
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMedicinal chemistry of the myeloid C-type lectin receptors Mincle, Langerin, and DC-SIGN
Jonathan
Cramer
 *
*
Institute for Pharmaceutical and Medicinal Chemistry, Heinrich-Heine-University of Düsseldorf, Universitätsstr. 1, 40225 Düsseldorf, Germany. E-mail: jonathan.cramer@hhu.de
First published on 16th September 2021
Abstract
In their role as pattern-recognition receptors on cells of the innate immune system, myeloid C-type lectin receptors (CLRs) assume important biological functions related to immunity, homeostasis, and cancer. As such, this family of receptors represents an appealing target for therapeutic interventions for modulating the outcome of many pathological processes, in particular related to infectious diseases. This review summarizes the current state of research into glycomimetic or drug-like small molecule ligands for the CLRs Mincle, Langerin, and DC-SIGN, which have potential therapeutic applications in vaccine research and anti-infective therapy.
 Jonathan Cramer | Jonathan Cramer obtained his PhD from the Philipps-Universität Marburg (Germany) in the group of Prof. Gerhard Klebe, studying the role of water networks in protein–ligand interactions. He then joined the laboratory of Prof. Beat Ernst at the University of Basel (Switzerland), where he worked on the development of glycomimetic drugs for the treatment of bacterial and viral infections. He was recently appointed to an assistant professorship at the Heinrich-Heine-University of Düsseldorf, where his group is focused on the medicinal chemistry and chemical biology of carbohydrate-derived drugs. |
Introduction
Myeloid C-type lectin receptors (CLRs) are pattern-recognition receptors (PRRs) expressed on cells of the innate immune system.1–5 These proteins have evolved to recognize carbohydrate epitopes on pathogens, such as viruses, bacteria, fungi, and parasites. After an encounter with a pathogen, CLR stimulation can trigger an immunological response on multiple levels (Fig. 1). Firstly, pathogen recognition by CLRs that are expressed by antigen-presenting cells, such as dendritic cells (DCs) and macrophages, is often followed by receptor-mediated endocytosis. After intracellular trafficking into lysosomal compartments, products of lysosomal degradation are then presented to CD4+ T cells on MHC II receptors. Alternatively, antigen presentation to CD8+ T cells on MHC I receptors can occur through cross-presentation pathways. Secondly, many CLRs can also trigger signalling cascades that ultimately result in a modulation of the cytokine inventory of the antigen-presenting cell.6–8 The composition of the secreted cytokine cocktail affects polarization of adjacent T cells and shapes immune responses depending on the encountered pathogen. Thus, myeloid CLRs are mediators between innate and adaptive immunity. Apart from their predominant role in antimicrobial immunity, myeloid CLRs can exert many other functions related to cell homeostasis or cancer.5,9 The fact that CLR signalling can potentiate and shape adaptive immune responses renders them highly attractive targets for the development of CLR-agonistic vaccine adjuvants.10,11 In addition, targeted delivery of antigens to specific myeloid cell subsets has the potential to generate very efficient vaccination responses.12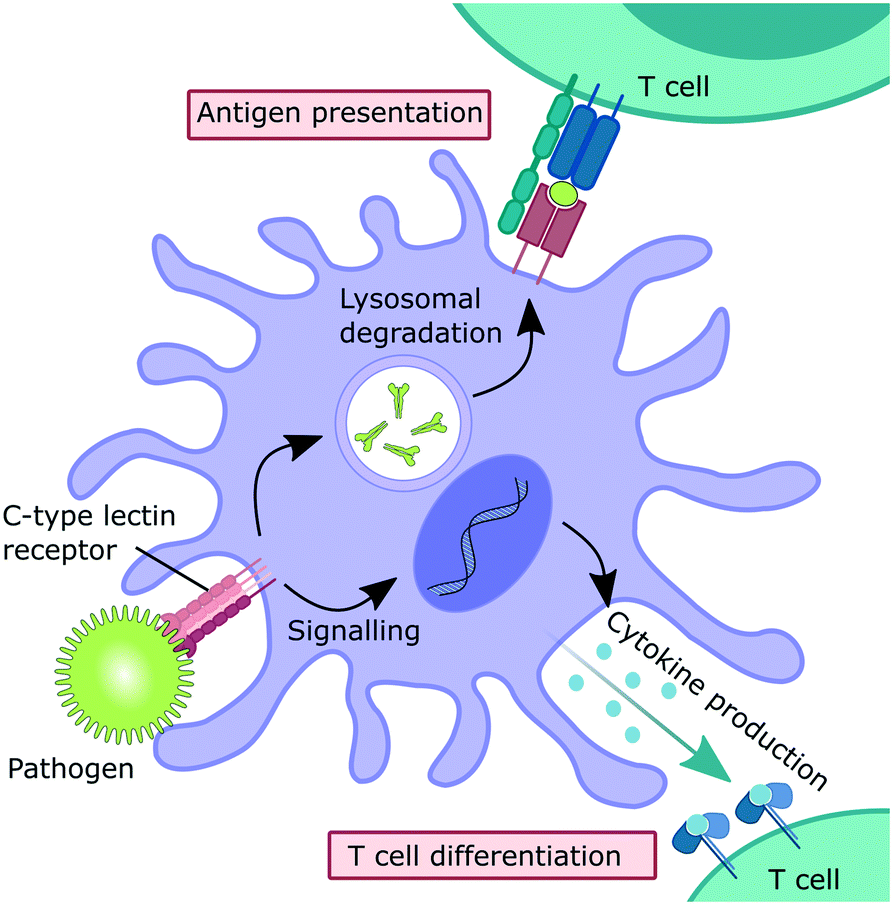 | ||
| Fig. 1 Immune functions mediated by myeloid CLRs. | ||
However, besides their protective role in innate immunity, CLRs represent potential entry receptors for various pathogens.2,3 Exploitation of CLRs has been described for many pathogenic viruses, such as coronaviruses,13,14 Ebola virus,15 HIV,16 and many others.2,3 In addition, CLRs can have a detrimental role in bacterial infections and parasitic diseases.17–22 Similarly, overstimulation of CLR signalling by pathogens can induce exacerbated immune reactions, so called “cytokine storm” pathologies.23–26 Thus, the inhibition of CLR–pathogen interactions represents a promising strategy for modulating disease progression in various infectious diseases, with the added benefit of addressing a target that is encoded in the host genome and not subject to frequent genomic mutations.
Due to broad and overlapping specificities for natural carbohydrate ligands, targeting individual receptors of the CLR family remains a challenging task and molecules that are able to specifically address certain CLRs are in high demand for functional biological studies, as well as various therapeutically-relevant applications. To achieve this goal, many efforts have focused on antibody-based strategies or the development of multivalent molecules presenting natural carbohydrate ligands. This review, however, focuses on the application of rational drug design strategies for the development of small molecule ligands targeting myeloid CLRs. Also, CLRs that are predominantly expressed on non-myeloid cell types, such as the selectins, Lox-1, or ASGPR, are not considered here. Since many myeloid CLRs have not yet been the target of drug discovery campaigns and research into glycomimetic dectin-2 and macrophage galactose lectin (MGL) ligands is still in its infancy,27,28 this review focuses on case studies of the most thoroughly investigated myeloid CLRs Mincle, Langerin, and DC-SIGN.
CLR architecture and carbohydrate binding
In distinction to other modes of carbohydrate binding, CLRs were historically defined as receptors that utilize Ca2+ cofactors to coordinate the hydroxyl groups of their natural ligands.29 In fact, many CLRs feature up to four Ca2+ binding sites. Besides their role in carbohydrate binding (Fig. 2A), some Ca2+ binding sites also regulate domain stability.30,31 However, certain CLR subgroups lack typical Ca2+-binding motifs. These receptors recognize carbohydrates by a different mechanism (e.g., β-glucan in the case of dectin-1) or have evolved to recognize different classes of ligands altogether (e.g., F-actin in the case of CLEC9A).32,33 Overall, the C-type lectin superfamily comprises more than 1000 members that are defined by their conserved C-type lectin-like domain (CTLD, Fig. 2A).30,31 CLRs have been categorized into 17 groups based on their domain organization and phylogenetic origin. CLRs expressed on myeloid cells generally belong to group 2 (asialoglycoprotein-like CLRs), group 5 (natural killer cell receptor-like CLRs), and group 6 (multi-CTLD endocytic receptors). Group 2 CLRs contain a single C-terminal carbohydrate recognition domain (CRD) at the end of a hydrophobic neck that separates it from the cell surface. These receptors predominantly exist as homooligomers,29,34–36 although heterooligomerization with other CLRs has also been described.31,37 A short transmembrane region connects the extracellular domain (ECD) to the N-terminal intracellular domain, which often contains signalling or internalization motifs. Notably, all CLRs discussed below belong to this group. Group 5 CLRs are characterized by a similar domain organization, but tend to form monomers or homo-/heterodimers.23,30,31,33 As described above, CLRs belonging to this group lack the canonical Ca2+-dependent carbohydrate binding site. The ECD of group 6 CLRs typically contains multiple CRD repeats. Importantly, not all of those CRDs contain residues necessary for the formation of Ca2+ binding sites. Those subdomains are incapable of binding carbohydrate ligands.38 | ||
| Fig. 2 A) Typical C-type lectin-like domain (CTLD) fold and close-up view of the carbohydrate binding site (DC-SIGN, PDB 2IT6). B) Domain organization and exemplary members of CLR groups expressed by myeloid cells. | ||
Carbohydrate binding sites and glycomimetic CLR ligands
In a typical CLR binding site, a carbohydrate ligand is bound via complexation of its hydroxyl groups by a Ca2+ cofactor. The primary carbohydrate binding site is enclosed by the so called long loop (Fig. 2A). This region is characterized by a high degree of flexibility and may be involved in an allosteric mechanism controlling Ca2+ affinity and, thus, ligand binding.39,40 Similarly, carbohydrate recognition can be tightly regulated by pH-sensors.40–42 In some cases, this mechanism mediates ligand release after endocytosis into acidic lysosomes.41 CLR binding sites are typically shallow and solvent exposed. Thus, polar carbohydrate ligands are in constant competition with water molecules, which can transiently engage in similar interactions as the hydroxyl groups of sugars. In addition, carbohydrate binding requires desolvation of those functionalities, which imposes further enthalpic costs on the free energy of binding.43 As a consequence, monosaccharide ligands are generally recognized with low binding affinities (KDs in millimolar concentrations) employing multiple transient binding modes. Oligosaccharide ligands achieve higher binding affinities through secondary interactions with nearby residues, beyond the canonical Ca2+ complexation. Nevertheless, intermediate affinities (KDs in sub millimolar concentration) are common. As a result, efficient pathogen recognition requires binding to a surface presenting large numbers of repeating units of low affinity carbohydrate ligands. Thus, high avidity is typically achieved by multivalent presentation. Apart from their low binding affinities, carbohydrate-derived molecules suffer from pharmacokinetic drawbacks related to their high polarity, quick metabolic turnover by glycosidases, and rapid renal clearance.44 Thus, glycomimetic ligands must be designed to overcome the inherent disadvantages of carbohydrates in drug design. The implementation of secondary hydrophobic interactions, in particular, has the potential to not only improve affinity, but also reduce polarity compared with the parent carbohydrate.Non-carbohydrate CLR ligands
Given the shallow and polar nature of their carbohydrate binding sites, it is not unreasonable to assume that CLRs constitute challenging targets for the development of drug-like small molecule ligands. In fact, computational tools for binding site prediction and druggability analysis, such as DoGSitescorer,45 categorize CLRs as “difficult” or even “undruggable”.46 However, when this prediction was probed in biophysical fragment screening campaigns, high hit rates were observed for multiple CLRs.46–48 It has since been shown that fragments often bind to secondary sites on the solvent exposed surface of CLRs, or that inherent flexibility in loop regions can open transient secondary binding pockets for fragment-sized ligands.48 These transient sites may exert allosteric control over ligand binding to the primary carbohydrate binding site.49 These insights open avenues for an alternative drug design strategy that avoids the use of potentially problematic carbohydrate substructures in prospective drugs.Case studies of medicinal chemistry efforts targeting CLRs
