
Stimuli-responsive dynamic pseudorotaxane crystals
Masaki
Horie
 * and
Chi-Hsien
Wang
* and
Chi-Hsien
Wang
Department of Chemical Engineering, National Tsing Hua University, 101, Sec. 2, Kuang-Fu Road, Hsinchu 30013, Taiwan. E-mail: mhorie@mx.nthu.edu.tw
First published on 2nd September 2019
Abstract
This review summarizes recent advances in stimuli-responsive dynamic molecular crystals. For the construction of dynamic molecular crystals with response to external stimuli, such as light and/or heat, a number of strategies are available, which are based on (i) metal–organic frameworks, (ii) molecular rotors, (iii) photochromic molecules including azobenzenes and diarylethenes, (iv) cycloaddition reactions, and (v) rotaxanes and pseudorotaxanes. In particular, this review focuses on our recent results on dynamic pseudorotaxane crystals. Pseudorotaxane molecules comprising a crown ether as a ring and an ammonium cation as an axle respond to light and/or heat changing their molecular structures in the single-crystal state. Such structural changes at a molecular level influence the macroscopic properties of pseudorotaxanes, including mechanical motions of the crystals. Consequently, they are potential candidates for application as light-sensing and harvesting materials in microdevices for photomechanical conversion.
 Masaki Horie | Masaki Horie studied for a doctorate at Tokyo Institute of Technology. After obtaining his PhD in 2004, he moved to RIKEN as a special postdoctoral researcher. In 2007, he moved to the UK and joined the School of Chemistry at the University of Manchester as a research associate. In 2010, he joined the Department of Chemical Engineering at the National Tsing Hua University in Taiwan. In 2018, he was promoted to full professor in the same department. His research interests are the synthesis of molecular machines and conjugated molecules for application as organic optoelectronic devices. |
 Chi-Hsien Wang | Chi-Hsien Wang is currently a PhD student under the guidance of Prof. Masaki Horie at the Department of Chemical Engineering, the National Tsing Hua University. She received her bachelor's degree in Chemical Engineering at the same school in 2018. Her research focuses on stimuli-responsive supramolecules in the solid state with potential applications as molecular machines. |
1. Introduction
Stimulus-responsive molecular machines and switches have been recently attracting the interest of scientists in the fields of chemistry and interdisciplinary research.1–20 This has produced interesting developments, such as the first nanocar race, in which single-molecule nanocars are controlled on a two-dimensional surface by scanning tunneling microscopes.21 Meanwhile, rotaxanes and pseudorotaxanes comprising interlocked ring and axle molecules exhibit translational and rotational motions of their components, which leads to changes in their relative positions.2–16 These inter- and intramolecular interactions and molecular motions can be activated and controlled by external stimuli, converting chemical, electrical, photochemical, and electrochemical energy into mechanical motion. The motion of rotaxanes and pseudorotaxanes is effectively controlled in various media, such as solution, aggregation, film, and crystal (Fig. 1). In solution and aggregation states, molecules have overall random alignment (isotropic), and the relative positions of the complexes can be changed. In contrast, molecules have well-defined two- and three-dimensional alignments in films and single crystals, respectively. In these cases, concerted molecular motions can be induced, leading to specific large crystal motions. In addition, the continuous minimization of these molecules has led to micro- and nanoscale applications via top-down approaches, whereas their self-assembled well-aligned architectures in the solid state provide large motions on a macroscopic level by bottom-up approaches.22–25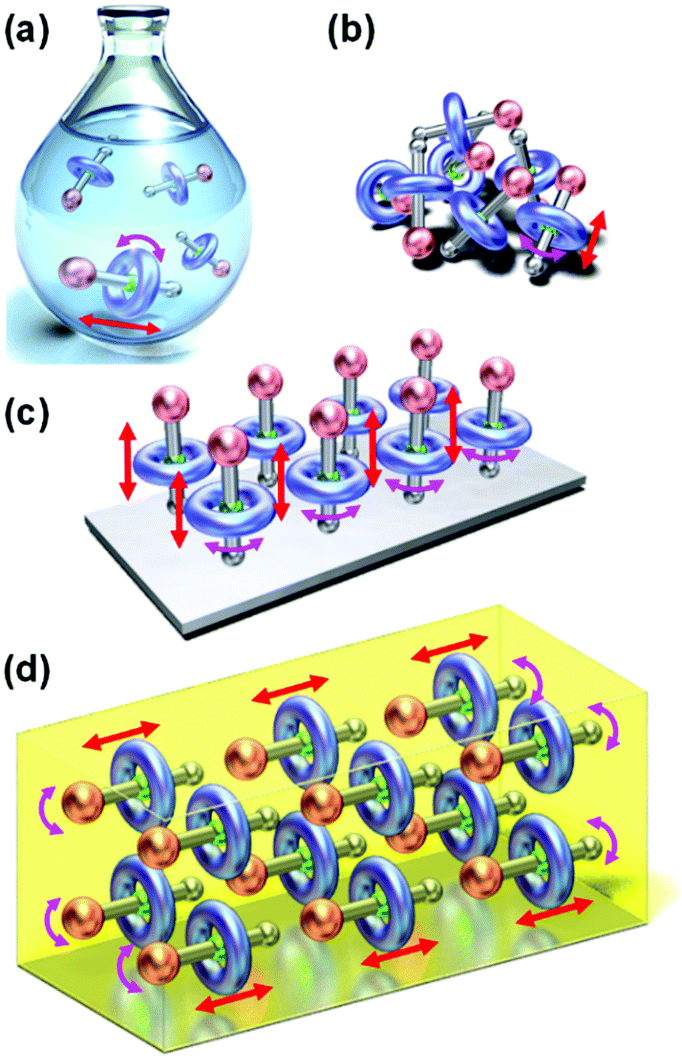 | ||
| Fig. 1 Conceptual illustrations of the motions of rotaxanes in (a) solution, (b) aggregation, (c) film, and (d) crystal. | ||
The number of detailed studies on the molecular motions of rotaxanes and pseudorotaxanes in the crystal state is limited partly due to the lack of investigations on the molecular dynamics in such state. This review article aims to summarize the current literature on stimuli-responsive molecules with molecular dynamics in the crystal state. Furthermore, our recent results on pseudorotaxane 1 (Fig. 2) and related molecules are described. The dynamic motions of these molecules can be activated by heat and/or photoirradiation in the crystal state and are related to macroscopic motions of the crystals. Finally, new insights into the relationship between molecular structure and mechanical motions of pseudorotaxane crystals are discussed.
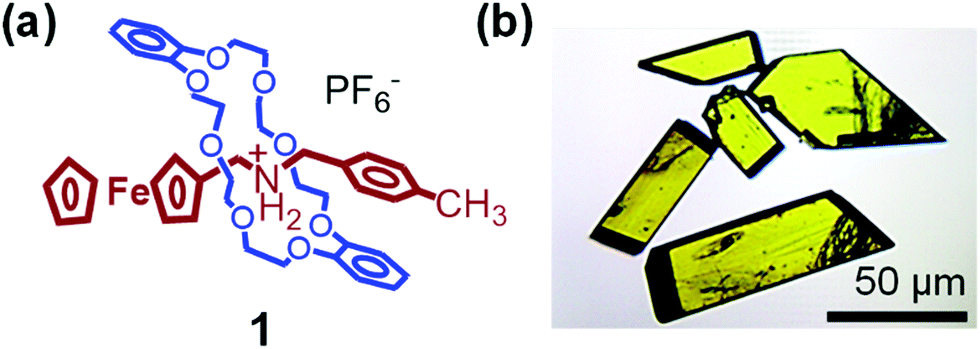 | ||
| Fig. 2 (a) Chemical structure of pseudorotaxane 1 and (b) its single crystals. | ||
2. Molecular motions induced in the solid state
This section describes various strategies that have been proposed to induce mechanical motions in the solid state, such as the construction of interlocked structures or the use of photochemical reactions.23,26–282.1 MOFs incorporating rotaxanes and molecular rotors
The first approach involves constructing interlocked molecules, such as rotaxanes and pseudorotaxanes, which consist of ring and axle components. These interlocked molecules have been introduced into metal–organic frameworks (MOFs) with sufficient porosity and internal volume to accommodate the movement of the bulky axle component.29–34Fig. 3 shows a conceptual illustration of these MOFs, in which rotaxanes are introduced by linking their ring component with the organic struts of the MOFs or by direct insertion of rotaxanes into the MOFs. For instance, Stoddart, Sauvage, Yaghi, et al. reported the incorporation of copper(I)-complexed [2]pseudorotaxane struts into a zinc oxide MOF.30 They synthesized a Sauvage-type organic strut composed of a phenanthroline axle and an oligo-ether tethered phenanthroline ring using a Cu(I) template, which was then removed by demetalation. The resulting MOF was analyzed by powder X-ray diffraction techniques. Schurko, Loeb, et al. also investigated the molecular dynamics of a rotaxane in a zinc oxide MOF.31–33 They synthesized an organic strut composed of an imidazolium axle and a crown ether ring by ring-closing metathesis. In addition to powder X-ray diffraction techniques, the molecular dynamics were analyzed by 13C and 2H solid-state NMR spectroscopy focusing on the motion of the ring component. Unfortunately, despite these advances, direct observation of molecular structural changes by in situ single-crystal X-ray crystallography in this kind of MOF is still challenging due to their complicated molecular structures.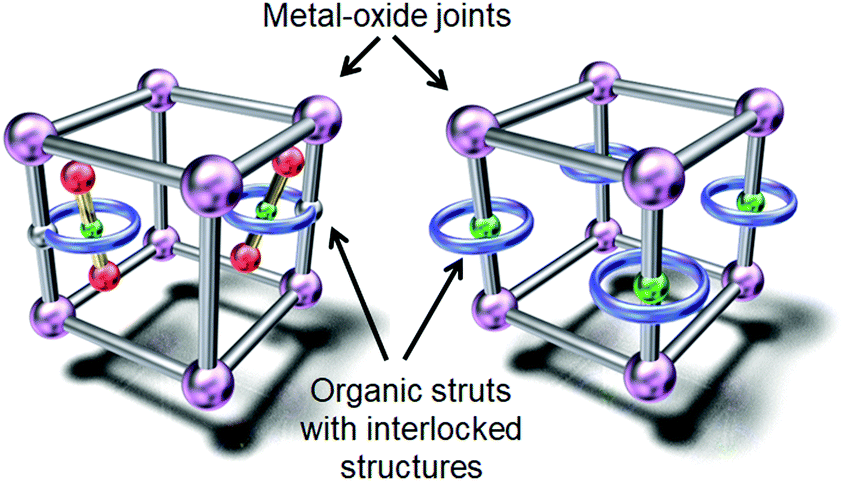 | ||
| Fig. 3 Conceptual illustration of MOFs incorporating rotaxanes. | ||
Molecular rotors and gyroscopes show dynamic rotational motions in the crystal state. Garcia-Garibay et al. reported various molecular rotators, in which typically a 1,4-diethynylbenzene axle is connected with two bulky moieties, such as triphenylmethyl (CPh3) groups, to produce free volume (2, Fig. 4).35,36 When the diethynylbenzene axle is capped with bulkier fragments (e.g., 3 in Fig. 4), the rotation of the axle is faster.35,36 In addition, two phenyl groups of 2 were connected with an azobenzene group to produce photochromic molecular gyroscope 4 (Fig. 4), in which the rotation rate of the axle part is controlled by photoisomerization of the azobenzene group in the solid state.37 Their as-synthesized molecular structures were determined by single-crystal X-ray crystallography, and their rotation rates were estimated by 2H solid-state NMR spectroscopy at various temperatures.
