
Effect of anions on the phase transition temperature of two structurally isomeric polymers: poly(N-isopropylacrylamide) and poly(2-isopropyl-2-oxazoline)†
Taehun
Chung
 a,
Jihoon
Han
a,
Young Jae
Kim
b,
Kyeong-Jun
Jeong
b,
Jun Mo
Koo
c,
Jemin
Lee
d,
Hyung Gyu
Park
d,
Taiha
Joo
b and
Youn Soo
Kim
*ae
a,
Jihoon
Han
a,
Young Jae
Kim
b,
Kyeong-Jun
Jeong
b,
Jun Mo
Koo
c,
Jemin
Lee
d,
Hyung Gyu
Park
d,
Taiha
Joo
b and
Youn Soo
Kim
*ae
aDepartment of Materials Science and Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-Ro, Nam-Gu, Pohang, Gyeongbuk 37673, Republic of Korea. E-mail: ysookim@postech.ac.kr
bDepartment of Chemistry, Pohang University of Science and Technology (POSTECH), 77 Cheongam-Ro, Nam-Gu, Pohang, Gyeongbuk 37673, Republic of Korea
cResearch Center for Bio-based Chemistry, Korea Research Institute of Chemical Technology (KRICT), Ulsan 44429, Republic of Korea
dDepartment of Mechanical Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-Ro, Nam-Gu, Pohang, Gyeongbuk 37673, Republic of Korea
eDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-Ro, Nam-Gu, Pohang, Gyeongbuk 37673, Republic of Korea
First published on 6th July 2022
Abstract
The effect of Hofmeister anions on the lower critical solution temperature (LCST) of two structural isomers, namely poly(N-isopropylacrylamide) (PNIPAAm) and poly(2-isopropyl-2-oxazoline) (PiPOx), is studied. Following the preparation of these two polymers with the same molecular weights and chain ends, their thermal hysteresis and LCST properties are observed in the aqueous and Hofmeister salt solutions. We investigate the effects of the salt type and salt concentration on the LCST for both polymers. More specifically, the LCST of PiPOx sharply increases as the concentration of SCN− increases, whereas that of PNIPAAm slightly increases prior to decreasing again. It is also confirmed that the boundary between the salting-out and salting-in effects is different for both polymers according to the type of salt present. Furthermore, Raman spectroscopy and two-dimensional correlation spectroscopy are used to examine the conformational changes in both polymers with increasing SCN− (chaotropes) and Cl− (kosmotropes) concentrations. Since the interaction mechanism between the polymer and anion is remarkably different even between isomeric polymers, it is necessary to rationally design the polymer structure to enhance the anion effect of the thermoresponsive polymer.
Introduction
Thermoresponsive polymers that exhibit a lower critical solution temperature (LCST) in aqueous solution have attracted considerable attention because of their potential application in controlled drug delivery,1,2 tissue engineering,3–5 and hydrogel actuators.6–8 For example, poly(N-isopropylacrylamide) (PNIPAAm) is considered a representative example of a thermoresponsive polymer, and it possesses an LCST of approximately 35 °C in deionized (DI) water.9–12 PNIPAAm possesses a unique hydrophilic–hydrophobic balance due to the presence of polar groups, such as amides, and apolar groups, such as methyl groups or main-chain hydrocarbons. The formation of hydrogen bonds between water molecules and the polar moieties of PNIPAAm therefore contributes to the negative change in the enthalpic term of the free energy of mixing.13–16 In contrast, the water molecules surrounding the apolar moieties tend to bind strongly to one another, forming a structured shell similar to an iceberg; the formation of this hydrated shell is known as hydrophobic hydration. At temperatures below the LCST, the favorable enthalpic term is dominant, and the polymer remains hydrated. However, at temperatures above the LCST, the unfavorable entropic term enforces phase separation, causing water molecules to be released from the hydrophobic hydration shell to increase their entropy. Phase separation via this LCST transition leads to the partial dehydration of the PNIPAAm chains, while PNIPAAm undergoes conformational changes from the coiled to the globular form via intra- and interchain hydrogen bonding between the amide groups.Similar to PNIPAAm, poly(2-isopropyl-2-oxazoline) (PiPOx), which was first reported as a thermoresponsive polymer by Uyama and Kobayashi in 1992,17 possesses an LCST of approximately 40 °C. Although PNIPAAm is typically synthesized using a radical polymerization approach, PiPOx is prepared by the living cationic ring-opening polymerization (CROP) of 2-oxazolines.18–20 PNIPAAm and PiPOx are structural isomers, in which the constituent atoms of the repeating units of each polymer are identical, and the two polymers possess the same isopropyl side-chain. The difference between these two structures is that the nitrogen atom exists in the main chain of PiPOx but is present in the side group of PNIPAAm. In addition, PNIPAAm can form intra- and interchain hydrogen bonds, whereas PiPOx is not capable of establishing such interactions, which makes the hydration states of the two polymers distinct.21–23 In addition, it should be noted that PiPOx has recently attracted particular attention in biological applications because of its rapid response to temperature changes and its nontoxic properties.18,24–27
The Hofmeister series, which was proposed in 1888,28 describes the ability of salts to solvate or precipitate certain proteins in aqueous solution. Salts that tend to stabilize or precipitate proteins are known as kosmotropes, whereas salts that tend to denature or solubilize proteins are referred to as chaotropes. In water, kosmotropes form thick and tight hydration shells, whereas chaotropes form thin and loose hydration shells. In this context, the effects of different salts on polymeric structures were also studied.29–31 More specifically, for thermoresponsive polymers, it has been determined that the salt environment of the polymer aqueous solution can control the LCST. Cremer et al. investigated the effects of these salts on the LCST of PNIPAAm.32 They revealed that three mechanisms could explain the effects of the Hofmeister anions on PNIPAAm solvation by considering the interactions between the anions, the polymer, and the hydration water molecules. Firstly, if the anion is hydrated, it interferes with the hydrogen bonding between the hydrated amide group and water molecules within the polymeric structure, thereby reducing the solubility of the polymer. Secondly, the hydration of the anion increases the surface tension of the cavity surrounding the main chain and the isopropyl group of the side group, thereby preventing the hydrophobic hydration of water and reducing the solubility of the polymer. Thirdly, the anion preferentially binds directly to the amide group of the polymer rather than being hydrated by water, which can polarize the polymer and increase its solubility. The first and second mechanisms mainly occur in kosmotropic anions that polarize water molecules, which leads to a salting-out effect and decreases the LCST. The third mechanism mainly occurs in chaotropic anions that polarize the polymer, leading to the salting-in of the polymer, which increases the LCST. Furthermore, the LCST changes caused by the incorporation of Hofmeister ions into PiPOx were studied by Schubert et al.33 They described the LCST shift of PiPOx using a mechanism similar to that of PNIPAAm. However, the LCST change in the Hofmeister salt solution was only investigated for each of the two polymers independently and was not compared for polymeric isomers with the same molecular weight. Moreover, studies on the effect of salts on the polymer architecture are limited. In fact, various factors, such as the hydrophilicity of the side group, molecular weight, end groups, and solution concentration, of the polymer may also influence the LCST changes.12,34,35 We therefore considered that it was necessary to compare these two isomeric polymers, namely PNIPAAm and PiPOx, under the same conditions. To the best of our knowledge, these two polymers with equal molecular weights and identical chain ends have never been compared.
