
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly fluorescent aryl-cyclopentadienyl ligands and their tetra-nuclear mixed metallic potassium–dysprosium clusters†
Selvakumar Arumugam‡
 a,
Pulikanti Guruprasad Reddy‡b,
Maria Francisb,
Aditya Kulkarnib,
Sudipta Roy*b and
Kartik Chandra Mondal*a
a,
Pulikanti Guruprasad Reddy‡b,
Maria Francisb,
Aditya Kulkarnib,
Sudipta Roy*b and
Kartik Chandra Mondal*a
aDepartment of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: csdkartik@iitm.ac.in
bDepartment of Chemistry, Indian Institute of Science Education and Research (IISER), Tirupati 517507, India. E-mail: roy.sudipta@iisertirupati.ac.in
First published on 30th October 2020
Abstract
Two alkyl substituted triaryl-cyclopentadienyl ligands [4,4′-(4-phenylcyclopenta-1,3-diene-1,2-diyl)bis(methylbenzene) (1) and 4,4′,4′′-(cyclopenta-1,3-diene-1,2,4-triyl)tris(methylbenzene) (2)] have been synthesized via cross-aldol condensation followed by Zn-dust mediated cyclization and acid catalyzed dehydration reactions. The fluorescence properties of 1 and 2 have been studied in solution and solid state. The ligands exhibited aggregation-induced emission enhancement (AIEE) in THF/water solution. 1 and 2 have been found to be significantly more fluorescent in the solid state than in their respective solutions. This phenomenon can be attributed to the strong intermolecular CH⋯π interactions present in 1 and 2 which leads to the tight packing of molecules in their solid-state. Both 1, 2 and their corresponding anions have been studied by theoretical calculations. Ligands 1 and 2 have been shown to react with anhydrous DyCl3 in the presence of potassium metal at high temperature to afford two fluorescent chloride-bridged tetra-nuclear mixed potassium–dysprosium metallocenes [(Me2Cp)4Dy2IIICl4K2]·3.5(C7H8) (5) and [(Me3Cp)4Dy2IIICl4K2]·3(C7H8) (6), respectively in good yields.
Introduction
The development of stable and π-conjugated organic luminescent materials has received significant interest for both fundamental research and practical applications due to their potential use as organic light-emitting diodes (OLEDs),1 biological probes,2 organic solid state lasers,3 fluorescent sensors,4 photonic devices,5 and flat panel displays and in illumination.6 Most of the reported organic luminescent materials are highly emissive in dilute solutions and poorly/non-emitting in the solid state/neat thin solid films due to the aggregation caused quenching (ACQ) effect resulting in their unsatisfactory applications in optoelectronic devices such as OLEDs and lasers.7 In this context, it is noteworthy to mention that to overcome the problems associated with ACQ, two interesting photoluminescence concepts, i.e., “Aggregation Induced Emission (AIE)”, and “Aggregation Induced Emission Enhancement (AIEE)” have been subsequently reported by Tang et al. and Park et al., respectively.8,9 However, the main mechanism behind the AIEE phenomenon is the restriction of the intramolecular rotation (RIR) in propeller-shaped molecules. RIR has helped to block the nonradiative pathways resulting in enhanced emission response in the solid and aggregated states. AIEE has opened a new channel towards the blooming of novel and versatile organic luminescent molecules with high emission efficiency and fluorescence quantum yields. A few examples of AIEE active molecules reported are: aromatic silole derivatives,10 tetraphenylethenes,11 diphenyldi-benzofulvenes,12 pyrene,13 and thiophene derivatives.14 Thus, AIEE active organic materials are highly essential for luminescent devices such as OLEDs and also for other applications in material sciences. Moreover, designing of blue-emitting organic fluorophores has particularly remained to be a challenge because of their low emission efficiency and poor stability as compared to red and green-fluorophores reported for their outstanding performance.15 A few traditional blue-emitting molecules reported in the literature are anthracene,16 fluorene,17 pyrene derivatives,18 and triphenylamine derivatives.19 However, these molecules largely suffer from the ACQ effect in the aggregated states owing to their planar structures. Subsequently, the alternative polyaryl cyclopentadiene (Cp)-derivatives, viz., 1,2,3,4-tetraphenyl-1,3-cyclopentadiene (TPC) and 1,2,3,4,5-pentaphenyl-1,3-cyclopentadiene (PPCp) have been developed as functional propeller-shaped molecules.20 Additionally, their utility has been explored in electroluminescent devices as blue light-emitting layers.20b–f As a part of the continuous efforts21–23 in finding the blue-emitting organic fluorophores, Ning et al.22 developed a series of triaryl substituted Cp ligands with improved photoluminescence (PL) and high quantum yields.Apart from their excellent photophysical properties that they exhibit the Cp ligands are well established for the synthesis of one of the very important class of organometallic complexes, known as the sandwich complexes or the metallocenes.24 Since the discovery of ferrocene,25 the quest for developing new synthetic routes for novel sandwich complexes are on great demand because of their potential applications in catalysis, medicinal-, electro-chemistry, and as fuel additives.26 In this regard, syntheses of lanthanide-based sandwich complexes27 are noteworthy in recent times, especially because of their applications as single molecule magnets (SMMs) and single ion magnets (SIMs).28,29 Herein, we report the syntheses of two new (AIEE active and blue light emitting) triaryl Cp ligands (Me2Cp–H) (1) [Me2Cp–H = 4,4′-(4-phenylcyclopenta-1,3-diene-1,2-diyl)bis(methylbenzene)], (Me3Cp–H) (2) [Me3Cp–H = 4,4′,4′′-(cyclopenta-1,3-diene-1,2,4-triyl)tris(methylbenzene)] and their photophysical properties in solution, solid state and as aggregates. Finally, 1 and 2 have been shown to afford the two novel chloride bridged, tetra-nuclear, mixed potassium–dysprosium sandwich complexes [(Me2Cp)4Dy2IIICl4K2]·3.5(C7H8) (5) and [(Me3Cp)4Dy2IIICl4K2]·3(C7H8) (6), respectively in very good yields.
