of dynamic networks
As a demonstration of the use of the nucleophile-mediated mechanism in vitrimer type materials, a thermoset network containing pendant alcohol groups and a dioxaborinane-based crosslinker was synthesized. In order to prepare the network a new dioxaborinane-based crosslinker (B6) was prepared and photopolymerized using butyl acrylate and 2-hydroxyethyl methacrylate as monomers. It was proposed that HEMA would play the role of an accelerator through the action of the nucleophilic alcohol group. Another commercial dimethacrylate crosslinker, ethylene glycol dimethacrylate, (M1) was used to prepare control materials as a non-dynamic network. The crosslinker was introduced as 3 mol% compared to BA in the monomer feed and a series of PBA + pHEMA networks were prepared, using 0, 0.6 and 1.5 mol% HEMA with respect to the main monomer in the formulation (see Table 2). Following photopolymerization, the vitrimers were obtained as free-standing films.| Name | % Crosslinker | % HEMA |
|---|---|---|
| VT0 | 3 | 0 |
| VT1 | 3 | 0.6 |
| VT2 | 3 | 1.5 |
Note that the network is prepared using monomers that contain methacrylate, acrylate and styrenic-type vinyl groups and therefore some composition drift is expected during the polymerization. Based on previously published values for acrylate/methacrylate/styrene terpolymerization systems,46 the extent of composition drift in terpolymerization systems was estimated by direct numerical integration using the method suggested by Kazemi et al.47 as shown in Fig. S35.† It can be seen that while the methacrylic components (HEMA and the methacrylate from the crosslinker) are largely reacted below 60% conversion, the styrenic double bond of the crosslinking agent is expected to be consumed fairly evenly throughout the polymerization. It may be noted that as a network is formed during the reaction, although there is some compositional heterogeneity in the individual chains that make the network, the final sample will be relatively homogeneous.
Characterization of the materials
The crosslinked nature of the vitrimers was confirmed by swelling measurements and DMTA. The swelling of the vitrimers measured in DCM was similar to the control sample indicating that the crosslinked network is structurally similar in all cases (see Table S1†). All samples showed a single glass transition in the DMTA above which the modulus was approximately constant, as would be expected for a crosslinked material (see Fig. S45†). Measurement of the Tg by DSC showed that all samples has a Tg of around −41 °C with no notable change upon incorporation of the HEMA (see Table S1†). The thermal properties were measured by TGA. Under a nitrogen atmosphere, all the networks were thermally stable up to 310 °C with the temperatures of 5% weight loss being above 335 °C (see Fig. S37†), which was much higher than the temperatures required for metathesis of the dioxaborinanes as shown in Fig. 4.Rheological properties
To confirm that the dynamics of the material were accelerated in the presence of the catalytic hydroxyl moieties, stress relaxation measurements of the dynamic vitrimer networks were performed at a number of temperatures (see Fig. S38–S40†). Representative measurements at 160 °C comparing the blank and the samples containing the dynamic crosslinker with or without HEMA are shown in Fig. 5. It can be seen that all the vitrimer networks prepared with the boronic ester crosslinker relax, with those containing higher concentrations of HEMA showing significantly reduced relaxation times. In contrast, the control sample showed no stress relaxation as expected for a non-dynamic crosslinked material.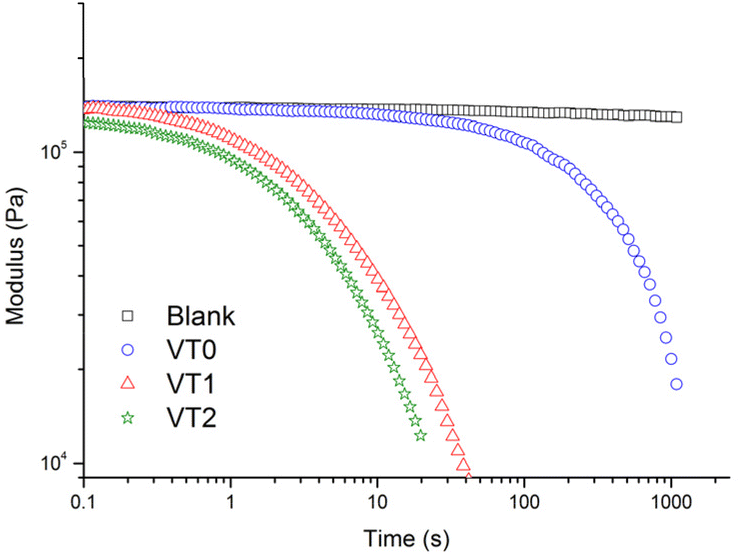 | ||
| Fig. 5 Stress relaxation analysis of control crosslinked network “blank”, vitrimer VT0, VT1 and VT2 at 160 °C as function of time. | ||
The characteristic relaxation times for the different samples were obtained by a fit of the stress relaxation data to a stretched exponential for all temperatures. The Arrhenius plot of the relaxation times at different temperatures are shown in Fig. 6. There are two important points that can be seen in this figure. The first is that at a given temperature there is an enormous decrease of the relaxation time. This decrease in the relaxation time is particularly remarkable given the relatively low concentrations of HEMA that were incorporated into the formulations. Note that the increased rate of stress relaxation could also be observed at low frequencies in frequency sweep experiments where vitrimers of sample VT2 could be seen to reach a G′, G′′ crossover point (see Fig. S41–44†). The second important point is that there is also a significant reduction in the activation energy when HEMA is incorporated into the copolymer.
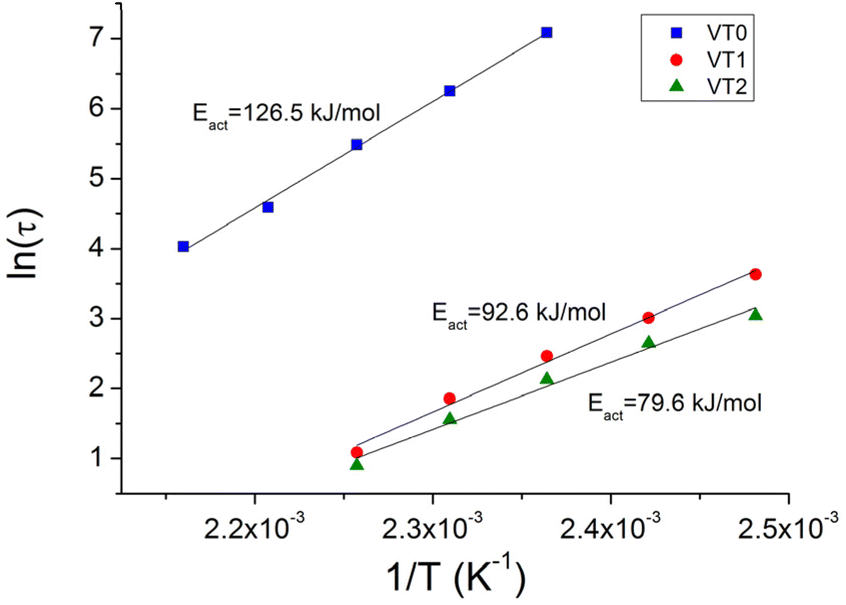 | ||
| Fig. 6 Arrhenius plot of relaxation times for vitrimer VT0, VT1 and VT2 to give the activation energy for bond exchange. | ||
These results are in line with the DFT studies, which show a smaller energy barrier for the rate determining step of the MeOH-mediated mechanism, compared to the rate determining step of the direct metathesis mechanism. This behaviour is also in agreement with the model studies, where the addition of MeOH increased the rate of the reaction. Moreover, the stress relaxation exhibited by vitrimer sample VT0 is further proof of the metathesis mechanism in the absence of catalyst.
Thermal healing and reprocessing
In order to assess the reprocessing of the vitrimer networks based on dioxaboranines, the prepared samples were cut into multiple pieces and hot-pressed for 10 minutes at 150 °C under 8 MPa. The networks comprising boronic esters were able to be reprocessed and intact specimens were recovered completely (see Fig. 7 and Fig. S46–S48†). In the case of control networks, the same processing protocol (150 °C) afforded samples of extremely poor quality, which were too weak to be characterized (Fig. S49†). Frequency sweep and stress relaxation measurements on the reprocessed samples showed that there were no significant changes in the mechanical properties of the reprocessed samples (see Fig. 7 and Fig. S50–S52†). | ||
| Fig. 7 (a) Reprocessing of vitrimer network VT1: step 1 consists in cutting in small pieces the pristine vitrimer material. Step 2 refers to the thermal reprocessing at 150 °C for 10 minutes to give the reprocessed material. (b) Storage and loss modulus of pristine and reprocessed VT1. (c) Stress relaxation behaviour of pristine and reprocessed VT1. | ||
4 Conclusions
In conclusion, we have performed a computational and experimental study to understand the mechanism of exchange in boronic ester-containing polymer networks. The quantum chemical calculations suggested two separate mechanistic pathways depending on the presence of a nucleophilic species. In the absence of a nucleophile, a direct metathesis pathway was found. Experimental evidence for this was demonstrated through the exchange of model dioxaborinanes in the absence of nucleophilic mediators. When a nucleophile (water or methanol) was added to the system, the exchange rate increased significantly. It was demonstrated that the increased rate of exchange in the presence of free alcohol groups can be used advantageously to drastically accelerate stress relaxation in vitrimers synthesized using dioxaborinane-based crosslinkers by inclusion of nucleophile containing comonomers.Conflicts of interest
There are no conflicts to declareAcknowledgements
Technical and human support provided by IZO-SGI, SGIker (UPV/EHU, MICINN, GV/EJ, ERDF and ESF) is gratefully acknowledged for assistance and generous allocation of computational resources. The authors would like to recognize funding provided by the Spanish government (Grant PID2019-107889GA-100 funded by MCIN/AEI/10.13039/501100011033a and RYC2021-031668-I funded by MCIN/AEI/10.13039/501100011033 (D. M.)) and from the Basque Government (IT-99916). D. M. also acknowledges funding provided by the European Union (NextGenerationEU/PRTR). MX acknowledges the grant from the Gipuzkoa Fellows Programme.References
- L. Lebreton and A. Andrady, Future Scenarios of Global Plastic Waste Generation and Disposal, Palgrave Commun., 2019, 5(1), 6, DOI:10.1057/s41599-018-0212-7.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Vitrimers, Directing Chemical Reactivity to Control Material Properties, Chem. Sci., 2020, 11(19), 4855–4870, 10.1039/D0SC01069C.
- A. M. Hubbard, Y. Ren, A. Sarvestani, D. Konkolewicz, C. R. Picu, A. K. Roy, V. Varshney and D. Nepal, Recyclability of Vitrimer Materials: Impact of Catalyst and Processing Conditions, ACS Omega, 2022, 7(33), 29125–29134, DOI:10.1021/acsomega.2c02677.
- F. Van Lijsebetten, K. De Bruycker, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, Suppressing Creep and Promoting Fast Reprocessing of Vitrimers with Reversibly Trapped Amines, Angew. Chem., Int. Ed., 2022, 61(9), e202113872/1–8, DOI:10.1002/anie.202113872.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Fast, Processing of Highly Crosslinked, Low-Viscosity Vitrimers, Mater. Horiz., 2020, 7(1), 104–110, 10.1039/C9MH01062A.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Covalent Adaptable Networks (CANs): A Unique Paradigm in Cross-Linked Polymers, Macromolecules, 2010, 43(6), 2643–2653, DOI:10.1021/ma902596s.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, Adaptable Crosslinks in Polymeric Materials: Resolving the Intersection of Thermoplastics and Thermosets, J. Am. Chem. Soc., 2019, 141(41), 16181–16196, DOI:10.1021/jacs.9b07922.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-Like Malleable Materials from Permanent Organic Networks, Science, 2011, 334(6058), 965–968, DOI:10.1126/science.1212648.
- V. Schenk, K. Labastie, M. Destarac, P. Olivier and M. Guerre, Vitrimer Composites: Current Status and Future Challenges, Mater. Adv., 2022, 3(22), 8012–8029, 10.1039/D2MA00654E.
- N. J. Van Zee and R. Nicolaÿ, Vitrimers: Permanently Crosslinked Polymers with Dynamic Network Topology, Prog. Polym. Sci., 2020, 104, 101233, DOI:10.1016/j.progpolymsci.2020.101233.
- W. Denissen, J. M. Winne and F. E. Du Prez, Vitrimers: Permanent Organic Networks with Glass-like Fluidity, Chem. Sci., 2016, 7(1), 30–38, 10.1039/C5SC02223A.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, Catalytic Control of the Vitrimer Glass Transition, ACS Macro Lett., 2012, 1(7), 789–792, DOI:10.1021/mz300239f.
- B. M. El-Zaatari, J. S. A. Ishibashi and J. A. Kalow, Cross-Linker Control of Vitrimer Flow, Polym. Chem., 2020, 11(33), 5339–5345, 10.1039/D0PY00233J.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Chemical, Control of the Viscoelastic Properties of Vinylogous Urethane Vitrimers, Nat. Commun., 2017, 8(1), 14857, DOI:10.1038/ncomms14857.
- A. Ruiz De Luzuriaga, G. Solera, I. Azcarate-Ascasua, V. Boucher, H.-J. Grande and A. Rekondo, Chemical Control of the Aromatic Disulfide Exchange Kinetics for Tailor-Made Epoxy Vitrimers, Polymer, 2022, 239, 124457, DOI:10.1016/j.polymer.2021.124457.
- S. K. Schoustra, J. A. Dijksman, H. Zuilhof and M. M. J. Smulders, Molecular Control over Vitrimer-like Mechanics – Tuneable Dynamic Motifs Based on the Hammett Equation in Polyimine Materials, Chem. Sci., 2021, 12(1), 293–302, 10.1039/D0SC05458E.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2002, 41(6), 898–952, DOI:10.1002/1521-3773(20020315)41:6<898::AID-ANIE898>3.0.CO;2-E.
- B. J. Adzima, H. A. Aguirre, C. J. Kloxin, T. F. Scott and C. N. Bowman, Rheological and Chemical Analysis of Reverse Gelation in a Covalently Cross-Linked Diels−Alder Polymer Network, Macromolecules, 2008, 41(23), 9112–9117, DOI:10.1021/ma801863d.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Dynamic Covalent Polymer Networks: From Old Chemistry to Modern Day Innovations, Adv. Mater., 2017, 29(14), 1606100, DOI:10.1002/adma.201606100.
- S. Huang, X. Kong, Y. Xiong, X. Zhang, H. Chen, W. Jiang, Y. Niu, W. Xu and C. Ren, An Overview of Dynamic Covalent Bonds in Polymer Material and Their Applications, Eur. Polym. J., 2020, 141, 110094, DOI:10.1016/j.eurpolymj.2020.110094.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Polymer Engineering Based on Reversible Covalent Chemistry: A Promising Innovative Pathway towards New Materials and New Functionalities, Prog. Polym. Sci., 2018, 80, 39–93, DOI:10.1016/j.progpolymsci.2018.03.002.
- W. L. A. Brooks and B. S. Sumerlin, Synthesis and Applications of Boronic Acid-Containing Polymers: From Materials to Medicine, Chem. Rev., 2016, 116(3), 1375–1397, DOI:10.1021/acs.chemrev.5b00300.
- M. Gosecki and M. Gosecka, Boronic Acid Esters and Anhydrates as Dynamic Cross-Links in Vitrimers, Polymers, 2022, 14(4), 842, DOI:10.3390/polym14040842.
- A. P. Bapat, D. Roy, J. G. Ray, D. A. Savin and B. S. Sumerlin, Dynamic-Covalent Macromolecular Stars with Boronic Ester Linkages, J. Am. Chem. Soc., 2011, 133(49), 19832–19838, DOI:10.1021/ja207005z.
- F. Jäkle, Advances in the Synthesis of Organoborane Polymers for Optical, Electronic, and Sensory Applications, Chem. Rev., 2010, 110(7), 3985–4022, DOI:10.1021/cr100026f.
- N. Fujita, S. Shinkai and T. D. James, Boronic Acids in Molecular Self–Assembly, Chem. – Asian J., 2008, 3(7), 1076–1091, DOI:10.1002/asia.200800069.
- W. Niu, C. O'Sullivan, B. M. Rambo, M. D. Smith and J. J. Lavigne, Self-Repairing Polymers: Poly(Dioxaborolane)s Containing Trigonal Planar Boron, Chem. Commun., 2005, 34, 4342, 10.1039/b504634c.
- T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Saccharide Sensing with Molecular Receptors Based on Boronic Acid, Angew. Chem., Int. Ed. Engl., 1996, 35(17), 1910–1922, DOI:10.1002/anie.199619101.
- L. He, D. E. Fullenkamp, J. G. Rivera and P. B. Messersmith, pH Responsive Self-Healing Hydrogels Formed by Boronate–Catechol Complexation, Chem. Commun., 2011, 47(26), 7497, 10.1039/c1cc11928a.
- F. Caffy and R. Nicolaÿ, Transformation of Polyethylene into a Vitrimer by Nitroxide Radical Coupling of a Bis-Dioxaborolane, Polym. Chem., 2019, 10(23), 3107–3115, 10.1039/C9PY00253G.
- Y. Chen, Z. Tang, Y. Liu, S. Wu and B. Guo, Mechanically Robust, Self-Healable, and Reprocessable Elastomers Enabled by Dynamic Dual Cross-Links, Macromolecules, 2019, 52(10), 3805–3812, DOI:10.1021/acs.macromol.9b00419.
- Y. Chen, Z. Tang, X. Zhang, Y. Liu, S. Wu and B. Guo, Covalently Cross-Linked Elastomers with Self-Healing and Malleable Abilities Enabled by Boronic Ester Bonds, ACS Appl. Mater. Interfaces, 2018, 10(28), 24224–24231, DOI:10.1021/acsami.8b09863.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Polybutadiene Vitrimers Based on Dioxaborolane Chemistry and Dual Networks with Static and Dynamic Cross-Links, Macromolecules, 2019, 52(18), 7102–7113, DOI:10.1021/acs.macromol.9b01288.
- M. Röttger, T. Domenech, R. Van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, High-Performance Vitrimers from Commodity Thermoplastics through Dioxaborolane Metathesis, Science, 2017, 356(6333), 62–65, DOI:10.1126/science.aah5281.
- J. M. Winne, L. Leibler and F. E. Du Prez, Dynamic, Covalent Chemistry in Polymer Networks: A Mechanistic Perspective, Polym. Chem., 2019, 10(45), 6091–6108, 10.1039/C9PY01260E.
- J. Brunet, F. Collas, M. Humbert, L. Perrin, F. Brunel, E. Lacôte, D. Montarnal and J. Raynaud, High Glass–Transition Temperature Polymer Networks Harnessing the Dynamic Ring Opening of Pinacol Boronates, Angew. Chem., Int. Ed., 2019, 58(35), 12216–12222, DOI:10.1002/anie.201904559.
- Y. Yang, F.-S. Du and Z.-C. Li, Thermally Healable and Reprocessable Polymethacrylate Networks Based on Diol-Mediated Metathesis of 6-Membered Boronic Esters, Polym. Chem., 2020, 11(11), 1860–1870, 10.1039/C9PY01546A.
- F. Siopa, V.-A. Ramis Cladera, C. A. M. Afonso, J. Oble and G. Poli, Ruthenium-Catalyzed C-H Arylation and Alkenylation of Furfural Imines with Boronates: Ruthenium-Catalyzed C-H Arylation and Alkenylation of Furfural Imines with Boronates, Eur. J. Org. Chem., 2018,(44), 6101–6106, DOI:10.1002/ejoc.201800767.
- N. Dastbaravardeh, M. Schnürch and M. D. Mihovilovic, Ruthenium(0)-Catalyzed Sp 3 C–H Bond Arylation of Benzylic Amines Using Arylboronates, Org. Lett., 2012, 14(7), 1930–1933, DOI:10.1021/ol300627p.
- S. B. Tailor, M. Manzotti, G. J. Smith, S. A. Davis and R. B. Bedford, Cobalt-Catalyzed Coupling of Aryl Chlorides with Aryl Boron Esters Activated by Alkoxides, ACS Catal., 2021, 11(7), 3856–3866, DOI:10.1021/acscatal.0c05557.
- J.-D. Chai and M. Head-Gordon, Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections, Phys. Chem. Chem. Phys., 2008, 10(44), 6615, 10.1039/b810189b.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self–consistent Molecular Orbital Methods, XX. A Basis Set for Correlated Wave Functions, J. Chem. Phys., 1980, 72(1), 650–654, DOI:10.1063/1.438955.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56(5), 2257–2261, DOI:10.1063/1.1677527.
- M. J. Frisch, G. W. Trucks, G. E. Schlegel, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, A. Petersonn and H. Nakatsujii, Gaussian 16 Revision C.01, Gaussian Inc, Wallingford CT, 2016 Search PubMed.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Room-Temperature Self-Healing Polymers Based on Dynamic-Covalent Boronic Esters, Macromolecules, 2015, 48(7), 2098–2106, DOI:10.1021/acs.macromol.5b00210.
- H. A. S. Schoonbrood, B. Van Den Reijen, J. B. L. De Kock, B. G. Manders, A. M. Van Herk and A. L. German, Pulsed Laser Terpolymerization of Styrene, Methyl Methacrylate and Methyl Acrylate, Macromol. Rapid Commun., 1995, 16(2), 119–124, DOI:10.1002/marc.1995.030160204.
- N. Kazemi, T. A. Duever and A. Penlidis, Demystifying the Estimation of Reactivity Ratios for Terpolymerization Systems, AIChE J., 2014, 60(5), 1752–1766, DOI:10.1002/aic.14439.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py01047c |
| This journal is © The Royal Society of Chemistry 2024 |