Thus, we herein report the synthesis of two isomeric polymers, namely PNIPAAm and PiPOx, with the same molecular weight and identical chain ends (i.e., methyl and azide groups). The thermal hysteresis properties and LCST changes of the two polymers are both observed in Cl− and SCN− solutions. Subsequently, to further investigate the effect of the Hofmeister anions on the thermal behavior of the two polymers, the LCST changes are investigated in the presence of different salt types (i.e., Na2SO4, NaCl, NaBr, NaI, NaSCN, KCl, and KSCN) and salt concentrations. Furthermore, the expected conformations of both polymers are analyzed in NaCl (kosmotrope) and NaSCN (chaotrope) solutions using Raman spectroscopy and two-dimensional correlation spectroscopy (2DCOS).
Experimental section
Materials
The chemicals and solvents employed herein were purchased from various suppliers and used as received, unless otherwise noted. N-isopropylacrylamide (NIPAAm; Sigma-Aldrich, St Louis, MO, United States; 97%) was recrystallized from toluene/n-hexane. To synthesize 2-isopropyl-2-oxazoline (iPOx), isobutyronitrile (TCI, Japan; 98.0%), 2-aminoethanol (TCI, Japan; >99.0%), and cadmium acetate dehydrate (Sigma-Aldrich, St Louis, MO, United States; 98.0%) were used. To synthesize the two polymers, methyl p-toluenesulfonate (TCI, Japan; >98.0%), sodium azide (NaN3; Sigma-Aldrich, St Louis, MO, United States; >99.5%), methyl-2-chloropropionate (MCP; TCI, Japan; >95.0%), tris[2-(dimethylamino)ethyl]amine (Me6TREN, TCI, Japan; >98.0%), acetonitrile (Samchun Chemical Co., Korea; 99.9%), 2-propanol (Samchun Chemical Co., Korea; 99.8%), diethyl ether (Samchun Chemical Co., Korea; 99.9%), n-hexane (Samchun Chemical Co., Korea; 95.0%), N,N-dimethylformamide (DMF; Samchun Chemical Co., Korea; 99.9%), copper(I) chloride (CuCl; Sigma-Aldrich, St Louis, MO, United States; 99%), aluminum oxide (Al2O3, activated, neutral; Alfa Aesar, United States), and sea sand (Samchun Chemical Co., Korea; 30–50 mesh) were used. For the characterization of the polymers, chloroform-d (CDCl3; Sigma-Aldrich, St Louis, MO, United States; 100%) and lithium bromide (LiBr; Sigma-Aldrich, St Louis, MO, United States; >99%) were used. To measure the cloud points (Tcp), sodium thiocyanate (NaSCN; Sigma-Aldrich, St Louis, MO, United States; >98.0%), sodium sulfate (Na2SO4; Sigma-Aldrich, St Louis, MO, United States; >99.0%), sodium bromide (Samchun Chemical Co., Korea; 99%), sodium iodide (Samchun Chemical Co., Korea; 98.5%), potassium thiocyanate (KSCN; Sigma-Aldrich, St Louis, MO, United States; 99.0%), sodium chloride (NaCl; Sigma-Aldrich, St Louis, MO, United States; 99.5%), and potassium chloride (KCl; Sigma-Aldrich, St Louis, MO, United States; 99.0%) were used. DI water with a resistivity of 18.2 MΩ cm (Direct-Q® 5UV; Merck Millipore) was used throughout the experiments.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), and this purification cycle was repeated three times. Finally, the obtained solid was dried in a vacuum oven for 24 h to obtain the desired PNIPAAm-Cl as a white powder (Mn,NMR = 5554 g mol−1, Mn,GPC = 7784 g mol−1, Mw/Mn = 1.16).
:1). This purification cycle was repeated three times. The obtained solid was dried in a vacuum oven for 24 h to obtain the desired PNIPAAm-N3 as a white powder (Mn,NMR = 5561 g mol−1, Mn,GPC = 8289 g mol−1, Mw/Mn = 1.17). 1H NMR (400 MHz, CDCl3, 25 °C) δ (ppm): 4.03 (broad s; –CH(CH3)2 on the polymer side group), 3.68 (broad s; terminal –O–CH3), 2.35–1.49 (broad m; –CH2– on the polymer main chain), 1.17 (strong broad s; –CH(CH3)2 on the polymer side group).
:1), and this purification cycle was repeated three times. Finally, the obtained solid was dried in a vacuum oven for 24 h to obtain the desired PNIPAAm-Cl as a white powder (Mn,NMR = 5554 g mol−1, Mn,GPC = 7784 g mol−1, Mw/Mn = 1.16).
:1). This purification cycle was repeated three times. The obtained solid was dried in a vacuum oven for 24 h to obtain the desired PNIPAAm-N3 as a white powder (Mn,NMR = 5561 g mol−1, Mn,GPC = 8289 g mol−1, Mw/Mn = 1.17). 1H NMR (400 MHz, CDCl3, 25 °C) δ (ppm): 4.03 (broad s; –CH(CH3)2 on the polymer side group), 3.68 (broad s; terminal –O–CH3), 2.35–1.49 (broad m; –CH2– on the polymer main chain), 1.17 (strong broad s; –CH(CH3)2 on the polymer side group).