Results and discussion
The triaryl substituted cyclopentadienyl ligands 1 and 2 were synthesized in a three-step synthetic process. Initially, the 1,5-di-keto compounds (1a, 1b) were synthesized through cross-aldol condensation between the corresponding aromatic aldehyde and acetophenone derivatives in the presence of sodium hydroxide as a base. Next, the 1,2-diol compounds (2a, 2b) were obtained by the reductive cyclization of the corresponding 1,5-di-keto compounds (1a, 1b) in presence of Zn-dust/acetic acid mixture at 90–100 °C.22 Finally, the targeted triaryl substituted Cp compounds (Me2Cp–H) (1) and (Me3Cp–H) (2) were obtained by the dehydration of the corresponding 1,2-diol compounds (2a, 2b) in presence of ethanol and hydrochloric acid under reflux conditions (Scheme 1, see ESI† for details).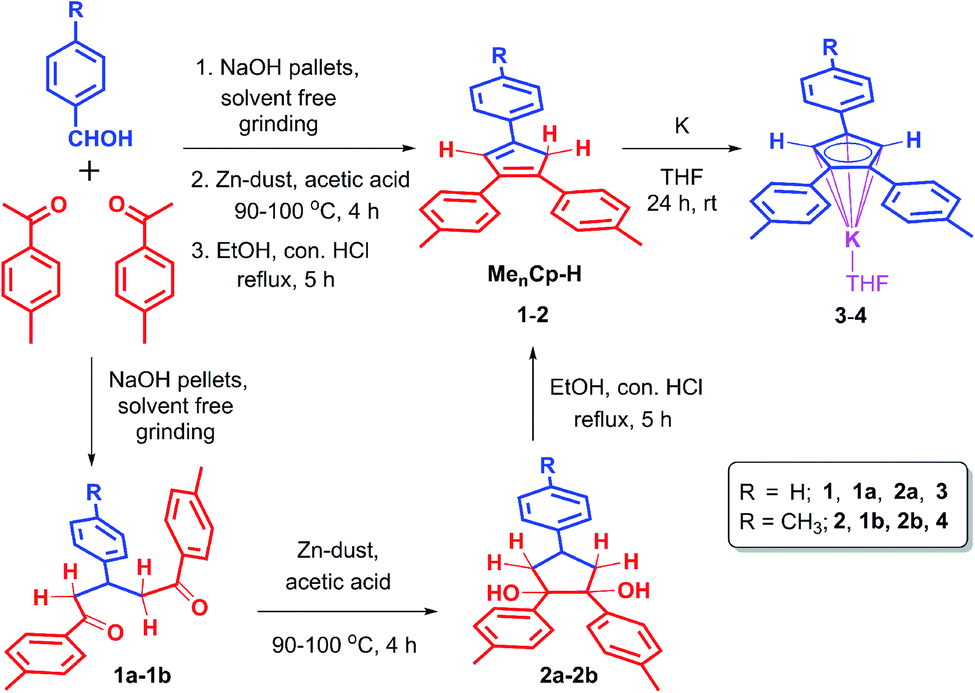 | ||
| Scheme 1 Synthetic routes for 4,4′-(4-phenylcyclopenta-1,3-diene-1,2-diyl)bis(methylbenzene) (1), 4,4′,4′′-(cyclopenta-1,3-diene-1,2,4-triyl)tris(methylbenzene) (2) and their potassium salts 3, 4. | ||
Compounds 1 and 2 are thoroughly characterized by IR, NMR, and mass spectrometric analysis (see ESI†). The molecular structures of 1 and 2 are determined by single-crystal X-ray diffraction studies. After successful syntheses and characterization of 1 and 2, we have investigated their photophysical properties in solution (Fig. 1) as well as in solid state using UV-visible (Fig. 1b), diffuse reflectance spectroscopy (DRS), and photoluminescence (PL) spectroscopic techniques. Fig. 1a shows UV-visible absorption and emission spectra of 1 and 2 in DCM, where both of these compounds display absorption at 359 nm (HOMO to LUMO). The π–π* transitions of the conjugated aromatic phenyl moieties (see ESI† for TD-DFT calculations) and core Cp units in 1 and 2 (ΔELUMO–HOMO = 5.32 (1), 5.29 (2) eV) are responsible for this absorption. The fluorescence spectra of 1 and 2 in DCM shows emission at 455 nm and 470 nm, respectively when excited at 359 nm (Fig. 1a).
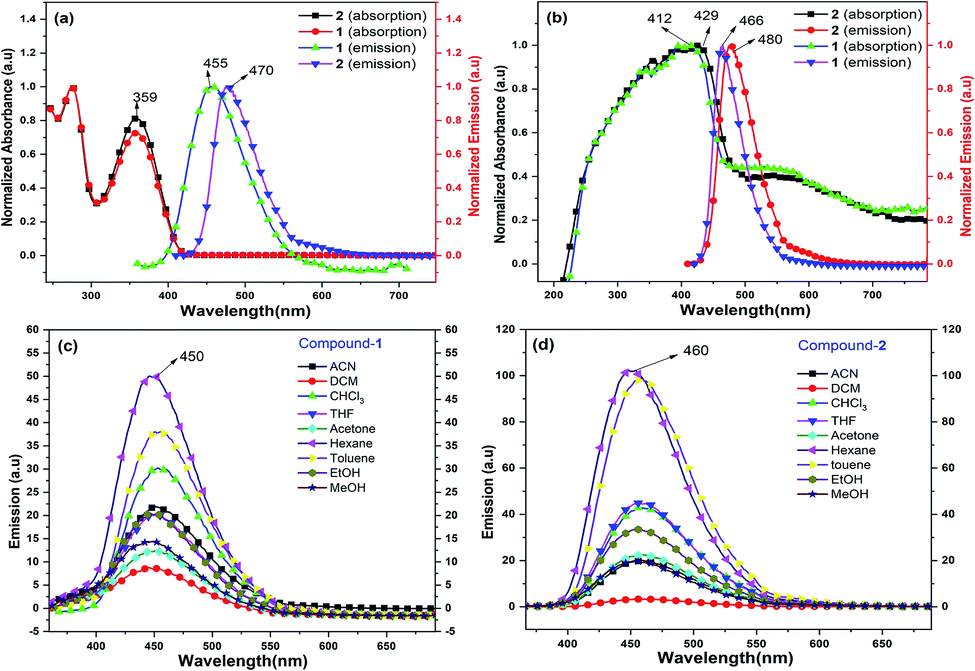 | ||
| Fig. 1 (a) UV-visible absorption (left) and emission spectra (right) of 1 and 2 in DCM; (b) solid-state DRS-UV (left) and emission spectra (right) of 1 and 2; (c) PL spectra of 1 (left); (d) PL spectra of 2 (right) in different solvents. | ||
The solid-state photoluminescence (PL) spectra of 1 and 2 display emissions at 466 nm and 480 nm, respectively (Fig. 1b). Moreover, 1 and 2 illuminate bright green-blue light in their solid-state upon irradiation with UV-365 nm light as shown in Fig. 2a. It is worth mentioning that the solid state absorption and emissions of compounds 1–2 are red-shifted with higher intensity than those observed in solution state PL spectra, attributing to their solid state packing (Fig. 2b), leading to AIEE property2,8,30 (see ESI†). Interestingly, the emission spectra of 1 and 2 in different solvents are influenced by the nature of the solvent polarity.22c Emissions with higher intensities were noticed in non-polar, non-coordinating solvents (n-hexane, toluene) than those in polar, coordinating (MeCN, THF, Me2CO, MeOH, EtOH) and polar, non-coordinating (DCM, CHCl3) solvents. For example, the emission of 1 in n-hexane and toluene are slightly red-shifted with high intensity when compared to the emission in polar solvents (Fig. 1c). Likewise, the similar trend was noticed for 2, but the emission in n-hexane is slightly blue-shifted with higher intensity than in toluene (Fig. 1d). Moreover, the quenching of fluorescence activity was noticed for both the compounds 1–2 in polar solvents like, DCM, acetone and methanol as shown in Fig. 1c and d (also see ESI, Fig. S6†). This is probably due to the unchanged singlet excited state of 1 and reduced non-emissive decay with decreasing solvent polarity (Fig. S6†).22c,29 In polar, coordinating solvents (MeOH, EtOH, MeCN, THF, Me2CO) (Fig. S4–S6†) the electrons in the excited states of molecules can undergo relaxation via radiative processes due to the possible Cp–H⋯solvent interactions (donor atom of solvent interacting with acidic Cp–H of ligands 1–2, see ESI†), leading to the observed decrease in PL intensity. In case of non-polar, non-coordinating solvents, like toluene and n-hexane; such solvent interactions with MenCp–H (1, 2) are not observed leading to high PL intensity. The AIEE properties of 1 and 2 were studied in acetone/water and THF/water mixtures which were further correlated with their respective solid state structures and 3D molecular packing diagrams (see ESI† for details).