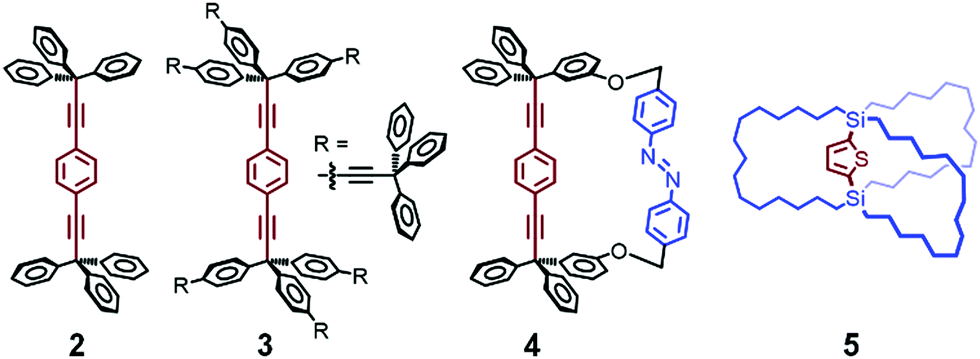 | ||
| Fig. 4 Molecular rotors and gyroscopes. | ||
Setaka et al. reported a crystalline molecular gyrotop 5 (Fig. 4), which was synthesized by ring-closing metathesis of 1,4-bis(tri-7-octenyl)silylthiophene.38 This molecule shows thermally induced order–disorder phase transition in a single-crystal state, as demonstrated by single-crystal X-ray crystallography, polarized optical microscopy, and 2H solid-state NMR spectroscopy. In addition, they replaced the 2,5-thienylene unit of the axle core with biphenylene39 or thiophene dioxide-diyl40 and modified the outer chain length to control the axle rotation in the crystal state.
2.2 Photochromic molecules
Mechanical motions in the solid state can also be induced by subjecting photochromic molecules, such as azobenzenes and diarylethenes, to photochemical reactions. Azobenzene-containing polymer films exhibit reversible and controllable bending based on cis–trans photoisomerization, which has been exploited for applications involving the bending of films and the rotation of a light-driven plastic motor.22,41–43Photoresponsive dynamic azobenzene crystals have also been described (Fig. 5). Koshima et al. reported that plate-shaped crystals of 4-dimethylaminoazobenzene 6 show reversible bending upon UV irradiation.44 Recently, the same group reported the photoinduced bending, twisting, and rolling of chiral azobenzene 7.45 This crystal shows crystal-to-crystal thermal phase transition upon heating at 145 °C and cooling at 143 °C, exhibiting flipping of the phenyl group, whose structural changes were analyzed by single-crystal X-ray crystallography.46
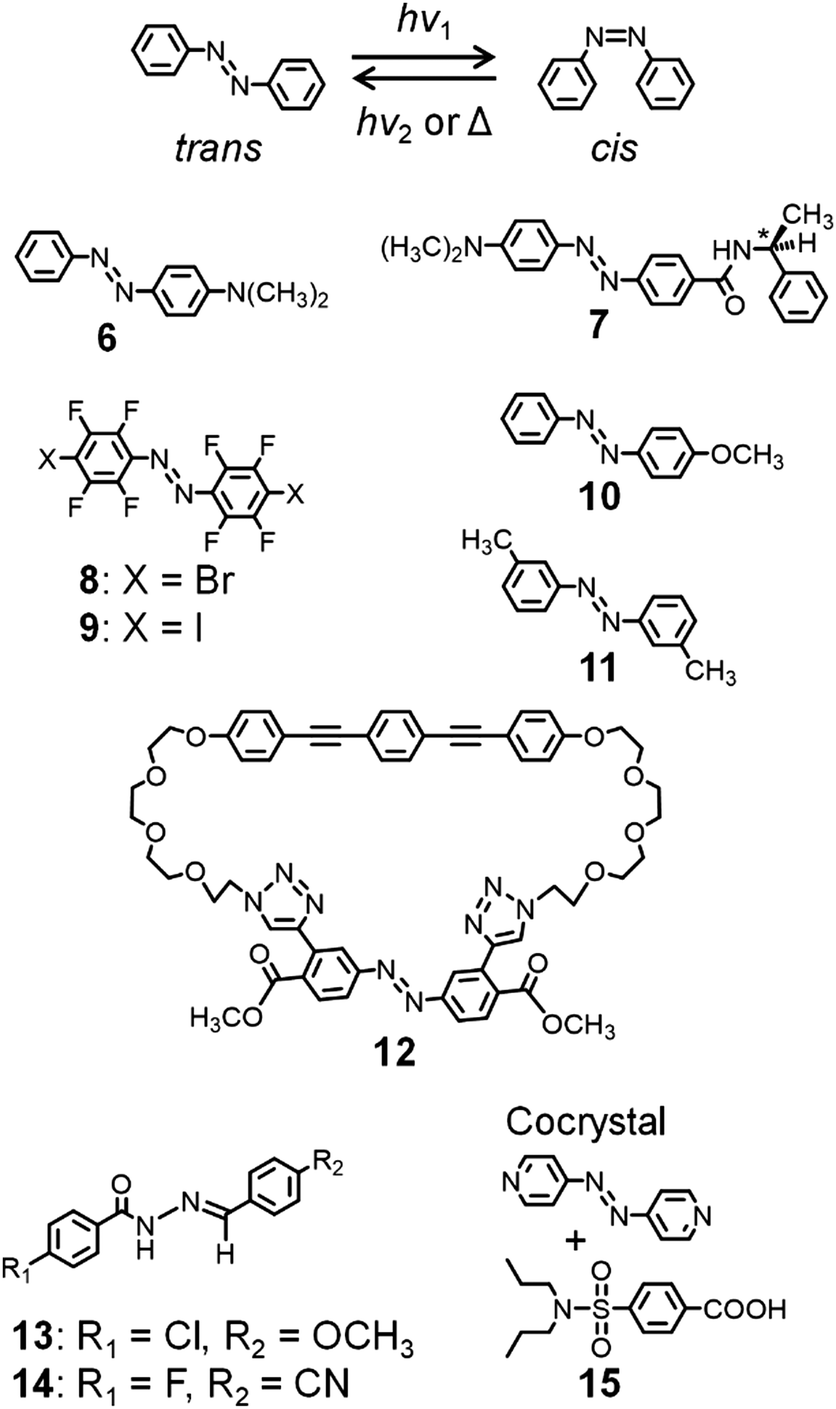 | ||
| Fig. 5 cis–trans isomerization of azobenzene and chemical structures of azo-molecules exhibiting dynamic crystal motions. | ||
Barrett and Friščić et al. reported the cis and trans forms of octafluorinated 4,4′-dihalogenated azobenzenes 8 (X = Br) and 9 (X = I) (Fig. 5), which were synthesized by oxidative coupling using MnO2.47 The cis forms show very long half-lives (ca. 2 months), allowing their isolation as crystals. In addition, their cis forms and cis–trans isomerization in the crystals were analyzed by single-crystal X-ray crystallography. These crystals show irreversible bending and crystal-to-crystal isomerization upon 457 nm irradiation. Furthermore, they cocrystallized these molecules with cis- and trans-1,2-bis(4-pyridyl)ethylene and with trans-4,4′-azopyridine, affording supramolecular structures based on halogen bond-driven cocrystallization.48
Norikane et al. demonstrated the photoinduced translation (so-called swimming) of a single crystal of 4-methoxyazobenzene 10 (Fig. 5) on the water surface, which is caused by its trans–cis photoisomerization.49 In addition, a crystal of 10 was used to carry a boat on the water surface under UV light. Furthermore, they observed photoinduced translational motions, also called crawling, of crystals of 3,3′-dimethylazobenzene 11 (Fig. 5), caused by retraction induced by UV light irradiation and crystal growth prompted by visible light.50
In contrast with the relatively simple chemical structures of the abovementioned molecules, Muraoka and Kinbara et al. reported a cyclic azobenzene tethered with two tetra(ethylene glycol) chains and a 1,4-bis(phenylethynyl)benzene group (12, Fig. 5).51 Interestingly, crystals of 12 show crystal-to-crystal thermal phase transition at 60 °C and 56 °C on heating and cooling, respectively, caused by structural changes in the tetra(ethylene glycol) chains due to their flexible features.
Naumov and Nath et al. described the occurrence of multiple mechanical responses of crystals of acylhydrazone derivatives 13 and 14 (Fig. 5).52 Thus, a needle-shaped crystal of 13 undergoes plastic bending by mechanical stress, with storing of the deformed shape after releasing the stress, whereas 14 undergoes elastic bending by mechanical stress. Furthermore, these crystals exhibit photoinduced bending, caused by E-to-Z photoisomerization upon exposure to 365 nm UV light. In addition, they reported that cocrystals of 4,4′-azopyridine and probenecid 15 (Fig. 5) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio respond to multiple external stimuli (heat, UV light, and mechanical pressure), exhibiting twisting, bending, and elastic deformation.53 The photo- and thermomechanical effects and self-healing properties of 15 result from the cis–trans photoisomerization of the azopyridine unit and crystal-to-crystal thermal phase transition. They proposed that such crystalline materials could find application as crystal actuators.
:2 molar ratio respond to multiple external stimuli (heat, UV light, and mechanical pressure), exhibiting twisting, bending, and elastic deformation.53 The photo- and thermomechanical effects and self-healing properties of 15 result from the cis–trans photoisomerization of the azopyridine unit and crystal-to-crystal thermal phase transition. They proposed that such crystalline materials could find application as crystal actuators.
Meanwhile, diarylethene derivatives have been shown to exhibit significant color changes accompanied by ring-closing and ring-opening photochemical reactions, being both processes photochemically reversible but thermally irreversible (Fig. 6).54 Irie and Kobatake et al. reported various diarylethene derivatives whose crystals show reversible dynamic motions based on photoisomerization induced by UV or visible light irradiation.54–60 Kobatake and Bardeen et al. reported the control of the bending direction of a ribbon-shaped crystal of 16 (Fig. 6) by irradiation direction.58 Furthermore, they prepared organic–inorganic hybrid materials as photon-powered actuators using nanocrystals of 16 and porous alumina, which was used for the alignment control of 16.59 A crystal of n-octyloxy-containing 17 (Fig. 6) was found to exhibit bending and crystal-to-crystal thermal phase transitions on heating and cooling, which is exceptional for the diarylethene series.57 Such thermally induced behavior of 17 was combined with photochromism to control crystal bending.