The following section summarizes the current state of research on the CLRs Mincle, Langerin, and DC-SIGN. As noted above, the focus of this review is on studies driven by medicinal chemistry principles, rather than multivalent strategies relying on the presentation of natural carbohydrates. In many cases, affinity values of lead compounds are given as half maximal inhibitory concentrations (IC50) or some other appropriate measure. It has to be noted that affinity values from different assays are not easily comparable and an evaluation of the relative potency of compounds characterized in different assays is difficult. This issue could be alleviated if more researchers implemented the use of unified controls, such as defined monosaccharide ligands.Monocyte-inducible C-type lectin (Mincle)
As its name suggests, Mincle is an inducible receptor on myeloid cells that is barely expressed in the resting state.50,51 Its expression can be induced upon activation of the cell, e.g., by stimulation of toll-like receptors (TLRs) or the constitutively expressed macrophage C-type lectin (MCL). The intracellular domain of Mincle associates with the Fc receptor gamma (FcγR) signalling chain, which contains an immunoreceptor tyrosine-based activation motif (ITAM). Stimulation of Mincle by an agonistic ligand ultimately results in the excretion of pro-inflammatory cytokines that induce proliferation of T helper cells towards a Th1 or Th17 phenotype. This renders Mincle as an attractive target for glycolipid-based vaccine adjuvants boosting the immunogenicity of protein antigens.Mincle has been identified as the main receptor for the potent mycobacterial cell wall antigen trehalose dimycolate (TDM, cord factor, 1) and its synthetic analogue trehalose dibehenate (TDB, 2, Fig. 3).52,53 Another Mincle ligand is the natural product brartemicin (3).54 Besides these trehalose-based molecules, other glycolipids containing mannose or glucose have been identified as Mincle activators.55–57 In addition, Mincle recognizes dead-cell associated self-antigens (SAP-130, β-glucosylceramide),58,59 as well as cholesterol crystals in atherosclerotic plaques and cholesterol sulfate as a damage-associated ligand in the skin.60,61 Established liposomal formulations of TDB (CAF01) have proven potent adjuvanticity in vaccines against C. trachomatis and M. tuberculosis.62,63
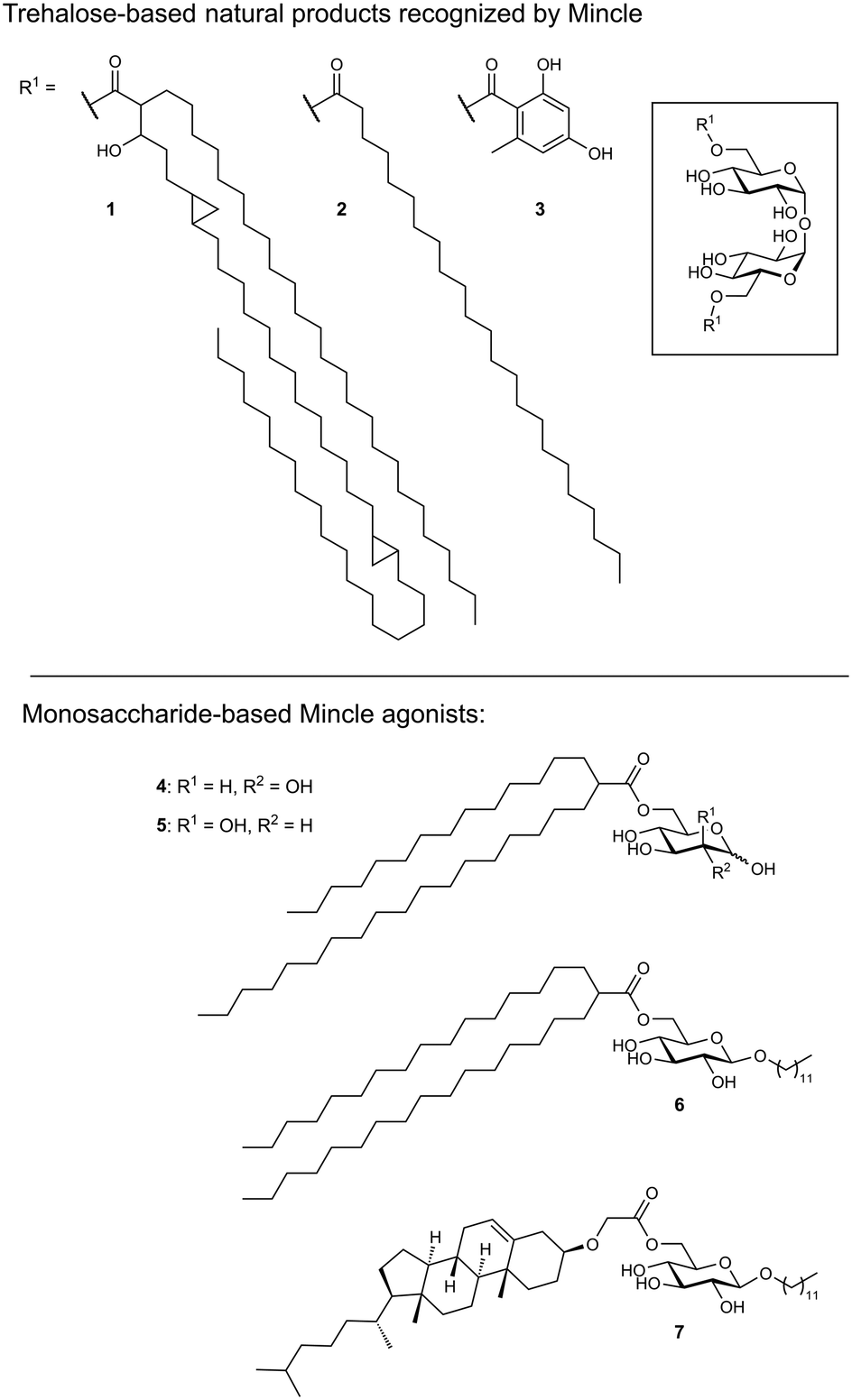 | ||
| Fig. 3 Structures of reported Mincle agonists based on fatty acid esters of trehalose, mannose or glucose. Structure of the mycolic acid side chains in the natural product TDM (1) may vary in length and composition. | ||
Several crystal structures of bovine and human Mincle are available that shed light into potential binding modes of glycolipid ligands (Fig. 4A).64–67 The Ca2+ cofactor in the primary carbohydrate binding site has been shown to coordinate the trans-diequatorial 3-OH and 4-OH of a glucose moiety in trehalose (Fig. 4). The 2-OH group of the second glucose is bound between Arg182 and Glu135 in a cooperative hydrogen bond network. Hydrophobic substituents in position 6 of the central glucose then extend towards a hydrophobic cleft on the protein surface that is formed by Leu172, Val173, Phe197, and Phe198.64 This region represents the main binding site for lipid substructures. However, secondary lipid binding modes have also been proposed.57,65
Ligand-based approaches for the design of pro-inflammatory Mincle agonists are commonly based on trehalose glycolipids as starting points. Early on, it was reported that the length of the lipid chain crucially determined efficacy, with C22 (TDB, 2) resulting in the most potent macrophage activation, whereas trehalose itself or its short-chain fatty esters showed no effect.68 Later, increasing evidence pointed to a striking dependence on the exact structure of the lipid chain.57,69,70 Whereas branched-chain gentiobiosyl diacylglycerides only evoked a weak response, truncated versions were found to be more active.69 Similarly, linear fatty acids of glucose did not activate Mincle reporter cell lines, whereas esters of mycolic acid derivatives gave a strong response.57 Based on these observations, branched fatty acid derivatives of glucose and mannose (4–5) were prepared, which efficiently activated Mincle reporter cell lines. These compounds also stimulated cytokine production in various primary cell lines and showed pro-inflammatory activity in mouse models, including a M. tuberculosis challenge experiment.57 Importantly, the hydroxyl group in position 3 of certain mycolic acids (corynemycolic acid, as in 1) did not contribute significantly to Mincle activation. From this detailed SAR study supported by molecular dynamics simulations, the authors derived a pharmacophore model for Mincle agonists that predicted receptor activation when at least three of four binding sites in the Mincle CRD are occupied by a ligand (Fig. 4B): 1.) the primary Ca2+-coordination site, 2.) a secondary carbohydrate binding site for trehalose derivatives formed by Glu135 and Arg182, 3.) the hydrophobic cleft that forms the primary lipid binding site, 4.) a secondary hydrophobic binding site on the CRD surface. Importantly, the hydrophobic sites can be either addressed by a single branched or two independent fatty acids.
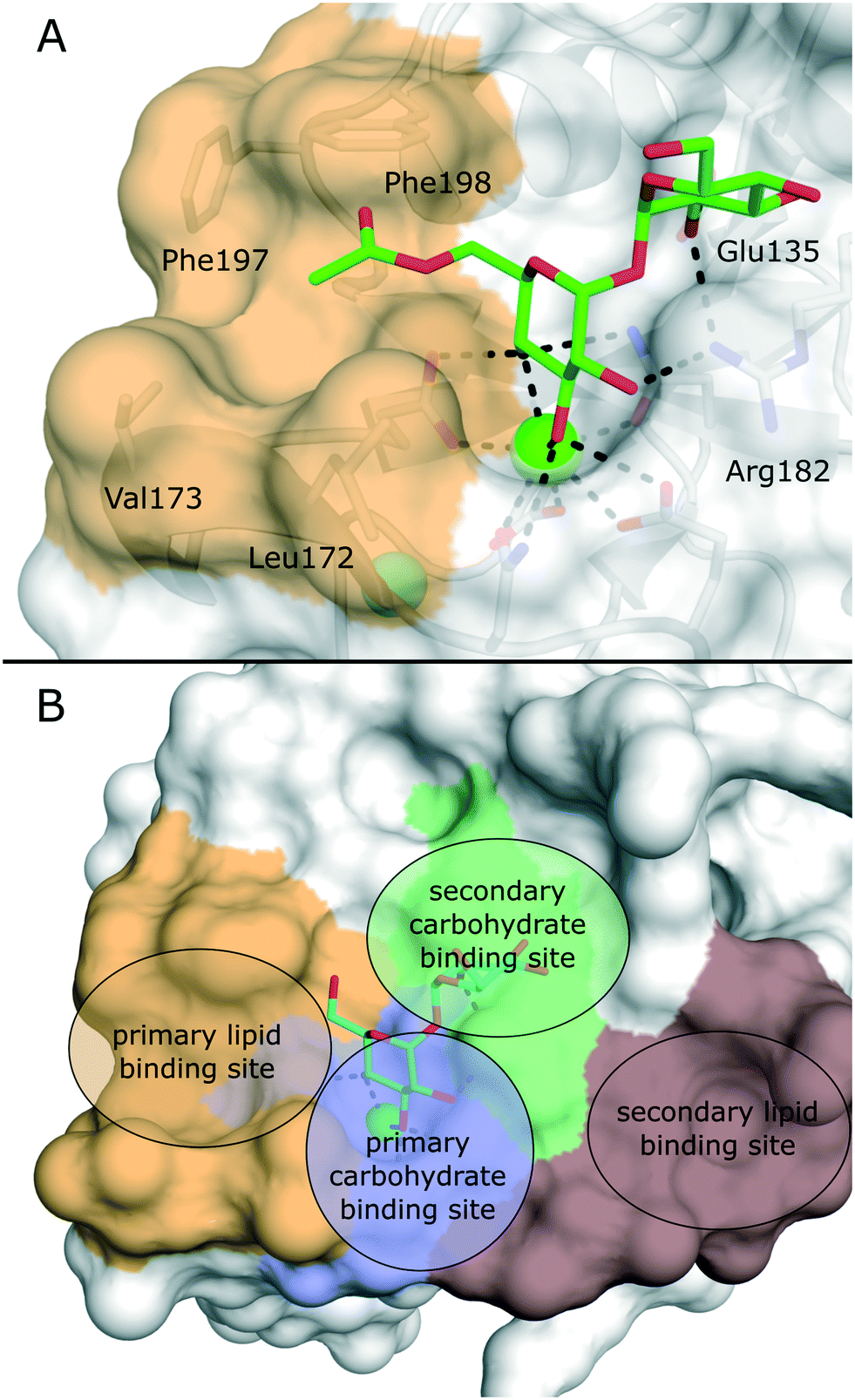 | ||
| Fig. 4 A) X-ray structure of bovine Mincle in complex with trehalose monobutyrate (PDB 4ZRV). The butyrate acyl chain addressing the hydrophobic cleft (orange surface) is not fully resolved. B) Pharmacophore model for Mincle agonists (PDB 4ZRW). Primary (blue) and secondary (green) carbohydrate binding sites are enclosed by the primary (light brown) and secondary (dark brown) lipid binding site. Whereas carbohydrate binding has been experimentally characterized by X-ray crystallography, prediction of the lipid binding sites mainly relied on computational studies.57 | ||
Interestingly, the dependence of a functional Mincle response on lipid structures seems to be partly species-specific. Whereas murine cells displayed a higher tolerance for variations in the structure of α-branched trehalose diesters, human cells showed strong preference for fatty acid chains of intermediate chain length.71 It was also demonstrated that Mincle signalling can be activated by structurally simple β-alkyl glycosides modified with an additional fatty acid.72 The most potent compounds from this series were octyl or lauryl β-glucosides featuring branched fatty acid esters or esters of a cholesterol derivative in position 6 (6–7). Considering the relative instability of ester linkages in vivo, Lynch et al. synthesized a series of trehalose diamide glycolipids.73 The authors hypothesized that an increased hydrolytic stability could result in an enhanced adjuvanticity of Mincle agonists. In cellular assay systems, matched amide and ester pairs showed no significant activity difference. However, when the compounds were applied in a mycobacterial vaccination challenge in mice and sheep, amide-based adjuvants elicited enhanced antigen-specific immune responses.
After identification of the natural product brartemicin (3) as a Mincle ligand,54 efforts for the rational optimization of Mincle agonists based on its structure were launched (Fig. 5). Initially, the natural product itself was reported to bind to bovine Mincle with a Ki of 5.5 μM.54 Later studies with murine and human isoforms, however, showed no binding.74 Whereas, these apparent discrepancies could potentially be attributable to species-specific polymorphisms in the binding sites, the authors hypothesized that also lack of comparability between the employed assays could be a factor. In cellular assays, however, brartemicin itself evoked no or a less effective Mincle-induced immune activation in peripheral blood mononuclear cells (PBMCs) or bone marrow-derived macrophages (BMDM).74,75 Synthetic brartemicin analogues containing aliphatic aryl ethers showed a much improved binding affinity in a direct ELISA-type binding assay.74 An intermediate chain length (C7, 8) was found to give a more potent interaction than longer variants (C18, 9). Surprisingly, the long-chain derivatives were found to be much more active in a functional NFAT-GFP reporter cell assay, revealing a stark disconnect between Mincle binding affinity and functional activation of pro-inflammatory signalling. In an extensive optimization campaign, brartemicin analogues with differently substituted aromatic moieties were synthesized.75 Introduction of short alkyl- or alkoxy residues in positions 3 and 5 of the benzoate (10–13) lead to increased activity in human PBMCs. Upon extension of the alkyl substitution pattern to a tert-butyl group (14), an agonist with remarkable activity was obtained. In concentrations below 1 μM, 14 potently induced expression of cytokines indicative of a Th17 response in PBMCs (IL-1β, IL-6, IL-23, TNF-α). Further SAR studies revealed that the tert-butyl group in position 3 was critical for activity, whereas the hydroxyl group in position 2 was not. In an in vivo vaccine challenge with the M. tuberculosis antigen M72 in mice, a liposomal formulation of 14 produced a strong IgG2a/humoral Th1-biased immune response.75 A follow-up experiment revealed a pronounced Th17-polarized T cell response, indicating efficient protection against M. tuberculosis infection after immunization. To improve the physicochemical properties of prospective Mincle ligands and extend secondary interactions in the vicinity of the trehalose binding site, Rasheed et al. investigated the brartemicin derivatives containing an amide or sulfonamide linkage.76 As observed for other brartemicin analogues, the introduction of bulky groups in benzamide substituents was required for induction of cytokine production in human PBMCs. Intriguingly, related compounds featuring a sulfonamide linkage were only moderately active in cellular assays.