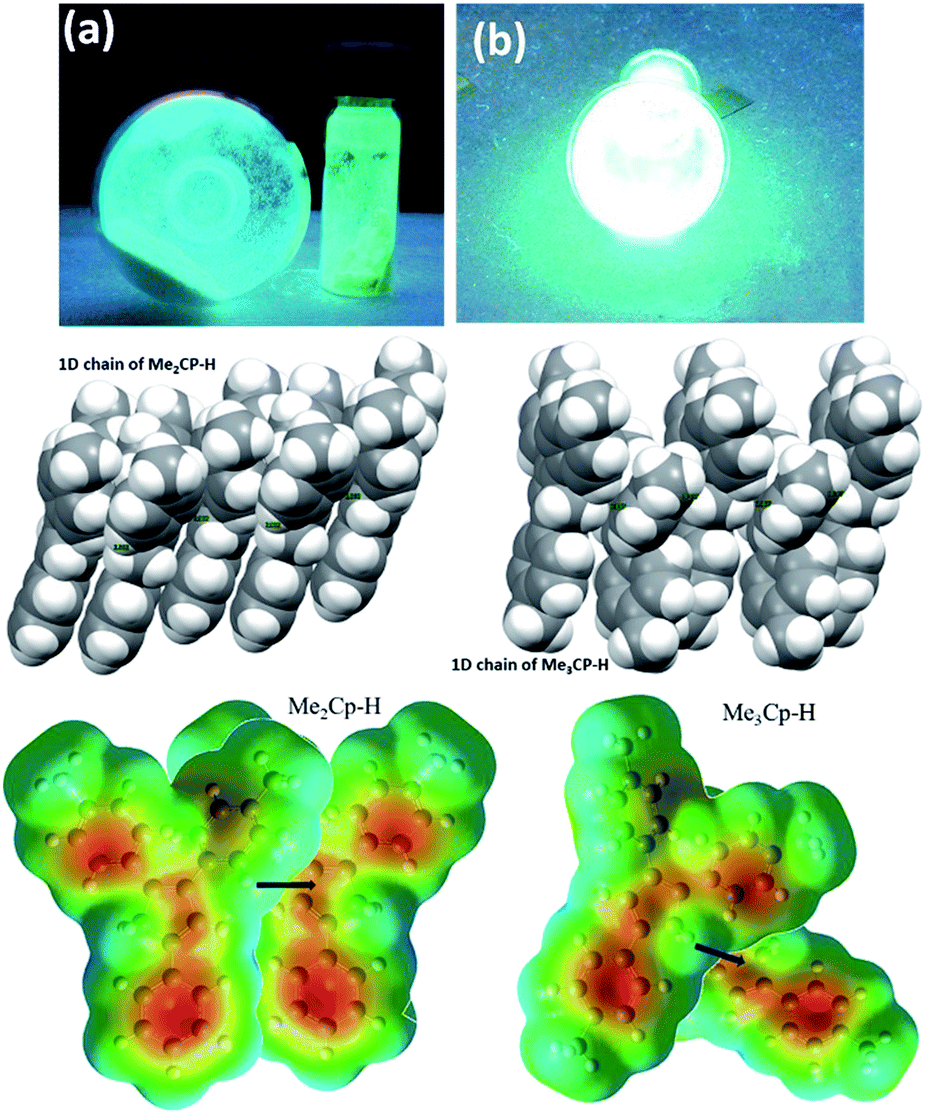 | ||
| Fig. 2 Ligand 1 with UV light (365 nm (a)) and its packing along 1D chain (top, left). Ligand 2 with UV light (365 nm (b)) and its packing along 1D chain (top, right). Intermolecular C–HCp⋯πCp interactions (middle) shown in space filling model along 1D chain leading to much brighter emission of 2 than that of 1. Molecular electrostatic potential (MEP) surfaces of two interacting neighbouring molecules of 1 (bottom, left) and 2 (bottom, right). | ||
The fluorescence lifetimes of 1–2 were measured both in solution- and solid states which were found to be nearly 7–8 times higher in the solid state (2.25 ns (1), 1.6 ns (2)) than those in solutions (0.3 ns (1), 0.2 ns (2)) (Fig. S2†). The quantum yields of 1 and 2 are found to be 4.5% and 10.3%, respectively in toluene (see ESI†).22
Moreover, a detailed AIEE studies of 1–2 were investigated in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water or acetone:water mixture by sequential addition of different fractions of water into THF or acetone. According to our findings, compounds 1 and 2 exhibited highest fluorescence emissions when the fractions of water were 60% (acetone:water mixture, see ESI†) and 80% (THF:water mixtures, Fig. 3), respectively. The increase of fluorescence intensity can be attributed to the AIEE effect caused by the formation of molecular nano-aggregates in the miscible solvent pair, which can restrict the intermolecular rotations of molecules resulting in increased fluorescence emission.8,22
:water or acetone:water mixture by sequential addition of different fractions of water into THF or acetone. According to our findings, compounds 1 and 2 exhibited highest fluorescence emissions when the fractions of water were 60% (acetone:water mixture, see ESI†) and 80% (THF:water mixtures, Fig. 3), respectively. The increase of fluorescence intensity can be attributed to the AIEE effect caused by the formation of molecular nano-aggregates in the miscible solvent pair, which can restrict the intermolecular rotations of molecules resulting in increased fluorescence emission.8,22
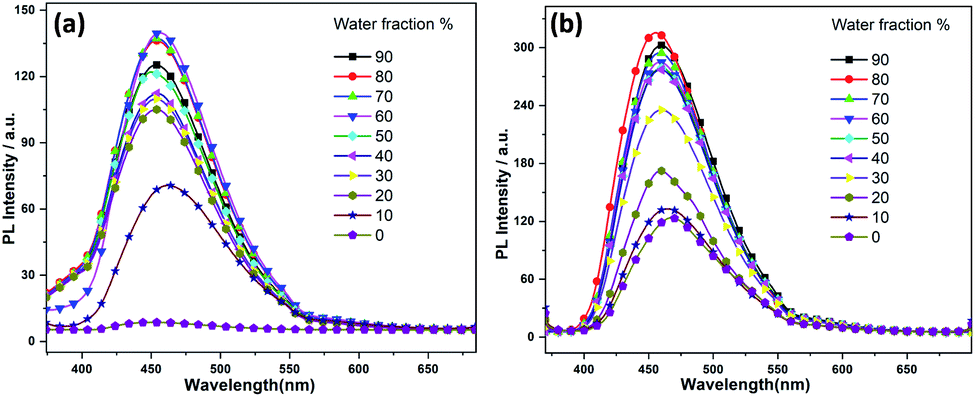 | ||
| Fig. 3 Fluorescence spectra of (a) 1 and (b) 2 in water/THF mixture with the increases of the water fractions, starting from 10–90%. | ||
The detailed AIEE investigations of these compounds are described in ESI.† In addition, to understand the mechanism for florescence emission of compounds 1 and 2 in the solid or aggregated states, we have investigated the relationship between their structure and photophysical properties. The molecular structures of compounds 1 and 2 adopt non-coplanar geometry and possess different packing patterns in the crystal lattice as shown in Fig. 4 (top) (also see Fig. S10–S14, ESI†). Briefly, due to strong C–H⋯π interactions exerted between the molecules of 1 or 2 can led to 1D J-type molecular aggregation of molecular columns in the crystal lattice (Fig. 2, middle), this means intermolecular motions are not free between the molecules hence enhanced PL intensity observed (for detailed mechanistic ESI†).