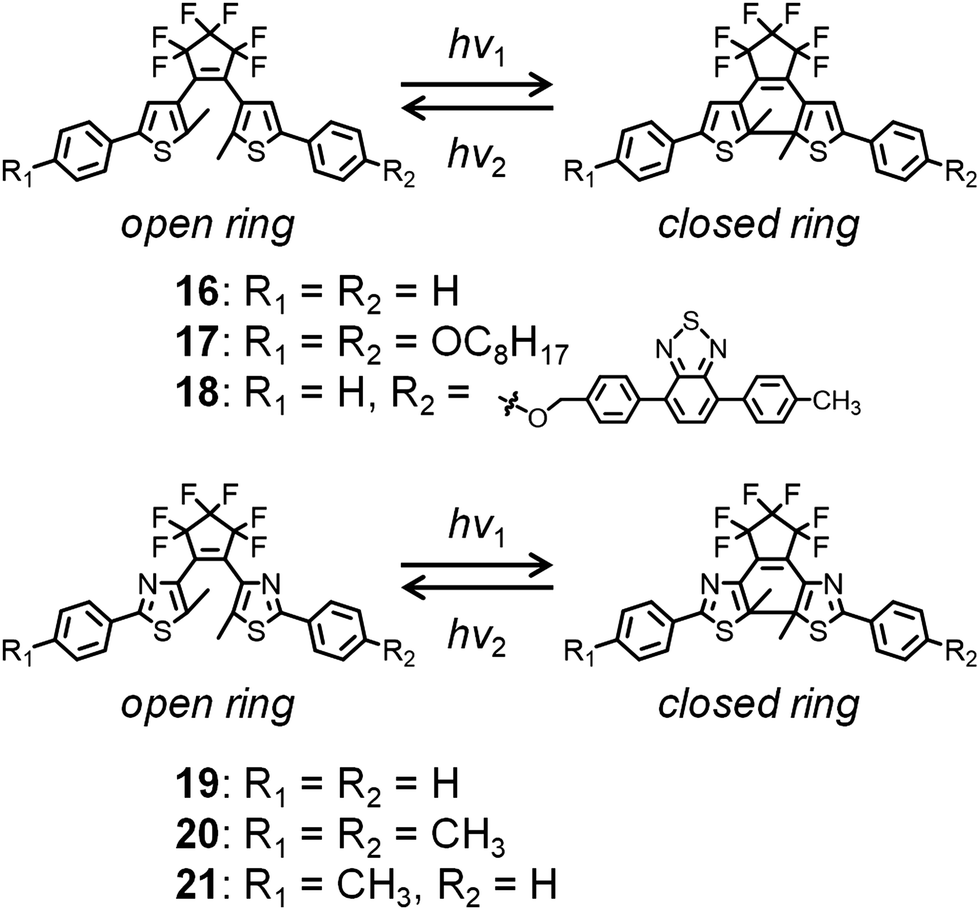 | ||
| Fig. 6 Photochromism of diarylethene derivatives exhibiting dynamic crystal motions. | ||
Fluorescence switching could also be controlled by irradiating a crystal of 18 tethered with a 4,7-diphenylbenzothiadiazole unit with UV and visible light.60 Due to intermolecular energy transfer in the dense molecular packing structure of 18, its open-ring state is fluorescent, whereas its closed-ring state is nonfluorescent.
Finally, a crystal of 19 and mixed crystals of 20 and 21 (Fig. 6) were exploited for applications involving an electric switch56 and rotation of gears, respectively.55
2.3 Cycloaddition in crystals
The [2+2] photocycloaddition of two aligned olefins (e.g., stilbene in Fig. 7) induced by UV or visible light provides carbocyclic products.61–63 In the crystal state, the regular alignment of the target molecules allows this [2+2] photocycloaddition to proceed with high regio- and stereocontrol. The photocycloaddition requires that the two reactive sites are located within a short distance, preferably shorter than 4.2 Å, from each other.61–63 To achieve this, various strategies using weak intermolecular interactions, hydrogen bonds, coordination complexes, external templates, and MOFs have been reported.61–68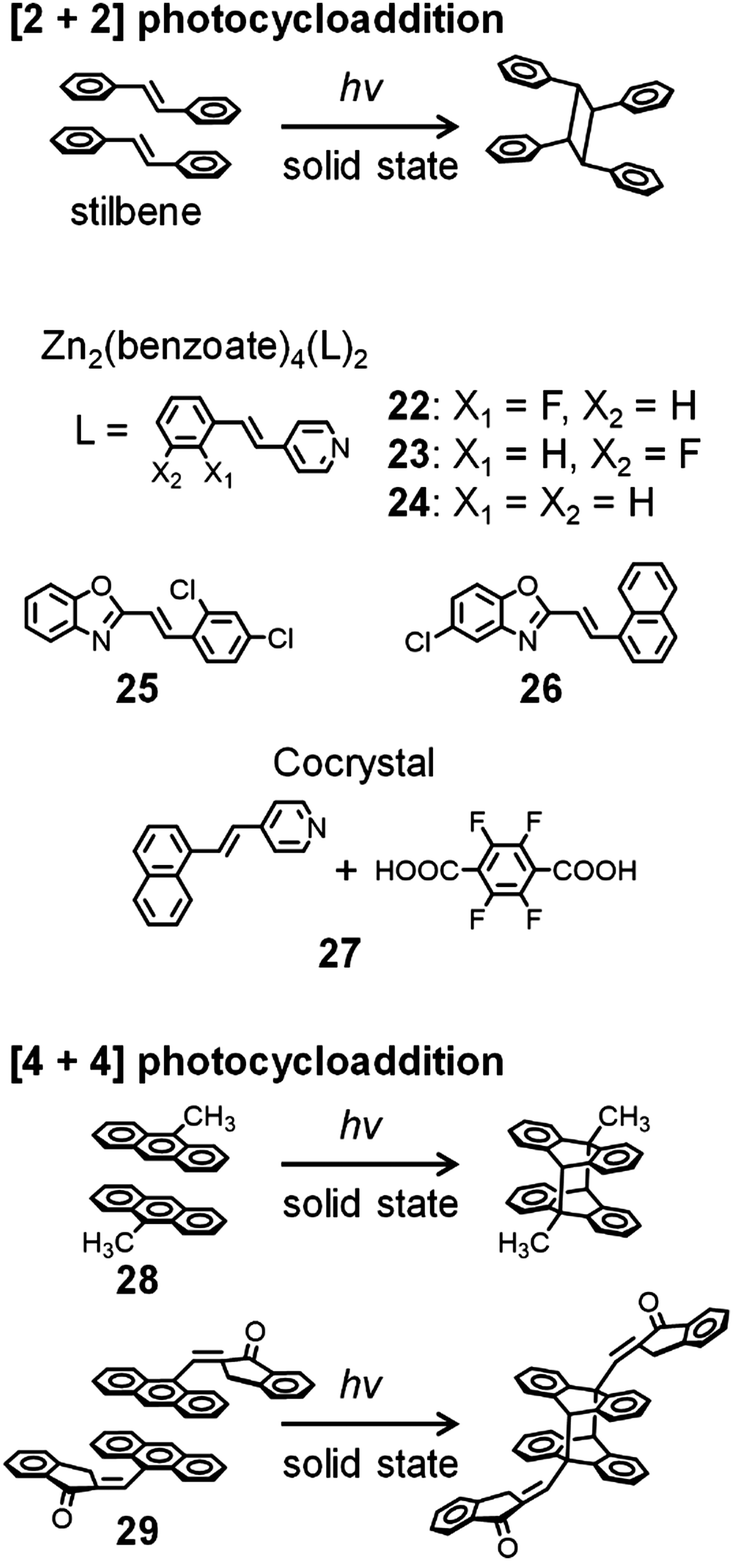 | ||
| Fig. 7 Photocycloaddition of stilbene and anthracene derivatives exhibiting dynamic crystal motions. | ||
In addition, photocycloaddition can induce dynamic motions, such as bending, curling, rolling, and salient behaviors in crystals. For instance, Vittal and Naumov et al. reported photosalient effects of Zn-benzoate (PhCOO−) complexes, [Zn2(benzoate)4(L)2] (22–24), where the ligand (L) is 2′-fluoro-4-styrylpyridine for 22, 3′-fluoro-4-styrylpyridine for 23, or 4-styrylpyridine for 24 (Fig. 7).69 In addition, using the same ligands, they synthesized Zn-cinnamate (PhCH = CHCOO−) complexes,70 Zn-isothiocyanate (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CS−) complexes,71 and silver complexes (AgL2X2) with various counteranions (X = BF4−, ClO4−, or NO3−).72 Upon exposure to weak UV light, with the concomitant [2+2] photocycloaddition, these silver crystals burst violently and are propelled several millimeters. The crystals of silver complexes exhibit photosalient, popped, hopped, and/or leaped motions, depending on their head-to-tail or head-to-head arrangement.
CS−) complexes,71 and silver complexes (AgL2X2) with various counteranions (X = BF4−, ClO4−, or NO3−).72 Upon exposure to weak UV light, with the concomitant [2+2] photocycloaddition, these silver crystals burst violently and are propelled several millimeters. The crystals of silver complexes exhibit photosalient, popped, hopped, and/or leaped motions, depending on their head-to-tail or head-to-head arrangement.
Lu et al. investigated the motion of (E)-2-(2,4-dichlorostyryl)benzo[d]oxazole 25 and (E)-5-chloro-2-(2-(naphthalen-1-yl)vinyl)benzo[d]oxazole 26 (Fig. 7). Their crystals show bending, curling, and rolling caused by [2+2] photocycloaddition.73,74 In particular, crystals of 26 exhibit green photoluminescence with a maximum photoluminescence wavelength (λPL) of 492 nm, which changes to blue with λPL = 454 nm after UV irradiation at 365 nm. They proposed that such photoluminescence color change could be useful for the remote detection of photomechanical reactions. Yan et al. reported that cocrystals of naphthylvinylpyridine and tetrafluoroterephthalic acid 27 with the molar ratio of 2:1 show photosalient phenomena resulting from [2+2] photocycloaddition, whereas the 1:1 derivative shows bending motions.75 The photochemical reaction also affects the photoluminescence intensity of these crystals.