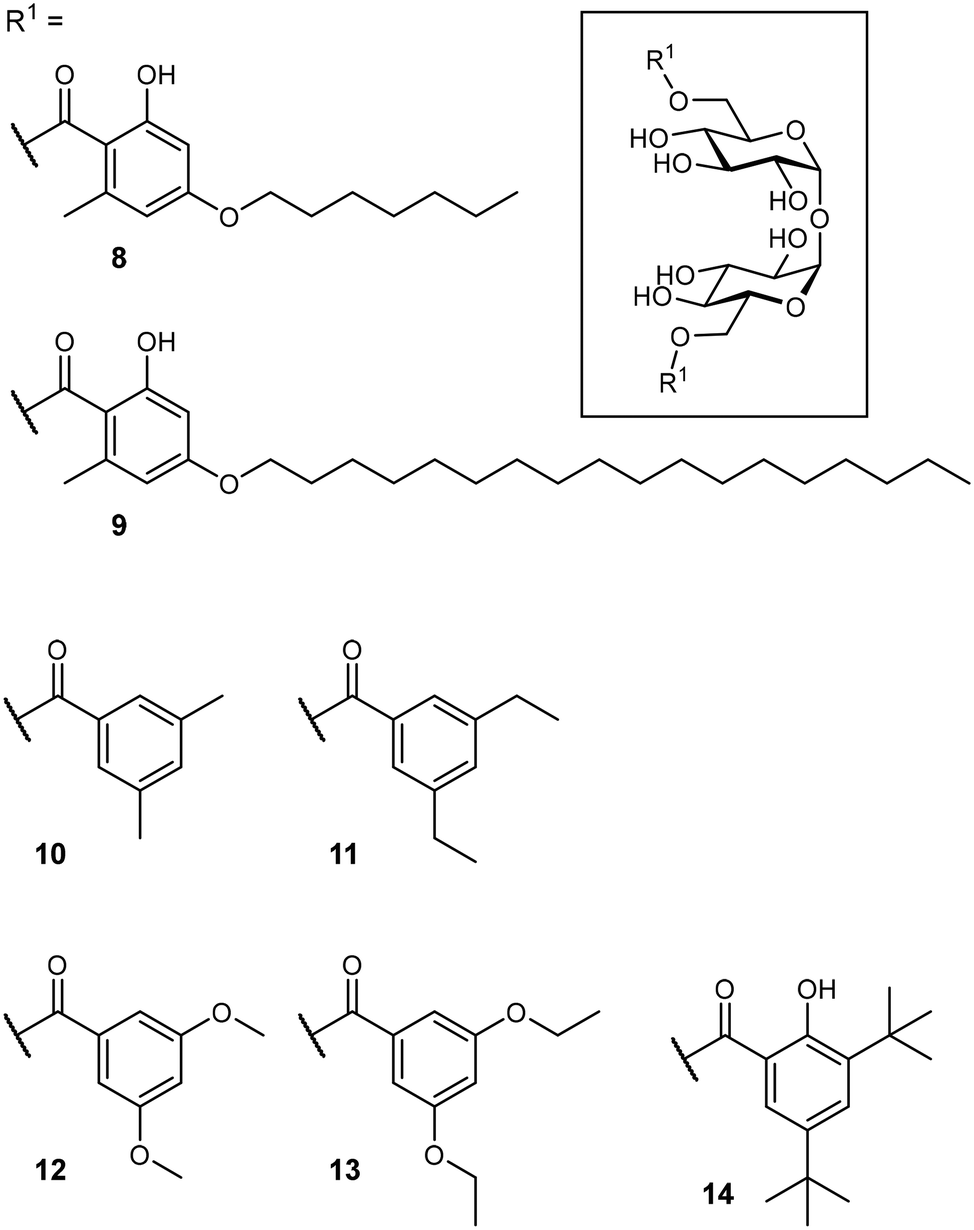 | ||
| Fig. 5 Mincle agonists based on the natural product brartemicin (3). | ||
It must be mentioned that most of the compounds developed as Mincle ligands are very hydrophobic. In a typical Mincle activation assay, compound stock solutions in volatile organic solvents are used to coat microtiter plates, which are in turn charged with cell culture medium. Alternatively, ligand stocks in DMSO are directly diluted in cell culture medium, which leads to compound precipitation.75 Fully soluble ligands, such as brartemicin itself or its short-chain analogues, are an exception. Thus, the general picture that emerges from the extensive SAR studies summarized here shows that glycolipids can only activate Mincle signalling in the solid state or when embedded into a cell membrane or liposomal formulation. Soluble ligands, however, do not seem to be able to activate receptor signalling, despite formidable binding affinity. It is yet unclear how information about ligand recognition is propagated across the cell membrane and which structural mechanisms contribute to receptor activation on a molecular level. Some authors even describe a dependence of the assay result on the solvent used for plate coating.76 Thus, Mincle activation might be dependent on the particular nanoscale properties of the precipitates generated in assay preparation. This hypothesis adds a new layer of complexity to the research into Mincle-targeted adjuvants. Further studies are required to gain a better understanding of the structural prerequisites, as well as supramolecular effects on Mincle activation.
Langerin
Langerin is exclusively expressed by a subset of skin-resident DCs called Langerhans cells (LCs).77 This surface bound CLR recognizes pathogens via their surface carbohydrates and mediates internalization, antigen processing and presentation. Because of its restricted expression pattern on powerful antigen-presenting cells, targeted antigen delivery to Langerin-expressing antigen-presenting cells has been identified as a promising strategy to boost vaccination responses.12 In particular, the slow release of antigens from endosomal compartments after Langerin-mediated antigen internalization promotes efficient cross-presentation and, in turn, stimulation of CD8+ T cells.12,77,78 In addition, specific LC targeting could also prove to be relevant for the treatment of Langerhans cell histiocytosis, a paediatric form of cancer characterized by the abnormal proliferation of Langerin+ myeloid progenitor cells.The extracellular domain of Langerin consists of a canonical CRD and a neck region promoting the formation of receptor trimers stabilized by a coiled-coil of α-helices.36,79 In the primary carbohydrate binding site, Langerin binds cell surface glycans with a specificity for mannose, fucose, N-acetyl mannosamine, and N-acetyl glucosamine epitopes, as well as heparin-derived, sulfated carbohydrates in a Ca2+-dependent manner.79–81 Langerin features two positively charged lysine residues in close proximity to the primary carbohydrate binding site, which provide a site of interaction for sulfated sugars (Fig. 6).82–84
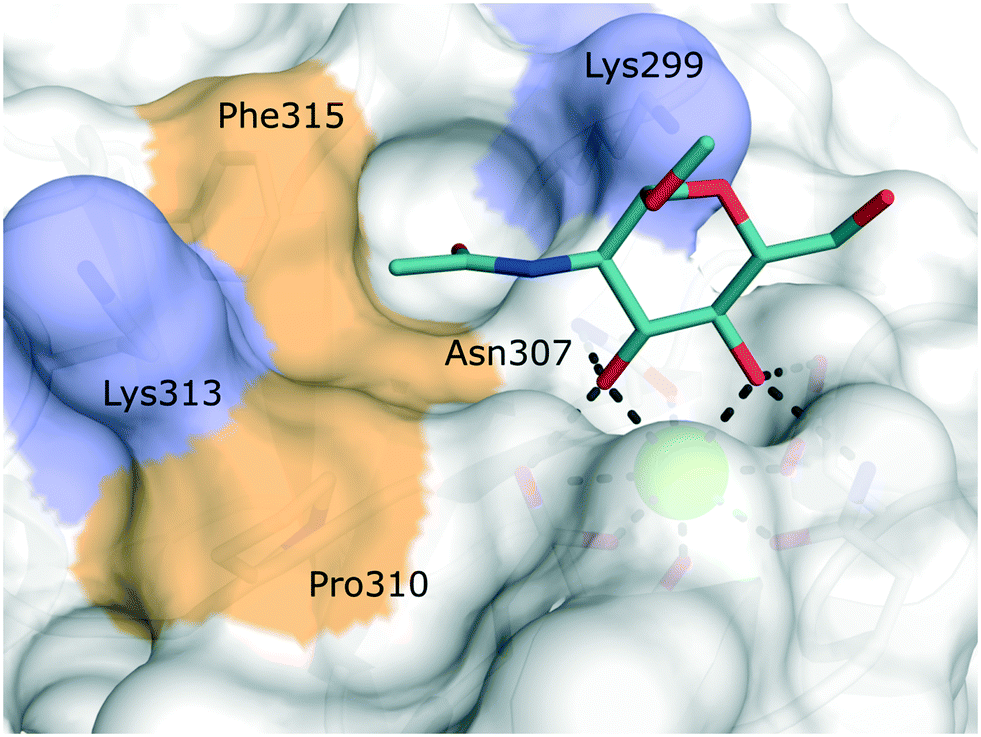 | ||
| Fig. 6 Carbohydrate binding site of human Langerin in complex with N-acetyl glucosamine (PDB 4N32). Positively charged lysine residues involved in the recognition of sulfated carbohydrates are coloured blue. Secondary hydrophobic patches implicated to accommodate aromatic substituents of glycomimetics are highlighted in light brown. | ||
Targeting approaches employing natural carbohydrate epitopes would undoubtedly face insurmountable issues with selectivity, as other myeloid cell subsets express many different receptors with similar specificities on a monosaccharide level. Thus, the identification of selective glycomimetic ligands tailored towards Langerin recognition is a significant milestone in the development of Langerin-targeted vaccines. Supported by a preliminary docking study, a series of compounds based on propargyl N-acetyl mannosaminide (15) were prepared and analysed for their Langerin affinity in a 19F R2-filtered NMR assay (Fig. 7).85 The most potent compounds from this series featured negatively charged substituents with moderate affinities (16: Ki = 7.6 mM, 17: Ki = 1.3 mM), which still represents an up to 36-fold affinity increase over the parent compound 15 (Ki = 48 mM). Based on data from an 1H–15N HSQC NMR experiment, the increased potency of 17 was attributed to an electrostatic interaction between its sulfonate group and the residues Lys299 and Asn307 (Fig. 6). Using a similar approach, Wamhoff et al. later developed glycomimetic Langerin ligands based on the heparin-derived ligand glucosamine-2-sulfate (18).86 Leveraging sulfonamide substituents as an isosteric replacement of the sulfate group in position 2 of the glucosamine core, the authors reported a 13-fold increase in binding affinity for the N-tosylated glucosamine derivative 19 compared to the reference N-acetyl glucosamine, with an NMR-derived Ki value of 0.32 mM. In the complex of 19 with Langerin, the electrostatic interactions between the negatively charged sulfate group of parent compound 18 and nearby lysine residues were presumably replaced by cation–π interactions with the same positively charged sites, as well as additional hydrophobic contacts to the apolar side chains of Phe315 and Pro310 (Fig. 6). Glycolipids presenting 19 on a flexible ethylene glycol linker were then incorporated into functionalized liposomes.86 Flow cytometric analysis showed specific interaction of the functionalized liposomes with Langerin-expressing cells, whereas no interaction was observed with cells expressing the related CLRs dectin-1 or DC-SIGN. Furthermore, when primary epidermal cell suspensions from skin biopsies were treated with the functionalized liposomes, targeting of LCs proved to be equally effective, as no interaction with keratinocytes or T cells was observed. The experiment was extended towards whole skin cell suspensions, in which only 3% of off-target myeloid cells interacted with functionalized liposomes. Importantly, liposomes containing doxorubicin as a cytotoxic payload were only lethal to Langerin+ cells, whereas no cytotoxicity was observed for Langerin− controls.
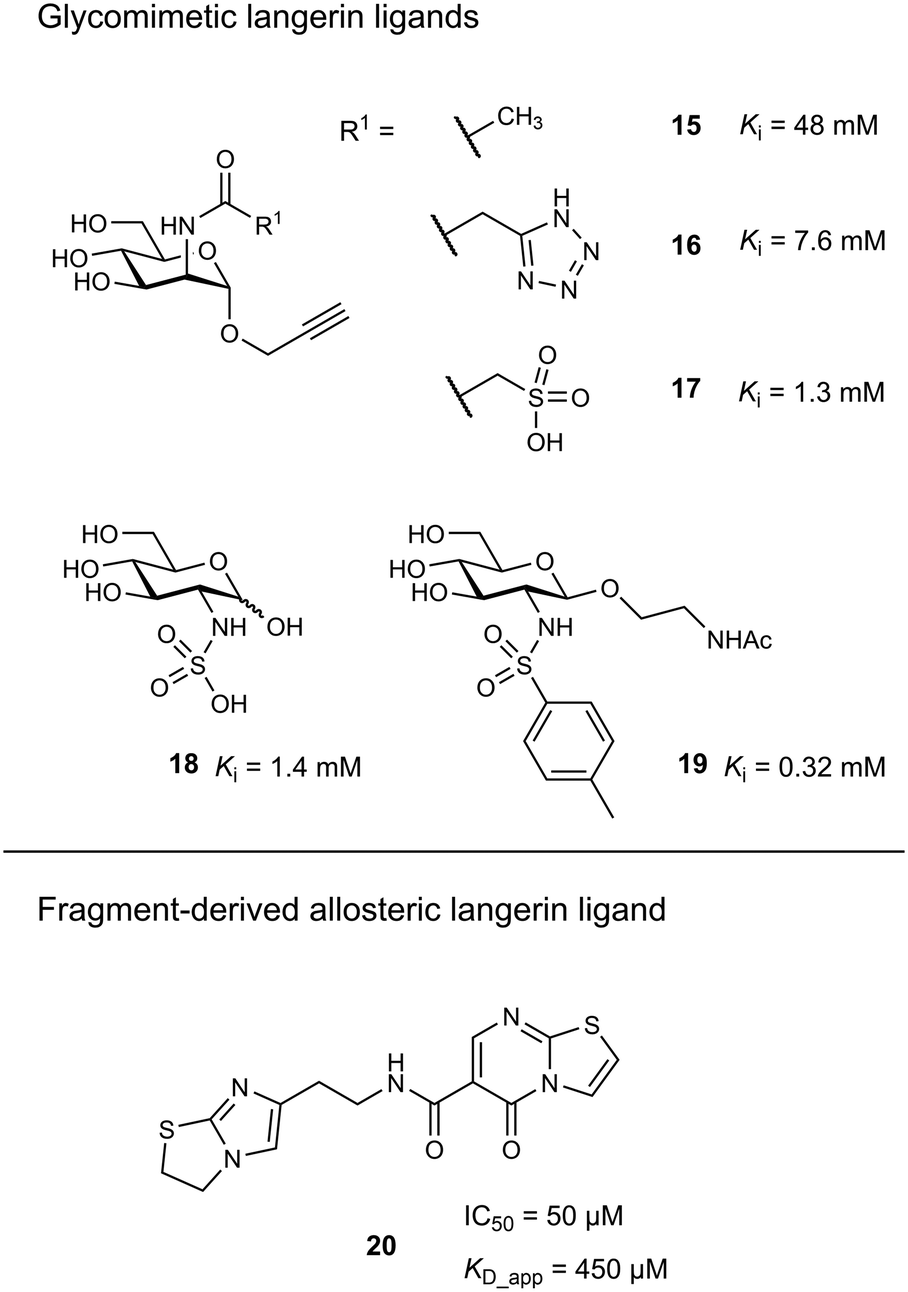 | ||
| Fig. 7 Exemplary structures of glycomimetic and fragment-derived Langerin ligands. | ||
As an alternative strategy to targeting CLRs with glycomimetic ligands, fragment-based approaches have been proven viable. Early reports revealed a high hit-rate of 15.7% from an NMR-based screening against Langerin.46 Following up on a pilot study with a smaller library,85 Aretz et al. evaluated 871 fragments for their ability to bind murine Langerin ECD by NMR.49 This effort revealed 78 hits (9% hit-rate). Hit validation by SPR confirmed 53 hits, however, only three compounds were found to compete with the carbohydrate-based ligand mannan in an enzyme-linked lectin assay (ELLA). Derivatives of the three respective fragment scaffolds were evaluated by STD-NMR and a fragment microarray assay to gain a first insight into SAR. Compounds based on a thiazolopyrimidine-5-one core were found to be most promising and highly selective for the targeted murine ortholog of Langerin. An extensive effort of fragment elaboration and rescaffolding revealed activity cliffs for certain substitution patterns in the functional mannan ELLA that were absent in the biophysical NMR binding assay, indicating that only a subset of physically interacting ligands was able to compete with carbohydrate binding. This was attributed to a switch of binding modes that induced shifts in the protein conformation towards an active or inactive state, respectively. Ultimately, the most potent thiazolopyrimidine derivative 20 inhibited mannan binding with an IC50 of 50 μM (KD_app_NMR = 450 μM). The apparent allosteric mechanism that controlled carbohydrate binding was further investigated by 1H–15N HSQC NMR experiments revealing that the putative allosteric ligand 20 and primary site ligand mannose perturbed different resonances. Further NMR titrations then showed that the allosteric ligand solely affected the affinity of carbohydrate ligands, whereas no effect on the capacity of the protein to bind the Ca2+ cofactor was observed. The presence of a carbohydrate ligand similarly reduced the affinity of 20, confirming a bidirectional allosteric “switch” mechanism between the fragment binding pocket and the primary carbohydrate binding site. Whereas the applicability of allosteric CLR ligands for therapeutic applications is still to be proven, the allosteric inhibition of carbohydrate binding by fragment-derived small molecules in Langerin is an important proof of concept. Because carbohydrate-derived molecules are likely to struggle with pharmacokinetic issues related to their comparably high polarity and metabolic instability, an allosteric modulation of CLR function with drug-like ligands holds great potential. Moreover, it was recently demonstrated that catechol-containing fragments can even directly compete with carbohydrate ligands in the primary binding site.87
Dendritic cell-specific intercellular adhesion molecule 3 grabbing non-integrin (DC-SIGN)
DC-SIGN is mainly expressed by immature dermal DCs, interstitial DCs in lungs, intestine, rectum, cervix, and placenta, as well as mature DCs in lymphoid tissues.9 Disregarding the implications of its name, however, the receptor is also expressed by other myeloid cells.88 DC-SIGN is involved in the capture of many different pathogens of viral, bacterial, fungal, and parasitic origin.89 The main function of the receptor is the internalization of captured pathogens into lysosomal compartments, which is usually followed by antigen processing and presentation. DC-SIGN gained a lot of attention as a potential drug target when its detrimental role in the pathology of HIV infections was reported.16 Studies showed that HIV particles were able to subvert intracellular routing into lysosomes, but were instead accommodated in subcellular compartments near the cell surface (“invaginated pockets”), where they could persist undetected by the host immune system during DC migration into lymph nodes.90 In a process called trans-infection, HIV-susceptible CD4+ T cells were, in turn, presented with infective virus particles. Similar cis- and trans-infection mechanisms involving DC-SIGN, or its close analogue DC-SIGNR, have since been demonstrated for many different pathogens, many of which of viral origin.2,3,89 In the context of the recent SARS-CoV-2 pandemic, preliminary reports from proteomic studies of COVID-19 patients drew a connection between DC-SIGN expression levels and disease severity.91,92 Overall, the link between DC-SIGN-mediated virus recognition and a detrimental outcome for the host predisposes this receptor as a target for a broad-spectrum, host-directed, antivirulent treatment strategy.13,93–95 Given the fact that both the receptor, as well as glycan structures decorating viral envelope glycoproteins are ultimately encoded in the host genome, targeting this system represents a promising strategy for the early response to newly emerging viruses with pandemic potential and problematic variants of existing pathogens. However, whereas these processes are well-documented in cellular in vitro models, the study of trans-infection phenomena and other DC-SIGN-related pathologies in animal models remains a challenge. In mice, eight homologous genes (SIGNR1–8) clustered in the same genomic region encode for receptors varying in ligand specificity, function, and domain organization.96–98 Thus, studies of DC-SIGN function in mouse models require the use of transgenic animals.99,100 The lack of meaningful and easily accessible in vivo models complicates the translation of DC-SIGN-targeted pharmacological interventions into the clinic.The canonical CRD of DC-SIGN is anchored to the cell membrane via a hydrophobic neck of variable length.88 The neck domain typically consists of five to seven consensus repeats that have been shown to mediate receptor tetramerization.35 However, soluble isoforms have also been described.101 Upon acidification, residues in the neck domain change their protonation state, which induces the separation of receptor tetramers.35 This pH-sensitive switch mechanism ultimately reduces the affinity towards multivalent ligands in lysosomal compartments and enables ligand release and recycling of the free receptor to the cell membrane. DC-SIGN is characterized by a broad specificity for glycans containing mannose and fucose epitopes. This includes mannose-based carbohydrate structures that are often found on pathogen cell surfaces (e.g., high-mannose glycans, mannan, mannosylated lipoarabinomannan), as well as fucose-containing “self”-glycans (e.g., blood group antigens, Lewis antigens). The latter category emphasizes the role of DC-SIGN in self-adhesion processes, such as the formation of the immunological synapse between DCs and T cells.9 The molecular details of carbohydrate binding to DC-SIGN have been particularly well studied. Canonically, the receptor binds fucose ligands via complexation of the cis-configured 3-OH and 4-OH and mannose ligands via complexation of the trans-configured 3-OH and 4-OH, resulting in a distinct binding mode for fucose- and mannose-based glycans.102 For small mono- or disaccharide mannose ligands, carbohydrate coordination can be achieved in two distinct orientations, which are related by a rotation of the ligand molecule.103 Based on the observed occupancy in the X-ray structure of the DC-SIGN/Man-α-1,2-Man-complex, the resulting binding modes have been dubbed “major” (reducing end oriented away from the binding site, Fig. 8A) and “minor” (reducing end oriented towards the binding site, Fig. 8B).103 However, binding modes employing other hydroxyl groups are also known. By employing fluorinated mannose derivatives and an NMR method that was able to report on low affinities and weakly populated states, Martínez et al. were able to uncover alternative binding modes employing the trans-configured 2-OH and 3-OH of mannose, thereby mimicking the fucose binding mode, albeit with much reduced affinity.104 This binding mode promiscuity has to be taken into account in the rational design of glycomimetic ligands, because sudden switches of the preferred mannose orientation may induce unexpected affinity cliffs in SAR studies. Many ligand optimization efforts have targeted partly hydrophobic subsites adjacent to the primary carbohydrate binding site. Notably, Phe313 and a shallow hydrophobic cleft in its vicinity have proven capable of binding hydrophobic ligands (Fig. 8C).48,105 In addition, a loop region towards the back of the binding site, the so called long loop (Fig. 2A), can accommodate secondary interactions. In particular, Val351, which is important for the recognition of fucose and fucose-based glycans,102 represents a possible target for glycomimetic compounds (Fig. 8C).106–108