 | ||
| Fig. 4 Molecular structures of 1–2 (top); color codes: carbon: grey; hydrogen: yellow. Laplacian contour plots of ligands 1–2 (bottom). | ||
The contour plots of the Laplacian for ligands 1–2 are shown in Fig. 4, (bottom). The electron density greater than 0.2, negative Laplacian ∇2ρ(r) and negative H(r) (Table S5†) at the bond critical point indicate an electron sharing covalent bond between Cp and the phenyl rings. The Bader charge on phenyl ring is slightly positive (+0.007), indicating a possible very weak charge transfer from Cp-ring to phenyl ring of 1–2. The contour plots show charge accumulation near to the C-atoms in both Cp and phenyl rings supporting the electron sharing interactions (Fig. 4, bottom). The extracted parameters (Table S5†) of each molecule of 1–2 are very similar, suggeting that the differences in their luminescence properties are solely due to their differences in the molecular packing in solid state (Fig. 2, also see ESI† for detailed structure descriptions (1–2) and correlation).
After completing the PL studies of 1 and 2, we explored their applicability as the stabilizing ligands for the syntheses of rare earth,31 Dy(III)-based sandwich24–27 complexes. Initially, we converted 1 and 2 into their corresponding potassium cyclopentadienyl salts, (Me2Cp)K(THF) (3) and (Me3Cp)K(THF) (4) by separately reacting 1 and 2 with K-metal (1.2 equiv) for 24 h in THF at room temperature (Scheme 1). In case of complex 3, the color of the reaction mixture changed from yellow to dark blue upon reaction completion as indicated by the complete consumption of the metallic potassium. This might be due to the possible, facile charge transfer from the central anionic-Cp ring to the adjacent phenyl ring of 3, leading to a quinone like bonding situation (partial CPh–CCp double bond) in certain rotamers of 3. Free rotation of phenyl ring around CPh–CCp bond can increase the delocalization of electron densities with a lower HOMO–LUMO energy gap of 3 producing a blue color. Such delocalization might have reduced in the most stable rotational conformation of 3, leading to the light-yellow color. Another possibility is that the reaction of 1 with potassium metal proceeds via a radical intermediate affording the potassium salt 3.
The filtration of the blue reaction mixture, followed by removal of THF under high vacuum yielded 3 as a pale-yellow solid. In contrast, 2 did not show any color change of the reaction mixture. Instead, the formation of yellow precipitate was observed after 24 h. The pure, yellow solid of 4 was obtained upon filtration, followed by the subsequent drying under high vacuum. These KCp salts (3–4) are found to be highly sensitive towards moisture and air, hence stored under inert atmosphere. The disappearance of the Cp–H resonances for KCp salts in the 1H-NMR spectra was indicative of the formation of the 6π electronic aromatic Cp core.
NBO analysis (Fig. 5) of Me2Cp− at M06-GD3/def2TZVP level of theory indicates that the HOMO consists of the π-cloud (Fig. 5c) in the central Cp-core and the phenyl rings, while the LUMO is composed of vacant π* orbitals (Fig. 5d). The NBO analysis indicates covalent C–C σ-bonds with high occupancy (1.96–1.97) for all five C–C bonds of the Cp-core, with WBI (Wiberg Bond Indices) values ranging from 1.22–1.41, indicating partial double bond character consistent with the aromaticity. The C–C double bonds carry a low occupation of 1.70–1.77e. The NBO analysis also indicates the existence of highly conjugated π-cloud in the central Cp-core in both the Cp− anions (see ESI†).
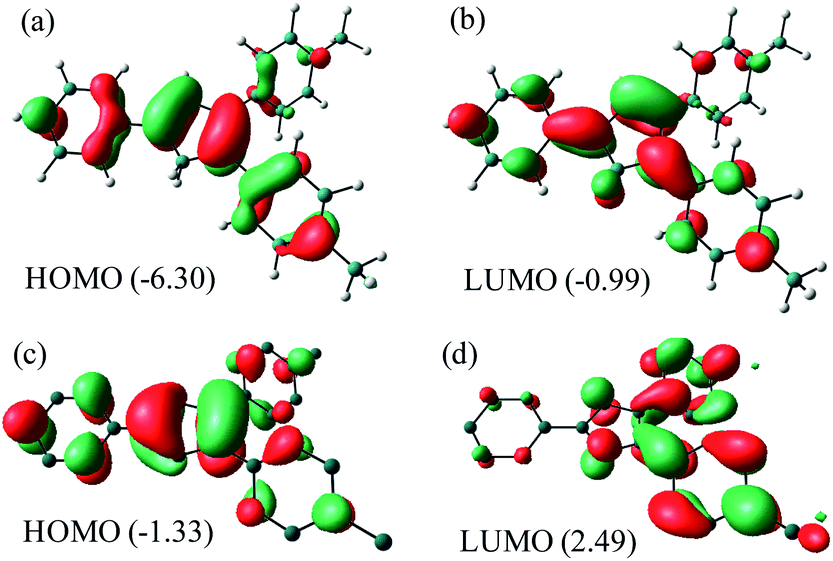 | ||
| Fig. 5 HOMO and LUMO of Me2Cp–H (1) (a and b) and Me2Cp− anion (c and d) calculated at M06-GD3/def2TZVP level of theory (in gas phase). Energies are in eV. | ||
The energies of HOMO and LUMO of the anions (MenCp−) are higher than those of their corresponding protonated neutral analogues (1–2). Moreover, the HOMO–LUMO energy gaps of the anions are found to be smaller than those of their neutral analogues (1–2) (Table S7†).
Me2Cp–H (1) was further optimized in singlet diradical excited state (1*), which is 3.06 eV (70.6 kcal mol−1) higher in energy than its closed shell singlet ground state. This difference in energy is very close to the energy of the photon (∼360 nm, Fig. 1) required to excite 1 → 1*. The α-SOMO and β-SOMO of 1* are given in Fig. 6.