Bardeen et al. reported photoinduced dynamics of crystalline 9-methylanthracene 28, whose motions are induced by the [4+4] photodimerization of two molecules in the crystal state (Fig. 7).76,77 A ribbon-like crystal of 28 shows photoinduced rolling and unrolling during UV exposure and during extended UV exposure, respectively, whereas a sheet-like crystal of 28 shows twisting and untwisting during UV exposure and during extended UV exposure, respectively. They demonstrated translational motion of the sheet-like crystal induced by a magnetic field in the presence of superparamagnetic colloidal nanocrystal clusters.77 Koshima et al. also reported photoinduced crystal bending motions induced by the [4+4] photodimerization of (E)-2-(9-anthrylmethylene)-1-indanone 29 (Fig. 7).78
Sureshan et al. reported the thermal dimerization of 30 (Fig. 8) in the crystal state by the intramolecular reaction of azide and alkyne groups, which releases strain that provides mechanical output.79 They observed instant explosion of the crystals upon fast heating. In contrast, a silent crystal-to-crystal pathway occurs during the absorption of water molecules at a slow rate. Such phenomena were detected by a unique method using an electric circuit. Furthermore, they reported the thermal oligomerization of dipeptide to triazole-linked oligopeptides in the crystal state by the intermolecular reaction of azide and alkyne groups, which causes twisting of crystals.80
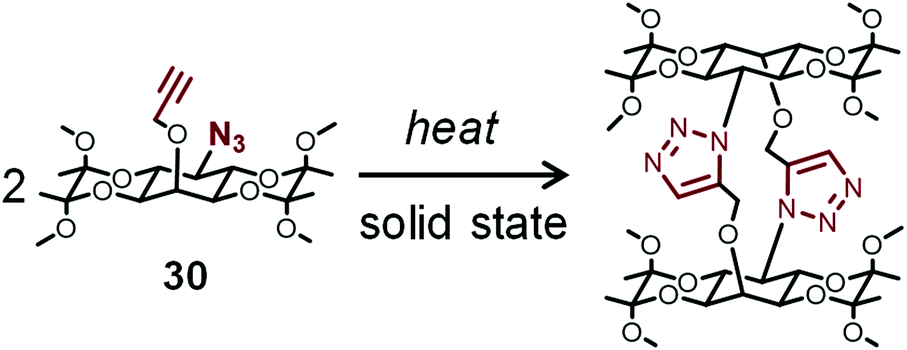 | ||
| Fig. 8 Thermal dimerization of molecules containing azide and alkyne in the crystal state. | ||
3. Pseudorotaxane crystals
3.1 Thermal phase transition
With the idea of controlling the formation of pseudorotaxane by an electrochemical reaction, we prepared [2]pseudorotaxane 1 (Fig. 2), which is composed of ferrocenylmethyl(4-methylphenyl)ammonium as an axle and dibenzo[24]crown-8 ether (DB24C8) as a ring molecule.81,82 In the electrochemical reaction, the ferrocenyl group in the neutral axle precursor is oxidized to form an ammonium cation that interacts ionically with DB24C8.81,82The crystal-to-crystal phase transition of 1 was serendipitously discovered when the crystals were analyzed by differential scanning calorimetry (DSC) and polarized optical microscopy (Fig. 9).83 The DSC scans of the crystals are shown in Fig. 9, in which endothermic and exothermic peaks at 128 °C and 115 °C, respectively, can be observed. Upon heating up to 190 °C, irreversible endothermic peaks appear around 160 °C and 170 °C, being the higher temperature peak assigned to the melting.84 The crystal exhibits interference color change under cross-polarized light at 128 °C. The color switches from green to orange above 128 °C, to return to green at 115 °C on cooling. Although an irreversible change before the melting seemed to occur, the instability of this state prevented its analysis by X-ray crystallography at 160 °C. On the other hand, we were interested in unveiling the nature of the first peak at 128 °C, which is not due to melting.
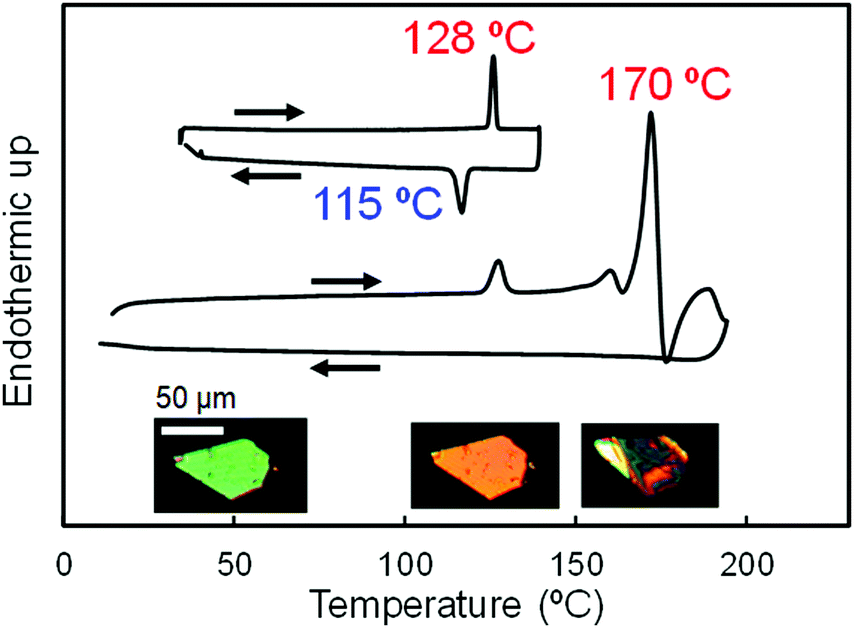 | ||
| Fig. 9 DSC heating and cooling scans of 1 up to 130 °C (top) and 190 °C (bottom). Optical micrographs of a single crystal at 30 °C, 128 °C, and 160 °C under cross-polarized light are inserted. | ||
Therefore, we conducted single-crystal X-ray crystallography to directly observe the molecular structure of 1.83,84 As shown in Fig. 10 (left), 1 is stabilized by intramolecular ionic interactions between oxygen atoms of the crown ether and the ammonium cation of the axle molecule. In addition, intra- and intermolecular π–π interactions between the catechol group of the crown ether and the tolyl group (PhCH3) in the axle molecule are also observed. At 128 °C, 1 shows a different structure from that at 30 °C and undergoes crystal-to-crystal thermal phase transition. This change in the molecular structure results from the twisting of a pair of tolyl groups of two axle molecules (Fig. 10, right), which changes the crystal anisotropy, leading to interference color changes (Fig. 9).
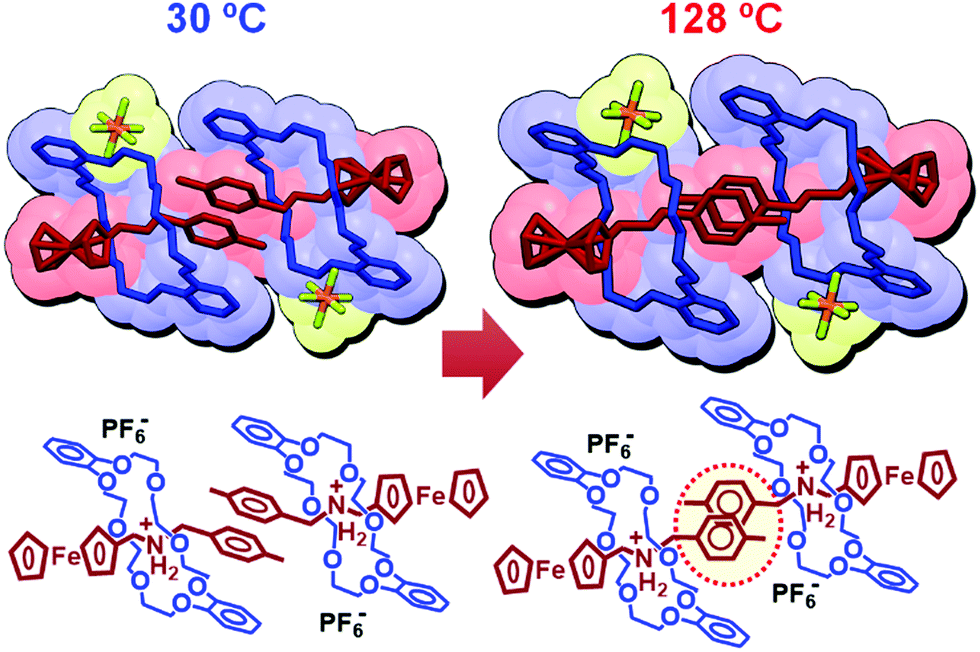 | ||
| Fig. 10 Molecular structures of 1 obtained by single-crystal X-ray crystallography at 30 °C and 128 °C. | ||
Such phase transition and reversible interference color changes were also observed in thin films of 1 deposited on substrates (Fig. 11).85 Highly ordered thin films of 1 were prepared on glass or SiO2 substrates via casting of chloroform solution followed by annealing. Grazing-incidence wide-angle X-ray scattering (GIWAXS) experiments indicated that 1 forms a highly ordered crystalline film during solvent evaporation. In addition, 1 has an edge-on orientation of the DB24C8 rings, which are perpendicular to the substrate, forming an interlocked pseudorotaxane structure with the axle molecules. Upon heating at 130 °C, the pseudorotaxane molecules reorganize to form large domains with sizes in the range of 50–100 μm. These crystal domains exhibit reversible crystal-to-crystal thermal phase transition upon heating and cooling, which were observed via in situ GIWAXS and polarized optical microscopy.
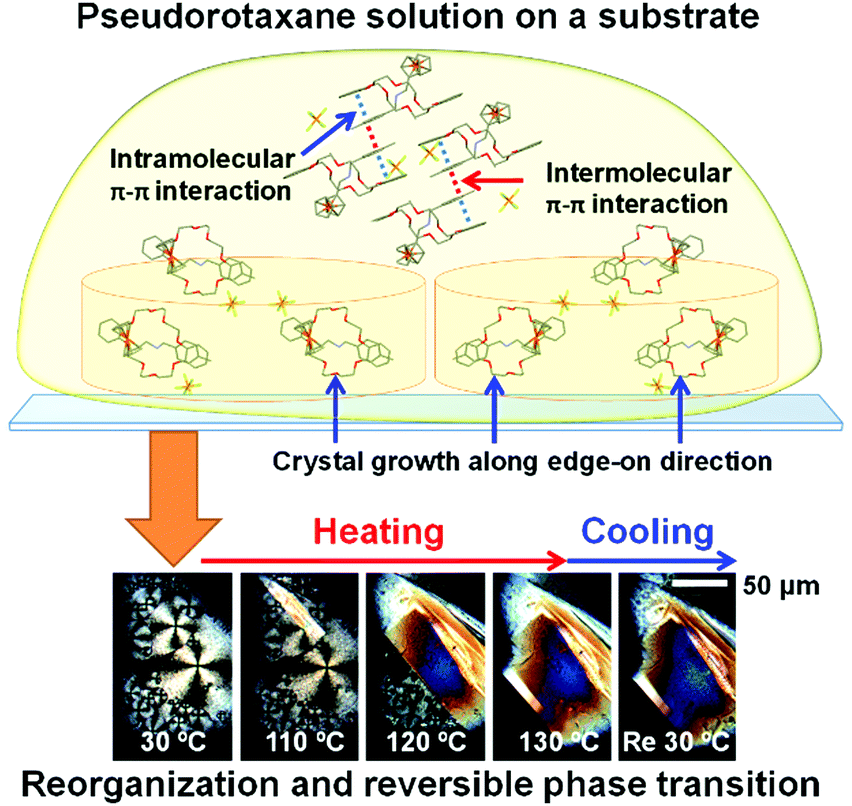 | ||
| Fig. 11 Schematic illustration of the film formation and optical micrographs of a film of 1 under polarized light. The figure was modified using ref. 85 and reprinted with permission from the Royal Society of Chemistry, Copyright (2014). | ||
We then explored the thermal properties of other ferrocene-containing pseudorotaxanes (Fig. 12), which are composed of AsF6− counter anions (31),84 phenyl (32),84 ethyl (33),84 tetrabromo (34),86 biferrocene (35),86 biferrocene and tetrabromo (36),86 [3]pseudorotaxane structure (37),82 or ruthenocene (38).87 Among them, only 38 shows crystal-to-crystal phase transition.87 In the crystal state, 38 has a similar alignment to that of 1 owing to π–π interactions between catechol and tolyl groups. The crystal-to-crystal thermal phase transition of 38 was observed at 86 °C and 55 °C on heating and cooling, respectively, which is significantly lower than for 1 by 42 °C and 60 °C, respectively. This is due to the strain provided by the bulky ruthenocene group in the unit cell. As a result, the formation of the high-temperature structure of the Ru complex occurs at lower temperature than for the Fe complex. Interestingly, these phase transition temperatures could be tailored by mixing 1 and 38 in different ratios during crystallization.87