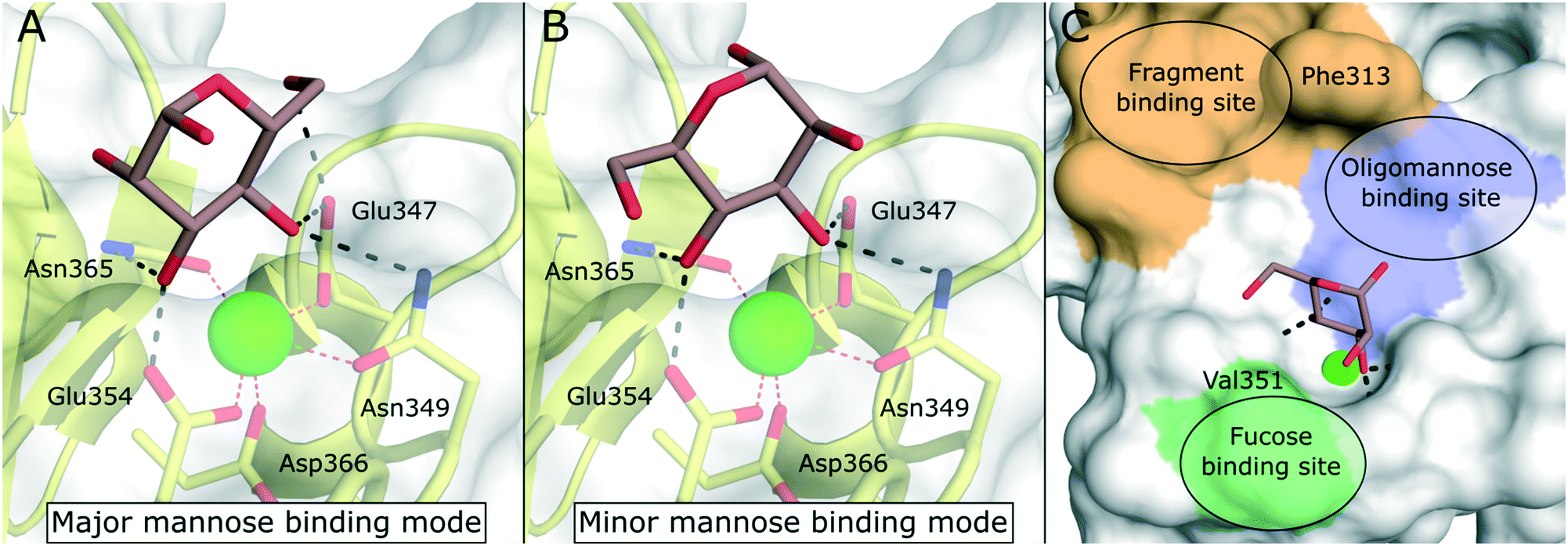 | ||
| Fig. 8 Carbohydrate recognition in DC-SIGN. A) Mannose binding in the major binding mode (PDB 2IT6, occupancy >50%). B) Mannose binding in the minor binding mode (PDB 2IT6, occupancy <50%). C) General pharmacophore model for glycomimetic DC-SIGN ligands. Secondary binding sites for fucose-based glycans (green), oligomannose-type glycans (blue), and shallow pocket implicated in binding of small molecule fragments (light brown) are highlighted. | ||
Mannose-based glycomimetics as ligands for DC-SIGN
To improve the binding affinity of the minimal binding epitope mannose, early glycomimetic approaches investigated C-2 branched mannose derivatives as DC-SIGN ligands (Fig. 9). These studies resulted in the discovery of 2-C-aminomethyl-D-mannose (21) that showed a sub millimolar activity (IC50 = 0.35 mM) in an SPR competition assay.109 The consideration that the hydroxyl group configuration of mannose is mimicked by the natural product shikimic acid, prompted the investigation of a series of shikimic acid derivatives in a solid-phase fluorescence assay.110 Although only moderately active (IC50 = 3.2 mM), the most potent compound from this study (22) proved amenable to incorporation into a multivalent polymeric scaffold. Multivalent presentation of the weakly binding epitope gave an approximately 1000-fold increase in affinity for the functionalized polymer (IC50 = 2.9 μM). By targeting the hydrophobic groove adjacent to Phe313, mannose derivatives bearing large hydrophobic aglycones have been designed.105 The most potent analogue 23 bearing two bulky naphthyl groups showed an IC50 value of 40 μM in a competitive solid-phase immunoassay. Docking studies initially supported the expected binding mode hypothesis, in which the mannose is coordinated in the minor binding mode and the aromatic aglycone is accommodated in two partly hydrophobic clefts on both sides of Phe313. However, molecular dynamics simulations starting from the proposed binding mode revealed residual flexibility in the hydrophobic moieties and hinted at various other potential binding modes. The Bernardi group pioneered a class of pseudo-dimannoside mimetics employing a carbasugar analogue.111 In comparison to Man-α-1,2-Man, pseudo-dimannoside mimetic 24 proved to be more resistant towards enzymatic digestion and was found to be three times more active in inhibiting the interaction of Ebola envelope glycoprotein-pseudotyped viruses to DC-SIGN+ cells (IC50 = 0.62 mM).112 Moreover, structural analysis by X-ray crystallography and NMR revealed that, in contrast to the natural disaccharide, pseudo-dimannoside 24 engaged the protein in a single binding mode.106 The binding mode of 24 was characterized by a hydrophobic contact of its cyclohexane moiety with Val351, which is accommodated in the long loop toward the back of the binding site (Fig. 10B). This interaction firmly locked the ligand into the major binding mode. Based on this finding, further structure-based optimization studies were initiated. With the goal of extending the Val351 interaction, two benzamide moieties were introduced into the cyclohexane core of the molecule.107 This yielded a series of compounds with improved potency up to IC50 = 0.3 mM for pseudo-dimannoside 25 in an SPR competition assay. Importantly, the authors were able to show a marked increase in the selectivity against Langerin binding, which is a supposed anti-target for the therapeutic application of DC-SIGN ligands in HIV infection. A further effort to improve selectivity capitalized on the fact that Langerin features a specific binding site for sulfated sugars, which is not present in DC-SIGN.84 In particular, Langerin recognizes 6-O-sulfated hexoses via interaction with charged lysine residues in a secondary binding site (Fig. 6). The introduction of a positively charged amino group in position 6 of pseudo-dimannoside 25 was expected to abrogate any off-target binding to Langerin. In fact, the validity of this approach could be demonstrated in an SPR assay, in which 26 proved to be fully selective for DC-SIGN over Langerin. Compared with pseudo-dimannoside 25, the positively charged analogue 26 even showed a slight improvement in affinity (SPRcompetitive: IC50 = 254 μM; SPRdirect: KD = 310 μM; ITC: KD = 171 μM). Further improvement of this class of DC-SIGN ligands was driven by a virtual fragment screening that aimed to identify viable secondary interactions in the vicinity of the pseudo-dimannoside binding site.108 These efforts revealed an ammonium binding pocket between Glu358 and Ser360. In the respective docking poses, an ammonium group of positively charged fragments assumed a position, which is often occupied by a conserved water molecule or a hydroxyl group of an oligomannoside ligand in other crystal structures.103,106 Besides engaging in charge-assisted hydrogen bond interactions to Glu358 and Ser360, aromatic fragments bound in this region were able to capitalize on π–π or CH–π interactions to Phe313. This observation prompted the authors to extend the mannose moiety in pseudo-dimannoside 24 with aminomethyl triazolyl substituents in position 2. The resulting compound series generally showed a markedly improved affinity over 24 in an SPR competition assay. A breakthrough was achieved upon combination of the benzamide substitution found in 25 with the additional engagement of the ammonium binding site. As a result, pseudo-dimannoside 27 showed a high affinity towards DC-SIGN (SPRcompetitive: IC50 = 76 μM; SPRdirect: KD = 53 μM; ITC: KD = 52 μM) and full selectivity against Langerin. The discovery of 27 exemplifies the additivity of secondary interactions observed for mannose- and fucose-type glycans. Whereas hydroxyl groups of Man-α-1,2-Man-based glycans assume the conserved water position between Glu358 and Ser360,103,106 fucose-based glycans commonly interact with Val351 (Fig. 10C).102,113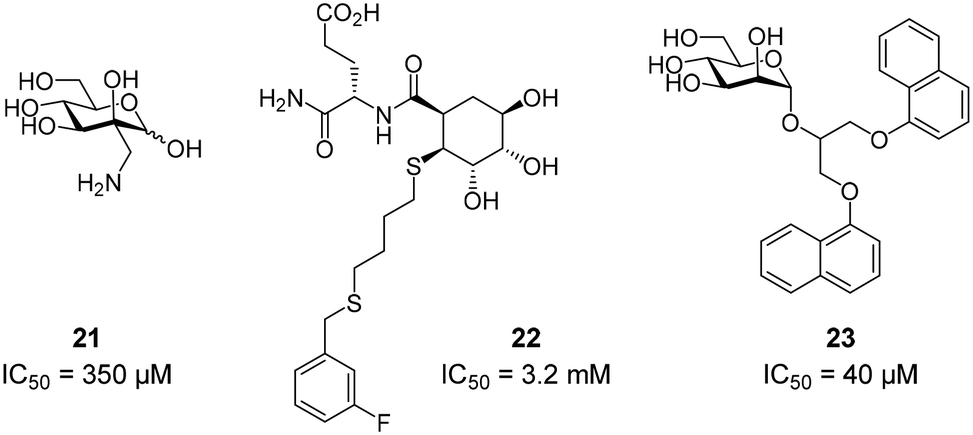 | ||
| Fig. 9 Exemplary structures of glycomimetic DC-SIGN ligands. | ||
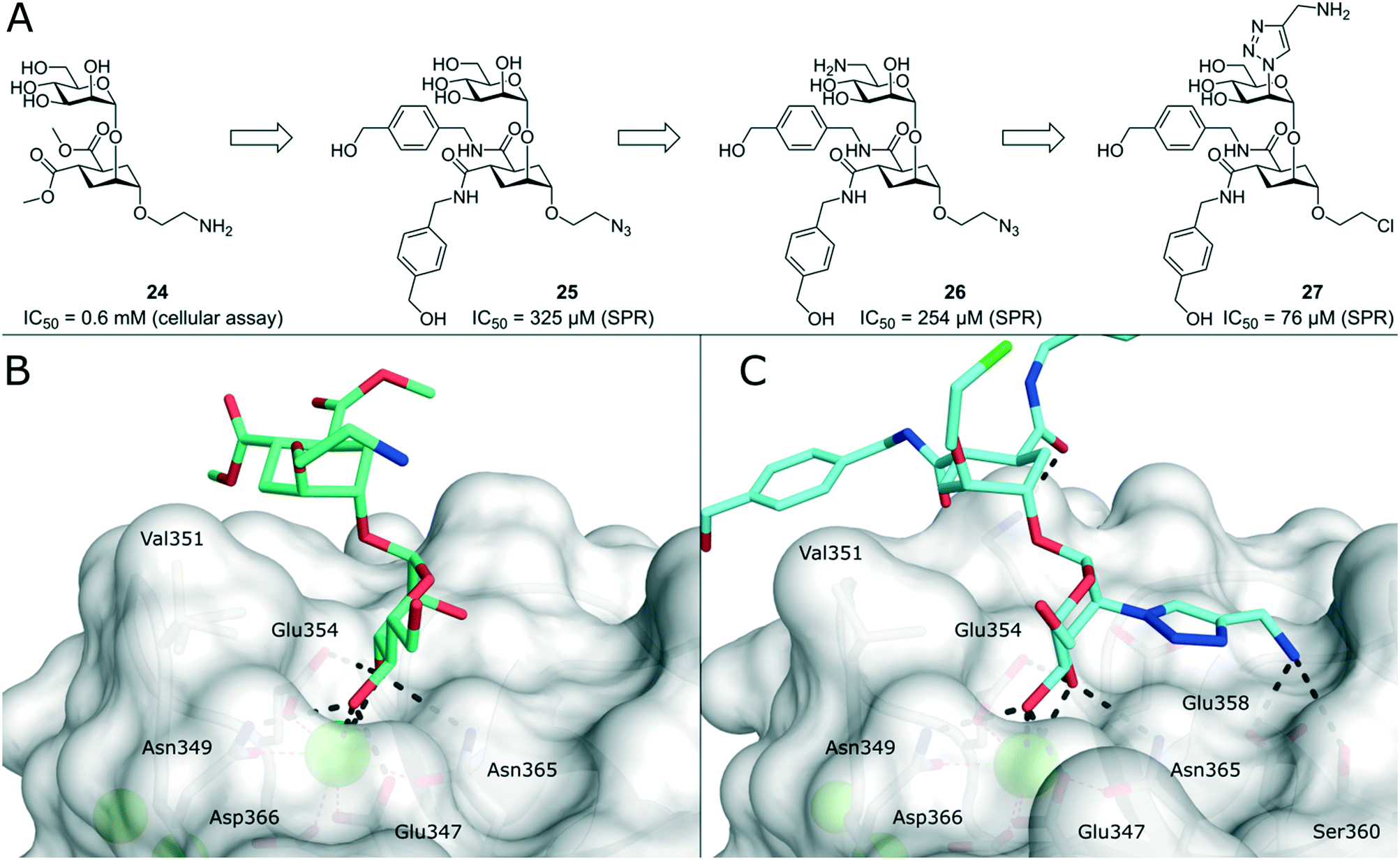 | ||
| Fig. 10 Evolution of pseudo-dimannoside ligands from Man-α-1,2-Man analogue 24 to potent and highly selective compound 27. A) Chemical structures of pseudo-dimannoside DC-SIGN ligands. B) X-ray crystal structure of 24 in complex with DC-SIGN CRD (PDB 2XR5). In addition to canonical mannose recognition in the major binding mode, the cyclohexane ring engages in hydrophobic interactions with Val351. C) X-ray crystal structure of 27 in complex with DC-SIGN CRD (PDB 6GHV). The increased affinity of optimized pseudo-dimannoside 27 is driven by extended hydrophobic contacts to Val351 and charge-assisted hydrogen bonds to Glu358 and Ser360. | ||
Several efforts for the multivalent presentation of pseudo-dimannoside glycomimetics were initiated. Unlike multivalent compounds employing natural carbohydrates, this strategy represents a direct extension of medicinal chemistry design principles to multivalency. Whereas higher valency dendrimers presenting 24 or 25 struggled with low solubility, less strongly modified analogues proved to be efficient DC-SIGN ligands with high potency in biophysical assays. In cellular models of Ebola virus infection, the dendrimers inhibited DC-SIGN-mediated cis- and trans-infection with IC50 values below 60 nM.114,115 An oligovalent display of compounds 24 or 25 on rigid molecular rods resulted in even more efficient ligands that inhibited trans-infection of CD4+ T cells in an HIV infection model with an IC50 as low as 24 nM.116 Moreover, the most potent compound Polyman26 (28, Fig. 11) demonstrated immunostimulatory properties in DCs.117 Just recently, the activity of 28 against SARS-CoV-2 has been investigated.118 In a competitive SPR setup, 28 inhibited the interaction between DC-SIGN and SARS-CoV-2 spike glycoprotein with an IC50 of 9.6 μM. In addition, the compound was active in a trans-infection assay.
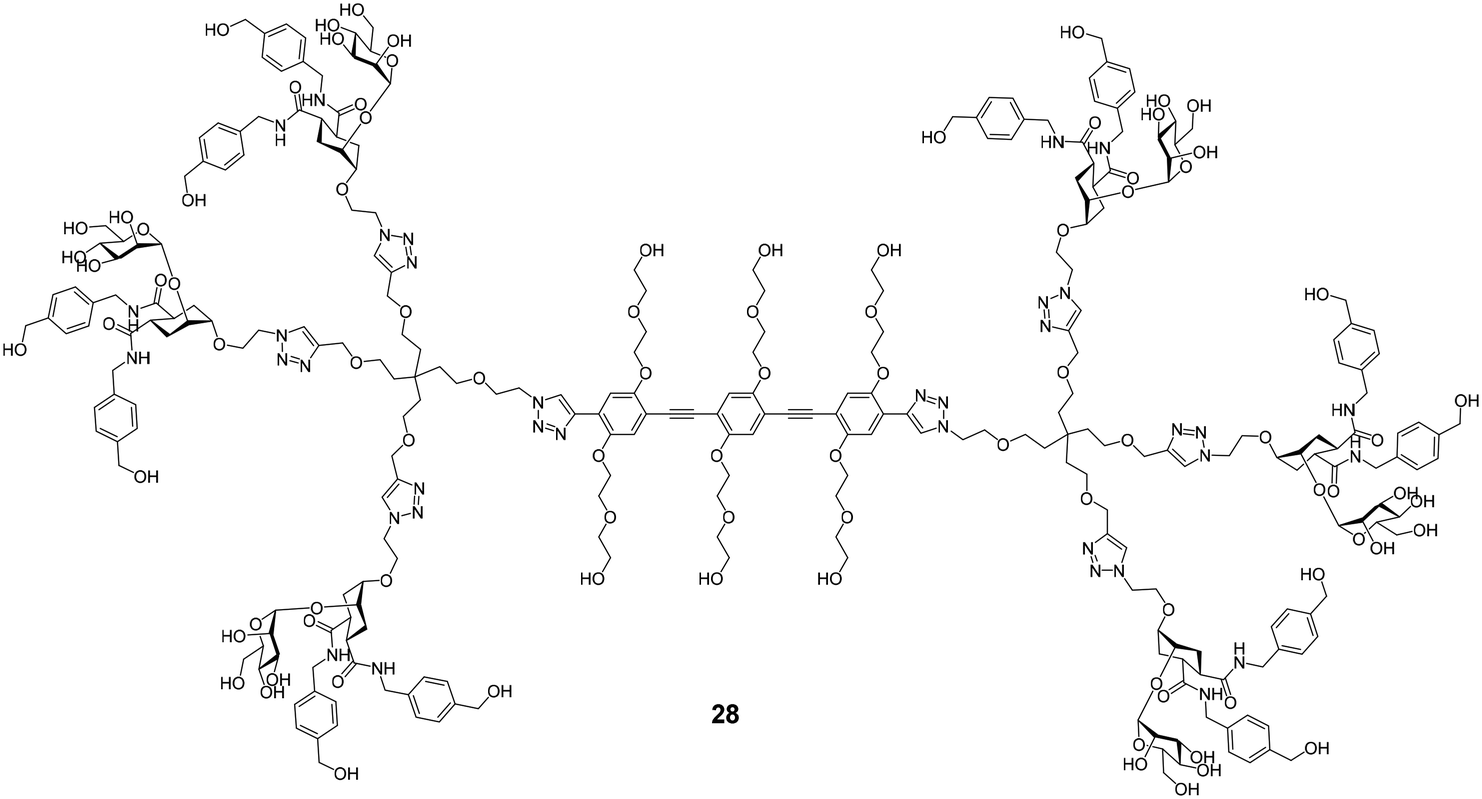 | ||
| Fig. 11 Structure of hexavalent molecular rod Polyman26 (28). | ||
Fucose-based glycomimetics as ligands for DC-SIGN
Efforts for the design of fucose-based glycomimetics have employed the natural glycan ligand Lewisx (29) as a design template (Fig. 12). By substituting the GlcNAc and/or galactose sugars in Lewisx with substituted (1S,2R)-2-aminocyclohexane-1-carboxylic acid, a glycomimetic based on a fucosylamide core (30) was obtained.119 These molecules bound DC-SIGN with an up to two-fold higher affinity compared with Lewisx. To improve synthetic accessibility and pharmacokinetic properties, further fucosylamide glycomimetics featuring aromatic amide substituents on the cyclohexane core were synthesized.120 However, these compounds and analogues, in which the rigid cyclohexane scaffold was replaced by a flexible β-alanine linker, showed reduced affinity, compared with pseudo-trisaccharide 30. Interestingly, variation of the cyclohexane stereochemistry yielded improved binders, with diastereomers based on the enantiomeric (1R,2S)-2-aminocyclohexane-1-carboxylic acid scaffold showing the highest affinity (31). Overall, optimization of fucose-based ligands was met with limited success and mostly abandoned in favour of more promising mannose-based glycomimetics, which offer more favourable exit vectors to engage in secondary interactions.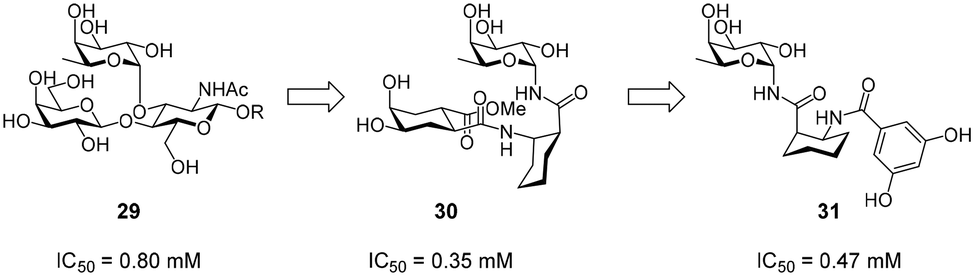 | ||
| Fig. 12 Development of fucose-based glycomimetics from Lewisx (29). | ||
Non-carbohydrate ligands of DC-SIGN
Due to their high polarity and unfavourable pharmacokinetic properties, carbohydrate substructures can be problematic in prospective drug molecules.44,121 In an effort to identify DC-SIGN ligands that were not based on a carbohydrate structure, commercially available compound libraries were screened for activity against DC-SIGN in a high-throughput ELLA-type assay.122 With an overall hit-rate of 0.6%, this effort provided seven hits that mainly belonged to two structural classes (Fig. 13). Compounds based on a quinoxazoline or pyrazolone scaffold (32–33) were able to inhibit the interaction of a fluorescently labelled, mannosylated BSA probe to a DC-SIGN-coated plate with IC50 values between 1.6 μM and 32 μM. In a cellular adhesion assay, 32 and 33 showed activity, albeit with decreased affinity by up to 100-fold. Following up on these initial results, the authors reported a study on the optimization of quinazoline-type DC-SIGN ligands.123 After the oxidizable thioether function in previous quinoxazoline ligands had been identified as a potential source of instability that precluded the use of 32 and 33 in cellular assays and in vivo studies, a chemical strategy for the substitution of the thioether linkage by a saturated aliphatic chain was devised. In particular, derivatives with aromatic substituents on the piperazine-heterocycle and the aliphatic linker chain were reported with high binding affinities for DC-SIGN ECD between 0.31 μM and 10 μM. The most potent compounds 35 and 36 featuring a heterocyclic substituent on the piperazine residue simultaneously showed improved aqueous solubility, which enabled an assessment of their ability to block DC-SIGN function in a cellular model. A respective flow cytometric assay employing a DC-SIGN-expressing Raji cell line showed that 35 and 36 inhibited DC-SIGN-mediated binding of a fluorescent probe (35: 53%/77% inhibition at 0.1/1 mM; 36: 79%/92% inhibition at 0.1/1 mM). Compared with glycomimetic ligands, these compounds were reported with unprecedented affinities for DC-SIGN. Accordingly, the aromatic and mostly planar quinoxazoline or pyrazolone ligands described above were able to interfere with carbohydrate binding to the primary binding site of DC-SIGN, despite the radically different chemical structure of the natural ligands. Unfortunately, no conclusive structural evidence for the binding mode of these compounds has been provided so far.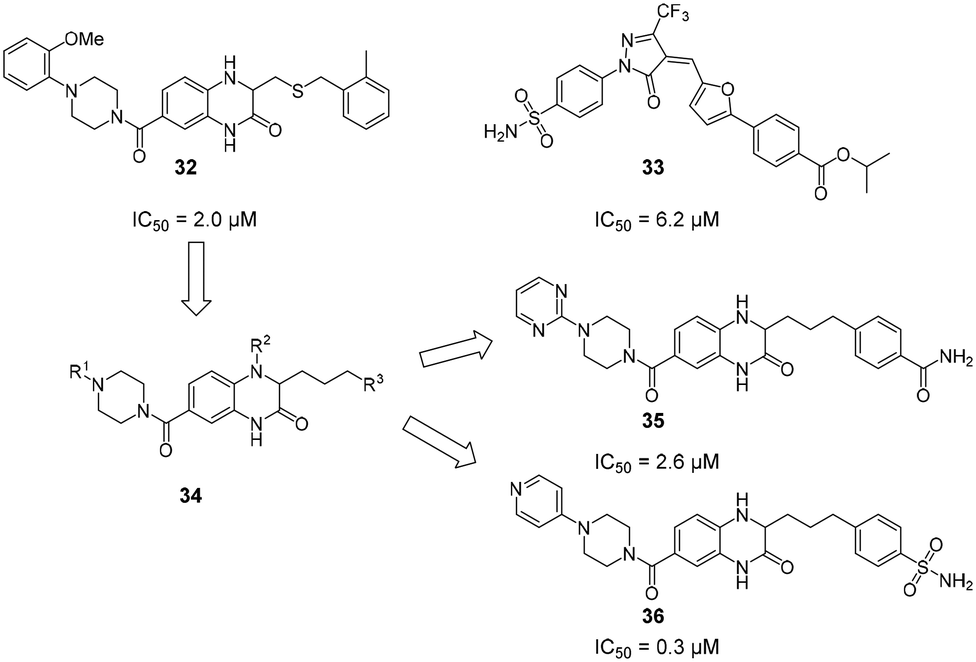 | ||
| Fig. 13 Non-carbohydrate ligands discovered in a high-throughput screening campaign with high reported affinities for DC-SIGN. | ||
Following up on their initial studies and previous successes with Langerin, the Rademacher group collected screening data for a library of 981 fragments from multiple orthogonal fragment screening methods against DC-SIGN.48 The resulting 95 hits were counter-screened by SPR and an ELLA-type assay. Interestingly, 61 compounds showed binding affinity for DC-SIGN by SPR, whereas only four of the initial hits actively interfered with carbohydrate binding to the active site in the functional ELLA assay. Besides a low druggability of the primary carbohydrate binding site, this result indicated the presence of peripheral secondary biding sites that were well-suited to bind fragment-like small molecules. The most potent fragments from this study showed affinities up to KD = 0.3 mM. Intrigued by this finding, the researchers employed extensive 1H–15N HSQC NMR studies that resulted in the discovery of five secondary binding pockets. In accordance with the screening data, it was found that all four dextran-competitive fragments bound to the same shallow pocket in the direct vicinity of the primary carbohydrate binding site. The authors proposed that the steric hindrance of carbohydrate binding from fragments bound to this position was more likely than a direct interaction with the central Ca2+ cofactor. Thus, an extension of a carbohydrate ligand toward this pocket was proposed as a promising approach for the development of potent glycomimetics. Considering reports on allostery in CLRs, however, targeting allosteric pockets with drug-like ligands remains a similarly attractive strategy.39,40,49
Conclusions
Because of their role in many biological processes related to innate immunity, homeostasis, and cancer, myeloid CLRs remain highly attractive targets for pharmacological interventions. The potential uses of selective ligands for myeloid CLRs include antiviral therapy, immune stimulation, or targeted delivery of vaccines or other therapeutics to specific myeloid cell subsets. In addition, potential applications in autoimmune diseases, fibrosis, and cancer have been proposed. The development of potent and selective compounds is complicated by the fact that the binding sites of most CLRs are shallow and solvent-exposed, resulting in low binding affinities and broad ligand specificities on a monovalent level. To overcome the low druggability of CLRs, two dominant strategies have been employed. Firstly, glycomimetic ligands have been developed that are able to engage in secondary interactions in the vicinity of the primary carbohydrate binding site. Whereas these compounds usually bind with modest affinities in the micromolar range, secondary interactions can elicit a high specificity for the target lectin. In combination with multivalent presentation platforms, highly potent and selective ligands for CLRs can be generated. Secondly, binding sites for non-carbohydrate-derived molecules have been described. As demonstrated for Langerin, molecules binding to those sites can exert allosteric control over ligand binding, which may be a general feature of ligand binding in CLRs.Many studies have provided SAR analyses of ligand binding to CLRs and generated a much-improved picture about the prerequisites for high affinity ligands for those receptors. However, much less is known about the relationship between ligand binding and receptor activation. Given that many CLRs possess signalling capabilities, information on ligand binding must be propagated from the extracellular CRD into the intracellular space. Yet, the specific mechanisms that promote signal transduction remain mostly elusive. As an example, the diverging effect of mannose- and fucose-based ligands in DC-SIGN recognition can currently not be explained mechanistically by the differences of the respective glycan binding modes in the active site.102 An improved understanding of these processes could, for example, greatly support the development of pro-inflammatory Mincle agonists.
Besides the receptors discussed in this review, other members of the myeloid CLR family represent interesting therapeutic targets for diverse applications. Yet, most of these proteins have not been targeted with glycomimetics or other small molecule-based approaches. The accumulated knowledge from research on Mincle, Langerin, and DC-SIGN should provide ample ground for further drug design efforts targeting this highly attractive class of proteins.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author would like to thank Gabriele Conti for diligently proofreading the manuscript.References
- M. J. Robinson, D. Sancho, E. C. Slack, S. LeibundGut-Landmann and C. R. e Sousa, Nat. Immunol., 2006, 7, 1258–1265 CrossRef CAS PubMed.
- J. Monteiro and B. Lepenies, Viruses, 2017, 9, 59 CrossRef PubMed.
- M. Bermejo-Jambrina, J. Eder, L. C. Helgers, N. Hertoghs, B. M. Nijmeijer, M. Stunnenberg and T. B. H. Geijtenbeek, Front. Immunol., 2018, 9, 590 CrossRef PubMed.
- S. Mayer, M.-K. Raulf and B. Lepenies, Histochem. Cell Biol., 2017, 147, 223–237 CrossRef CAS PubMed.
- G. D. Brown, J. A. Willment and L. Whitehead, Nat. Rev. Immunol., 2018, 18, 374–389 CrossRef CAS PubMed.
- S. LeibundGut-Landmann, O. Groß, M. J. Robinson, F. Osorio, E. C. Slack, S. V. Tsoni, E. Schweighoffer, V. Tybulewicz, G. D. Brown, J. Ruland and C. R. e Sousa, Nat. Immunol., 2007, 8, 630–638 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek and S. I. Gringhuis, Nat. Rev. Immunol., 2016, 16, 433–448 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek and S. I. Gringhuis, Nat. Rev. Immunol., 2009, 9, 465–479 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek, R. Torensma, S. J. Van Vliet, G. C. F. Van Duijnhoven, G. J. Adema, Y. Van Kooyk and C. G. Figdor, Cell, 2000, 100, 575–585 CrossRef CAS PubMed.
- M. Wagener, J. C. Hoving, H. Ndlovu and M. J. Marakalala, Front. Immunol., 2018, 9, 225 CrossRef PubMed.
- R. Lang, H. Schoenen and C. Desel, Immunobiology, 2011, 216, 1184–1191 CrossRef CAS PubMed.
- W. Kastenmüller, K. Kastenmüller, C. Kurts and R. A. Seder, Nat. Rev. Immunol., 2014, 14, 705–711 CrossRef PubMed.
- Q. Lu, J. Liu, S. Zhao, M. F. Gomez Castro, M. Laurent-Rolle, J. Dong, X. Ran, P. Damani-Yokota, H. Tang, T. Karakousi, J. Son, M. E. Kaczmarek, Z. Zhang, S. T. Yeung, B. T. McCune, R. E. Chen, F. Tang, X. Ren, X. Chen, J. C. C. Hsu, M. Teplova, B. Huang, H. Deng, Z. Long, T. Mudianto, S. Jin, P. Lin, J. Du, R. Zang, T. T. Su, A. Herrera, M. Zhou, R. Yan, J. Cui, J. Zhu, Q. Zhou, T. Wang, J. Ma, S. B. Koralov, Z. Zhang, I. Aifantis, L. N. Segal, M. S. Diamond, K. M. Khanna, K. A. Stapleford, P. Cresswell, Y. Liu, S. Ding, Q. Xie and J. Wang, Immunity, 2021, 54, 1304–1319 CrossRef CAS PubMed.
- A. Marzi, T. Gramberg, G. Simmons, P. Moller, A. J. Rennekamp, M. Krumbiegel, M. Geier, J. Eisemann, N. Turza, B. Saunier, A. Steinkasserer, S. Becker, P. Bates, H. Hofmann and S. Pohlmann, J. Virol., 2004, 78, 12090–12095 CrossRef CAS PubMed.
- G. Simmons, J. D. Reeves, C. C. Grogan, L. H. Vandenberghe, F. Baribaud, J. C. Whitbeck, E. Burke, M. J. Buchmeier, E. J. Soilleux, J. L. Riley, R. W. Doms, P. Bates and S. Pöhlmann, Virology, 2003, 305, 115–123 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek, D. S. Kwon, R. Torensma, S. J. Van Vliet, G. C. F. Van Duijnhoven, J. Middel, I. L. M. H. A. Cornelissen, H. S. L. M. Nottet, V. N. KewalRamani, D. R. Littman, C. G. Figdor and Y. Van Kooyk, Cell, 2000, 100, 587–597 CrossRef CAS PubMed.
- A. K. Azad, M. V. S. Rajaram and L. S. Schlesinger, J. Cytol. Mol. Biol., 2014, 1, 1000003 Search PubMed.
- L. Tailleux, O. Schwartz, J.-L. Herrmann, E. Pivert, M. Jackson, A. Amara, L. Legres, D. Dreher, L. P. Nicod, J. C. Gluckman, P. H. Lagrange, B. Gicquel and O. Neyrolles, J. Exp. Med., 2003, 197, 121–127 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek, S. J. Van Vliet, E. A. Koppel, M. Sanchez-Hernandez, C. M. J. E. Vandenbroucke-Grauls, B. Appelmelk and Y. Van Kooyk, J. Exp. Med., 2003, 197, 7–17 CrossRef CAS PubMed.
- P. Kalantari, S. C. Bunnell and M. J. Stadecker, Front. Immunol., 2019, 10, 26 CrossRef CAS PubMed.
- M. Colmenares, A. Puig-Kröger, O. M. Pello, A. L. Corbí and L. Rivas, J. Biol. Chem., 2002, 277, 36766–36769 CrossRef CAS PubMed.
- S. H. Lee, M. Charmoy, A. Romano, A. Paun, M. M. Chaves, F. O. Cope, D. A. Ralph and D. L. Sacks, J. Exp. Med., 2018, 215, 357–375 CrossRef CAS PubMed.
- P.-S. Sung and S.-L. Hsieh, Front. Immunol., 2019, 10, 2867 CrossRef CAS PubMed.
- Y.-L. Huang, S.-T. Chen, R.-S. Liu, Y.-H. Chen, C.-Y. Lin, C.-H. Huang, P.-Y. Shu, C.-L. Liao and S.-L. Hsieh, J. Mol. Med., 2016, 94, 1025–1037 CrossRef CAS PubMed.
- S. L. Londrigan, M. D. Tate, A. G. Brooks and P. C. Reading, J. Leukocyte Biol., 2012, 92, 97–106 CrossRef CAS PubMed.
- O. Teng, S.-T. Chen, T.-L. Hsu, S. F. Sia, S. Cole, S. A. Valkenburg, T.-Y. Hsu, J. T. Zheng, W. Tu, R. Bruzzone, J. S. M. Peiris, S.-L. Hsieh and H.-L. Yen, J. Virol., 2017, 91, e01813–e01816 CrossRef CAS PubMed.
- A. Cioce, M. Thépaut, F. Fieschi and N. Reichardt, Chem. – Eur. J., 2020, 26, 12809–12817 CrossRef CAS PubMed.
- L. Medve, S. Achilli, S. Serna, F. Zuccotto, N. Varga, M. Thépaut, M. Civera, C. Vivès, F. Fieschi, N. Reichardt and A. Bernardi, Chem. – Eur. J., 2018, 24, 14448–14460 CrossRef CAS PubMed.
- K. Drickamer, Curr. Opin. Struct. Biol., 1999, 9, 585–590 CrossRef CAS PubMed.
- A. N. Zelensky and J. E. Gready, FEBS J., 2005, 272, 6179–6217 CrossRef CAS PubMed.
- K. Drickamer and M. E. Taylor, Curr. Opin. Struct. Biol., 2015, 34, 26–34 CrossRef CAS PubMed.
- I. Caminschi, A. I. Proietto, F. Ahmet, S. Kitsoulis, J. S. Teh, J. C. Y. Lo, A. Rizzitelli, L. Wu, D. Vremec, S. L. H. Van Dommelen, I. K. Campbell, E. Maraskovsky, H. Braley, G. M. Davey, P. Mottram, N. Van De Velde, K. Jensen, A. M. Lew, M. D. Wright, W. R. Heath, K. Shortman and M. H. Lahoud, Blood, 2008, 112, 3264–3273 CrossRef CAS PubMed.
- G. D. Brown, Nat. Rev. Immunol., 2006, 6, 33–43 CrossRef CAS PubMed.
- B. S. Gully, H. Venugopal, A. J. Fulcher, Z. Fu, J. Li, F. A. Deuss, C. Llerena, W. R. Heath, M. H. Lahoud, I. Caminschi, J. Rossjohn and R. Berry, J. Biol. Chem., 2021, 296, 100127 CrossRef CAS PubMed.
- G. Tabarani, M. Thépaut, D. Stroebel, C. Ebei, C. Vivès, P. Vachette, D. Durand and F. Fieschi, J. Biol. Chem., 2009, 284, 21229–21240 CrossRef CAS PubMed.
- H. Feinberg, A. S. Powlesland, M. E. Taylor and W. I. Weis, J. Biol. Chem., 2010, 285, 13285–13293 CrossRef CAS PubMed.
- M. B. Richardson and S. J. Williams, Front. Immunol., 2014, 5, 288 Search PubMed.
- M. E. Taylor, K. Bezouska and K. Drickamer, J. Biol. Chem., 1992, 267, 1719–1726 CrossRef CAS.
- B. G. Keller and C. Rademacher, Curr. Opin. Struct. Biol., 2020, 62, 31–38 CrossRef CAS PubMed.
- J. Hanske, S. Aleksić, M. Ballaschk, M. Jurk, E. Shanina, M. Beerbaum, P. Schmieder, B. G. Keller and C. Rademacher, J. Am. Chem. Soc., 2016, 138, 12176–12186 CrossRef CAS PubMed.
- G. Tabarani, M. Thépaut, D. Stroebel, C. Ebel, C. Vivès, P. Vachette, D. Durand and F. Fieschi, J. Biol. Chem., 2009, 284, 21229–21240 CrossRef CAS PubMed.
- Z. Hu, Y. Wang, C. Cheng and Y. He, J. Struct. Biol., 2019, 208, 107384 CrossRef CAS PubMed.
- J. Cramer, C. P. Sager and B. Ernst, J. Med. Chem., 2019, 62, 8915–8930 CrossRef CAS PubMed.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661–677 CrossRef CAS PubMed.
- A. Volkamer, D. Kuhn, T. Grombacher, F. Rippmann and M. Rarey, J. Chem. Inf. Model., 2012, 52, 360–372 CrossRef CAS PubMed.
- J. Aretz, E.-C. Wamhoff, J. Hanske, D. Heymann and C. Rademacher, Front. Immunol., 2014, 5, 323 Search PubMed.
- J. Aretz, Y. Kondoh, C. K. Honda, U. R. Anumala, M. Nazaré, N. Watanabe, H. Osada and C. Rademacher, Chem. Commun., 2016, 52, 9067–9070 RSC.
- J. Aretz, H. Baukmann, E. Shanina, J. Hanske, R. Wawrzinek, V. A. Zapol'skii, P. H. Seeberger, D. E. Kaufmann and C. Rademacher, Angew. Chem., Int. Ed., 2017, 56, 7292–7296 CrossRef CAS PubMed.
- J. Aretz, U. R. Anumala, F. F. Fuchsberger, N. Molavi, N. Ziebart, H. Zhang, M. Nazaré and C. Rademacher, J. Am. Chem. Soc., 2018, 140, 14915–14925 CrossRef CAS PubMed.
- X. Lu, M. Nagata and S. Yamasaki, Int. Immunol., 2018, 30, 233–239 CrossRef CAS PubMed.
- E. C. Patin, S. J. Orr and U. E. Schaible, Front. Immunol., 2017, 8, 861 CrossRef PubMed.
- H. Schoenen, B. Bodendorfer, K. Hitchens, S. Manzanero, K. Werninghaus, F. Nimmerjahn, E. M. Agger, S. Stenger, P. Andersen, J. Ruland, G. D. Brown, C. Wells and R. Lang, J. Immunol., 2010, 184, 2756–2760 CrossRef CAS PubMed.
- E. Ishikawa, T. Ishikawa, Y. S. Morita, K. Toyonaga, H. Yamada, O. Takeuchi, T. Kinoshita, S. Akira, Y. Yoshikai and S. Yamasaki, J. Exp. Med., 2009, 206, 2879–2888 CrossRef CAS PubMed.
- K. M. Jacobsen, U. B. Keiding, L. L. Clement, E. S. Schaffert, N. D. S. Rambaruth, M. Johannsen, K. Drickamer and T. B. Poulsen, MedChemComm, 2015, 6, 647–652 RSC.
- Y. Hattori, D. Morita, N. Fujiwara, D. Mori, T. Nakamura, H. Harashima, S. Yamasaki and M. Sugita, J. Biol. Chem., 2014, 289, 15405–15412 CrossRef CAS PubMed.
- T. Ishikawa, F. Itoh, S. Yoshida, S. Saijo, T. Matsuzawa, T. Gonoi, T. Saito, Y. Okawa, N. Shibata, T. Miyamoto and S. Yamasaki, Cell Host Microbe, 2013, 13, 477–488 CrossRef CAS PubMed.
- A. Decout, S. Silva-Gomes, D. Drocourt, S. Barbe, I. André, F. J. Cueto, T. Lioux, D. Sancho, E. Pérouzel, A. Vercellone, J. Prandi, M. Gilleron, G. Tiraby and J. Nigou, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2675–2680 CrossRef CAS PubMed.
- M. Nagata, Y. Izumi, E. Ishikawa, R. Kiyotake, R. Doi, S. Iwai, Z. Omahdi, T. Yamaji, T. Miyamoto, T. Bamba and S. Yamasaki, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3285–E3294 CrossRef CAS PubMed.
- S. Yamasaki, E. Ishikawa, M. Sakuma, H. Hara, K. Ogata and T. Saito, Nat. Immunol., 2008, 9, 1179–1188 CrossRef CAS PubMed.
- A. V. Kostarnoy, P. G. Gancheva, B. Lepenies, A. I. Tukhvatulin, A. S. Dzharullaeva, N. B. Polyakov, D. A. Grumov, D. A. Egorova, A. Y. Kulibin, M. A. Bobrov, E. A. Malolina, P. A. Zykin, A. I. Soloviev, E. Riabenko, D. V. Maltseva, D. A. Sakharov, A. G. Tonevitsky, L. V. Verkhovskaya, D. Y. Logunov, B. S. Naroditsky and A. L. Gintsburg, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E2758–E2765 CrossRef CAS PubMed.
- R. Kiyotake, M. Oh-Hora, E. Ishikawa, T. Miyamoto, T. Ishibashi and S. Yamasaki, J. Biol. Chem., 2015, 290, 25322–25332 CrossRef CAS PubMed.
- C. Aagaard, T. Hoang, J. Dietrich, P. J. Cardona, A. Izzo, G. Dolganov, G. K. Schoolnik, J. P. Cassidy, R. Billeskov and P. Andersen, Nat. Med., 2011, 17, 189–195 CrossRef CAS PubMed.
- S. Abraham, H. B. Juel, P. Bang, H. M. Cheeseman, R. B. Dohn, T. Cole, M. P. Kristiansen, K. S. Korsholm, D. Lewis, A. W. Olsen, L. R. McFarlane, S. Day, S. Knudsen, K. Moen, M. Ruhwald, I. Kromann, P. Andersen, R. J. Shattock and F. Follmann, Lancet Infect. Dis., 2019, 19, 1091–1100 CrossRef CAS PubMed.
- H. Feinberg, S. A. F. Jégouzo, T. J. W. Rowntree, Y. Guan, M. A. Brash, M. E. Taylor, W. I. Weis and K. Drickamer, J. Biol. Chem., 2013, 288, 28457–28465 CrossRef CAS.
- H. Feinberg, N. D. S. Rambaruth, S. A. F. Jégouzo, K. M. Jacobsen, R. Djurhuus, T. B. Poulsen, W. I. Weis, M. E. Taylor and K. Drickamer, J. Biol. Chem., 2016, 291, 21222–21233 CrossRef CAS.
- A. Furukawa, J. Kamishikiryo, D. Mori, K. Toyonaga, Y. Okabe, A. Toji, R. Kanda, Y. Miyake, T. Ose, S. Yamasaki and K. Maenaka, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17438–17443 CrossRef CAS.
- S. A. F. Jégouzo, E. C. Harding, O. Acton, M. J. Rex, A. J. Fadden, M. E. Taylor and K. Drickamer, Glycobiology, 2014, 24, 1291–1300 CrossRef PubMed.
- A. A. Khan, S. H. Chee, R. J. McLaughlin, J. L. Harper, F. Kamena, M. S. M. Timmer and B. L. Stocker, ChemBioChem, 2011, 12, 2572–2576 CrossRef CAS PubMed.
- M. B. Richardson, S. Torigoe, S. Yamasaki and S. J. Williams, Chem. Commun., 2015, 51, 15027–15030 RSC.
- A. Khan, K. Kodar, M. S. M. Timmer and B. L. Stocker, Tetrahedron, 2018, 74, 1269–1277 CrossRef CAS.
- A. J. Smith, S. M. Miller, C. Buhl, R. Child, M. Whitacre, R. Schoener, G. Ettenger, D. Burkhart, K. Ryter and J. T. Evans, Front. Immunol., 2019, 10, 338 CrossRef CAS PubMed.
- D. G. M. Smith, Y. Hosono, M. Nagata, S. Yamasaki and S. J. Williams, Chem. Commun., 2020, 56, 4292–4295 RSC.
- A. T. Lynch, C. Motozono, A. J. Foster, K. Kodar, E. M. Dangerfield, S. Yamasaki, D. N. Wedlock, M. S. M. Timmer and B. L. Stocker, Bioorg. Chem., 2021, 110, 104747 CrossRef CAS PubMed.
- A. J. Foster, M. Nagata, X. Lu, A. T. Lynch, Z. Omahdi, E. Ishikawa, S. Yamasaki, M. S. M. Timmer and B. L. Stocker, J. Med. Chem., 2018, 61, 1045–1060 CrossRef CAS PubMed.
- K. T. Ryter, G. Ettenger, O. K. Rasheed, C. Buhl, R. Child, S. M. Miller, D. Holley, A. J. Smith and J. T. Evans, J. Med. Chem., 2020, 63, 309–320 CrossRef CAS PubMed.
- O. K. Rasheed, C. Buhl, J. T. Evans and K. T. Ryter, ChemMedChem, 2021, 16, 1246–1251 CrossRef CAS PubMed.
- C. M. Fehres, S. Duinkerken, S. C. Bruijns, H. Kalay, S. J. Van Vliet, M. Ambrosini, T. D. De Gruijl, W. W. Unger, J. J. Garcia-Vallejo and Y. Van Kooyk, Cell. Mol. Immunol., 2017, 14, 360–370 CrossRef CAS PubMed.
- C. M. Fehres, H. Kalay, S. C. M. Bruijns, S. A. M. Musaafir, M. Ambrosini, L. Van Bloois, S. J. Van Vliet, G. Storm, J. J. Garcia-Vallejo and Y. Van Kooyk, J. Controlled Release, 2015, 203, 67–76 CrossRef CAS PubMed.
- N. S. Stambach and M. E. Taylor, Glycobiology, 2003, 13, 401–410 CrossRef CAS PubMed.
- E. Chabrol, A. Nurisso, A. Daina, E. Vassal-Stermann, M. Thepaut, E. Girard, R. R. Vivès and F. Fieschi, PLoS One, 2012, 7, e50722 CrossRef CAS PubMed.
- J. C. Muñoz-García, E. Chabrol, R. R. Vivès, A. Thomas, J. L. De Paz, J. Rojo, A. Imberty, F. Fieschi, P. M. Nieto and J. Angulo, J. Am. Chem. Soc., 2015, 137, 4100–4110 CrossRef PubMed.
- H. Feinberg, M. E. Taylor, N. Razi, R. McBride, Y. A. Knirel, S. A. Graham, K. Drickamer and W. I. Weis, J. Mol. Biol., 2011, 405, 1027–1039 CrossRef CAS PubMed.
- H. Feinberg, T. J. W. Rowntree, S. L. W. Tan, K. Drickamer, W. I. Weis and M. E. Taylor, J. Biol. Chem., 2013, 288, 36762–36771 CrossRef CAS PubMed.
- V. Porkolab, E. Chabrol, N. Varga, S. Ordanini, I. Sutkevičiute, M. Thépaut, M. J. García-Jiménez, E. Girard, P. M. Nieto, A. Bernardi and F. Fieschi, ACS Chem. Biol., 2018, 13, 600–608 CrossRef CAS PubMed.
- E. C. Wamhoff, J. Hanske, L. Schnirch, J. Aretz, M. Grube, D. Varón Silva and C. Rademacher, ACS Chem. Biol., 2016, 11, 2407–2413 CrossRef CAS PubMed.
- E. C. Wamhoff, J. Schulze, L. Bellmann, M. Rentzsch, G. Bachem, F. F. Fuchsberger, J. Rademacher, M. Hermann, B. Del Frari, R. Van Dalen, D. Hartmann, N. M. Van Sorge, O. Seitz, P. Stoitzner and C. Rademacher, ACS Cent. Sci., 2019, 5, 808–820 CrossRef CAS PubMed.
- S. Kuhaudomlarp, E. Siebs, E. Shanina, J. Topin, I. Joachim, P. Silva Figueiredo Celestino Gomes, A. Varrot, D. Rognan, C. Rademacher, A. Imberty and A. Titz, Angew. Chem., Int. Ed., 2021, 60, 8104–8114 CrossRef CAS PubMed.
- S. Mummidi, G. Catano, L. A. Lam, A. Hoefle, V. Telles, K. Begum, F. Jimenez, S. S. Ahuja and S. K. Ahuja, J. Biol. Chem., 2001, 276, 33196–33212 CrossRef CAS PubMed.
- Y. van Kooyk and T. B. H. Geijtenbeek, Nat. Rev. Immunol., 2003, 3, 697–709 CrossRef CAS PubMed.
- H. J. Yu, M. A. Reuter and D. McDonald, PLoS Pathog., 2008, 4, e1000134 CrossRef PubMed.
- D. H. Katz, U. A. Tahir, D. Ngo, M. D. Benson, A. G. Bick, A. Pampana, Y. Gao, M. J. Keyes, A. Correa, S. Sinha, D. Shen, Q. Yang, J. M. Robbins, Z.-Z. Chen, D. E. Cruz, B. Peterson, P. Natarajan, R. S. Vasan, G. Smith, T. J. Wang and R. E. Gerszten, Medrxiv, 2020, DOI:10.1101/2020.06.09.20125690.
- L. Wang, T. J. Balmat, A. L. Antonia, F. J. Constantine, R. Henao, T. W. Burke, A. Ingham, M. T. McClain, E. L. Tsalik, E. R. Ko, G. S. Ginsburg, M. R. DeLong, X. Shen, C. W. Woods, E. R. Hauser and D. C. Ko, Genome Med., 2021, 13, 83 CrossRef CAS PubMed.
- F. Chiodo, S. C. M. Bruijns, E. Rodriguez, R. J. E. Li, A. Molinaro, A. Silipo, F. Di Lorenzo, D. Garcia-Rivera, Y. Valdes-Balbin, V. Verez-Bencomo and Y. van Kooyk, Biorxiv, 2020, DOI:10.1101/2020.05.13.092478.
- R. Amraei, W. Yin, M. A. Napoleon, E. L. Suder, J. Berrigan, Q. Zhao, J. Olejnik, K. B. Chandler, C. Xia, J. Feldman, B. M. Hauser, T. M. Caradonna, A. G. Schmidt, S. Gummuluru, E. Mühlberger, V. Chitalia, C. E. Costello and N. Rahimi, ACS Cent. Sci., 2021, 7, 1156–1165 CrossRef CAS PubMed.
- F. A. Lempp, L. Soriaga, M. Montiel-Ruiz, F. Benigni, J. Noack, Y.-J. Park, S. Bianchi, A. C. Walls, J. E. Bowen, J. Zhou, H. Kaiser, A. Joshi, M. Agostini, M. Meury, E. Dellota, S. Jaconi, E. Cameroni, J. Martinez-Picado, J. Vergara-Alert, N. Izquierdo-Useros, H. W. Virgin, A. Lanzavecchia, D. Veesler, L. Purcell, A. Telenti and D. Corti, Nature, 2021 DOI:10.1038/s41586-021-03925-1.
- C. G. Park, K. Takahara, E. Umemoto, Y. Yashima, K. Matsubara, Y. Matsuda, B. E. Clausen, K. Inaba and R. M. Steinman, Int. Immunol., 2001, 13, 1283–1290 CrossRef CAS PubMed.
- A. S. Powlesland, E. M. Ward, S. K. Sadhu, Y. Guo, M. E. Taylor and K. Drickamer, J. Biol. Chem., 2006, 281, 20440–20449 CrossRef CAS PubMed.
- J. J. Garcia-Vallejo and Y. van Kooyk, Trends Immunol., 2013, 34, 482–486 CrossRef CAS PubMed.
- M. Schaefer, N. Reiling, C. Fessler, J. Stephani, I. Taniuchi, F. Hatam, A. O. Yildirim, H. Fehrenbach, K. Walter, J. Ruland, H. Wagner, S. Ehlers and T. Sparwasser, J. Immunol., 2008, 180, 6836–6845 CrossRef CAS PubMed.
- R. M. Anthony, T. Kobayashi, F. Wermeling and J. V. Ravetch, Nature, 2011, 475, 110–114 CrossRef CAS PubMed.
- L. H. Sales Pereira, A. C. do Alves, J. M. Siqueira Ferreira and L. L. dos Santos, Microb. Pathog., 2021, 150, 104731 CrossRef CAS PubMed.
- Y. Guo, H. Feinberg, E. Conroy, D. A. Mitchell, R. Alvarez, O. Blixt, M. E. Taylor, W. I. Weis and K. Drickamer, Nat. Struct. Mol. Biol., 2004, 11, 591–598 CrossRef CAS PubMed.
- H. Feinberg, R. Castelli, K. Drickamer, P. H. Seeberger and W. I. Weis, J. Biol. Chem., 2007, 282, 4202–4209 CrossRef CAS PubMed.
- J. D. Martínez, P. Valverde, S. Delgado, C. Romanò, B. Linclau, N. C. Reichardt, S. Oscarson, A. Ardá, J. Jiménez-Barbero and F. J. Cañada, Molecules, 2019, 24, 2337 CrossRef PubMed.
- T. Tomašić, D. Hajšek, U. Švajger, J. Luzar, N. Obermajer, I. Petit-Haertlein, F. Fieschi and M. Anderluh, Eur. J. Med. Chem., 2014, 75, 308–326 CrossRef PubMed.
- M. Thépaut, C. Guzzi, I. Sutkeviciute, S. Sattin, R. Ribeiro-Viana, N. Varga, E. Chabrol, J. Rojo, A. Bernardi, J. Angulo, P. M. Nieto and F. Fieschi, J. Am. Chem. Soc., 2013, 135, 2518–2529 CrossRef PubMed.
- N. Varga, I. Sutkeviciute, C. Guzzi, J. McGeagh, I. Petit-Haertlein, S. Gugliotta, J. Weiser, J. Angulo, F. Fieschi and A. Bernardi, Chem. – Eur. J., 2013, 19, 4786–4797 CrossRef CAS.
- L. Medve, S. Achilli, J. Guzman-Caldentey, M. Thépaut, L. Senaldi, A. Le Roy, S. Sattin, C. Ebel, C. Vivès, S. Martin-Santamaria, A. Bernardi and F. Fieschi, Chem. – Eur. J., 2019, 25, 14659–14668 CrossRef CAS.
- D. A. Mitchell, N. A. Jones, S. J. Hunter, J. M. D. Cook, S. F. Jenkinson, M. R. Wormald, R. A. Dwek and G. W. J. Fleet, Tetrahedron: Asymmetry, 2007, 18, 1502–1510 CrossRef CAS.
- K. C. A. Garber, K. Wangkanont, E. E. Carlson and L. L. Kiessling, Chem. Commun., 2010, 46, 6747–6749 RSC.
- A. Bernardi, D. Arosio, L. Manzoni, F. Micheli, A. Pasquarello and P. Seneci, J. Org. Chem., 2001, 66, 6209–6216 CrossRef CAS.
- J. J. Reina, S. Sattin, D. Invernizzi, S. Mari, L. Martínez-Prats, G. Tabarani, F. Fieschi, R. Delgado, P. M. Nieto, J. Rojo and A. Bernardi, ChemMedChem, 2007, 2, 1030–1036 CrossRef CAS.
- P. Valverde, S. Delgado, J. D. Martínez, J. B. Vendeville, J. Malassis, B. Linclau, N. C. Reichardt, F. J. Cañada, J. Jiménez-Barbero and A. Ardá, ACS Chem. Biol., 2019, 14, 1660–1671 CrossRef CAS.
- N. Varga, I. Sutkeviciute, R. Ribeiro-Viana, A. Berzi, R. Ramdasi, A. Daghetti, G. Vettoretti, A. Amara, M. Clerici, J. Rojo, F. Fieschi and A. Bernardi, Biomaterials, 2014, 35, 4175–4184 CrossRef CAS.
- J. Luczkowiak, S. Sattin, I. Sutkevičiute, J. J. Reina, M. Sánchez-Navarro, M. Thépaut, L. Martínez-Prats, A. Daghetti, F. Fieschi, R. Delgado, A. Bernardi and J. Rojo, Bioconjugate Chem., 2011, 22, 1354–1365 CrossRef CAS PubMed.
- S. Ordanini, N. Varga, V. Porkolab, M. Thépaut, L. Belvisi, A. Bertaglia, A. Palmioli, A. Berzi, D. Trabattoni, M. Clerici, F. Fieschi and A. Bernardi, Chem. Commun., 2015, 51, 3816–3819 RSC.
- A. Berzi, S. Ordanini, B. Joosten, D. Trabattoni, A. Cambi, A. Bernardi and M. Clerici, Sci. Rep., 2016, 6, 35373 CrossRef CAS PubMed.
- M. Thépaut, J. Luczkowiak, C. Vivès, N. Labiod, I. Bally, F. Lasala, Y. Grimoire, D. Fenel, S. Sattin, N. Thielens, G. Schoehn, A. Bernardi, R. Delgado and F. Fieschi, PLoS Pathog., 2021, 17, e1009576 CrossRef PubMed.
- G. Timpano, G. Tabarani, M. Anderluh, D. Invernizzi, F. Vasile, D. Potenza, P. M. Nieto, J. Rojo, F. Fieschi and A. Bernardi, ChemBioChem, 2008, 9, 1921–1930 CrossRef CAS PubMed.
- M. Andreini, D. Doknic, I. Sutkeviciute, J. J. Reina, J. Duan, E. Chabrol, M. Thepaut, E. Moroni, F. Doro, L. Belvisi, J. Weiser, J. Rojo, F. Fieschi and A. Bernardi, Org. Biomol. Chem., 2011, 9, 5778 RSC.
- J. Cramer, C. P. Sager and B. Ernst, J. Med. Chem., 2019, 62, 8915–8930 CrossRef CAS PubMed.
- M. J. Borrok and L. L. Kiessling, J. Am. Chem. Soc., 2007, 129, 12780–12785 CrossRef CAS PubMed.
- S. L. Mangold, L. R. Prost and L. L. Kiessling, Chem. Sci., 2012, 3, 772–777 RSC.
| This journal is © The Royal Society of Chemistry 2021 |