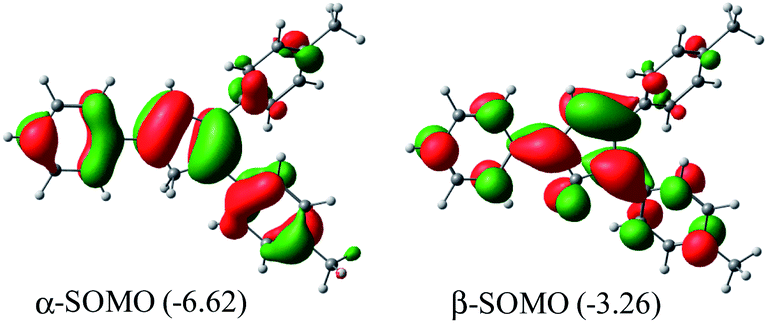 | ||
| Fig. 6 α- and β-SOMO of Me2Cp–H (1*) singlet biradical state (in gas phase) calculated at UM062x(GD3)/Def2TZVPP//BP86(GD3)/Def2TZVPP level of theory. | ||
The reaction between (MenCp)K(THF) salts (3–4) and anhydrous DyCl3 either in toluene or THF at high temperature in a pressure tube at 110 °C does not lead to the isolation of desired MenCp-containing Dy-complexes. However, the one pot synthetic approach became successful (Scheme 2). A 1:2:2 molar mixture of anhydrous DyCl3, MenCp–H and K-metal in toluene at 110 °C stirred for two days under argon atmosphere leading to the formation of light-yellow turbid reaction mixture. Upon filtration of the reaction mixture, the clear filtrate was concentrated and stored at room temperature for 4 days to produce the light yellow blocks of [(Me2Cp)4Dy2IIICl4K2]·3.5(C7H8) (5) or [(Me3Cp)4Dy2IIICl4K2]·3(C7H8) (6) in 71–72% yields, respectively. The molecular structures of complexes 5 and 6 were determined by X-ray single crystal diffraction at 200 K. The structure refinement and selected bond parameters for complexes 5 and 6 are given in the ESI.†
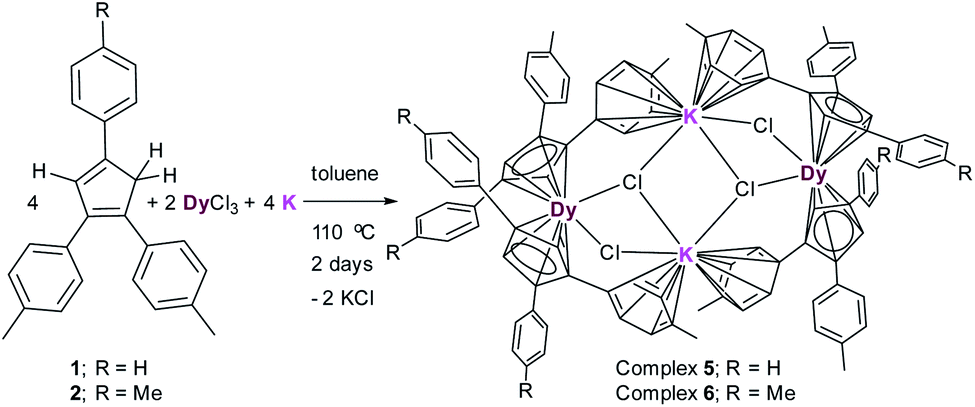 | ||
| Scheme 2 Syntheses of complexes [(Me2Cp)4Dy2IIICl4K2]·3.5(C7H8) (5) and [(Me3Cp)4Dy2IIICl4K2]·3(C7H8) (6). | ||
Structural description of complex 5
Complexes 5 and 6 are iso-structural; hence, only the structure of 5 (Fig. 7) is described here (see ESI† for molecular structure of 6). Complex [(Me2Cp)4Dy2IIICl4K2]·3.5(C7H8) (5) crystallizes in triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. It possesses a butterfly like Dy2K2Cl4 core (Fig. 8) with two μ3-bridged Cl-atoms (Cl3, Cl1) below (1.02 and 1.43 Å) and other two μ-bridged Cl-atoms (Cl4, Cl2) above (1.76 and 1.80 Å) the Dy2K2-plane (Fig. 7). Two K+ and Dy3+ ions occupy the body and wing positions, respectively. Each K+ ion is centered between two phenyl rings showing η6 coordination with K–C distance in the range of 3.0729(12)–3.502(3) Å. On the hand, each Dy3+ ion is sandwiched between two cyclopentadienyl rings showing η5 coordination with Dy–C distances in the range of 2.644(3)–2.768(3) Å. This adopts a bent metallocene structure with the corresponding angles (Cp–Dy–Cp centroid) in the range of 129.73–130.85° (5). These bending angles are significantly lower than those in the previously reported neutral [(Cpttt)2DyCl] (147.59°)28b and cationic complexes [Dy(Cp*)2{μ-(Ph)2BPh2}] (134.00°),28g [Dy(Cpttt)2][B(C6F5)4] (152.56(7)°)28d and [(η5-CpiPr5)Dy(η5-Cp*) (BC6F5)4] (162.507(1)°).28c In addition, the Dy–Cpcen distances in 5 vary in the range of 2.391–2.423 Å which is very close to that of [(Cpttt)2DyCl] (2.413 Å)28b and greater than that of the corresponding cationic complex [(Cpttt)2Dy]+ (2.309–2.324 Å).28b