Characterization
Results and discussion
Polymer synthesis and characterization
The PNIPAAm sample was synthesized by atom transfer radical polymerization (ATRP) under controlled conditions, as outlined in Scheme 1a. It has been previously reported that the ATRP of acrylamides suffers from low conversion, mainly due to the competitive coordination of the amide group with the metal catalyst.37–39 However, several groups have reported that the use of an alcohol-based polymerization solvent can preserve the terminal halogen atoms while also increasing the conversion.40,41 Thus, similar protocols were employed in this study to synthesize the PNIPAAm sample. Initially, PNIPAAm-Cl was prepared using an MCP/CuCl/Me6TREN system at 25 °C in IPA; subsequently, the terminal chlorine atom was transformed into an azide group via a simple nucleophilic substitution reaction in the presence of excess NaN3 in DMF.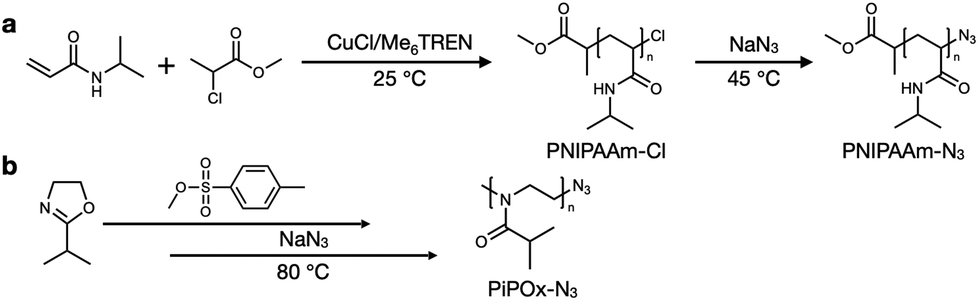 | ||
| Scheme 1 Synthesis of (a) poly(N-isopropylacrylamide) (PNIPAAm) and (b) poly(2-isopropyl-2-oxazoline) (PiPOx). | ||
The synthetic process employed to obtain PiPOx is summarized in Scheme 1b. More specifically, the PiPOx samples were synthesized by cationic ring-opening polymerization (CROP), which has been demonstrated to provide fine control of the average molar mass while ensuring a narrow molar mass distribution.42 Importantly, it is possible to functionalize the end group of this system because of its living nature.43–46 Thus, we used methyl p-toluenesulfonate as the CROP initiator, introducing the same methyl group at one chain end of both PiPOx and PNIPAAm. To terminate the reaction, sodium azide was added to the reaction terminal end of the living PiPOx chain to obtain the corresponding azide-terminated chain end. Following the purification of the two polymers, they were characterized by 1H NMR spectroscopy (see Fig. 1). Due to the presence of a polar C–N bond, the proton signal corresponding to the b-position of the isopropyl group of PNIPAAm appears at a more downfield position than that of PiPOx. Comparing the main chains of the two polymers using the same logic, the proton signal corresponding to the a-position of PiPOx appears more downfield than that of PNIPAAm. It was therefore confirmed from these NMR spectra that in PNIPAAm, the side group is partially polar, whereas in PiPOx, the main chain is polar. In contrast, the proton signal corresponding to the c-position of the isopropyl methyl groups appears at the same position in both polymers; therefore, it can be inferred that the polarities of the two polymers are similar with respect to the isopropyl group.
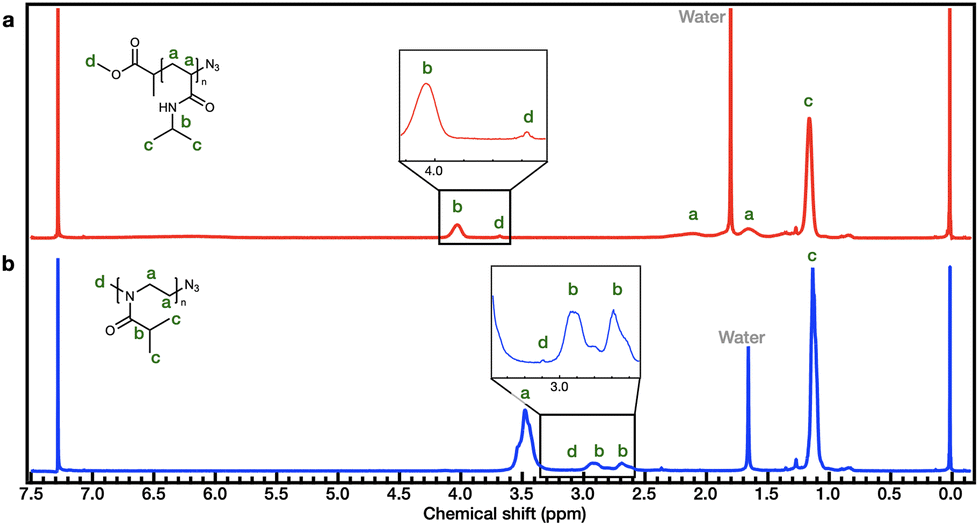 | ||
| Fig. 1 1H NMR spectra of (a) PNIPAAm and (b) PiPOx after purification in CDCl3. | ||
Furthermore, the molecular weight of PNIPAAm was determined by comparing the peak areas of the isopropyl group (–CH(CH3)2, 4.03 ppm) with those of the terminal methoxy group (–O–CH3, 3.68 ppm) arising from the MCP initiator, resulting in a Mn,NMR value of 5561 g mol−1 for PNIPAAm. In the case of PiPOx, the broad peak of the isopropyl group (–CH(CH3)2, 3.05–2.55 ppm) and the peak of the methyl group (–N–CH3, 3.10 ppm) at the chain end are used to determine the Mn,NMR value, which was 5489 g mol−1.
The GPC traces of PNIPAAm and PiPOx in the DMF eluent containing 10 mM LiBr are shown in Fig. 2, wherein a symmetric and sharp peak can be observed. The absence of tailing or shoulders indicates the lack of premature chain termination. Based on the GPC results, Mn,GPC of PNIPAAm was determined to be 8289 g mol−1, while its Đ was calculated as 1.17. Similarly, PiPOx was found to possess a Mn,GPC of 6990 g mol−1 and a Đ of 1.19.
 | ||
| Fig. 2 GPC traces obtained for PNIPAAm (red line) and PiPOx (blue line) in DMF containing 10 mM LiBr. | ||
Moreover, the Fourier transform infrared (FT-IR) spectra confirmed the conversion of PNIPAAm-Cl to PNIPAAm-N3 through the emergence of a new absorbance peak at 2120 cm−1, which corresponded to the terminal azide group (Fig. S1†) (Table 1).