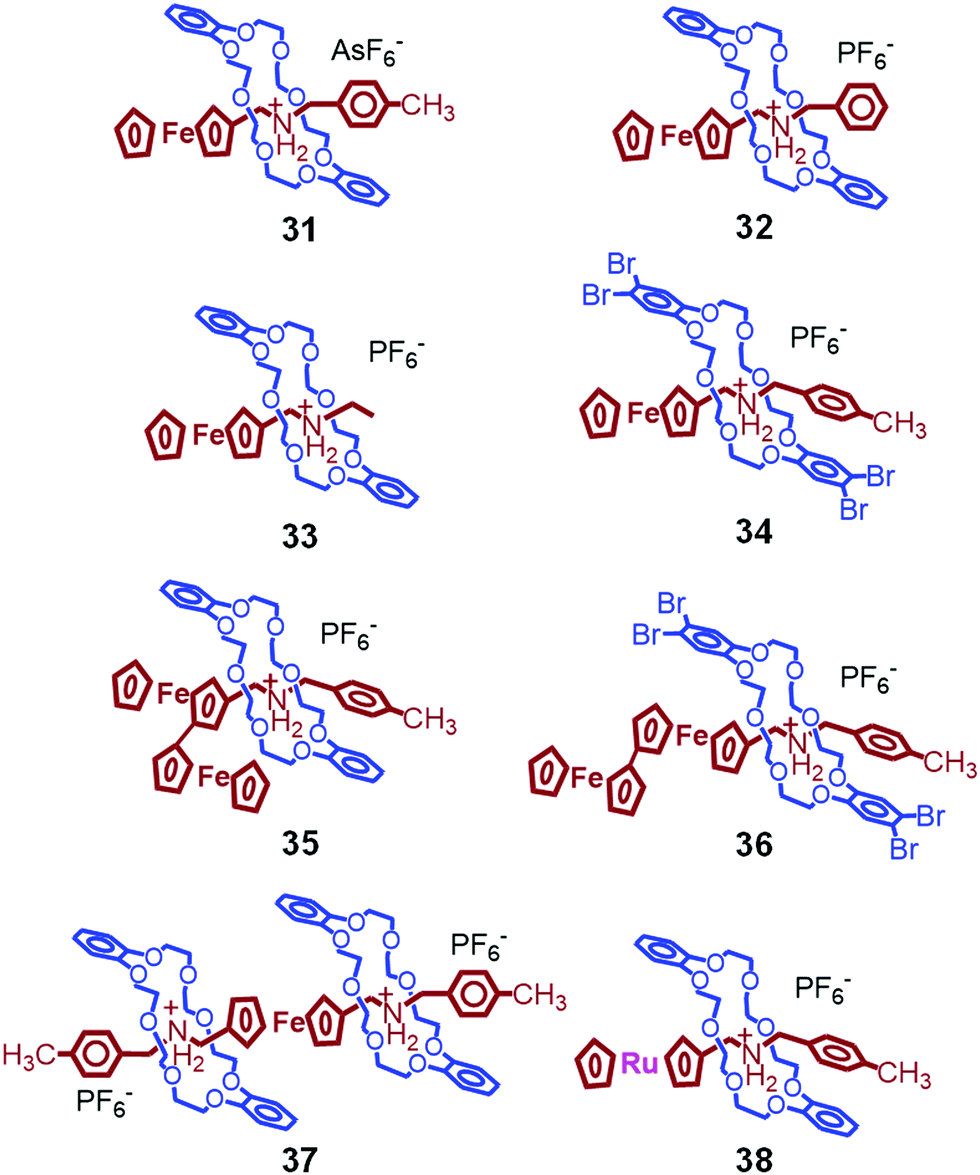 | ||
| Fig. 12 Chemical structures of 31–38. Only 38 shows crystal-to-crystal thermal phase transition. | ||
Mixed crystals of 1 and 31 also show crystal-to-crystal phase transition induced by the presence of π–π interactions between catechol and tolyl groups, whereas the twisted conformation of the tolyl group in pure 31 hinders such phase transition.84 Crystals of 33–37 melt upon heating without the occurrence of crystal-to-crystal phase transition. In the case of 32 and 37, this is most likely due to the twisted conformation of phenyl and tolyl groups, respectively. On the other hand, 34 and 36 have tight intermolecular π–π interactions between tetrabromocatechol groups, possibly restricting the molecular motions for the thermal phase transition.
In addition, we investigated the thermal properties of crystals of various Stoddart-type pseudorotaxanes. Some examples are shown in Fig. 13.88 However, these crystals only melt, and no crystal-to-crystal thermal phase transitions are observed. From these results, we concluded that the presence of (i) a ferrocenyl group, (ii) π–π interactions between the catechol and tolyl groups, (iii) PF6− anions, and (iv) DB24C8 (not tetrabromo-DB24C8) is required for the crystal-to-crystal thermal phase transition to occur.
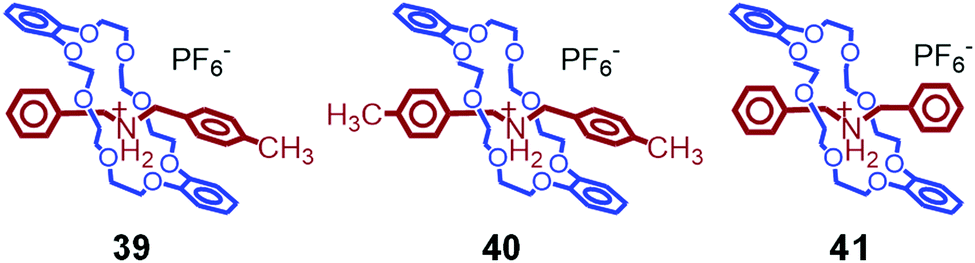 | ||
| Fig. 13 Chemical structures of 39–41. | ||
3.2 Photoinduced molecular motions
Next, in our investigation of pseudorotaxane crystals, we applied photoirradiation to induce molecular motions in the crystal state. These experiments were conducted using a polarized optical microscope equipped with a ×20 objective lens, a continuous-wave 445 nm diode laser, a hot stage, top- and side-view CCD cameras, and a force detector (Fig. 14).87,89 The crystal dimensions were measured by a confocal laser microscope with/without laser irradiation. | ||
| Fig. 14 Photographs of the setup for the photoirradiation experiments. (a) Overall view and (b) expanded view around the crystal sample. A laser beam line is illustrated. | ||
Upon subjecting a single crystal of 1 to irradiation by a focused 445 nm laser at 30 °C, the crystal exhibits slightly subtractive interference color compared with the nonirradiated crystal (Fig. 15a, left and middle).89 This change is accompanied by a quick anisotropic deformation of the crystal; its length, width, and thickness increase by 0.2%, 1.0%, and 0.6%, respectively. After turning off the laser, the crystal immediately returns to the original interference color and size. When the crystal is heated at 115 °C, a much lower temperature than that of the thermal phase transition temperature (128 °C), a photo-triggered crystal-to-crystal phase transition is observed under focused laser irradiation at 445 nm (Fig. 15a, right). The significant interference color change from green to red is indicative of the formation of a high-temperature phase. Since the laser beam spot (ca. 1 μm diameter) was much smaller than the top face of the crystal, the local excitation is rapidly propagated by concerted molecular structural changes in the entire crystal.
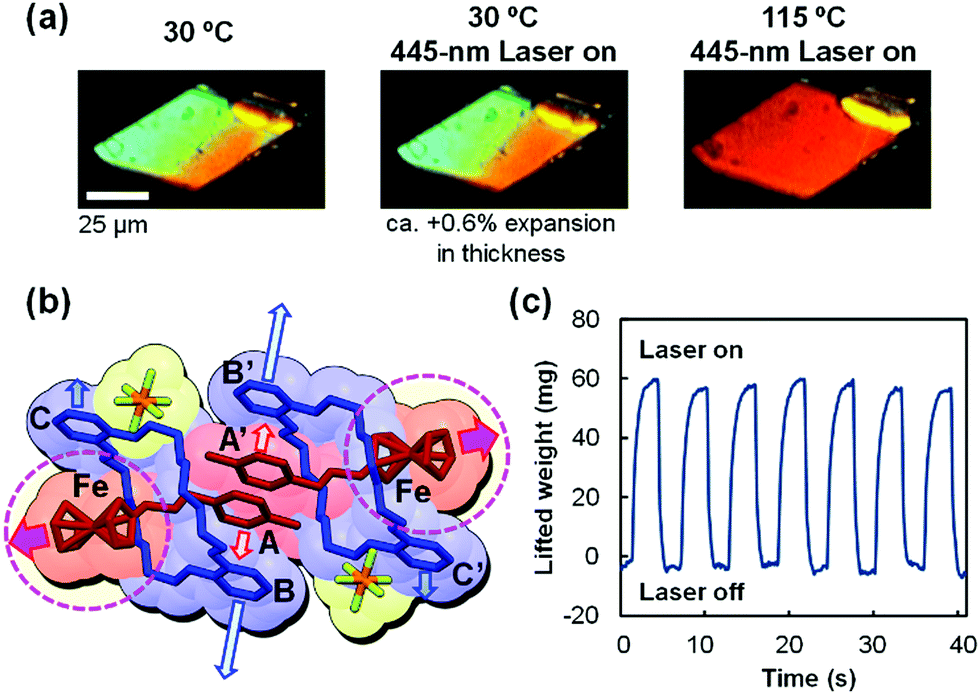 | ||
| Fig. 15 (a) Optical micrographs of a single crystal of 1 under polarized light. (b) Molecular structure of 1 and its photoinduced motion by 445 nm laser irradiation. (c) Time dependence of lifting weight of the crystal induced by 445 nm focused laser irradiation at 30 °C. | ||
The photoinduced molecular structural changes in 1 were analyzed by single-crystal X-ray crystallography with 445 nm laser irradiation at 30 °C (Fig. 15b).89 We found that the photoinduced three-dimensional expansion of the crystal is related to the elongation of the distances A–A′, B–B′, and A–Fe. These molecular structural changes are different from the heat-induced motions of 1. Upon heating at 105 °C in the absence of laser irradiation, the distances A–A′ and B–B′ increase, whereas A–Fe slightly decreases. Thus, we concluded that the elongation of the A–Fe distance is due to the photoactivation of the ferrocenyl group leading to the expansion of the whole crystal.
The force produced by the photoinduced expansion of the crystal of 1 was monitored using a microforce analyzer containing two strain gauges. As shown in Fig. 15c, the crystal can lift ca. 68 mg under laser irradiation. The crystal weight was estimated to be 41 μg from its volume by confocal laser microscopy and from its density by single-crystal X-ray crystallography. Therefore, the ratio (lifted weight)/(crystal weight) was estimated to be 1650.89 Such mechanical force produced by a crystal was used for the demonstration of transport of silica microparticles (Fig. 16a). According to this experiment, silica microparticles placed on a crystal of 1 jump from the crystal upon irradiation by a 445 nm laser, due to the quick expansion of the crystal. In addition, a crystal of 1 can be used as a mechanical microswitch to control an electric circuit (Fig. 16b).89 Thus, a gold-coated crystal of 1 and a nearby copper plate are introduced in an electric circuit, and the current is remotely controlled by turning the laser on and off. This is accompanied by connection and disconnection of the crystal with the copper plate due to the expansion/contraction motion of the crystal. The current switching is robust and repeated 300 times in 330 s (0.91 times s−1, Fig. 16b) or 560 times in 60 s (9.3 times s−1).89 This switching is much faster and more facile than the current switching using diarylethene crystals,56 because the expansion/contraction motion of the crystal of 1 only requires one laser wavelength to change the state; moreover, the backward process is spontaneous.
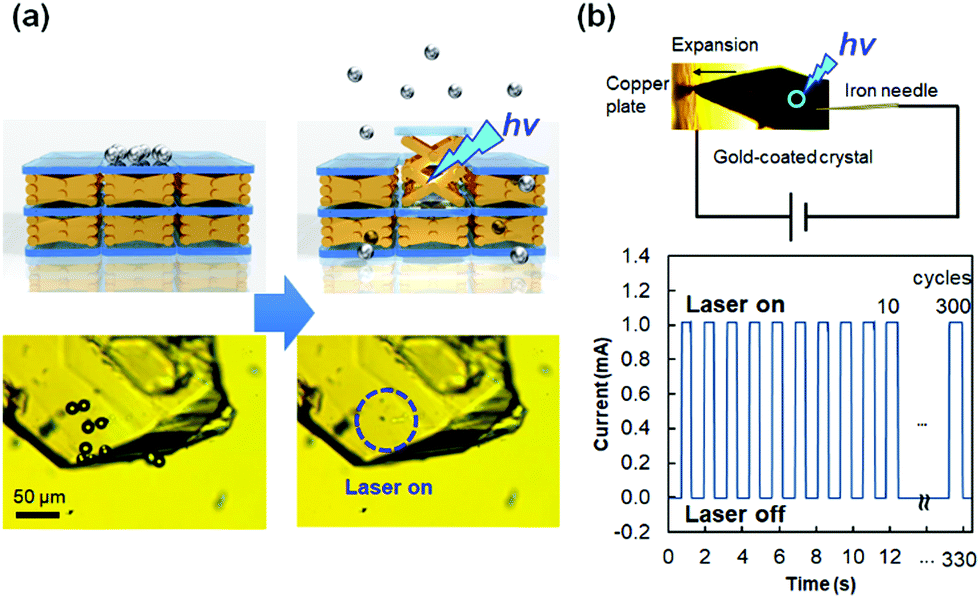 | ||
| Fig. 16 (a) Transport of silica microparticles induced by expansion of a crystal of 1 triggered by focused 445 nm laser irradiation. (b) Electric current switching controlled by deformation of a gold-coated crystal of 1 with/without 445 nm laser irradiation. | ||
Next, the magnitude of the photoinduced crystal expansion of 1 was compared with that of other molecules. The laser power dependence of the relative area change of the top face of crystals of 1, 35, and 38 and a mixed crystal 1:38 in a 7:3 molar ratio is plotted in Fig. 17a. As can be seen, the crystal of biferrocenyl pseudorotaxane 35 shows the highest sensitivity to the 445 nm laser light (+1.2% expansion at 10 mW), whereas 1 shows +0.35% expansion at the same laser power.86 The mixed crystal shows a slightly higher magnitude of expansion (+0.8%) than 1 at a laser power of 10 mW.87 In contrast, the change of 38 is negligible (<+0.1%).87
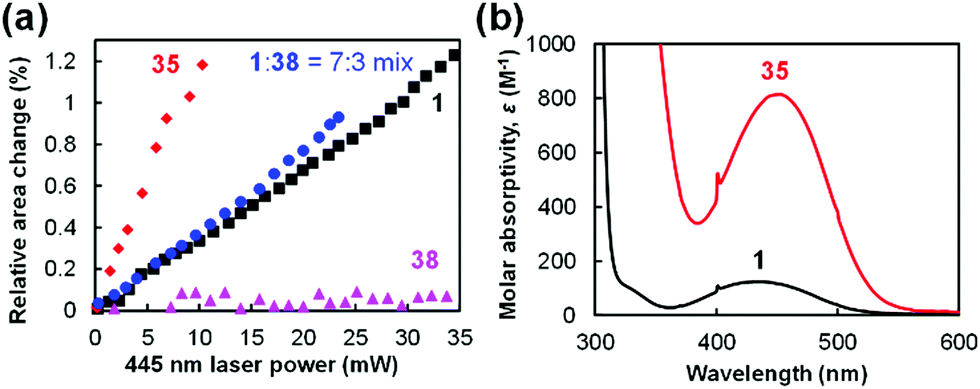 | ||
| Fig. 17 (a) Laser power dependence of the change in the relative area of the top face of the single crystals induced by focused 445 nm laser irradiation. (b) UV-visible absorption spectra of mixed crystals in chloroform solutions. | ||
The magnitude of the crystal expansion can be accounted for by the photoabsorption of the molecules. Fig. 17b shows the UV-vis absorption spectra of 1 and 35. The maximum absorption wavelength (λmax) of 1 is observed at 433 nm with a molar absorptivity (ε) of 125 M−1 cm−1, whereas 35 shows λmax = 453 nm with ε = 810 M−1 cm−1 (Fig. 17b).86 These peaks are attributed to the absorption of the ferrocenyl or biferrocenyl group. Therefore, 35 shows a much higher sensitivity than 1 in the relative area changes. On the other hand, no photoabsorption around 400–500 nm is observed for ruthenocene complex 38, leading to significantly low photosensitivity in the relative area changes.87
Interestingly, the mixed crystal 1:38 shows larger expansion of the crystal than 1, even though the absorbance of the former crystal is lower than that of the latter.87 This might be explained by the crystal packing structures. The structures of the mixed crystal are less packed than those of the crystals of pure 1 and 38, which results in the pseudorotaxane molecules in the mixed crystal having larger free volume, molecular flexibility, and higher dynamics.
The photoinduced weight lifting by crystals of biferrocene and tetrabromo complexes 34–36 and ruthenocene complex 38 was also measured by using the microforce analyzer. When a crystal of 35 is irradiated by a 445 nm laser, the expansion of the crystal produces a ratio (lifted weight)/(crystal weight) of 2900, which is nearly two times higher than that of 1.86 However, crystals of tetrabromo complexes 34 and 36 afford a much lower weight lifted, possibly due to the tight molecular packing derived from strong π–π interactions between the tetrabromocatechol rings.86
Similar to 1, the mixed crystal 1:38 produces a ratio (lifted weight)/(crystal weight) of 1600.87 In contrast, a negative force is observed for this crystal when it is irradiated by a 405 nm laser at 85 °C, due to a decrease in the thickness of the crystal that accompanied the laser-assisted thermal phase transition. When the laser is switched off, the crystal returns to the low-temperature phase producing a strong force corresponding to a ratio (lifted weight)/(crystal weight) of 6400.87 Under the same conditions, 1 does not undergo such phase transition due to its much higher thermal phase transition temperature than that of the mixed crystal. The observed (lifted weight)/(crystal weight) ratio resulting from the laser-assisted thermal phase transition is 1–2 orders of magnitude higher than those of previously reported materials, such as diarylethene crystals,55,90 azobenzene polymer films,91,92 polymer composite actuators,93 and electro-triggered hydrogels.94
On the other hand, crystals of 39–41 show insignificant changes by photoirradiation at 445 nm due to the lack of photoabsorbing groups in their molecules.
3.3 Azobenzene-containing pseudorotaxanes
Recently, we introduced azobenzene groups into ferrocene-containing [2]pseudorotaxanes.95Fig. 18 shows a conceptual illustration. The azobenzene group undergoes reversible cis–trans photoisomerization, transducing photoenergy into mechanical bending motion. Using pseudorotaxanes, such motions are expected to be flexible and diverse. In addition, pseudorotaxanes introduce structural diversity because they consist of various components.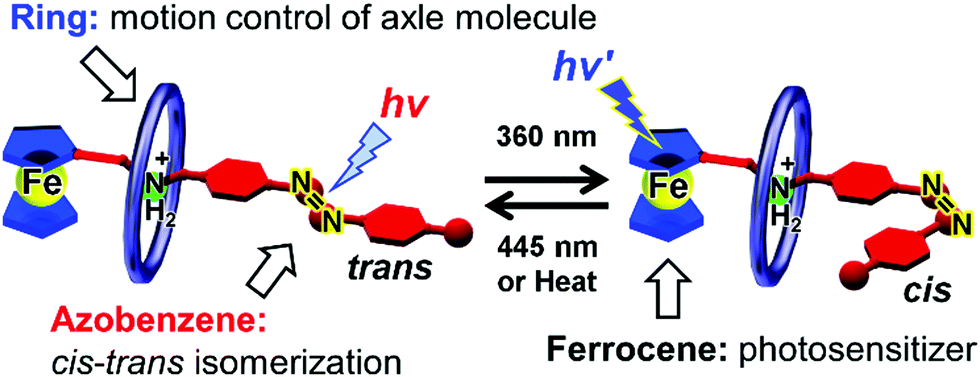 | ||
| Fig. 18 Illustration of cis–trans isomerization of azobenzene- and ferrocene-containing pseudorotaxane. | ||
Fig. 19a shows the chemical structures of azobenzene- and ferrocene-containing [2]pseudorotaxanes 42–44.95 Their molecular structures in a crystal state were determined by single-crystal X-ray crystallography. Despite having very similar chemical structures, their intra- and intermolecular interactions in the crystal state differ largely from one another. Thus, 42, which lacks a substituent on the 4-position of the azobenzene group, adopts strong intermolecular π–π interactions between a pair of aromatic rings of the azobenzene groups. These azobenzene groups have a planar trans-azobenzene structure. In contrast, methyl-substituted 43 has intramolecular π–π interactions between azobenzene and catechol groups. Therefore, one of the aromatic rings of the azobenzene group is relatively free, and thus, the azobenzene group has a twisted structure. On the other hand, 44, which contains tetrabromo-substituted DB24C8, shows only one intermolecular π–π interaction between one of the aromatic rings of the azobenzene group. As a result, the azobenzene group in 44 is also expected to be flexible.