space group. It possesses a butterfly like Dy2K2Cl4 core (Fig. 8) with two μ3-bridged Cl-atoms (Cl3, Cl1) below (1.02 and 1.43 Å) and other two μ-bridged Cl-atoms (Cl4, Cl2) above (1.76 and 1.80 Å) the Dy2K2-plane (Fig. 7). Two K+ and Dy3+ ions occupy the body and wing positions, respectively. Each K+ ion is centered between two phenyl rings showing η6 coordination with K–C distance in the range of 3.0729(12)–3.502(3) Å. On the hand, each Dy3+ ion is sandwiched between two cyclopentadienyl rings showing η5 coordination with Dy–C distances in the range of 2.644(3)–2.768(3) Å. This adopts a bent metallocene structure with the corresponding angles (Cp–Dy–Cp centroid) in the range of 129.73–130.85° (5). These bending angles are significantly lower than those in the previously reported neutral [(Cpttt)2DyCl] (147.59°)28b and cationic complexes [Dy(Cp*)2{μ-(Ph)2BPh2}] (134.00°),28g [Dy(Cpttt)2][B(C6F5)4] (152.56(7)°)28d and [(η5-CpiPr5)Dy(η5-Cp*) (BC6F5)4] (162.507(1)°).28c In addition, the Dy–Cpcen distances in 5 vary in the range of 2.391–2.423 Å which is very close to that of [(Cpttt)2DyCl] (2.413 Å)28b and greater than that of the corresponding cationic complex [(Cpttt)2Dy]+ (2.309–2.324 Å).28b
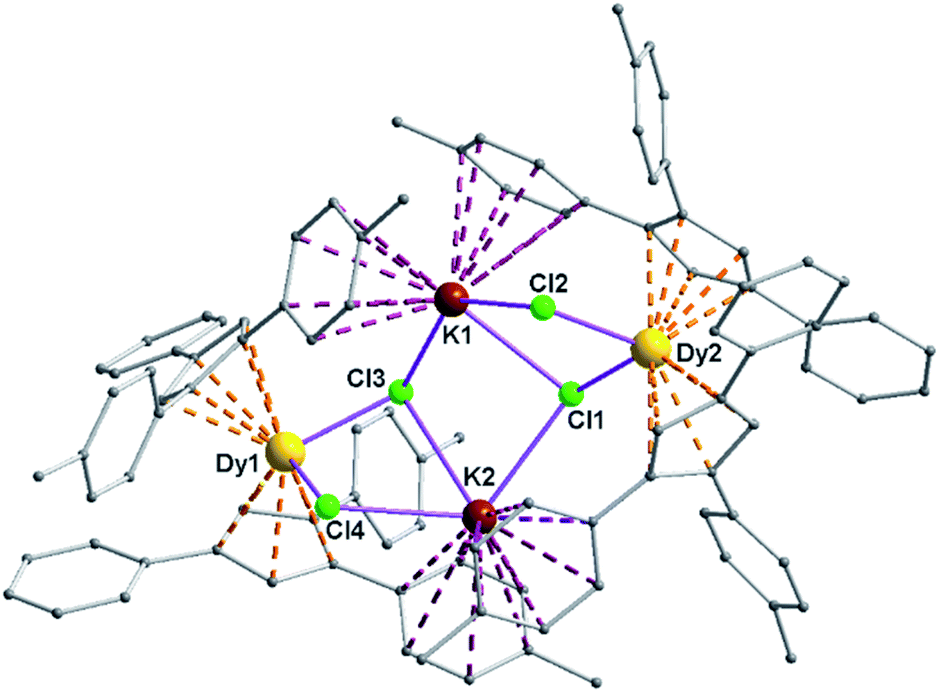 | ||
| Fig. 7 Molecular structure of 5; the hydrogen atoms are omitted for the clarity. Selected bond lengths (Å) and bond angles (°) are as follows: Cl1–Dy2 2.6185(8), Cl2–Dy2 2.5874(8), Cl3–Dy1 2.5993(8), Cl4–Dy1 2.5903(8); Cl1–K2 3.0279 (12), Cl1–K1 3.0559(11), Cl2–K1 3.1805(11), Cl3–K1 3.1404(12); Dy2–Cl1–K2 129.24 (3), Dy2–Cl1–K1 97.95 (3), K2–Cl1–K1 91.61 (3). | ||
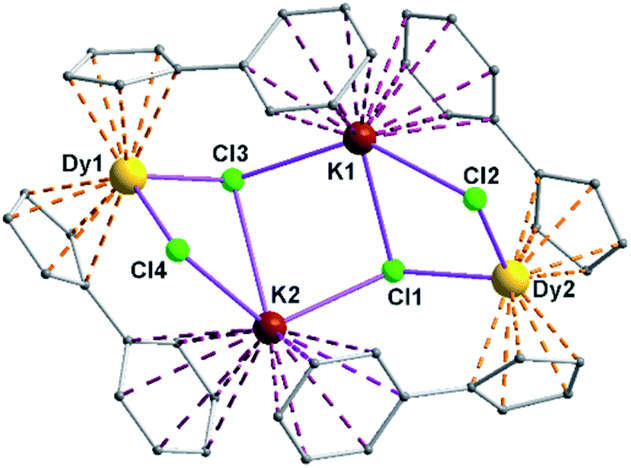 | ||
| Fig. 8 Non planar Dy2K2Cl4 core along with chelating aromatic units of complex 5. | ||
The Arcen1⋯K⋯Arcen2 (Ar = 4-methylphenyl unit) angles are found to be in the range of 113.97–116.84° (5–6). The K⋯K and Dy⋯Dy distances in the K2Cl4Dy2 core are 4.363 Å and 8.233 Å, respectively. Moreover, the Dy–Cl and K–Cl bond distances in 5 are in the range of 2.5903(8)–2.6185(12) Å and 3.0279(12)–3.4657(12) Å, respectively.
It is to be noted that the Dy–Cl distances reported here fall in the similar range of those reported previously in the complex, [Dy(Cpttt)2Cl] (2.5400(13) Å).28b The weak C–H⋯π interactions exerted between the molecules of 5 led to the supramolecular 2D network in the crystal lattice (see ESI†).
The UV/vis spectrum of 5, recorded in acetonitrile (MeCN) solution shows three absorption maxima at 232, 262 and 354 nm. The excitations of MeCN solution of 5 at 262, 320 and 354 nm lead to the ligand-centered emissions in the visible range (426 nm) of spectrum (Fig. 9). The emission band of 5 is blue shifted by 30 nm when it is compared with that of neutral ligand 1. The quantum yield of complex 5 is 0.8% in MeCN solution which is approximately six times lower than that of its neutral ligand (Me2Cp–H, 1), suggesting the significant quenching of fluorescence emission in 5. The UV/vis/NIR measurement of complex 5 in the range of 200–2000 nm did not show absorption bands other than those seen in the UV/vis region (262, 320 and 354 nm).
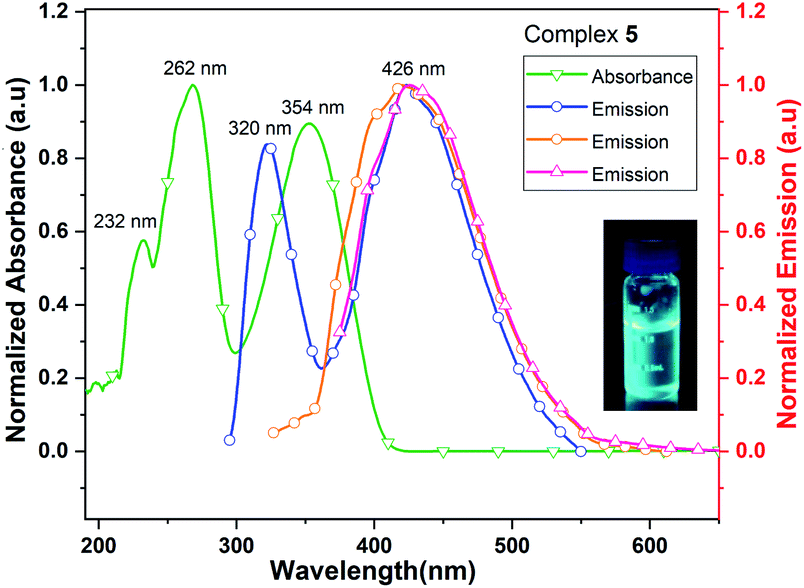 | ||
| Fig. 9 UV/vis absorption (green line) and emission (blue, orange and red lines when excited at 262, 320 and 354 nm, respectively) spectra of complex 5 in MeCN solution. | ||
Complex 4 (Me3CpK(THF)) also exhibits fluorescence emission at 456 nm when it is excited at 241 and 350 nm (Fig. 10). The quantum yield of complex 4 is calculated as 4.5% in MeCN solution. The signatures of UV/vis absorption spectra of all the metal (K and K/Dy) complexes (4–5) are found to be significantly different.