| Sample name | Conv.a [%] | M n,theo [g mol−1] | DPc | M n,NMR [g mol−1] | M n,GPC [g mol−1] | M w,GPC [g mol−1] | M w/Mn (Đ)d |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy of the crude samples. b Determined from the initiator/monomer ratio and the conversion. c Determined by 1H NMR spectroscopy after purification. d Determined by GPC in a DMF eluent containing 10 mM LiBr using a RI detector and a PMMA standard. | |||||||
| PNIPAAm-Cl | 94 | 5850 | 48 | 5554 | 7784 | 9033 | 1.16 |
| PNIPAAm-N3 | — | — | 48 | 5561 | 8289 | 9699 | 1.17 |
| PiPOx-N3 | 98 | 5774 | 48 | 5489 | 6990 | 8325 | 1.19 |
Thermal hysteresis and cloud point (Tcp) determination
We investigated the coil–globule transition and the association/dissociation properties of both polymers upon heating and cooling in DI water and in aqueous solutions of NaCl and NaSCN. Although PNIPAAm and PiPOx are structural isomers, one of the noticeable differences between their properties lies in their thermal hysteresis behavior. Thus, the thermal hysteresis was recorded for both polymers (1 wt%) after three cycles of heating and cooling at a rate of 1 °C min−1. Before comparing the thermal hysteresis, the LCST behavior was determined from the temperature of the cloud point (Tcp) in the solution. Firstly, the Tcp values of two polymers were determined in the absence of salts. As shown in Fig. 3a, the values of Tcp for PNIPAAm and PiPOx in DI water are 36.4 and 40.7 °C, respectively. This was attributed to the higher polarity of the PiPOx main chain (see section Polymer synthesis and characterization), thereby leading to a slightly higher LCST. | ||
| Fig. 3 (a) Transmittance plots of PNIPAAm (dotted line) and PiPOx (solid line) as a function of temperature obtained upon heating and cooling in DI water. (b) Transmittance plots of PNIPAAm (dotted line) and PiPOx (solid line) as a function of temperature obtained upon heating and cooling in a 1 M NaCl solution. (c) Transmittance plots of PNIPAAm (dotted line) and PiPOx (solid line) as a function of temperature obtained upon heating and cooling in a 2 M NaSCN solution. Red lines; heating cycles, blue lines; cooling cycles. In all experiments, both the heating and cooling rates were set at 1 °C min−1. | ||
Returning to the hysteresis, it is noted that this behavior is mainly due to the kinetically slow dissolution process of polymer chains from aggregates (i.e., the globular state) during cooling.47,48 As shown in Fig. 3a, PNIPAAm exhibits a distinct hysteresis (ΔTcp = 2.1 °C) in DI water. As the coil–globule transition of PNIPAAm is an irreversible process,47,49–52 our results indicate that intra- and interchain hydrogen bonds are formed in the globular state. In contrast, PiPOx shows almost identical transmittance curves during heating and cooling, indicating that little hysteresis occurs (ΔTcp = 0.5 °C, see Fig. 3a). Furthermore, we examined the effect of the heating rate on hysteresis (Fig. S2†). At a rate of 0.5 °C min−1, the ΔTcp values of PNIPAAm and PiPOx were determined to be 1.9 and 0.2 °C, respectively. Although the hysteresis decreased slightly when the rate was reduced, apparent hysteresis was still observed for PNIPAAm; however, negligible hysteresis was detected for PiPOx. In the case of the PNIPAAm aqueous solution, stable and dense aggregates are formed due to the strong polymer–polymer hydrogen bonding (i.e., N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) at temperatures above the LCST. This results in distinct hysteresis due to the slow dissolution of the polymer chain aggregates in the heating–cooling process. It is also known that weak polymer–water–polymer hydrogen bonds (i.e., CO⋯H–O–H⋯OC) are formed in aqueous solutions of PiPOx at temperatures above the LCST.21 This is attributed to the absence of a secondary amino group in PiPOx, due to which the hydrogen bond donor is absent from the polymer chain, thereby preventing the formation of intra- and interchain hydrogen bonds in DI water. Therefore, the weak and loose physical polymer–water–polymer hydrogen bonds of PiPOx formed at high temperatures can be readily dissociated during the cooling process, resulting in little hysteresis during the heating–cooling process.23,33
C) at temperatures above the LCST. This results in distinct hysteresis due to the slow dissolution of the polymer chain aggregates in the heating–cooling process. It is also known that weak polymer–water–polymer hydrogen bonds (i.e., CO⋯H–O–H⋯OC) are formed in aqueous solutions of PiPOx at temperatures above the LCST.21 This is attributed to the absence of a secondary amino group in PiPOx, due to which the hydrogen bond donor is absent from the polymer chain, thereby preventing the formation of intra- and interchain hydrogen bonds in DI water. Therefore, the weak and loose physical polymer–water–polymer hydrogen bonds of PiPOx formed at high temperatures can be readily dissociated during the cooling process, resulting in little hysteresis during the heating–cooling process.23,33
Additionally, the differences in the bond polarities were examined for the two polymers due to their role in imparting different hysteresis properties. For this purpose, density functional theory (DFT) calculations were used to probe the electron densities in terms of the molecular electrostatic potential (ESP) distributions. As illustrated in Fig. S3,† it was confirmed that the electron densities of PNIPAAm and PiPOx did not show any significant differences in the CO and N components. In contrast, the H atom in the N–H moiety of PNIPAAm possessed a distinctly stronger positive charge than those of the other moieties, thereby effectively making it the only available hydrogen bond donor. Since the polarity of the CO moiety was similar for both polymers, it was anticipated that the hydrogen atom of the strongly polar amide in PNIPAAm will exclusively boost its propensity to form hydrogen bonds between polymer chains. This polymer–polymer hydrogen bond was therefore considered to be the source of the kinetic trap, which is responsible for impeding additional polymer hydration during the dissolution stage that takes place upon cooling, ultimately resulting in a broader thermal hysteresis for PNIPAAm.49–52
We also investigated the effect of the Hofmeister ions on the thermal hysteresis of both polymers. More specifically, the Tcp values of PNIPAAm and PiPOx in 1.0 M NaCl solutions were determined to be 24.9 and 30.8 °C, respectively. This result indicates that the Tcp values of both polymers in NaCl solution are lower than those in DI water. In addition, as shown in Fig. 3b, PNIPAAm exhibits a broader hysteresis (ΔTcp = 6.2 °C) than PiPOx (ΔTcp = 2.3 °C). In a 2.0 M NaSCN solution, the Tcp of PNIPAAm was found to be 36.4 °C, which is comparable to that obtained in DI water, whereas the Tcp of PiPOx increased to 68.0 °C. A broader hysteresis can also be observed in PNIPAAm (ΔTcp = 4.7 °C) than in PiPOx (ΔTcp = 2.0 °C) (Fig. 3c), indicating that the large ΔTcp of PNIPAAm and the small ΔTcp of PiPOx observed in DI water persist in the presence of both kosmotropic and chaotropic anions.
Effects of the salt type and concentration on Tcp
Subsequently, the changes in the Tcp values of PNIPAAm and PiPOx upon varying the concentrations of the five Hofmeister anions (i.e., SO42−, Cl−, Br−, I−, and SCN−) were investigated (Fig. 4 and Fig. S4–S6†).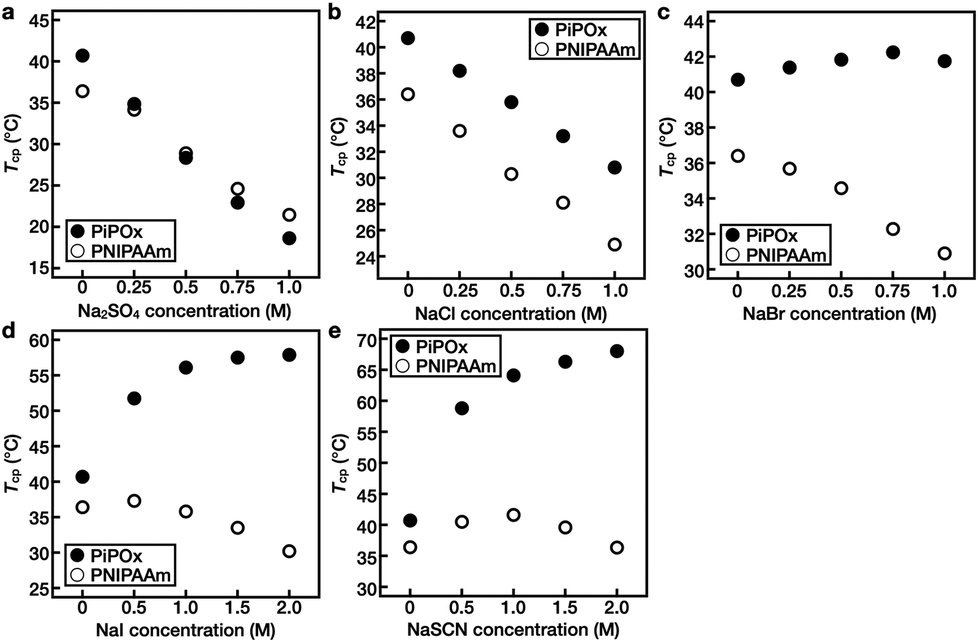 | ||
| Fig. 4 T cp profiles of PNIPAAm (open circles) and PiPOx (closed circles) in salt solutions as a function of the (a) Na2SO4, (b) NaCl, (c) NaBr, (d) NaI, and (e) NaSCN concentrations. The concentration of the polymer was 1 wt%. | ||
In the case of PNIPAAm, Tcp decreased linearly upon increasing the salt concentrations for the SO42−, Cl−, and Br− systems, while no such relationship was observed for the I− and SCN− solutions. Instead, upon increasing the concentrations of these salts, the Tcp first increased and then decreased, representing the turnover. For example, when the concentration of NaI was increased from 0 to 0.5 M, Tcp increased from 36.4 to 37.3 °C prior to decreasing at higher concentrations; at a concentration of 2.0 M, it reached a value of 30.2 °C (Fig. 4d). For the NaSCN system, the value of Tcp first increased to 40.6 °C at a concentration of 1.0 M and then decreased to 36.4 °C at a concentration of 2.0 M (Fig. 4e).
In the case of PiPOx, Tcp decreased linearly upon increasing the salt concentration in the SO42− and Cl− systems. In contrast, in the Br− solution, Tcp increased from 40.7 to 42.2 °C upon increasing the NaBr concentration from 0 to 0.75 M. Upon increasing the concentration of this salt further to 1.0 M, Tcp decreased to 41.7 °C (Fig. 4c). In addition, it was observed that PiPOx was particularly sensitive to I− and SCN−, and as a result, the value of Tcp increased rapidly even at a low salt concentration of 0.5 M. Furthermore, as the ion concentration was increased further, Tcp continued to increase. More specifically, the Tcp values of PiPOx in 2.0 M NaI and NaSCN solutions were 57.9 and 68.0 °C, respectively (Fig. 4d and e).
These results indicate that in aqueous solutions of the PNIPAAm and PiPOx polymers, kosmotropic anions lead to salting-out, which results in a linear decrease in Tcp upon increasing the anion concentration. As kosmotropic anions are hard anions that prefer to bind strongly to water rather than to polymers, a thick hydration layer is formed, which effectively excludes hydrated salts from the polymer surface (Fig. S7†). These properties of kosmotropic anions are dependent on the anion itself, thereby accounting for the varying degrees of reduction observed for Tcp in the presence of different anions. In contrast, chaotropic anions lead to salting-in, and the change in Tcp is non-linearly dependent on the salt concentration. If excess chaotropic anions are added, a second salting-out process occurs due to excess ion hydration, thereby resulting in a maximum Tcp being observed at a specific salt concentration. This turnover behavior results from the saturation of polar sites, such as amide groups, which are capable of binding anions within the polymer structure. In the same polymer, it was confirmed that the concentration of the salt representing the turnover of Tcp differed depending on the anion type. In addition, it was observed that the boundary of the salt transitioning from the salting-out effect to the salting-in effect was different for the two polymers.
As noted above, PiPOx is particularly sensitive to SCN−. Although the polar sites of PNIPAAm and PiPOx are identical, the differences lie in the fact that the polar sites of PiPOx are present throughout the main polymer chain, whereas in PNIPAAm, they are present only in the side groups. It is therefore considered that the salting-in effect is sensitive to the location of the polar binding site. Furthermore, upon comparison of the Tcp results obtained for KSCN and NaSCN over the same concentration range, the Tcp tendencies of the two polymers were found to be identical, thereby indicating that the effect of the cation is insignificant (Fig. S6†).
Based on the aforementioned observations, we considered the fact that a greater salting-in effect (i.e., in the case of PiPOx) results in greater expansion of the polymer chain, which can be observed via changes in the bonds constituting the main chain. Raman spectroscopy and 2DCOS were therefore employed to characterize and analyze the conformational changes of each polymer caused by the binding of anions, as discussed in the following subsections.
Two-dimensional correlation spectroscopy (2DCOS)
The two polymers investigated here showed the same trend in the presence of kosmotropic anions; however, because they showed different responses to the chaotropic anions, 2DCOS was performed based on the recorded Raman spectra. Typically, 2DCOS is used to interpret the transition of an observed system upon perturbation, such as a change in the concentration, time, or temperature. Hence, for the purpose of this study, an increasing concentration of salts was employed as a perturbation factor to verify the conformational changes of the main chain53,54 and non-covalent bonds (e.g., hydrogen bonding and ionic interactions) in the prepared polymers.21,55–57 As a result, synchronous and asynchronous 2DCOS spectra were obtained from the 1D Raman spectra, and the sequential order of the representative bands was confirmed.More specifically, in the synchronous 2DCOS spectrum, the red peak indicates that the two correlated peaks change in the same direction (either increasing or decreasing with increasing salt concentration), whereas the blue peak indicates that the two correlated peaks change in the opposite direction. According to Noda's rule,58 in the synchronous 2DCOS spectrum, when two correlated peaks have the same color at the same location as in the asynchronous spectrum, the peak on the x-axis changes before that on the y-axis as the salt concentration is increased; when the colors are different, the peak on the y-axis changes before that on the x-axis. In general, in the 2DCOS analysis of polymers, bonds with a fast sequence are either a bond constituting the main chain of a polymer that undergoes a significant conformational change53,54 or a polar bond with non-covalent bonds (hydrogen or ionic interactions) that increase or decrease significantly.21,55–57
As shown in Fig. S8 and S9,† it was initially confirmed that the trend of Tcp at a polymer concentration of 10 wt% was comparable to the results presented in Fig. 4b and e; hence, all experiments were performed at a concentration of 10 wt% to ensure the high sensitivity of the Raman measurements. The Raman spectrum of each polymer solution was obtained in the presence of 0–0.250 M NaCl at 25 °C. For PNIPAAm, the representative Raman bands were observed at wavenumbers of 1633 (CO), 1205 (C–N), and 938 cm−1 (C–C) (Fig. S10a†). Upon comparison of the 2DCOS results, it was found that the sequential order of the bands upon increasing the salt concentration was as follows: C–C < C–N < CO (Fig. S10b–S10d†). In the case of PiPOx, the representative Raman bands were observed at wavenumbers of 1606 (CO), 1205 (C–N), and 1090 cm−1 (C–C) (Fig. S11a†). Upon comparison of the 2DCOS results, it was found that the sequential order of the bands upon increasing the salt concentration was as follows: C–C < C–N < CO (Fig. S11b–S11d†). For both PNIPAAm and PiPOx, the sequential order of the bands was identical due to the fact that these polymers are structural isomers. It therefore appears that as the concentration of Cl− increases, Cl− dehydrates the polymer, interfering with the strong interactions between the polar amide bonds and water molecules. During this dehydration process, the vibration of the CO bond is the first to change since it is more polar than the C–N bond. This result is independent of the type of polymer, since the presence of a polar group, such as an amide, results in kosmotropic anions promoting salting-out along with the corresponding decrease in Tcp. Since the measurement temperature (25 °C) of the Raman spectra is lower than the Tcp values of the two polymers, the main chain does not completely collapse during the measurement, and so the C–C bond of the main chain appears to be the least insensitive of the bonds compared above.
Subsequently, the effects of SCN− on both PNIPAAm and PiPOx were analyzed. As shown in Fig. 4e and Fig. S9c,† the Tcp of PNIPAAm showed a tendency to increase and then decrease as the NaSCN concentration increased. More specifically, at the concentration before turnover (i.e., 0–1.0 M NaSCN), the representative bands were observed at wavenumbers of 1633 (CO), 1205 (C–N), and 938 cm−1 (C–C) (Fig. 5a). Upon comparison of the 2DCOS results, the sequential order of the bands upon increasing the salt concentration was determined to be as follows: C–C < CO < C–N (Fig. 5b–d). Beyond the turnover concentration (i.e., from 1.0–2.0 M NaSCN), the representative bands were observed at wavenumbers of 1638 (CO), 1211 (C–N), and 938 cm−1 (C–C) (Fig. 6a). In this case, upon comparison of the 2DCOS results, the sequential order of the bands upon increasing the salt concentration was as follows: C–C < CO < C–N (Fig. 6b–d). In the case of PiPOx, the value of Tcp continued to increase as the NaSCN concentration was increased. Accordingly, 2DCOS analysis was performed over the range of 0–2.0 M, and the representative bands were observed at wavenumbers of 1606 (CO), 1205 (C–N), and 1090 cm−1 (C–C) (Fig. 7a), resulting in a sequential order of: CO < C–N < C–C (Fig. 7b–d); this order is different from that observed for PNIPAAm. Thus, considering the resonance structure of the amide bond, it would be assumed that the SCN− ions bond directly to the N atoms of both polymers, which bear partial positive charges. As a result, the change in the C–N bond will be most pronounced upon increasing the NaSCN concentration. Indeed, in PNIPAAm, C–N bonding appears to be the most sensitive sequence, indicating that SCN− binds to the C–N bond present in the side group of PNIPAAm to generate the salting-in effect; however, since the effect is limited to the side group, the increase in Tcp is small, and the turnover phenomenon is observed. In contrast, for PiPOx, changes occurred more rapidly in the C–C bond than the C–N bond, thereby indicating that considerable structural changes occurred in the main chain. More specifically, the SCN− ions appeared to bind to the C–N bonds of the PiPOx main chain to polarize the main chain, and as a result, the degree of polymer hydration was increased to form an expanded conformation. As such, the polarization of the main chain maximized the salting-in effect of the polymer and resulted in a sharp increase in the value of Tcp.
 | ||
| Fig. 5 (a) Raman spectra of 10 wt% PNIPAAm solutions in D2O (black line), 0.5 M NaSCN (red line), and 1 M NaSCN (green line). 2DCOS (b) synchronous and (c) asynchronous spectra of PNIPAAm (10 wt%) with variation in the NaSCN concentration. (d) Peak descriptions for the 2DCOS results, where red is defined as a positive intensity, while blue is defined as a negative intensity. | ||
 | ||
| Fig. 6 (a) Raman spectra of 10 wt% PNIPAAm solutions in 1.0 M NaSCN (green line), 1.5 M NaSCN (blue line), and 2.0 M NaSCN (purple line). 2DCOS (b) synchronous and (c) asynchronous spectra of PNIPAAm (10 wt%) with variation in the NaSCN concentration. (d) Peak descriptions for the 2DCOS results, where red is defined as a positive intensity, while blue is defined as a negative intensity. | ||
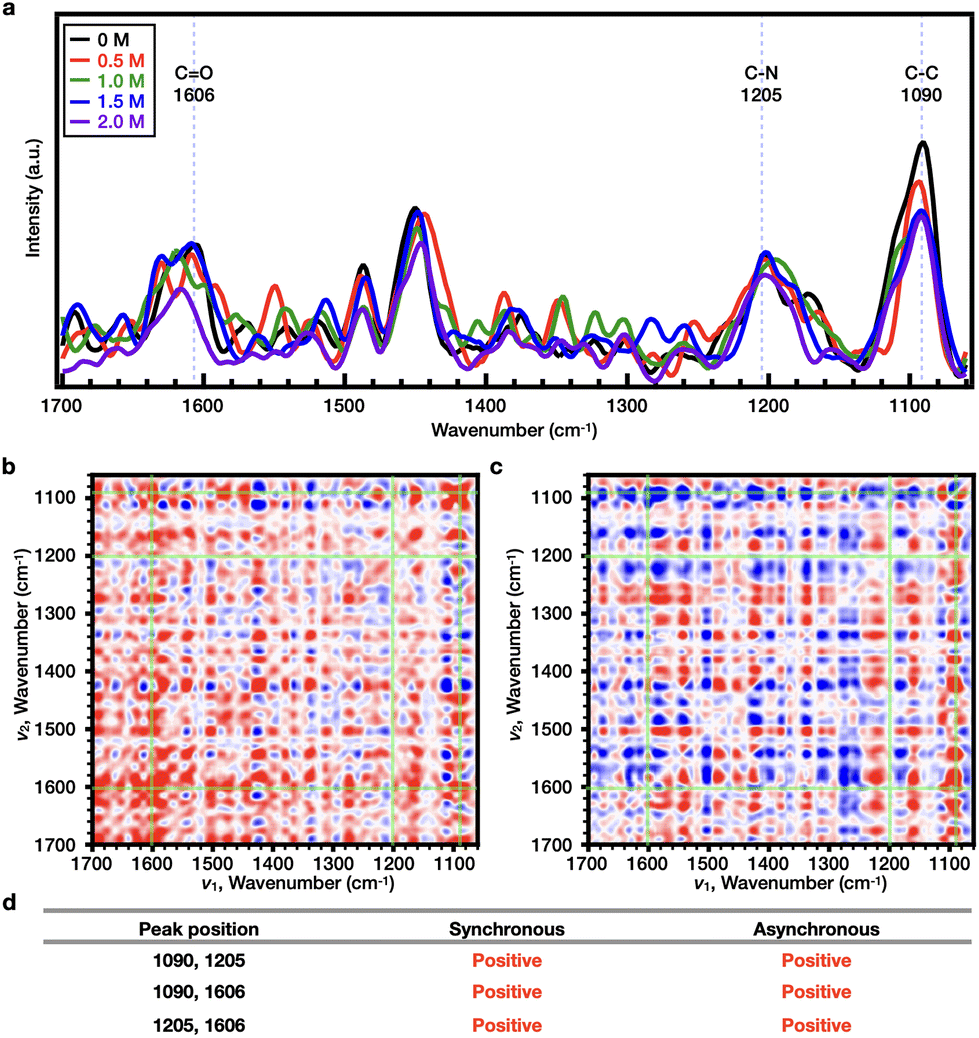 | ||
| Fig. 7 (a) Raman spectra of 10 wt% of PiPOx in D2O (black line), NaSCN 0.5 M (red line), NaSCN 1 M (green line), NaSCN 1.5 M (blue line), and NaSCN 2.0 M (purple line). 2DCOS (b) synchronous and (c) asynchronous spectra of PiPOx (10 wt%) depending on the NaSCN concentration. (d) Peak description of the 2DCOS result. Herein, red color is defined as a positive intensity, while blue color is defined as a negative one. | ||
Conclusions
Herein, we reported our observations regarding the effect of Hofmeister anions on the lower critical solution temperature (LCST) of two structural isomers, namely poly(N-isopropylacrylamide) (PNIPAAm) and poly(2-isopropyl-2-oxazoline) (PiPOx). To ensure a precise comparison, we synthesized these two polymers with the same degree of polymerization and the same functional groups at both chain ends. Subsequently, their thermal hysteresis and LCST properties were observed in aqueous solution and in the presence of various Hofmeister salts (i.e., Na2SO4, NaCl, NaBr, NaI, NaSCN, KCl, and KSCN). It was found that PNIPAAm exhibits a distinct hysteresis, whereas PiPOx shows little or no hysteresis upon heating and cooling. These observations are associated with the corresponding abilities of the two polymers to form intra- and interchain hydrogen bonds; PNIPAAm is able to form such bonds, whereas PiPOx is not. In addition, we observed the effect of the salt concentration on Tcp for both polymers. For the chaotropic anion (SCN−), the LCST increase was more remarkable in PiPOx than in PNIPAAm. More specifically, the cloud point (Tcp) of PiPOx was 40.7 °C in deionized water but 68.0 °C in a 2.0 M NaSCN solution. This can be accounted for by considering that in PiPOx, the polarization of the main chain occurs through the direct binding of the chaotrope to the polar region of the polymer. From the results of Raman spectroscopy and two-dimensional correlation spectroscopy, it was confirmed that as the concentration of SCN− increased, the PiPOx chain showed a more expanded structure, and thus, the C–C bond present in the main chain reacted sensitively; this accounts for the increase in the LCST at higher concentrations of SCN−. In contrast, for PNIPAAm, the polarization of the polymer through SCN− binding mainly occurs in the side groups. Therefore, only minimal conformational changes were found to occur in the main chain, resulting in relatively insensitive changes of the C–C bonds present in the main chain. As a result, the LCST remained relatively constant, even at higher SCN− concentrations. Furthermore, upon increasing the concentration of Cl−, both polymers showed the same sequential order due to Tcp decreasing in both cases. Based on these results, the effect of the Hofmeister anions on the two isomeric polymers was confirmed; Cl− causes a salting-out effect regardless of the type of polymer, while SCN− causes a salting-in effect, wherein the degree of this effect depends on the polymer architecture. The results presented herein therefore suggest that, to increase the sensitivity of thermoresponsive polymers to ions, it is necessary to structurally design the main chain of the polymer to allow direct ion binding. This in-depth exploration of thermoresponsive polymer solutions and the ion–polymer relationship during the phase transition process is therefore essential to provide a more theoretical basis for further applied studies.Author contributions
Conceptualization, T.C.; methodology, T.C.; validation, T.C., J.H., and J.M.K.; formal analysis, T.C., J.H., K.-J.J., J.L., and H.G.P.; investigation, T.C., J.H., K.-J.J., J.L., and H.G.P.; data curation, T.C., J.H., Y.J.K., and K.-J.J.; writing—original draft preparation, T.C.; writing—review and editing, T.C., J.H., K.-J.J., J.M.K., T.J., and Y.S.K.; visualization, T.C.; supervision, Y.S.K.; project administration, Y.S.K.; funding acquisition, Y.S.K. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by a National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (No. 2019R1C1C1002836). This research was also supported by the Basic Research Program through a NRF grant funded by the MSIT (No. 2020R1A4A3079853). The authors would like to thank Prof. Woo-Dong Jang, Jeong Heon Lee, and Jeongmin Lee for their help with obtaining the transmittance plots. The authors would also like to thank Prof. Moon Jeong Park and Jihoon Kim for their assistance in obtaining the FT-IR spectra. The authors would also like to thank Prof. Chang Yun Son for assistance in DFT calculation.Notes and references
- G. W. Ashley, J. Henise, R. Reid and D. V. Santi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2318–2323 CrossRef CAS PubMed.
- X. Dong, C. Wei, J. Liang, T. Liu, D. Kong and F. Lv, Colloids Surf., B, 2017, 154, 253–262 CrossRef CAS PubMed.
- A. Choi, K. D. Seo, H. Yoon, S. J. Han and D. S. Kim, Biomater. Sci., 2019, 7, 2277–2287 RSC.
- O. H. Kwon, A. Kikuchi, M. Yamato, Y. Sakurai and T. Okano, J. Biomed. Mater. Res., 2000, 50, 82–89 CrossRef CAS PubMed.
- K. Nagase, M. Yamato, H. Kanazawa and T. Okano, Biomaterials, 2018, 153, 27–48 CrossRef CAS PubMed.
- Y. Cheng, K. Ren, D. Yang and J. Wei, Sens. Actuators, B, 2018, 255, 3117–3126 CrossRef CAS.
- D. Kim, H. S. Lee and J. Yoon, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- Y. S. Kim, M. Liu, Y. Ishida, Y. Ebina, M. Osada, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nat. Mater., 2015, 14, 1002–1007 CrossRef CAS PubMed.
- S. Fujishige, K. Kubota and I. Ando, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef CAS.
- Y. Maeda, T. Higuchi and I. Ikeda, Langmuir, 2000, 16, 7503–7509 CrossRef CAS.
- T. Binkert, J. Oberreich, M. Meewes, R. Nyffenegger and J. Ricka, Macromolecules, 1991, 24, 5806–5810 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- P. H. Corkhill, A. M. Jolly, C. O. Ng and B. J. Tighe, Polymer, 1987, 28, 1758–1766 CrossRef CAS.
- A. Barnes, P. H. Corkhill and B. J. Tighe, Polymer, 1988, 29, 2191–2202 CrossRef CAS.
- Y. Ono and T. Shikata, J. Am. Chem. Soc., 2006, 128, 10030–10031 CrossRef CAS PubMed.
- H. Uyama and S. Kobayashi, Chem. Lett., 1992, 21, 1643–1646 CrossRef.
- R. Hoogenboom and H. Schlaad, Polymers, 2011, 3, 467–488 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- B. Verbraeken, B. D. Monnery, K. Lava and R. Hoogenboom, Eur. Polym. J., 2017, 88, 451–469 CrossRef CAS.
- T. Li, H. Tang and P. Wu, Langmuir, 2015, 31, 6870–6878 CrossRef CAS PubMed.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- R. Hoogenboom, H. M. Thijs, M. J. Jochems, B. M. van Lankvelt, M. W. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- N. Oleszko-Torbus, Polym. Rev., 2021, 1–20 Search PubMed.
- M. Haktaniyan, S. Atilla, E. Cagli and I. Erel-Goktepe, Polym. Int., 2017, 66, 1851–1863 CrossRef CAS.
- M. Ashjari, M. Kazemi, M. N. Abi, M. Mohammadi and S. Rafiezadeh, J. Drug Delivery Sci. Technol., 2020, 59, 101914 CrossRef CAS.
- V. R. de la Rosa, J. Mater. Sci.: Mater. Med., 2014, 25, 1211–1225 CrossRef CAS PubMed.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260 CrossRef.
- I. K. Han, J. Han and Y. S. Kim, Chem. – Asian J., 2021, 16, 1897–1900 CrossRef CAS PubMed.
- J. Paterová, K. B. Rembert, J. Heyda, Y. Kurra, H. I. Okur, W. R. Liu, C. Hilty, P. S. Cremer and P. Jungwirth, J. Phys. Chem. B, 2013, 117, 8150–8158 CrossRef PubMed.
- H. I. Okur, J. Hladílková, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, J. Phys. Chem. B, 2017, 121, 1997–2014 CrossRef CAS PubMed.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS PubMed.
- M. M. Bloksma, D. J. Bakker, C. Weber, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 724–728 CrossRef CAS PubMed.
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS PubMed.
- Q. Duan, A. Narumi, Y. Miura, X. Shen, S.-I. Sato, T. Satoh and T. Kakuchi, Polym. J., 2006, 38, 306–310 CrossRef CAS.
- H. Witte and W. Seeliger, Justus Liebigs Ann. Chem., 1974, 1974, 996–1009 CrossRef.
- J. T. Rademacher, M. Baum, M. E. Pallack, W. J. Brittain and W. J. Simonsick, Macromolecules, 2000, 33, 284–288 CrossRef CAS.
- M. Teodorescu and K. Matyjaszewski, Macromol. Rapid Commun., 2000, 21, 190–194 CrossRef CAS.
- D. Neugebauer and K. Matyjaszewski, Macromolecules, 2003, 36, 2598–2603 CrossRef CAS.
- Y. Xia, X. Yin, N. A. Burke and H. D. Stöver, Macromolecules, 2005, 38, 5937–5943 CrossRef CAS.
- Y. Xia, N. A. Burke and H. D. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- A. Makino and S. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1251–1270 CrossRef CAS.
- M. Glassner, M. Vergaelen and R. Hoogenboom, Polym. Int., 2018, 67, 32–45 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, M. Leenen, S. F. van Nispen, M. van der Loop, C. H. Abeln, A. M. van den Berg and U. S. Schubert, Macromolecules, 2005, 38, 7957–7966 CrossRef CAS.
- M. Glassner, D. R. D′hooge, J. Y. Park, P. H. Van Steenberge, B. D. Monnery, M.-F. Reyniers and R. Hoogenboom, Eur. Polym. J., 2015, 65, 298–304 CrossRef CAS.
- F. J. Arraez, X. Xu, M. Edeleva, P. H. M. Van Steenberge, Y. W. Marien, V.-V. Jerca, R. Hoogenboom and D. R. D'Hooge, Polym. Chem., 2022, 13, 861–876 RSC.
- Y. Ding, X. Ye and G. Zhang, Macromolecules, 2005, 38, 904–908 CrossRef CAS.
- M. R. Berber, H. Mori, I. H. Hafez, K. Minagawa, M. Tanaka, T. Niidome, Y. Katayama, A. Maruyama, T. Hirano, Y. Maeda and T. Mori, J. Phys. Chem. B, 2010, 114, 7784–7790 CrossRef CAS PubMed.
- H. Cheng, L. Shen and C. Wu, Macromolecules, 2006, 39, 2325–2329 CrossRef CAS.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- Y. Lu, K. Zhou, Y. Ding, G. Zhang and C. Wu, Phys. Chem. Chem. Phys., 2010, 12, 3188–3194 RSC.
- X. Wang, X. Qiu and C. Wu, Macromolecules, 1998, 31, 2972–2976 CrossRef CAS.
- Y. Eom, Y. Park, Y. M. Jung and B. C. Kim, Polymer, 2017, 108, 193–205 CrossRef CAS.
- W. Li and P. Wu, Polym. Chem., 2014, 5, 5578–5590 RSC.
- R. Geitner, S. Götz, R. Stach, M. Siegmann, P. Krebs, S. Zechel, K. Schreyer, A. Winter, M. D. Hager and U. S. Schubert, J. Phys. Chem. A, 2018, 122, 2677–2687 CrossRef CAS PubMed.
- J. M. Koo, H. Kim, M. Lee, S.-A. Park, H. Jeon, S.-H. Shin, S.-M. Kim, H. G. Cha, J. Jegal and B.-S. Kim, Macromolecules, 2019, 52, 923–934 CrossRef.
- S. Sun and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 32232–32240 RSC.
- I. Noda, Appl. Spectrosc., 2000, 54, 994–999 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00543c |
| This journal is © The Royal Society of Chemistry 2022 |