
Synergetic improvement in the mechanical properties of polyurethanes with movable crosslinking and hydrogen bonds†
Changming
Jin
 a,
Junsu
Park
ab,
Hidenori
Shirakawa
c,
Motofumi
Osaki
ab,
Yuka
Ikemoto
d,
Hiroyasu
Yamaguchi
abe,
Hiroaki
Takahashi
c,
Yasumasa
Ohashi
c,
Akira
Harada
f,
Go
Matsuba
*g and
Yoshinori
Takashima
*abeh
a,
Junsu
Park
ab,
Hidenori
Shirakawa
c,
Motofumi
Osaki
ab,
Yuka
Ikemoto
d,
Hiroyasu
Yamaguchi
abe,
Hiroaki
Takahashi
c,
Yasumasa
Ohashi
c,
Akira
Harada
f,
Go
Matsuba
*g and
Yoshinori
Takashima
*abeh
aDepartment of Macromolecular Science, Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka, 560-0043, Japan. E-mail: takasima@chem.sci.osaka-u.ac.jp
bForefront Research Center, Graduate School of Science, Osaka University, 1-1 Machikaneyama, Toyonaka, Osaka, 560-0043, Japan
cKanagawa Technical Center, Yushiro Chemical Industry Co., Ltd., 1580 Tabata, Samukawa, Koza, Kanagawa, 253-0193, Japan
dJapan Synchrotron Radiation Research Institute (SPring-8) Kouto, Sayo, Hyogo, 679-5198, Japan
eInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (OTRI), Osaka University, 1-1 Yamadaoka, Suita, Osaka, 565-0871, Japan
fSANKEN (The Institute of Scientific and Industrial Research), Osaka University, 8-1 Mihogaoka, Ibaraki, Osaka, 567-0047, Japan
gGraduate School of Organic Material Engineering, Yamagata University, 4-3-16 Jonan, Yonezawa, Yamagata, 992-8510, Japan. E-mail: gmatsuba@yz.yamagata-u.ac.jp
hInstitute for Advanced Co-Creation Studies, Osaka University, 1-1 Yamadaoka, Suita, Osaka, 565-0871, Japan
First published on 6th June 2022
Abstract
Polyurethane (PU) materials with movable crosslinking were prepared by a typical two-step synthetic process using an acetylated γ-cyclodextrin (TAcγCD) diol compound. The soft segment of PU is polytetrahydrofuran (PTHF), and the hard segment consists of hexamethylene diisocyanate (HDI) and 1,3-propylene glycol (POD). The synthesized PU materials exhibited the typical mechanical characteristics of a movable crosslinking network, and the presence of hydrogen bonds from the urethane bonds resulted in a synergistic effect. Two kinds of noncovalent bond crosslinking increased the Young's modulus of the material without affecting its toughness. Fourier transform infrared spectroscopy and X-ray scattering measurements were performed to analyze the effect of introducing movable crosslinking on the internal hydrogen bond and the microphase separation structure of PU, and the results showed that the carbonyl groups on TAcγCD could form hydrogen bonds with the PU chains and that the introduction of movable crosslinking weakened the hydrogen bonds between the hard segments of PU. When stretched, the movable crosslinking of the PU materials suppressed the orientation of polymer chains (shish-kebab orientation) in the tensile direction. The mechanical properties of the movable crosslinked PU materials show promise for future application in the industrial field.
1. Introduction
In recent years, research on polymer elastomers has received increasing attention and has had an irreplaceable effect on many fields, especially regarding wearable devices,1–3 flexible robots,4–6 and responsive materials.7–9 The applications of polymer elastomers are closely related to their mechanical properties. Therefore, many methods have been used to improve the mechanical properties of elastomers, such as adding fillers,10–13 polymer blending,14–16 minimizing structural defects17,18 and introducing a crosslinking network with reversible covalent19–21 or noncovalent bonds (such as hydrogen bonds,22–32 metal coordination bonds,33–36 π–π stacking,37–39 ionic interactions,40–43 and host–guest complexes44–53). However, it is still a challenge to fabricate a polymer with high toughness and a high Young's modulus (E) simultaneously.Previously, our group prepared movable crosslinking elastomers (called single movable cross-network (SC) elastomers) using acrylate-based or acrylamide-based polymers as polymer main chains.53,54 SC elastomers have higher toughness than chemically crosslinked elastomers due to their unique movable crosslinking structures. However, because the toughness and Young's modulus of a material are essentially contradictory properties, the preparation of elastomers with a high modulus as well as high toughness based on SC elastomer design remains a challenge. Adjusting the main-chain polymer type, as well as regulating the crosslinking density, is very useful for controlling the E of SC elastomers.
Herein, we successfully prepared polyurethane (PU) elastomers containing movable crosslinking based on the design of SC elastomers. We used PU as the main chain, and the physical crosslinking formed by the abundant hydrogen bond interactions in PU synergistically interacted with the introduced cyclodextrin (CD) movable crosslinker. Our design enables the modification of polyurethane networks using inexpensive modified CDs without the addition of other compounds. Compared to other designs that directly introduced βCD into PU,55,56 the PU prepared in this work formed movable crosslinks. The PU materials with movable crosslinking had higher E values than the linear PU elastomer without crosslinking (LPU) as well as the chemically crosslinked PU elastomer with no decrease in toughness.