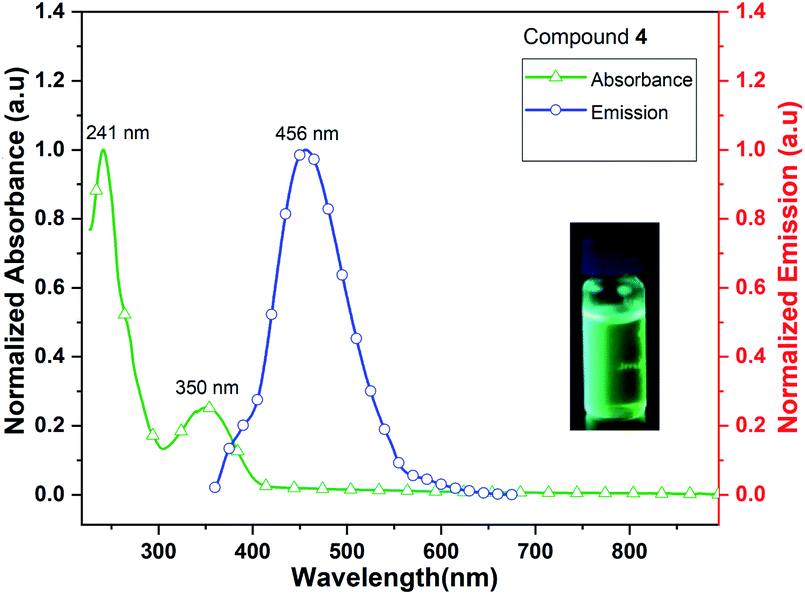 | ||
| Fig. 10 UV/vis absorption and emission spectra of Me3CpK(THF) (4). | ||
Conclusions
In conclusion, two alkyl-substituted triaryl-cyclopentadiene derivatives 1 and 2 have been synthesized and shown to display captivating fluorescence glow in the solid state upon irradiation with UV-lamp. The central five membered ring to ring direct electrostatic interaction along the 1D-chain has been found to be crucial for exhibiting a better fluorescence property by 2 than 1. Theoretical calculations and structure–property correlations have shown that the molecular electrostatic potential (MEP) maps along with the QTAIM analysis are helpful to rationalize the luminescent properties of 1–2. Deprotonation of 1–2 has afforded the corresponding potassium cyclopentadienide salts 3–4, respectively. Both the ligands 1, 2 and their corresponding cyclopentadienide anions have been studied by theoretical calculations. Most importantly, we have shown that the ligands 1 and 2 could be effectively utilized to obtain the novel chloride bridged, mixed-metallic tetra-nuclear potassium–dysprosium sandwich complexes (5–6). The metal (K and K/Dy) complexes (4–5) display fluorescence emission in the visible region of the wavelength. Utilization of these complexes as precursors for the lower coordinate Dy-cations are underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SR and KCM gratefully acknowledge SERB, New Delhi for their individual ECR grants (ECR/2016/000733 for SR; ECR/2016/000890 for KCM). SA, MF thank CSIR for SRF and JRF, respectively. PGR thanks IISER Tirupati for Postdoctoral Fellowship. AK thanks KYPY for financial support.References
- N. T. Kalyani and S. J. Dhoble, Renewable Sustainable Energy Rev., 2012, 16, 2696 CrossRef.
- F. F. Wang, J. Y. Wen, L. Y. Huang, J. J. Huang and J. Ouyang, Chem. Commun., 2012, 7395 RSC.
- W. Z. Yuan, S. Chen, J. W. Y. Lam, C. Deng, P. Lu, H. H. Y. Sung, I. D. Williams, H. S. Kwok, Y. Zhang and B. Z. Tang, Chem. Commun., 2011, 47, 11216 RSC.
- R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lama and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228 RSC.
- Photonic devices: S. Chen, Y. Hong, Y. Liu, J. Liu, C. W. T. Leung, M. Li, R. T. K. Kwok, E. G. Zhao, J. W. Y. Lam, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 4926 CrossRef CAS.
- M. A. Baldo, M. E. Thomson and S. R. Forrest, Nature, 2000, 403, 750 CrossRef CAS.
- (a) T. Y. Han, X. Feng, B. Tong, J. B. Shi, L. Chen, J. G. Zhi and Y. P. Dong, Chem. Commun., 2012, 48, 416 RSC; (b) J. Huang, X. Yang, J. Y. Wang, C. Zhong, L. Wang, J. G. Qin and Z. Li, J. Mater. Chem., 2012, 22, 2478 RSC.
- G. Yu, S. W. Yin, Y. Q. Liu, J. S. Chen, X. J. Xu, X. B. Sun, D. G. Ma, X. W. Zhan, Q. Peng, Z. G. Shuai, B. Z. Tang, D. B. Zhu, W. H. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335 CrossRef CAS.
- AIE mechanism: (a) B. K. An, D. S. Lee, J. S. Lee, Y. S. Park, H. S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232 CrossRef CAS; (b) B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS.
- L. Chen, Y. Jiang, H. Nie, P. Lu, H. H. Y. Sung, I. D. Williams, H. S. Kwok, F. Huang, A. Qin, Z. Zhao and E. Al, Adv. Funct. Mater., 2014, 24, 3621 CrossRef CAS.
- Tetraphenyl-ethylenes: (a) Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726 RSC; (b) J. Yang, J. Huang, Q. Q. Li and Z. Li, J. Mater. Chem. C, 2016, 4, 2663 RSC.
- Diphenyl-dibenzofulvenes: (a) X. Zhang, Z. Chi, B. Xu, L. Jiang, X. Zhou, Y. Zhang, S. Liu and J. Xu, Chem. Commun., 2012, 48, 10895 RSC; (b) J. He, B. Xu, F. Chen, H. Xia, K. Li, L. Ye and W. Tian, J. Phys. Chem. C, 2009, 113, 9892 CrossRef CAS.
- Pyrene derivatives: V. Kachwal, M. Joshi, V. Mittal, A. R. Choudhury and I. R. Laskar, Sensing and Bio-Sensing Research, 2019, 23, 100267 CrossRef.
- Pyrene-thiophene derivatives: P. Yadav, S. Gond, A. K. Singh and V. P. Singh, J. Lumin., 2019, 215, 116704 CrossRef CAS.
- Red and green-fluorophores: (a) S. Lamansky, P. Djurovich, D. Murphy, F. A. Razzaq, H. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS; (b) D. H. Kim, N. S. Cho, H. Oh, J. H. Yang, W. S. Jeon, J. S. Park, M. C. Suh and J. H. Kwon, Adv. Mater., 2011, 23, 2721 CrossRef CAS.
- Anthracene Derivatives: C. Wu, C. Chang, Y. Chang, C. Chen, C. Chen and C. Su, J. Mater. Chem. C, 2014, 2, 7188 RSC.
- Fluorene Derivatives: (a) A. Frazer, A. R. Morales, A. W. Woodward, P. Tongwa, T. Timofeeva and K. D. Belfield, J. Fluoresc., 2014, 24, 239 CrossRef CAS; (b) X. J. Feng, J. Peng, Z. Xu, R. Fang, H. Zhang, X. Xu, L. Li, J. Gao and M. S. Wong, ACS Appl. Mater. Interfaces, 2015, 7, 28156 CrossRef CAS.