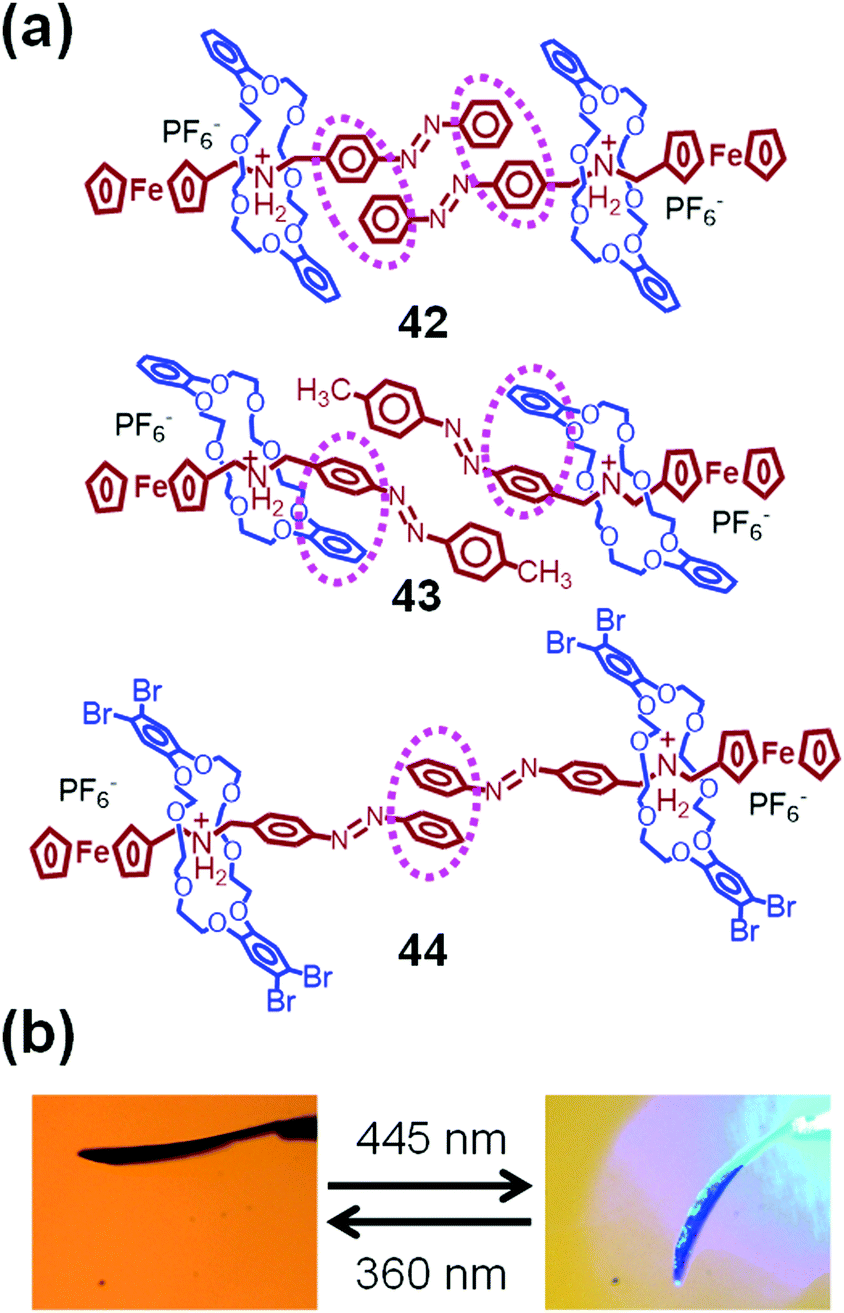 | ||
| Fig. 19 (a) Chemical structures of 42–44. (b) Optical micrographs of 44 under 360 nm (left) and 445 nm (right) laser irradiation. | ||
The structures of these crystals have a significant influence on their molecular motions and, consequently, on their crystal motions. Thus, a crystal of 42 remains immobile when it is irradiated at 445 nm, whereas it vibrates slightly upon irradiation by a 360 nm laser. In contrast, crystals of 43 and 44 show significant downward bending under 445 nm irradiation (Fig. 19b).95 These bended crystals undergo slow upward bending under 360 nm laser irradiation.
These crystal bending motions are related to the cis–trans photoisomerization of the azobenzene groups. In CH2Cl2 solutions, all [2]pseudorotaxanes 42–44 show a strong absorption peak at 324 nm and a weak peak around 450 nm attributable to trans-azobenzene and ferrocene, respectively. The peak at 324 nm decreases significantly after irradiation at 360 nm due to the formation of cis-azobenzene. Thus, the photoisomerization of the azobenzene group is very efficient for all [2]pseudorotaxanes 42–44 in solution. To estimate their photoisomerization, single crystals were irradiated at 360 nm and dissolved in CH2Cl2 to measure the corresponding UV-vis spectra. On the basis of the absorption intensities, the photoisomerization conversion was estimated to be 4% for 42, 28% for 43, and 40% for 44 in their crystal state.95
The low photoisomerization conversion for 42 is due to the stable planar trans-azobenzene group with strong intermolecular π–π interactions, which restricts the molecular motions and immobilizes the crystals. In contrast, the decent degrees of photoisomerization conversion for 43 and 44 can be explained in terms of the twisted azobenzene structures and weak π–π interactions providing significant flexibility of the azobenzene group, which results in macroscopic crystal bending.
It is worth mentioning that the crystals undergo a fast cis–trans backward process within 0.3 s, which contrasts with the fast forward and slow backward isomerizations previously reported for azobenzene derivatives.45,96 Such fast backward process is probably due to the presence of the ferrocenyl group, which absorbs at 445 nm converting photoenergy into heat, thereby accelerating the cis–trans backward process.
4. Conclusions and perspectives
This review covers recent literature on stimuli-responsive dynamic molecular crystals comprising MOFs, molecular rotors, photochromic molecules such as azobenzenes and diarylethenes, cycloaddition reactive units, and pseudorotaxanes. To achieve light- and/or heat-responsive dynamic crystal motions, efficient reactive sites, free volume, and mobile parts are needed. Rotaxanes and pseudorotaxanes have complicated interlocked structures composed of ring and axle molecules. Consequently, reports on the direct observation of molecular structural changes in rotaxanes and pseudorotaxanes by single-crystal X-ray crystallography are limited. The pseudorotaxanes investigated in our group overcome such limitation.The development of dynamic rotaxane and pseudorotaxane crystals is still challenging and would be a pioneering step in the study of solid-state molecular motions, compared with other sophisticated dynamic molecular systems. To develop sophisticated crystal molecular machines using rotaxanes and pseudorotaxanes, fundamental studies of the systematic tailoring of the molecular structures and packing properties of these molecules in the crystal state are required.
Fig. 20 summarizes the molecular motions and functions of our pseudorotaxanes in the crystal state. The ring molecule in the pseudorotaxane structure provides molecular flexibility by adjusting its structure with the structural changes of the axle molecule. The latter, in turn, undergoes dynamics, such as up-down, flipping, and cis–trans isomerizations induced by heat and/or light. Meanwhile, photoabsorption by the organometallic ferrocenyl group converts photoenergy into thermal energy, leading to photomechanical conversion in the crystals. In addition, the bulkiness of the ferrocenyl group is an important factor to provide molecular dynamics in the crystal state, since it provides flexible molecular packing structures.
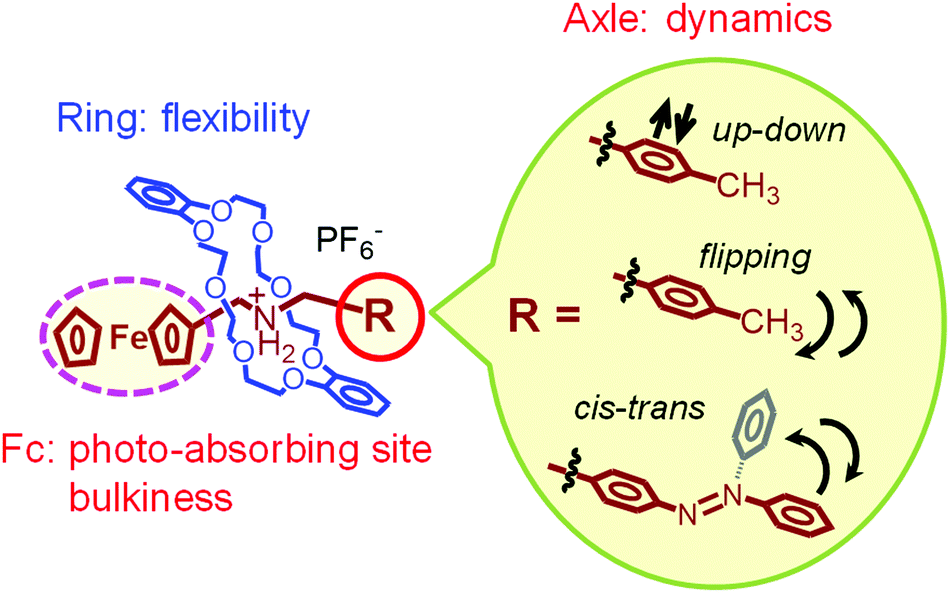 | ||
| Fig. 20 Schematic illustration of the molecular motions and functions of ferrocene-containing pseudorotaxanes. | ||
We believe that this review paves the way for new findings in molecular machines and rotaxanes with interesting novel motions in the crystal state. In addition, we expect that such stimuli-responsive rotaxane crystals will be used as light-sensing and harvesting materials in photomechanical microdevices and switches, such as miniaturized optical circuit elements, miniaturized chemical and biosensing tools, and future crystal actuators.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. K. Osakada of Tokyo Institute of Technology for enlightening discussion. The authors would like to acknowledge the financial support from the National Tsing Hua University and Ministry of Science and Technology Taiwan.References
- S. Shinkai, Pure Appl. Chem., 1987, 59, 425–430 CAS.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS.
- B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS.
- J. P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS.
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC.
- C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef CAS.
- R. Barat, T. Legigan, I. Tranoy-Opalinski, B. Renoux, E. Péraudeau, J. Clarhaut, P. Poinot, A. E. Fernandes, V. Aucagne, D. A. Leigh and S. Papot, Chem. Sci., 2015, 6, 2608–2613 RSC.
- C. Y. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. F. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2016, 116, 15089–15166 CrossRef CAS.
- S. Erbas-Cakmak, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Science, 2017, 358, 340–343 CrossRef CAS.
- A. Martinez-Cuezva, A. Saura-Sanmartin, T. Nicolas-Garcia, C. Navarro, R.-A. Orenes, M. Alajarin and J. Berna, Chem. Sci., 2017, 8, 3775–3780 RSC.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- K. Zhu, G. Baggi and S. J. Loeb, Nat. Chem., 2018, 10, 625–630 CrossRef CAS.
- D. Sluysmans, F. Devaux, C. J. Bruns, J. F. Stoddart and A.-S. Duwez, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9362–9366 CrossRef CAS.
- J. J. Yu, L. Y. Zhao, Z. T. Shi, Q. Zhang, G. London, W. J. Liang, C. Gao, M. M. Li, X. M. Cao, H. Tian, B. L. Feringa and D. H. Qu, J. Org. Chem., 2019, 84, 5790–5802 CrossRef CAS.
- K.-Y. Chen, O. Ivashenko, G. T. Carroll, J. Robertus, J. C. M. Kistemaker, G. London, W. R. Browne, P. Rudolf and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 3219–3224 CrossRef CAS.
- L. Greb and J.-M. Lehn, J. Am. Chem. Soc., 2014, 136, 13114–13117 CrossRef CAS.
- S. Kassem, A. T. L. Lee, D. A. Leigh, A. Markevicius and J. Sola, Nat. Chem., 2016, 8, 138–143 CrossRef CAS.
- S. K. M. Nalluri, J. W. Zhou, T. Cheng, Z. C. Liu, M. T. Nguyen, T. Y. Chen, H. A. Patel, M. D. Krzyaniak, W. A. Goddard, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 1290–1303 CrossRef.
- G. Rapenne and C. Joachim, Nat. Rev. Mater., 2017, 2, 17040 CrossRef.
- T. Ikeda and T. Ube, Mater. Today, 2011, 14, 480–487 CrossRef CAS.
- M. D. Manrique-Juárez, S. Rat, L. Salmon, G. Molnár, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef.
- Y. Hu, Z. Li, T. Lan and W. Chen, Adv. Mater., 2016, 28, 10548–10556 CrossRef CAS.
- M. A. Watson and S. L. Cockroft, Chem. Soc. Rev., 2016, 45, 6118–6129 RSC.
- N. K. Nath, M. K. Panda, S. C. Sahoo and P. Naumov, CrystEngComm, 2014, 16, 1850–1858 RSC.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS.
- O. S. Bushuyev, T. Friščić and C. J. Barrett, CrystEngComm, 2016, 18, 7204–7211 RSC.
- H. X. Deng, M. A. Olson, J. F. Stoddart and O. M. Yaghi, Nat. Chem., 2010, 2, 439–443 CrossRef CAS.
- A. Coskun, M. Hmadeh, G. Barin, F. Gándara, Q. Li, E. Choi, N. L. Strutt, D. B. Cordes, A. M. Z. Slawin, J. F. Stoddart, J.-P. Sauvage and O. M. Yaghi, Angew. Chem., Int. Ed., 2012, 51, 2160–2163 CrossRef CAS.
- K. Zhu, C. A. O'Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514 CrossRef CAS.
- N. Farahani, K. L. Zhu, C. A. O'Keefe, R. W. Schurko and S. J. Loeb, ChemPlusChem, 2016, 81, 836–841 CrossRef CAS.
- V. N. Vukotic, K. Zhu, G. Baggi and S. J. Loeb, Angew. Chem., 2017, 129, 6232–6237 CrossRef.
- D. K. Shen, G. Wang, Z. C. Liu, P. Li, K. Cai, C. Y. Cheng, Y. Shi, J. M. Han, C. W. Kung, X. R. Gong, Q. H. Guo, H. L. Chen, A. C. H. Sue, Y. Y. Botros, A. Facchetti, O. K. Farha, T. J. Marks and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 11402–11407 CrossRef CAS.
- X. Jiang, Z. J. O'Brien, S. Yang, L. H. Lai, J. Buenaflor, C. Tan, S. Khan, K. N. Houk and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2016, 138, 4650–4656 CrossRef CAS.
- M. E. Howe and M. A. Garcia-Garibay, J. Org. Chem., 2019, 84, 9835–9849 CrossRef CAS.
- P. Commins and M. A. Garcia-Garibay, J. Org. Chem., 2014, 79, 1611–1619 CrossRef CAS.
- W. Setaka and K. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 14560–14563 CrossRef CAS.
- A. Fujiwara, Y. Inagaki, H. Momma, E. Kwon, K. Yamaguchi, M. Kanno, H. Kono and W. Setaka, CrystEngComm, 2017, 19, 6049–6056 RSC.
- M. Tsurunaga, Y. Inagaki, H. Momma, E. Kwon, K. Yamaguchi, K. Yoza and W. Setaka, Org. Lett., 2018, 20, 6934–6937 CrossRef CAS.
- Y. L. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed.
- N. Hosono, T. Kajitani, T. Fukushima, K. Ito, S. Sasaki, M. Takata and T. Aida, Science, 2010, 330, 808–811 CrossRef CAS PubMed.
- T. Ube and T. Ikeda, Angew. Chem., Int. Ed., 2014, 53, 10290–10299 CrossRef CAS.
- H. Koshima, N. Ojima and H. Uchimoto, J. Am. Chem. Soc., 2009, 131, 6890–6891 CrossRef CAS.
- T. Taniguchi, J. Fujisawa, M. Shiro, H. Koshima and T. Asahi, Chem. – Eur. J., 2016, 22, 7950–7958 CrossRef CAS.
- T. Taniguchi, H. Sugiyama, H. Uekusa, M. Shiro, T. Asahi and H. Koshima, Nat. Commun., 2018, 9, 538 CrossRef.
- O. S. Bushuyev, T. C. Corkery, C. J. Barrett and T. Friščić, Chem. Sci., 2014, 5, 3158–3164 RSC.
- O. S. Bushuyev, A. Tomberg, T. Friščić and C. J. Barrett, J. Am. Chem. Soc., 2013, 135, 12556–12559 CrossRef CAS.
- Y. Norikane, S. Tanaka and E. Uchida, CrystEngComm, 2016, 18, 7225–7228 RSC.
- E. Uchida, R. Azumi and Y. Norikane, Nat. Commun., 2015, 6, 7310 CrossRef CAS PubMed.
- T. Shima, T. Muraoka, N. Hoshino, T. Akutagawa, Y. Kobayashi and K. Kinbara, Angew. Chem., Int. Ed., 2014, 53, 7173–7178 CrossRef CAS.
- P. Gupta, T. Panda, S. Allu, S. Borah, A. Baishya, A. Gunnam, A. Nangia, P. Naumov and N. K. Nath, Cryst. Growth Des., 2019, 19, 3039–3044 CrossRef CAS.
- P. Gupta, D. P. Karothu, E. Ahmed, P. Naumov and N. K. Nath, Angew. Chem., 2018, 130, 8634–8638 CrossRef.
- M. Irie, T. Fulcaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS.
- F. Terao, M. Morimoto and M. Irie, Angew. Chem., Int. Ed., 2012, 51, 901–904 CrossRef CAS.
- D. Kitagawa and S. Kobatake, Chem. Commun., 2015, 51, 4421–4424 RSC.
- D. Kitagawa, K. Kawasaki, R. Tanaka and S. Kobatake, Chem. Mater., 2017, 29, 7524–7532 CrossRef CAS.
- D. Kitagawa, H. Tsujioka, F. Tong, X. N. Dong, C. J. Bardeen and S. Kobatake, J. Am. Chem. Soc., 2018, 140, 4208–4212 CrossRef CAS.
- X. M. Dong, F. Tong, K. M. Hanson, R. O. Al-Kaysi, D. Kitagawa, S. Kobatake and C. J. Bardeen, Chem. Mater., 2019, 31, 1016–1022 CrossRef CAS.
- S. Ishida, D. Kitagawa, S. Kobatake, S. Kim, S. Kurihara and T. Fukaminato, Chem. Commun., 2019, 55, 5681–5684 RSC.
- K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950–967 RSC.
- V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS.
- J. J. Vittal and H. S. Quah, Dalton Trans., 2017, 46, 7120–7140 RSC.
- H. Ying-Feng, J. Guo-Xin, C. G. Daniliuc and H. F. Ekkehardt, Angew. Chem., Int. Ed., 2015, 54, 4958–4962 CrossRef.
- R. Medishetty, Z. Bai, H. Yang, M. W. Wong and J. J. Vittal, Cryst. Growth Des., 2015, 15, 4055–4061 CrossRef CAS.
- P. In-Hyeok, M. Raghavender, L. Hyeong-Hwan, M. C. Evania, Q. H. Sheng, L. S. Sung and J. J. Vittal, Angew. Chem., Int. Ed., 2015, 54, 7313–7317 CrossRef.
- J.-M. Chen, Y.-X. Hou, Q.-K. Zhou, H. Zhang and D. Liu, CrystEngComm, 2017, 19, 2603–2607 RSC.
- Y. X. Shi, W. H. Zhang, B. F. Abrahams, P. Braunstein and J. P. Lang, Angew. Chem., Int. Ed., 2019, 58, 9453–9458 CrossRef CAS.
- R. Medishetty, A. Husain, Z. Bai, T. Runčevski, R. E. Dinnebier, P. Naumov and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 5907–5911 CrossRef CAS.
- K. Yadava and J. J. Vittal, Cryst. Growth Des., 2019, 19, 2542–2547 CrossRef CAS.
- C. E. Mulijanto, H. S. Quah, G. K. Tan, B. Donnadieu and J. J. Vittal, IUCrJ, 2017, 4, 65–71 CrossRef CAS.
- R. Medishetty, S. C. Sahoo, C. E. Mulijanto, P. Naumov and J. J. Vittal, Chem. Mater., 2015, 27, 1821–1829 CrossRef CAS.
- H. Wang, P. Chen, Z. Wu, J. Zhao, J. Sun and R. Lu, Angew. Chem., Int. Ed., 2017, 56, 9463–9467 CrossRef CAS.
- J. Peng, K. Q. Ye, C. Liu, J. B. Sun and R. Lu, J. Mater. Chem. C, 2019, 7, 5433–5441 RSC.
- S. Z. Li and D. P. Yan, ACS Appl. Mater. Interfaces, 2018, 10, 22703–22710 CrossRef CAS.
- T. Kim, L. Zhu, L. J. Mueller and C. J. Bardeen, J. Am. Chem. Soc., 2014, 136, 6617–6625 CrossRef CAS.
- F. Tong, W. J. Xu, M. Al-Haidar, D. Kitagawa, R. O. Al-Kaysi and C. J. Bardeen, Angew. Chem., Int. Ed., 2018, 57, 7080–7084 CrossRef CAS.
- H. Koshima, H. Uchimoto, T. Taniguchi, J. Nakamura, T. Asahi and T. Asahi, CrystEngComm, 2016, 18, 7305–7310 RSC.
- A. Ravi and K. M. Sureshan, Angew. Chem., Int. Ed., 2018, 57, 9362–9366 CrossRef CAS.
- R. Rai, B. P. Krishnan and K. M. Sureshan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2896–2901 CrossRef CAS.
- M. Horie, Y. Suzaki and K. Osakada, J. Am. Chem. Soc., 2004, 126, 3684–3685 CrossRef CAS PubMed.
- M. Horie, Y. Suzaki and K. Osakada, Inorg. Chem., 2005, 44, 5844–5853 CrossRef CAS.
- M. Horie, T. Sassa, D. Hashizume, Y. Suzaki, K. Osakada and T. Wada, Angew. Chem., Int. Ed., 2007, 46, 4983–4986 CrossRef CAS.
- M. Horie, Y. Suzaki, D. Hashizume, T. Abe, T. Wu, T. Sassa, T. Hosokai and K. Osakada, J. Am. Chem. Soc., 2012, 134, 17932–17944 CrossRef CAS.
- Y.-C. Tsai, K.-J. Chen, C.-J. Su, W.-R. Wu, U. S. Jeng and M. Horie, J. Mater. Chem. C, 2014, 2, 2061–2068 RSC.
- K. J. Chen, A. C. Tan, C. H. Wang, T. S. Kuo, P. L. Chen and M. Horie, Cryst. Growth Des., 2019, 19, 17–22 CrossRef CAS.
- K.-J. Chen, P.-L. Chen and M. Horie, Sci. Rep., 2017, 7, 14195 CrossRef PubMed.
- P. R. Ashton, M. C. T. Fyfe, S. K. Hickingbottom, J. F. Stoddart, A. J. P. White and D. J. Williams, J. Chem. Soc., Perkin Trans. 2, 1998, 2117–2128 RSC.
- K.-J. Chen, Y.-C. Tsai, Y. Suzaki, K. Osakada, A. Miura and M. Horie, Nat. Commun., 2016, 7, 13321 CrossRef CAS.
- M. Morimoto and M. Irie, J. Am. Chem. Soc., 2010, 132, 14172–14178 CrossRef CAS.
- F. T. Cheng, R. Y. Yin, Y. Y. Zhang, C. C. Yen and Y. L. Yu, Soft Matter, 2010, 6, 3447–3449 RSC.
- L. D. Zhang, H. R. Liang, J. Jacob and P. Naumov, Nat. Commun., 2015, 6, 7429 CrossRef CAS.
- M. M. Ma, L. Guo, D. G. Anderson and R. Langer, Science, 2013, 339, 186–189 CrossRef CAS.
- C. Yang, W. Wang, C. Yao, R. Xie, X. J. Ju, Z. Liu and L. Y. Chu, Sci. Rep., 2015, 5, 13622 CrossRef.
- S.-C. Cheng, K.-J. Chen, Y. Suzaki, Y. Tsuchido, T.-S. Kuo, K. Osakada and M. Horie, J. Am. Chem. Soc., 2018, 140, 90–93 CrossRef CAS.
- S. Guo, K. Matsukawa, T. Miyata, T. Okubo, K. Kuroda and A. Shimojima, J. Am. Chem. Soc., 2015, 137, 15434–15440 CrossRef CAS.
| This journal is © the Partner Organisations 2019 |