2. Results and discussion
2.1. Preparation of PUs with movable crosslinking
Fig. 1(a) shows the structural formula of PUs with movable crosslinking introduced by acetylated γ-cyclodextrin (TAcγCD) (γCDMe(x)PU), where x refers to mol% of crosslinking (here TAcγCD), prepared by a two-step polymerization method (Scheme S2 and Table S1, ESI†). Dried TAcγCD diol monomer (Scheme S1 and Fig. S1–S3, ESI†) and poly(tetrahydrofuran) (PTHF: Mn = 1000)) were first mixed in dichloromethane (DCM). Then, excess hexamethylene diisocyanate (HDI) and dibutyltindiacetate (DBTDA) were added to the DCM solution for prepolymerization to obtain isocyanate-terminated chains. Subsequently, the chains were expanded by the addition of a diol chain extender, propan-1,3-diol (POD), at 25 °C. The polymerization reaction was continued for 24 hours until the peak attributed to isocyanate at 2270 cm−1 in the attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy disappeared. The polymer solution was poured into a mould and dried at room temperature overnight to obtain the elastomers. LPU (Fig. 1(b) and Scheme S3, Fig. S4, Tables S2, S3, ESI†) and PUs crosslinked by trifunctional triethanolamine (TEA) (C(x)PU, where x refers to mol% of TEA units; Fig. 1(c) and Scheme S4, Table S4, ESI†) were obtained in a similar manner. In the ATR-FTIR spectra, the isocyanate absorption peak at 2270 cm−1 disappeared, and the absorption peaks at approximately 1683 cm−1 and 1722 cm−1, attributed to the carbonyl groups, appeared after polymerization for all PUs (Fig. S5, ESI†). | ||
| Fig. 1 (a) Synthesis of γCDMe(x)PU and chemical structures of (b) LPU and (c) C(x)PU, where x refers to the molar ratio of TAcγCD diol monomer or TEA monomer. | ||
The formation of movable crosslinking between TAcγCD and the PU chain was confirmed by 2D nuclear Overhauser effect spectroscopy (NOESY) NMR spectroscopy. NMR spectroscopy of γCDMe(13)PU was performed after sufficient swelling in chloroform-d (24 hours of swelling). Fig. 2 exhibits the NOE correlation signals between the protons located on the internal side of the TAcγCD rings (C(3)H 5.3 ppm) and the protons in the PU chain (a 1.5 ppm). In contrast, a NOE signal of C(3)H was not observed in the spectrum of the reference sample (Fig. S6, ESI†). These results suggest that the PU chains penetrated the TAcγCD rings to form movable crosslinking.
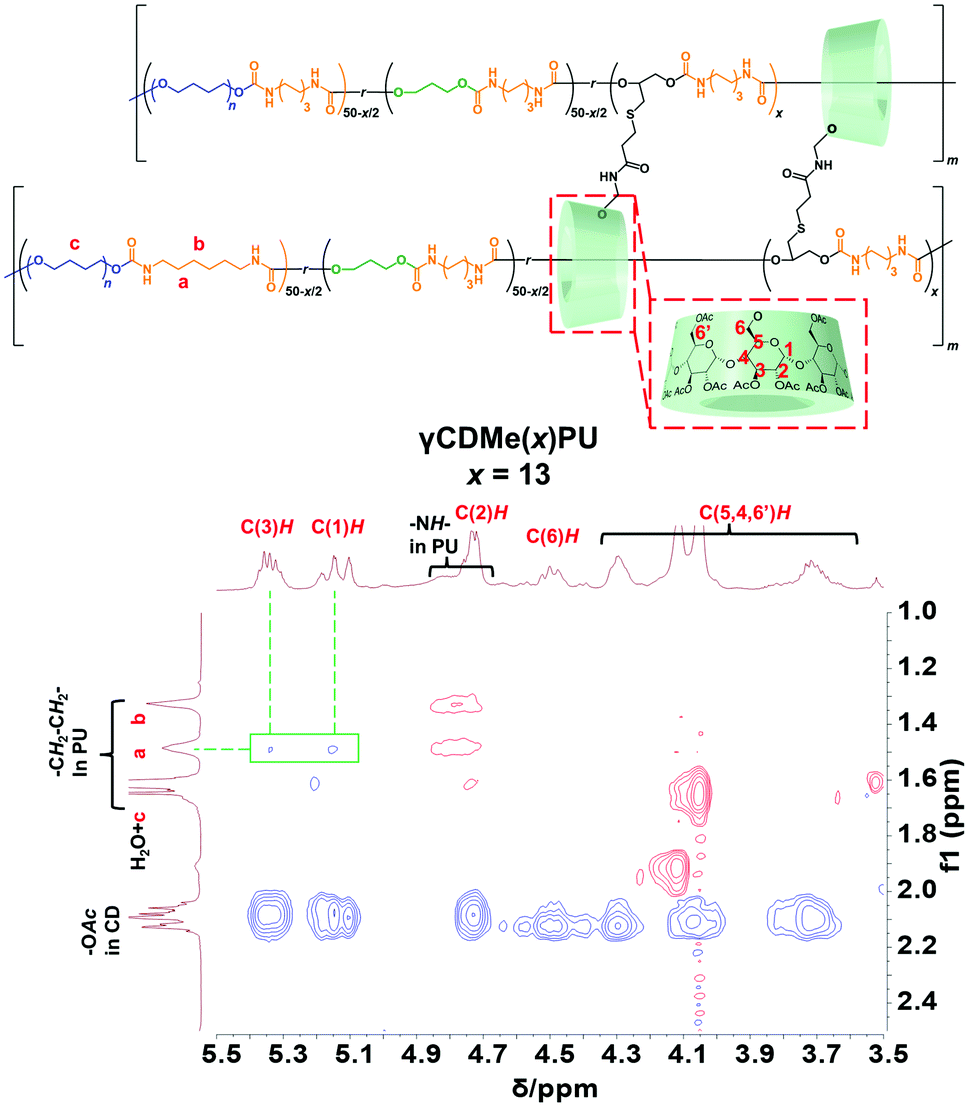 | ||
| Fig. 2 600 MHz 2D NOESY NMR spectrum of γCDMe(13)PU in chloroform-d. The NOE correlation signals between the TAcγCD units and the PU main chains are highlighted. | ||
2.2. Network structure of PU materials characterized by swelling tests
We performed swelling tests to study the amount of the movable crosslinking in the obtained PUs by immersing them in excess DCM (Fig. S7, ESI†). The swelling ratios were determined by the following equation.where W is the weight of the swollen PU and W0 is the initial weight of PU before the immersion. LPU with no crosslinking was dissolved, while γCDMe(x)PU and C(x)PU were swollen. Moreover, the swelling ratio of γCDMe(x)PU was larger than that of C(x)PU containing the same amount of crosslinking, which is a feature of movable crosslinking.59 The swelling ratios of both chemical and movable crosslinking PU materials decreased with increasing contents of the TEA and TAcγCD units, indicative of the formation of crosslinking by TEA and TAcγCD. The DSC data obtained after eliminating the effect of processing history demonstrated that the glass transition temperature (Tg) increased upon the formation of crosslinking (Fig. S8, ESI†). The thermal decomposition temperatures of γCDMe(x)PU and C(x)PU were higher than that of LPU, as determined by thermogravimetric analysis (TGA). The thermal decomposition temperatures of γCDMe(x)PU and C(x)PU increased with the mol% of TAcγCD and TEA units (Fig. S10, ESI†).
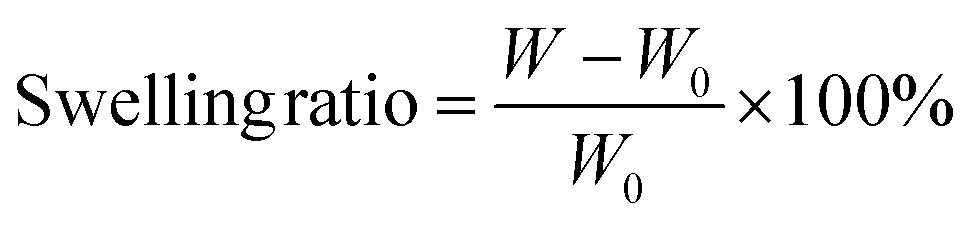
2.3. Mechanical properties of movable crosslinking PU
We investigated the mechanical properties of PUs by tensile tests. Fig. 3(a) and Fig. S11 in the ESI† show the stress–strain curves of γCDMe(x)PU, C(x)PU, and LPU. γCDMe(9)PU and γCDMe(13)PU showed higher fracture stress (42 ± 4 MPa and 46 ± 3 MPa, respectively) than LPU (24 ± 2 MPa). The fracture stress of C(9)PU (42 ± 1 MPa) was close to that of γCDMe(9)PU. When the amount of crosslinking units was increased to 13 mol%, the fracture stress of C(13)PU was increased to 44 ± 1 MPa. Fig. 3(b) shows the toughness and E values. The E values were determined from the slope at 0–5% strain of the curve. The E of γCDMe(x)PU was enhanced with increasing the x mol% of TAcγCD units. The E values of γCDMe(9)PU (67 ± 1 MPa) and γCDMe(13)PU (94 ± 1 MPa) were higher than that of LPU (57 ± 4 MPa), while the E values of C(9)PU (48 ± 2 MPa) and C(13)PU (50 ± 4 MPa) were lower than that of LPU. This result suggests that the formation of the movable crosslinking resulted in higher E values than the formation of covalent crosslinking. | ||
| Fig. 3 (a) Stress–strain curves, (b) plots of toughness versus Young's modulus, (c) plots of toughness divided by Young's modulus and Young's modulus, and (d) relaxation time curves of LPU, γCDMe(x)PU, and C(x)PU. The γCDMe(x)PU showed higher fracture stress, Young's modulus, and shorter relaxation times than the LPU and C(x)PU. | ||
The toughness was calculated from the integral of the stress–strain curve from the tensile test. C(9)PU and C(13)PU showed toughness values of 194 ± 5 MJ m−3 and 144 ± 4 MJ m−3, respectively, with a low E, while γCDMe(9)PU (171 ± 18 MJ m−3) and γCDMe(13)PU (146 ± 19 MJ m−3) showed higher toughness values than LPU (145 ± 14 MJ m−3) with a high E. This result suggests that the introduction of TAcγCD into γCDMe(x)PU to form movable crosslinking played an important role in simultaneously improving the fracture stress, toughness, and E value.
Fig. 3(c) shows plots of toughness/E versus E for γCDMe(x)PU, C(x)PU, and LPU. The toughness/E of C(x)PU decreased drastically with increasing E. On the other hand, the slope of the decrease in toughness/E was moderate for γCDMe(x)PU. The reason for these different changes depends on the nanostructure of the C(x)PU and γCDMe(x)PU materials. The nanostructure depends on the hydrogen bonding in the polyurethane materials and is an important factor affecting the Young's modulus. The introduction of chemical crosslinking may affect the formation of hydrogen bonds, resulting in no significant increase in the Young's modulus. We speculated that the effect would also occur with the introduction of movable crosslinking, but the Young's modulus of γCDMe(x)PU showed a significant increase. This indicated that the movable crosslinking improved the overall mechanical properties of the materials more than chemical crosslinking.
The relaxation time of PUs was investigated by stress relaxation tests (Fig. 3(d)). The γCDMe(x)PU, C(x)PU, and LPU materials were stretched to 100% strain at a rate of 10 mm min−1, and the strain was maintained for an hour. The stresses of the PU materials were normalized to focus on the behaviour of crosslinking. The results showed that γCDMe(x)PU relaxed earlier than C(x)PU and LPU. To determine the relaxation time of the relaxable components and the ratio of relaxable components to the residual components of γCDMe(x)PU, C(x)PU, and LPU, we carried out curve fitting using the Kohlrausch–Williams–Watts (KWW) models, as described by the following equation. All stress σ versus the stress relaxation time t curves of the samples were well fitted (R2 > 0.99).
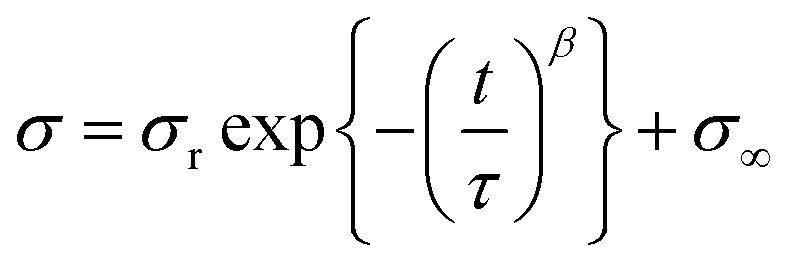
| PU materials | Relaxable components | Residual components | ||
|---|---|---|---|---|
| σ r /σ0e | τ /s | β | σ ∞ /σ0e | |
| a Relaxable stress. b Relaxation time. c Stretching exponent. d Residual stress. e Initial stress. | ||||
| γCDMe(9)PU | 0.52 | 96 | 0.27 | 0.48 |
| γCDMe(13)PU | 0.50 | 59 | 0.31 | 0.50 |
| C(9)PU | 0.30 | 86 | 0.29 | 0.70 |
| C(13)PU | 0.42 | 1047 | 0.26 | 0.58 |
| LPU | 0.29 | 148 | 0.35 | 0.71 |
The hysteresis of the PU materials was also investigated by cyclic stretching tests. γCDMe(x)PU, C(x)PU, and LPU were stretched to 50% strain and then returned to 0% strain 5 times. Fig. 4(a–c) and Fig. S12 in the ESI† show the results of the cyclic tensile tests. Based on the area of the stress–strain curves during stretching and recovery, we calculated the hysteresis loss (Fig. 4(d)). C(9)PU, C(13)PU, and LPU produced almost the same hysteresis loss in the first cycle. In contrast, the hysteresis loss of γCDMe(x)PU increased with increasing x mol%. This result suggests that the movable crosslinking of γCDMe(x)PU effectively dissipates energy and prevents stress concentration when the chains undergo large rearrangements in the first cycle. In the second cycle, the hysteresis losses of C(9)PU, C(13)PU, and LPU decreased rapidly, and after five cycles, the hysteresis losses were low. However, the hysteresis losses of both γCDMe(9)PU and γCDMe(13)PU remained constant from the second cycle, and the hysteresis loss of γCDMe(13)PU was higher than that of γCDMe(9)PU after 5 cycles. γCDMe(x)PU maintains a higher hysteresis loss than C(x)PU and LPU due to the presence of movable crosslinking.
 | ||
| Fig. 4 The cyclic stress–strain curve with fixed strain (50%) of (a) γCDMe(9)PU, (b) C(9)PU and (c) LPU. (d) Hysteresis loss calculated from the cyclic stress–strain curve. The cycles were performed five times. | ||
Furthermore, according to the DSC data of the PU materials before and after the cyclic stretching tests (Fig. S9, ESI†), the melting points (Tm) of C(9)PU, C(13)PU, and LPU increased slightly, and in contrast, the Tm of γCDMe(9)PU and γCDMe(13)PU decreased. The crystals of C(9)PU, C(13)PU, and LPU required much thermal energies after stretching, while the crystals of γCDMe(9)PU and γCDMe(13)PU required less. After stretching, the Tg of γCDMe(9)PU, γCDMe(13)PU, and LPU decreased. γCDMe(9)PU, γCDMe(13)PU, and LPU tended to soften after cyclic stretching. These results indicate that the hysteresis, Tm, and Tg of γCDMe(x)PU are different from those of C(x)PU and LPU.
2.4. Stretching effect of hydrogen bonds in γCDMe(9)PU observed by in situ FT-IR testing
We performed in situ FT-IR spectroscopy with tensile testing, as shown in Fig. 5. Fig. 5(a, c and e) show the stretching mode of the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group (black line) of C(9)PU, LPU, and γCDMe(9)PU before tensile stretching. After baseline calibration and normalization (based on the hydrocarbon band at 2856 cm−1), the bands of C(9)PU, LPU, and γCDMe(9)PU (black line) were separated into three, three, and five bands, respectively (red lines in Fig. 5(a, c and e)).
O) group (black line) of C(9)PU, LPU, and γCDMe(9)PU before tensile stretching. After baseline calibration and normalization (based on the hydrocarbon band at 2856 cm−1), the bands of C(9)PU, LPU, and γCDMe(9)PU (black line) were separated into three, three, and five bands, respectively (red lines in Fig. 5(a, c and e)).
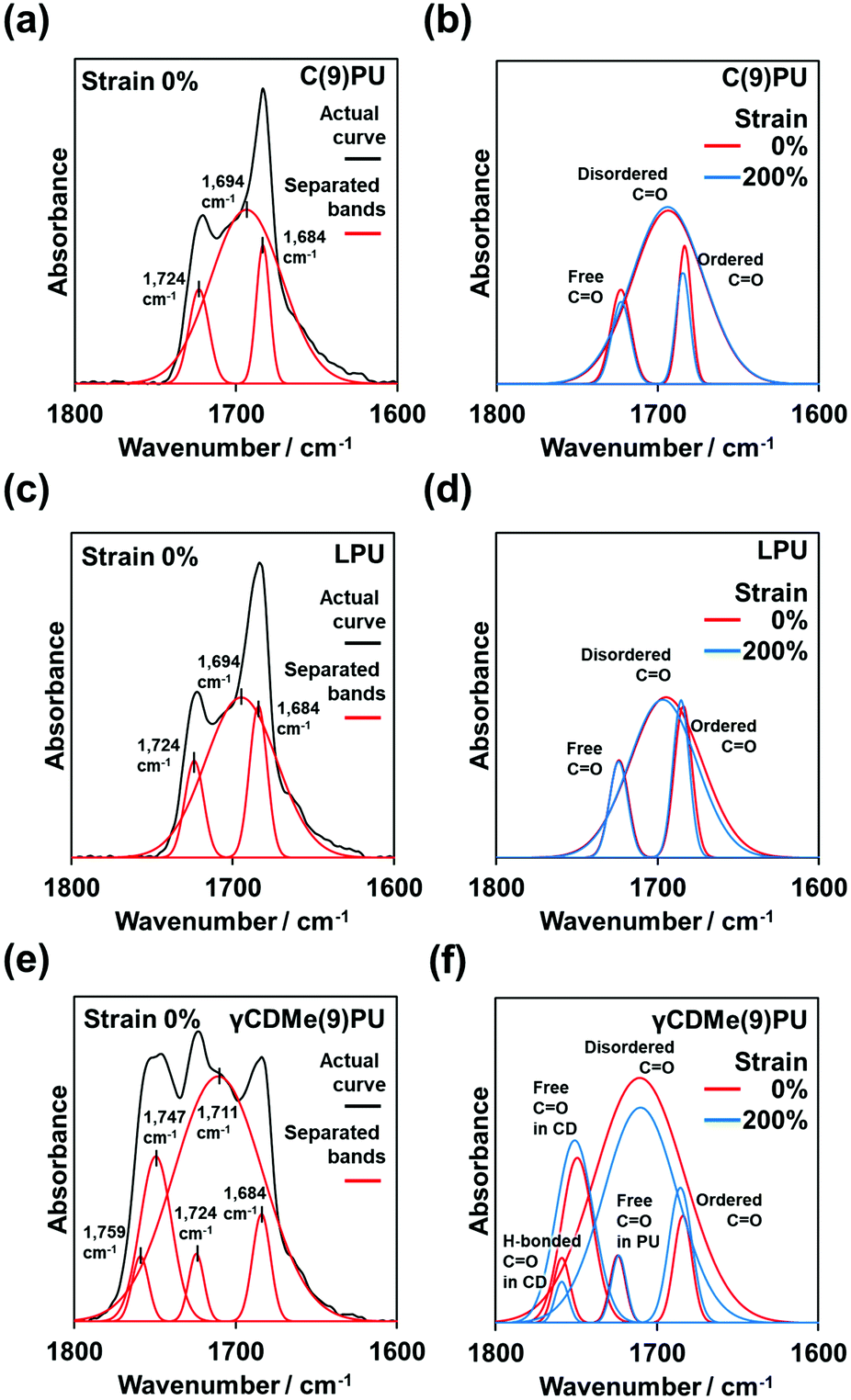 | ||
| Fig. 5 Actual FT-IR spectra and separated bands after peak deconvolution of C(9)PU (a), LPU (c), and γCDMe(9)PU (e) before tensile stretching. Changes in the separated bands of C(9)PU (b), LPU (d), and γCDMe(9)PU (f) during tensile stretching from 0% to 200% strain. | ||
The long diol chain segment (here, PTHF) is usually called the “soft segment”. The short diol chain with the isocyanate (here is POD + HDI) is called the “hard segment”.57 Therefore, the CO groups localized in the hard segments.58
Fig. 5 shows the ordered CO groups band in the ordered hard segments at 1684 cm−1 and the disordered CO groups band in the disordered hard segments at 1694 cm−1. The bands at 1684 cm−1 and 1694 cm−1 were assigned to the CO groups with hydrogen bonds between the hard–hard segments. The band at 1724 cm−1 was assigned to the CO groups without hydrogen bonds.59Fig. 5(b) shows the changes in the relevant bands upon stretching the C(9)PU specimen from 0% to 200% strain. The band intensity of the ordered CO groups decreased after stretching, and the band intensity of CO groups without hydrogen bonds also decreased after stretching, but the band intensity of the disordered CO groups almost no changes (or increased slightly). We postulated too large area of the disordered CO groups band caused the no (or small) change to observe. Namely, the ordered hard segments would dissociate into disordered segments after tensile stretching. The total number of hydrogen bonds in the stretched C(9)PU increased.
The positions of the bands in the LPU spectrum are similar to those in the C(9)PU spectrum. The band intensity of the ordered CO groups increased after tensile stretching, the band intensity of the disordered CO groups decreased, and the band intensity of CO groups without hydrogen bonds showed almost no changes (Fig. 5(d)). This indicated that the disordered hard segment transformed into the ordered hard segment in the stretched LPU.
In the γCDMe(9)PU spectrum, additional two bands were observed in addition to the ordered CO groups bands, the disordered CO groups bands, and the CO groups bands without hydrogen bonds (Fig. 5(e)). Because the TAcγCD unit also has the CO groups, these two additional bands should be associated with the TAcγCD unit. As our previous studies have shown that the CO group band without hydrogen bonds of a TAcγCD derivative can be observed at 1747 cm−1,51,53 the other band at 1759 cm−1 was assigned to the CO groups with hydrogen bonds between the CO groups of the TAcγCD units and the NH groups of the hard segments.
The position of the disordered CO group band in the γCDMe(9)PU spectrum is different from the position of this band in the C(9)PU and LPU spectra, blueshifting to 1711 cm−1. After stretching to 200% strain, the band intensity of the ordered CO groups increased, the band intensity of the disordered CO groups decreased, and the band intensity of the CO groups without hydrogen bonds showed almost no changes (Fig. 5(f)). The above trends of band changes in γCDMe(9)PU are similar to those in LPU but more pronounced. The band intensity of CO group band of the TAcγCD units with hydrogen bonds decreased, but that of the TAcγCD units without hydrogen bonds increased after stretching (Fig. 5(f)). γCDMe(9)PU had movable crosslinking and hydrogen bonds between the CO groups of the TAcγCD units and the hard segments. The hydrogen bonds of γCDMe(9)PU weakened, as indicated by the blueshift of the disordered CO group band. The changes in the hydrogen bonds of γCDMe(9)PU are different from those of C(x)PU but similar to those of LPU after stretching.
We calculated the band area changes of each separation before stretching and at 200% strain for each material to better visually describe the hydrogen bonding changes. The results are shown in Tables 2 and 3.
| PU materials | Band area with hydrogen bonding: A′ | Band area without hydrogen bonding: A′′ | A′/A′′ |
|---|---|---|---|
|
A′: the band area of the ordered CO band, disordered CO band and H-bonded CO band in CD/the band area at 2860 cm−1 (internal normalization peak: C–H band). A′′: the band area of the free CO band in PU and CD/the band area at 2860 cm−1 (internal normalization band: C–H band) |
|||
| C(9)PU | 1.09 | 0.14 | 7.54 |
| LPU | 0.95 | 0.12 | 7.69 |
| γCDMe(9)PU | 1.34 | 0.36 | 3.77 |
| PU materials | Band area with hydrogen bonding: A′ | Band area without hydrogen bonding: A′′ | A′/A′′ |
|---|---|---|---|
|
A′: the band area of the ordered CO band, disordered CO band and H-bonded CO band in CD/the band area at 2860 cm−1 (internal normalization peak: C–H band). A′′: the band area of the free CO band in PU and CD/the band area at 2860 cm−1 (internal normalization band: C–H band) |
|||
| C(9)PU | 1.12 | 0.13 | 8.52 |
| LPU | 0.86 | 0.11 | 7.71 |
| γCDMe(9)PU | 1.28 | 0.36 | 3.52 |
LPU shows the highest value of A′/A′′ (the ratio of the CO band area including hydrogen bonds to the CO band area not including hydrogen bonds) in Table 2. The A′/A′′ at 200% strain is essentially unchanged (slightly increased) in Table 3. This is because LPU does not contain fixed crosslinking points, and the hydrogen-bonded aggregation region can be rapidly dissociated and reorganized.
C(9)PU shows a lower A′/A′′ than LPU in Table 2 due to the introduction of chemical crosslinking that inhibits the formation of hydrogen bonds. This ratio increases significantly at 200% strain (Table 3). We speculate that the reason for these findings is that the molecular chains between the crosslinking points anchored in C(9)PU were drawn closer together, resulting in the formation of more hydrogen bonds.
γCDMe(9)PU shows the lowest A′/A′′ in Table 2 because the large size of TAcγCD further inhibits the formation of hydrogen bonds. This ratio decreases at 200% strain (Table 3). The hydrogen bonds from hard–hard segments or between the CO groups of the TAcγCD units and the NH groups of the hard segments are both broken when the TAcγCD movable crosslinking points are stretched.
The variation in the in situ FT-IR data shows the toughening mechanism of γCDMe(x)PU. The toughening mechanism is more due to the stress dispersion effect of the movable crosslinking rather than the strengthening of hydrogen bonding during stretching.
2.5. Stretching effect of the microphase separation structure of γCDMe(9)PU and C(9)PU by in situ small angle X-ray scattering (SAXS) and wide angle X-ray scattering (WAXS) measurements
In situ SAXS measurements with stretching were used to investigate the changes in the internal structure of γCDMe(9)PU and C(9)PU. Fig. 6 shows the 2D SAXS patterns of C(9)PU and γCDMe(9)PU when stretched from 0% to 200% strain. The scattered light intensity of C(9)PU decreased upon stretching to 100% strain (Fig. 6(a and b)). Then the intensity in the tensile direction increased upon stretching to 200% strain (Fig. 6(c)). This result indicates that C(9)PU undergoes orientation (shish-kebab orientation) along the tensile direction during stretching. The soft segments of C(9)PU are pulled and form the “shish”, and the hard segments are closely aligned to form the “kebab”.60 The orientation increased the microphase separation of C(9)PU. For γCDMe(9)PU, however, no significant change in the 2D SAXS patterns was observed during stretching (Fig. 6(d–f)). The 2D SAXS patterns of LPU were similar to those of C(9)PU. The scattered light intensity of LPU in the tensile direction diminished upon stretching to 100% strain but enhanced upon stretching to 200% strain due to the shish-kebab orientation (Fig. S13(a–c), ESI†). | ||
| Fig. 6 2D small-angle X-ray scattering (SAXS) patterns of (a–c) C(9)PU and (d–f) γCDMe(9)PU with 0%, 100% and 200% strain. The direction of tensile testing is shown by the arrow. | ||
The SAXS profiles of C(9)PU showed peaks caused by microphase separation in the tensile direction and vertical direction at 0% strain (Fig. 7(a and b)). When C(9)PU was stretched to 100% strain, the intensities of the peaks in both directions decreased. When the samples were stretched to 200% strain, the intensity of the peak in the vertical direction continued to decrease, while that of the peak in the tensile direction increased again. This result proves that the shish-kebab orientation is formed in the tensile direction of C(9)PU and increases the degree of microphase separation. The microphase separation caused by the shish-kebab orientation can increase the stress of C(9)PU; however, we speculate that when the action of this orientation is too strong, the toughness of the material decreases, leading to the steep slope observed in Fig. 3(c).
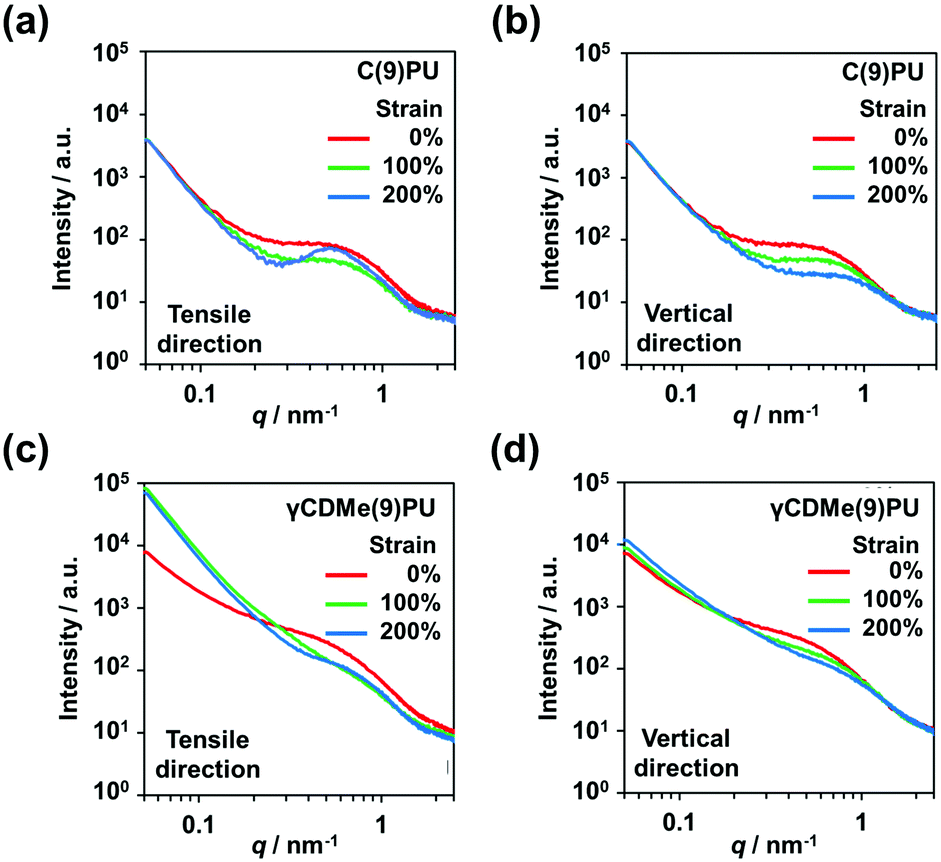 | ||
| Fig. 7 SAXS profiles of C(9)PU with 0%, 100% and 200% strains in the (a) tensile and (b) vertical directions. SAXS profiles of γCDMe(9)PU with 0%, 100% and 200% strains in the (c) tensile and (d) vertical directions. | ||
The SAXS profiles of LPU were also similar to those of C(9)PU. The scattered light intensity of LPU diminished when the sample was stretched to 100% strain but enhanced due to shish-kebab orientation along the tensile direction when the sample was stretched to 200% strain (Fig. S13(d and e), ESI†).
The SAXS profiles of γCDMe(9)PU also showed peaks in the tensile direction and vertical direction at 0% strain (Fig. 7(c and d)). When the strain increased, the intensities of peaks in both directions decreased and no peak was generated in the tensile direction, similar to that of C(9)PU, at 200% strain. This result indicates that the introduction of movable crosslinking hinders the shish-kebab orientation during stretching and is an important reason for its mechanical properties to maintain high toughness at a high Young's modulus. In addition, the scattered light intensity of γCDMe(9)PU was significantly enhanced upon stretching the sample from 0% to 100% in the tensile direction when the scattering vector (q) < approximately 0.2 nm−1 (Fig. 7(c)). This result indicates that the movable crosslinking units were pulled in γCDMe(9)PU.
The formation of the shish-kebab orientation can also account for the change in hydrogen bonding described in the previous section. The aggregation of hard segments due to orientation promotes the enhanced hydrogen bonding of C(9)PU during stretching. In contrast, the movable crosslinking suppressed the formation of this orientation. γCDMe(9)PU showed a decrease in hydrogen bonding during stretching.
In situ WAXS measurements with stretching were used to investigate the changes in the crystallinity of C(9)PU, γCDMe(9)PU, and LPU. The scattered light intensity of C(9)PU, γCDMe(9)PU, and LPU decreased with stretching, and these decreases were slow in the vertical direction, as shown in the 2D WAXS patterns (Fig. S14, ESI†). The new shoulder peaks appeared with stretching at approximately q = 17 nm−1 in the vertical direction of the WAXS profiles of C(9)PU and LPU (Fig. S15(a and c), ESI†). New shoulder peaks were not observed in the WAXS profiles of γCDMe(9)PU (Fig. S15(e), ESI†). In the tensile direction, the peak intensities of C(9)PU, γCDMe(9)PU, and LPU were decreased (Fig. S15(b, d, and f), ESI†).
These results indicate that the crystallinity of C(9)PU and LPU that stems from the shish-kebab orientation along the tensile direction increases with stretching. No relevant changes were observed in γCDMe(9)PU, which indicates that the presence of movable crosslinking makes the polyurethane chain less susceptible to shish-kebab orientation.
3. Conclusions
Here, a PU elastomer with movable crosslinking was successfully prepared via the introduction of TAcγCD and was named γCDMe(x)PU. γCDMe(x)PU showed similar fracture stresses and strains to C(x)PU with the same number of crosslinking units but higher E values, as determined by the tensile tests. The results of the cyclic tensile tests showed that the movable crosslinking of γCDMe(x)PU effectively dissipated energy and prevented stress concentration. The tensile tests of the relaxation time demonstrated that γCDMe(x)PU achieved both rapid relaxation and effective reduction of residual stress. Hydrogen bonds formed between the CO groups of the TAcγCD units and the hard segments in γCDMe(x)PU, according to the in situ FT-IR measurements. The synergetic effect of the movable crosslinking and the hydrogen bonds in γCDMe(x)PU contributed to the simultaneous increase in the mechanical properties such as Young's modulus and toughness. Moreover, the two components suppressed the shish-kebab orientation along the tensile direction, according to the in situ SAXS and WAXS measurements. We believe that this work will provide a design guidance for the fabrication of new tough polymer elastomers. γCDMe(x)PU with both high toughness and high Young's modulus are expected to become more important and be used in practical applications in the future.
Author contributions
C. J. performed the syntheses and spectroscopic studies. All authors contributed to the characterizations and discussion. Y. I. discussed and explained the results of the FT-IR spectroscopy. G. M. discussed and explained the results of the SAXS and WAXS measurements. C. J., J. P., and Y. T. co-wrote the paper. M. O., A. H., and H. Y. provided valuable suggestions. H. S., H. T., and Y. O. supported the syntheses of the monomer. Y. T. conceived and directed the study. Y. T. oversaw the project and contributed to the execution of the experiments and interpretation of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by a Grant-in-Aid for Scientific Research (B) (No. JP26288062) from JSPS of Japan and Scientific Research on Innovative Area JP19H05717 and JP19H05721 from MEXT of Japan, International Polyurethane Technology Foundation, JST SPRING, JPMJSP2138. The authors also appreciate the Analytical Instrument Faculty of Graduate School of Science, Osaka University, for supporting the NMR, ATR-FTIR and DSC measurements. The authors would like to thank Dr Noboru Ohta (SPring-8, JASRI) for the synchrotron radiation scattering measurements. The synchrotron radiation experiments were performed at BL40B2 (Proposal No. 2021B1830) and BL43IR (Proposal No. 2021B1814 and 2022A1505) of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI).References
- Z. Liu and G. Chen, Adv. Mater. Technol., 2020, 5, 2000049 CrossRef CAS.
- X. Chen, J. A. Rogers, S. P. Lacour, W. Hu and D.-H. Kim, Chem. Soc. Rev., 2019, 48, 1431–1433 RSC.
- S. Wang, J. Xu, W. Wang, G.-J. N. Wang, R. Rastak, F. Molina-Lopez, J. W. Chung, S. Niu, V. R. Feig, J. Lopez, T. Lei, S.-K. Kwon, Y. Kim, A. M. Foudeh, A. Ehrlich, A. Gasperini, Y. Yun, B. Murmann, J. B.-H. Tok and Z. Bao, Nature, 2018, 555, 83–88 CrossRef CAS PubMed.
- J. C. Yang, J. Mun, S. Y. Kwon, S. Park, Z. Bao and S. Park, Adv. Mater., 2019, 31, 1904765 CrossRef CAS PubMed.
- Y. Kim, H. Yuk, R. Zhao, S. A. Chester and X. Zhao, Nature, 2018, 558, 274–279 CrossRef CAS PubMed.
- Y. Qiu, E. Zhang, R. Plamthottam and Q. Pei, Acc. Chem. Res., 2019, 52, 316–325 CrossRef CAS PubMed.
- H. Liu, H. Xiang, Y. Wang, Z. Li, L. Qian, P. Li, Y. Ma, H. Zhou and W. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 40613–40619 CrossRef CAS PubMed.
- S. M. Mirvakili and I. W. Hunter, Adv. Mater., 2018, 30, 1704407 CrossRef PubMed.
- S. Babaee, S. Pajovic, A. R. Kirtane, J. Shi, E. Caffarel-Salvador, K. Hess, J. E. Collins, S. Tamang, A. V. Wahane, A. M. Hayward, H. Mazdiyasni, R. Langer and G. Traverso, Sci. Transl. Med., 2019, 11, eaau8581 CrossRef PubMed.
- J. N. Coleman, U. Khan, W. J. Blau and Y. K. Gun’ko, Carbon, 2006, 44, 1624–1652 CrossRef CAS.
- G. Tibbetts, M. Lake, K. Strong and B. Rice, Compos. Sci. Technol., 2007, 67, 1709–1718 CrossRef CAS.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud’Homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS.
- M. Nogi, S. Iwamoto, A. N. Nakagaito and H. Yano, Adv. Mater., 2009, 21, 1595–1598 CrossRef CAS.
- S. Wu, J. Appl. Polym. Sci., 1988, 35, 549–561 CrossRef CAS.
- C. Lee, Polymer, 2000, 41, 1337–1344 CrossRef CAS.
- T. H. Courtney, Mechanical Behavior of Materials, Waveland Press Inc., 2016 Search PubMed.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef CAS.
- S. Kondo, T. Hiroi, Y.-S. Han, T.-H. Kim, M. Shibayama, U. Chung and T. Sakai, Adv. Mater., 2015, 27, 7407–7411 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS.
- D.-D. Zhang, Y.-B. Ruan, B.-Q. Zhang, X. Qiao, G. Deng, Y. Chen and C.-Y. Liu, Polymer, 2017, 120, 189–196 CrossRef CAS.
- P. J. Woodward, D. Hermida Merino, B. W. Greenland, I. W. Hamley, Z. Light, A. T. Slark and W. Hayes, Macromolecules, 2010, 43, 2512–2517 CrossRef CAS.
- J. L. Lutkenhaus, K. D. Hrabak, K. McEnnis and P. T. Hammond, J. Am. Chem. Soc., 2005, 127, 17228–17234 CrossRef CAS PubMed.
- Y. Eom, S.-M. Kim, M. Lee, H. Jeon, J. Park, E. S. Lee, S. Y. Hwang, J. Park and D. X. Oh, Nat. Commun., 2021, 12, 621 CrossRef CAS PubMed.
- R. Du, Z. Xu, C. Zhu, Y. Jiang, H. Yan, H. Wu, O. Vardoulis, Y. Cai, X. Zhu, Z. Bao, Q. Zhang and X. Jia, Adv. Funct. Mater., 2020, 30, 1907139 CrossRef CAS.
- H. Chen, J. J. Koh, M. Liu, P. Li, X. Fan, S. Liu, J. C. C. Yeo, Y. Tan, B. C. K. Tee and C. He, ACS Appl. Mater. Interfaces, 2020, 12, 31975–31983 CrossRef CAS PubMed.
- Z. Cao, H. Liu and L. Jiang, Mater. Horiz., 2020, 7, 912–918 RSC.
- C.-F. Chow, S. Fujii and J.-M. Lehn, Angew. Chem., 2007, 119, 5095–5098 CrossRef.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717 RSC.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- J. Fox, J. J. Wie, B. W. Greenland, S. Burattini, W. Hayes, H. M. Colquhoun, M. E. Mackay and S. J. Rowan, J. Am. Chem. Soc., 2012, 134, 5362–5368 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, Md. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- J.-C. Lai, L. Li, D.-P. Wang, M.-H. Zhang, S.-R. Mo, X. Wang, K.-Y. Zeng, C.-H. Li, Q. Jiang, X.-Z. You and J.-L. Zuo, Nat. Commun., 2018, 9, 2725 CrossRef.
- Y. Miwa, K. Taira, J. Kurachi, T. Udagawa and S. Kutsumizu, Nat. Commun., 2019, 10, 1828 CrossRef.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 51, 7011–7015 CrossRef CAS PubMed.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270 CrossRef.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS PubMed.
- K. Miyamae, M. Nakahata, Y. Takashima and A. Harada, Angew. Chem., Int. Ed., 2015, 54, 8984–8987 CrossRef CAS PubMed.
- V. Kardelis, K. Li, I. Nierengarten, M. Holler, J.-F. Nierengarten and A. Adronov, Macromolecules, 2017, 50, 9144–9150 CrossRef CAS.
- J. Liu, C. S. Y. Tan, Z. Yu, N. Li, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1605325 CrossRef PubMed.
- S. Nomimura, M. Osaki, J. Park, R. Ikura, Y. Takashima, H. Yamaguchi and A. Harada, Macromolecules, 2019, 52, 2659–2668 CrossRef CAS.
- H. Aramoto, M. Osaki, S. Konishi, C. Ueda, Y. Kobayashi, Y. Takashima, A. Harada and H. Yamaguchi, Chem. Sci., 2020, 11, 4322–4331 RSC.
- R. Ikura, J. Park, M. Osaki, H. Yamaguchi, A. Harada and Y. Takashima, Macromolecules, 2019, 52, 6953–6962 CrossRef CAS.
- R. Ikura, Y. Ikemoto, M. Osaki, H. Yamaguchi, A. Harada and Y. Takashima, Polymer, 2020, 196, 122465 CrossRef CAS.
- A. Xie, M. Zhang and S.-I. Inoue, Open J. Org. Polym. Mater., 2016, 06, 99–111 CrossRef CAS.
- S. Oprea and V. O. Potolinca, Polym.-Plast. Technol. Eng., 2013, 52, 1550–1556 CrossRef CAS.
- I. Yilgör, E. Yilgör and G. L. Wilkes, Polymer, 2015, 58, A1–A36 CrossRef.
- L. Bistričić, G. Baranović, M. Leskovac and E. G. Bajsić, Eur. Polym. J., 2010, 46, 1975–1987 CrossRef.
- I. Yilgor, E. Yilgor, I. G. Guler, T. C. Ward and G. L. Wilkes, Polymer, 2006, 47, 4105–4114 CrossRef CAS.
- T. Kanaya, M. Murakami, T. Maede, H. Ogawa, R. Inoue, K. Nishida, G. Matsuba, N. Ohta, S. Takata, T. Tominaga, J. Suzuki, Y.-S. Han and T.-H. Kim, Polym. J., 2017, 49, 831–837 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm00408a |
| This journal is © The Royal Society of Chemistry 2022 |