- Pyrene Derivatives: Z. Zhao, S. Chen, J. W. Y. Lam, Z. Wang, P. Lu, F. Mahtab, H. H. Y. Sung, I. D. Williams, Y. Ma, H. S. Kwok and E. Al, J. Mater. Chem., 2011, 21, 7210 RSC.
- Tri-phenyl amine derivatives: F. Hao, D. Li, Q. Zhang, S. Li, S. Zhang, H. Zhou, J. Wu and Y. Tian, Spectrochim. Acta, Part A, 2015, 150, 867 CrossRef CAS.
- TPC and PPCp derivatives: (a) X. C. Gao, H. Cao, L. Huang, Y. Y. Huang, B. W. Zhang and C. H. Huang, Appl. Surf. Sci., 2003, 210, 183 CrossRef CAS; (b) Y. Ohmori, Y. Hironaka, M. Yoshida, A. Fujii and K. Yoshino, Jpn. J. Appl. Phys., 1996, 35, 4105 CrossRef CAS; (c) K. Sugiyama, D. Yoshimura, T. Miyamae, T. Miyazaki and H. Ishii, J. Appl. Phys., 1998, 83, 4928 CrossRef CAS; (d) N. Tada, A. Fujii, Y. Ohmori and K. Yoshino, IEEE Trans. Electron Devices, 1997, 44, 1234 CrossRef CAS; (e) Y. Ohmori, N. Tada, A. Fujii, H. Ueta, T. Sawatani and K. Yoshino, Thin Solid Films, 1998, 331, 89 CrossRef; (f) Y. S. Zhao, H. B. Fu, F. Q. Hu, A. D. Peng and J. N. Yao, Adv. Mater., 2007, 19, 3554 CrossRef CAS.
- Synthetic Strategy for TPC: L. Stojanović and R. C. Otero, ChemPhotoChem, 2019, 3, 907 CrossRef.
- Triaryl-Cp derivatives: (a) J. Ye, X. Huang, Y. Li, T. Zheng, G. Ning, J. Liang, Y. Liu and Y. Wang, Dyes Pigm., 2017, 147, 465 CrossRef CAS; (b) X. Zhang, J. Ye, L. Xu, L. Yang, D. Deng and G. Ning, J. Lumin., 2013, 139, 28 CrossRef CAS; (c) J. Ye, D. Deng, Y. Gao, X. Wang, L. Yang, Y. Lin and G. Ning, Spectrochim. Acta, Part A, 2015, 134, 22 CrossRef CAS; (d) L. Yang, J. Ye, L. Xu, X. Yang, W. Gong, Y. Lin and G. Ning, RSC Adv., 2012, 2, 11529 RSC.
- E. Orselli, G. S. Kottas, A. E. Konradsson, P. Coppo, R. Fröhlich, L. D. Cola, A. V. Dijken, M. Büchel and H. Börner, Inorg. Chem., 2007, 46(26), 11082 CrossRef CAS.
- (a) C. Elschenbroich, Organometallics, Wiley-VCH, Weinheim, 3rd edn, 2006 Search PubMed; (b) A. Streitwieser and U. Müller-Westerhoff, J. Am. Chem. Soc., 1968, 90, 7364 CrossRef CAS; (c) E. O. Fischer and W. Hafner, Z. Naturforsch., B: Chem. Sci., 1955, 10, 665 Search PubMed; (d) F. Mares, K. Hodgson and A. Streitwieser, J. Organomet. Chem., 1970, 24, C68 CrossRef CAS; (e) M. D. Walter, G. Wolmershäuser and H. Sitzmann, J. Am. Chem. Soc., 2005, 127, 17494 CrossRef CAS; (f) K. Kawasaki, et al., Chem. Commun., 2017, 53, 6557 RSC; (g) M. Xémard, et al., J. Am. Chem. Soc., 2018, 140, 14433 CrossRef; (h) P. E. Riley and R. E. Davis, J. Organomet. Chem., 1976, 113, 157 CrossRef CAS; (i) H. Braunschweig, et al., Chem. Eur.–J., 2013, 19, 270 CrossRef CAS.
- (a) T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039 CrossRef CAS; (b) G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125 CrossRef CAS; (c) E. O. Fischer and W. Pfab, Z. Naturforsch., B: Chem. Sci., 1952, 7, 377 Search PubMed.
- P. Štěpnička, Ferrocenes: Ligands, Materials and Biomolecules, John Wiley & Sons, Ltd, Chichester, 2008 Search PubMed.
- (a) W. J. Evans, C. A. Seibel and J. W. Ziller, J. Am. Chem. Soc., 1998, 120(27), 6745 CrossRef CAS; (b) R. R. Langeslay, C. J. Windorff, M. T. Dumas, J. W. Ziller and W. J. Evans, Organometallics, 2018, 37(3), 454 CrossRef CAS; (c) C. A. Gould, K. R. McClain, J. M. Yu, T. J. Groshens, F. Furche, B. G. Harvey and J. R. Long, J. Am. Chem. Soc., 2019, 141, 12967 CrossRef CAS; (d) C. A. P. Goodwin, D. Reta, F. Ortu, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2017, 139(51), 18714 CrossRef CAS; (e) D. H. Woen, C. M. Kotyk, T. J. Mueller, J. W. Ziller and W. J. Evans, Organometallics, 2017, 36(23), 4558 CrossRef CAS; (f) W. J. Evans, Organometallics, 2016, 35(18), 3088 CrossRef CAS.
- Lanthanide based SMMs: (a) J. Wu, S. Demeshko, S. Decherta and F. Meyer, Chem. Commun., 2020, 56, 3887 RSC; (b) F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. M. ki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445 CrossRef CAS; (c) F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400 CrossRef CAS; (d) C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439 CrossRef CAS; (e) L. E. Moreno, J. J. Baldoví, A. G. Ariño and E. Coronado, Inorg. Chem., 2019, 58, 11883 CrossRef; (f) K. R. Meihaus, M. E. Fieser, J. F. Corbey, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2015, 137, 9855 CrossRef CAS; (g) S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546 CrossRef CAS and the references therein; (h) L. Münzfeld, C. Schoo, S. Bestgen, E. Moreno-Pineda, R. Köppe, M. Ruben and P. W. Roesky, Nat. Commun., 2019, 10, 3135 CrossRef.
- QTM effect: D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, 2006 Search PubMed.
- R. Hu, S. Li, Y. Zeng, J. Chen, S. Wang, Y. Li and G. Yang, Phys. Chem. Chem. Phys., 2011, 13, 2044 RSC.
- D. M. Roitershtein, L. N. Puntus, A. A. Vinogradov, K. A. Lyssenko, M. E. Minyaev, M. D. Dobrokhodov, I. V. Taidakov, E. A. Varaksina, A. V. Churakov and I. E. Nifant'ev, Inorg. Chem., 2018, 57, 10199 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Single crystal X-ray structures of 1, 2, 5, 6, NMR spectroscopic data, and computational details. CCDC 2006636, 2006639 for compounds 1, 2; 2006659, 2007965 for complexes 5, 6, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra05316c |
| ‡ SA and PGPR contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |