DOI:
10.1039/C9QM00468H
(Research Article)
Mater. Chem. Front., 2019,
3, 2380-2387
Molecular design to increase the photosensitivity of photochromic phenoxyl–imidazolyl radical complexes†
Received
17th July 2019
, Accepted 9th September 2019
First published on 10th September 2019
Abstract
Photochromic molecules have received a lot of interest in potential applications. The development of the phenoxyl–imidazolyl radical complex (PIC), which generates two nonequivalent radicals upon light irradiation along with the fast photochemical reaction, is crucial for photoresponsive materials. However, as a novel photochromic compound, the low photosensitivity of PIC derivatives has limited their practical applications. Herein, increased photosensitivity and absorption coefficients of PIC derivatives were achieved while keeping the thermal back reaction rate by introducing the substitution of aryl groups. This strategy gives significant insight into the development of desirable fast photochromic compounds based on the PIC framework.
Introduction
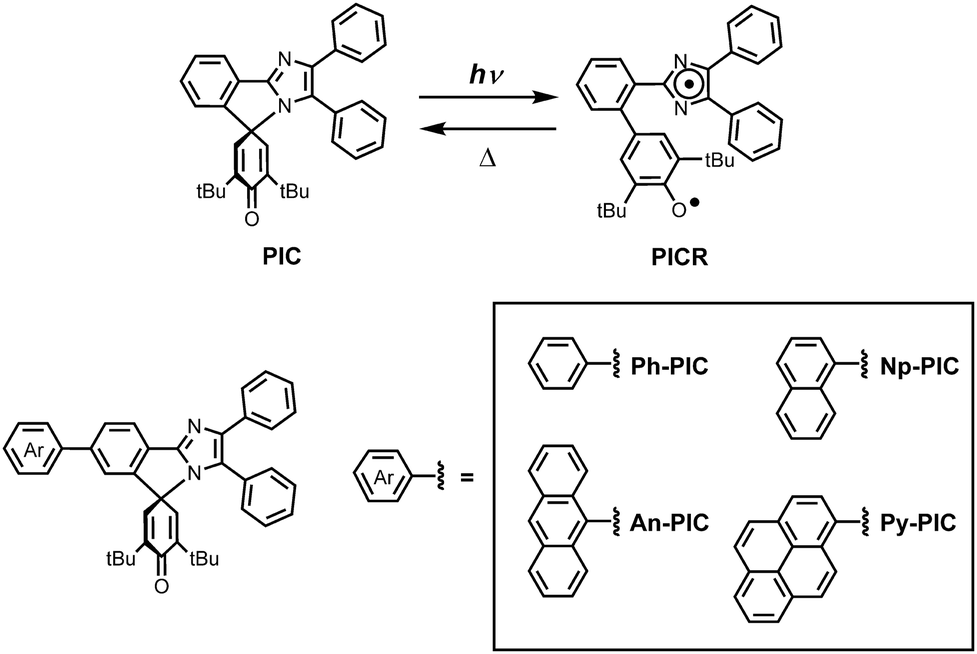
Photoresponsive molecular materials have attracted much interest because their switching properties can be spatiotemporally controlled in a remote and noninvasive manner.1–4 Photochromic molecules with reversible isomerization of chemical species upon light irradiation have been applied to various fields of science and technology. The organic photochromic molecules can be categorized by the type of chemical reaction during the photochromic reaction process: derivatives of salicylideneaniline as proton transfer processes,5–7 azobenene derivatives as trans–cis photoisomerization processes,8,9 hexaarylbiimizadole (HABI) derivatives as homolytic bond-cleavage processes,10–12 and spiropyrans,13 fulgides14 and diarylethenes15,16 as ring-opening and closing reactions. Photochromic compounds can also be divided into P-type and T-type according to the potential energy barrier between the photoinduced metastable species and the original stable species. The T-type photochromic compounds, in which the photogenerated colored species thermally reverts to the initial colorless form, such as bridged imidazole dimers,17,18 naphthopyrans,19,20 hexaarylbiimidazoles10–12 and azobenzene derivatives8,9 have been extensively studied due to their potential applications for ophthalmic lenses,21 dynamic holographies,22 fluorescent switches23–25 and security materials.26 We developed the phenoxyl–imidazolyl radical complex (PIC), which generates two different radical species when irradiated with light.27 The photochromic response of the PIC is the result of a homolytic cleavage of the C–N bond, i.e., the bond between the C-4 of the 4H-cyclohexadienone ring and the N-1 of the imidazole ring (Scheme 1). The photochemical bond cleavage causes the formation of a couple of nonequivalent phenoxyl and imidazolyl radicals. The decoloration speed from the photogenerated colored species to the colorless species can be controlled from the nanosecond to the second time scale by appropriate substitutions to the two radical units. However, because the PIC has no absorption band longer than 360 nm, increasing the photosensitivity of the photochromic reaction of PIC derivatives is required for increasing the conversion ratio at the photostationary state.28,29
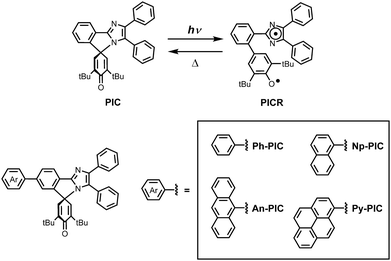 |
| | Scheme 1 Photochromic reaction scheme of PIC and the molecular structures of the PIC derivatives. | |
Therefore, the increase in the efficiency of the forward coloration reaction along with the relatively high photosensitivity of PIC compounds is necessary to achieve an efficient fast photochromic reaction and practical applications.
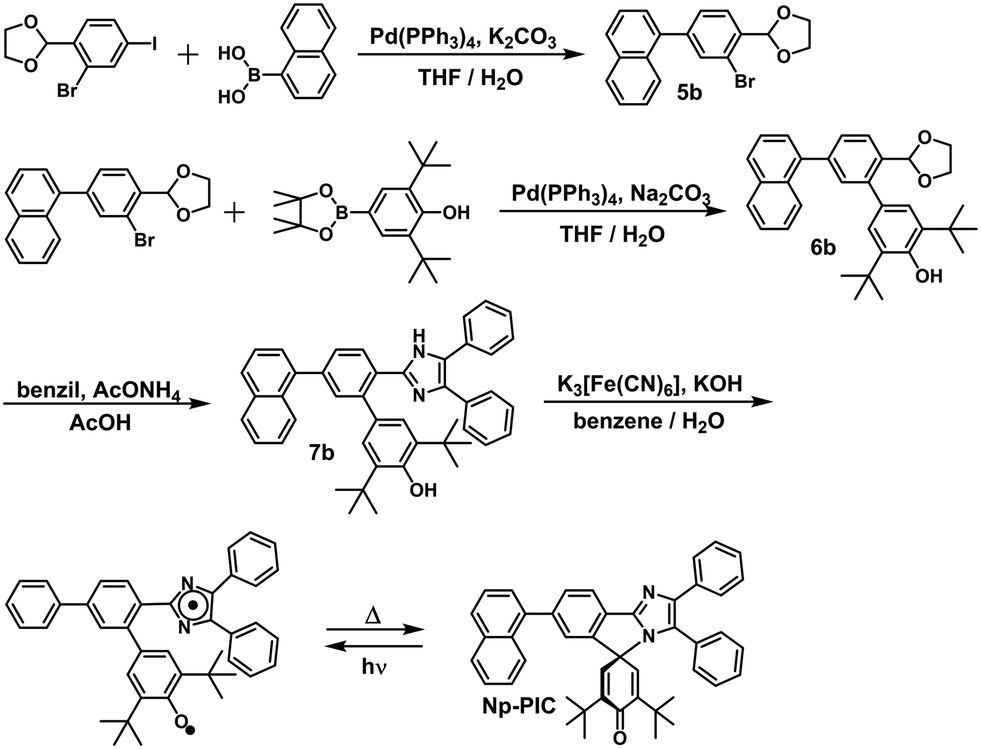
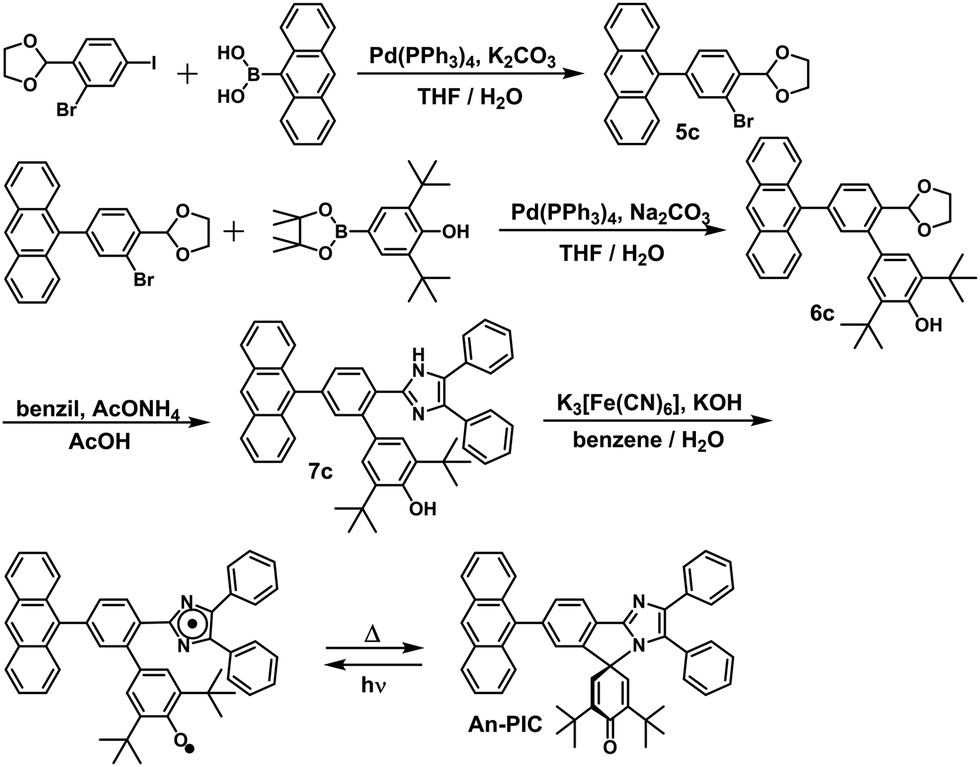
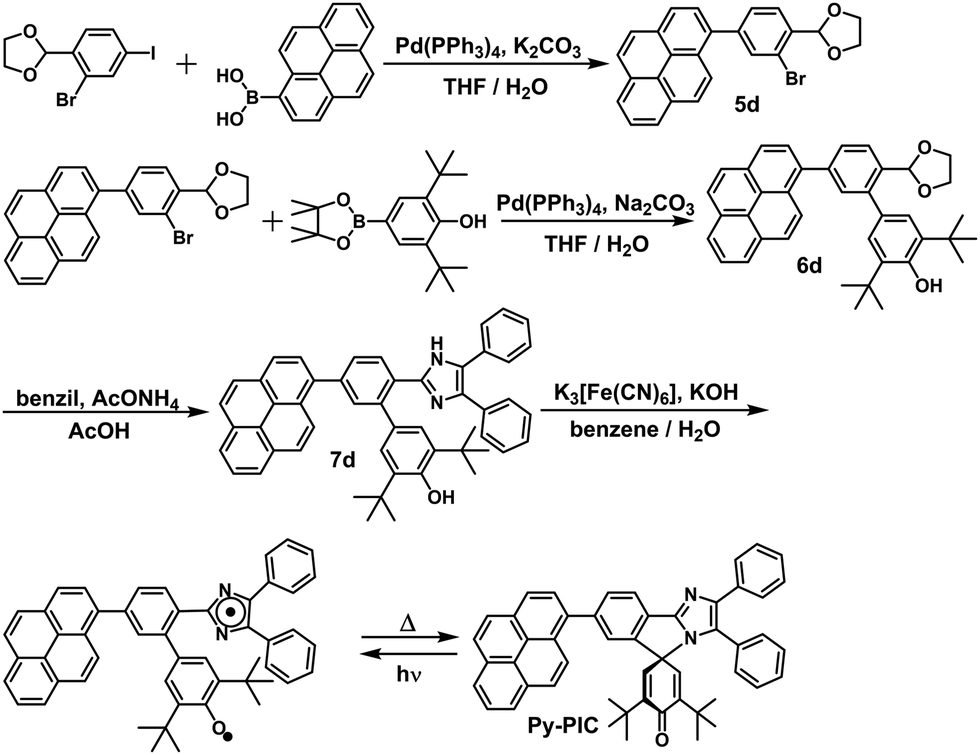
In this study, the PIC derivatives, Ph-PIC, Np-PIC, An-PIC and Py-PIC, in which an aromatic ring was introduced into the bridging phenyl ring at the 2-position of the imidazole ring, were designed and synthesized. The synthetic schemes for all the PIC derivatives are shown in the experimental section (Schemes 2–5).
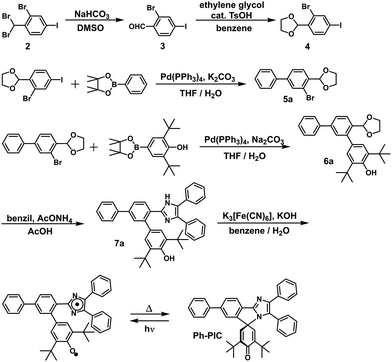 |
| | Scheme 2 Synthetic scheme of Ph-PIC. | |
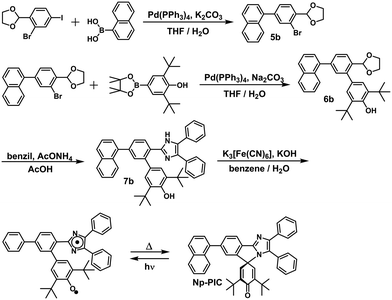 |
| | Scheme 3 Synthetic scheme of Np-PIC. | |
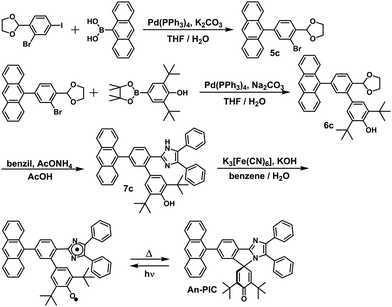 |
| | Scheme 4 Synthetic scheme of An-PIC. | |
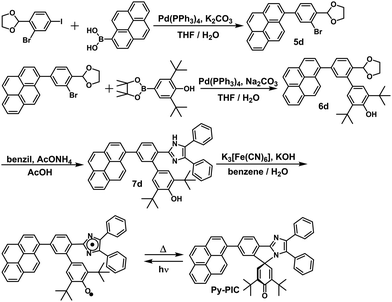 |
| | Scheme 5 Synthetic scheme of Py-PIC. | |
Results and discussion
The photochromic reaction of the PIC proceeds from the S1 state, which is characterized by the bond-dissociation potential energy surface. The S0 → S1 transition is assigned to the highest occupied molecular orbital (HOMO)-lowest unoccupied molecular orbital (LUMO) transition.30 The HOMO and LUMO are localized on the perpendicularly oriented imidazole ring and the phenoxyl ring, respectively. The small size of the molecule and the small overlap between the HOMO and LUMO cause the small oscillator strength of the S0 → S1 transition, resulting in the small molar extinction coefficient of the PIC at 365 nm, which is the wavelength of common UV-LEDs. In order to improve the photosensitivity of PIC derivatives, the introduction of a singlet sensitizer is efficient because the energy transfer from the excited state of the sensitizer to the optically forbidden S1 state of the PIC can be expected.
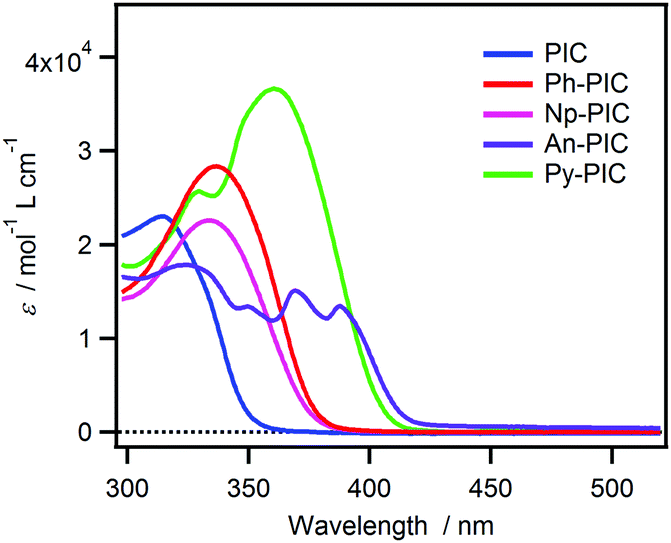
The steady-state absorption spectra of all the derivatives in benzene solution are shown in Fig. 1. The maximum absorption wavelength of the PIC is located at 320 nm, whereas the absorption bands of Ph-PIC, Np-PIC, An-PIC and Py-PIC are red-shifted by the introduction of the large aromatic ring as compared to the parent compound PIC. Especially for Py-PIC, the molar extinction coefficient at 365 nm increased distinctly (ε365nm = 3.60 × 104 M−1 cm−1). This phenomenon can be well explained by using the time-dependent density functional theory (TDDFT) calculation (MPW1PW91/6-31+G(d,p)//M05-2X/6-31G(d) level of the theory). The transition in the UVA region is attributable to the HOMO → LUMO+1 transition. Because the HOMO and LUMO+1 are delocalized over the diphenyl imidazole ring and the introduced aromatic ring, respectively, the overlap between the molecular orbitals is larger than that of the original PIC. This also guarantees the large molar extinction coefficient of these PIC derivatives (Fig. S36–S39 and Tables S2, S4, S6, S8, ESI†). By introducing the aromatic rings to the phenyl ring at the 2-position of the imidazole ring, the high photosensitivity and molar extinction coefficient at 365 nm were achieved for the PIC derivatives.
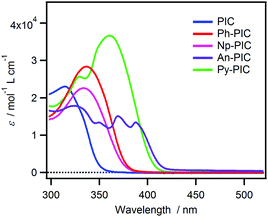 |
| | Fig. 1 Steady-state absorption spectra of PIC, Ph-PIC, Np-PIC, An-PIC and Py-PIC in benzene solution. | |
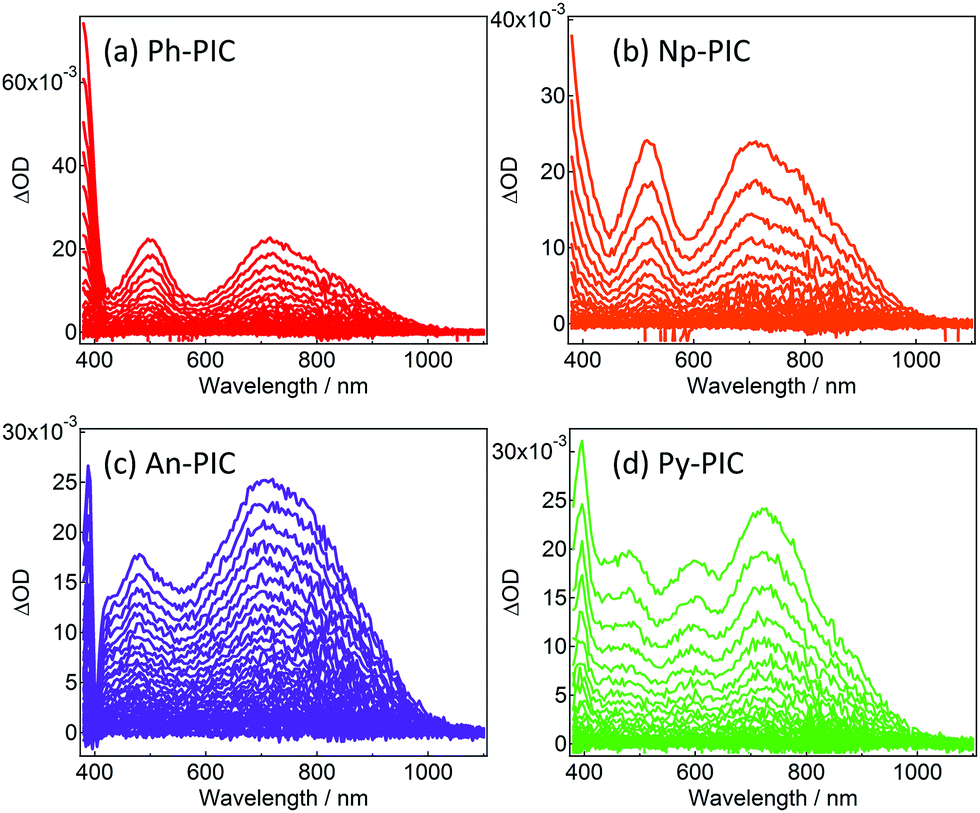
Fig. 2 shows the transient absorption spectra of Ph-PIC, Np-PIC, An-PIC and Py-PIC in toluene (6.7 × 10−5, 9.2 × 10−5, 4.9 × 10−5, and 6.0 × 10−5 M, respectively) upon 355 nm nanosecond laser pulse irradiation at 183 K. The transient absorption spectra assigned to the open-ring isomers (PICR) of Ph-PIC, Np-PIC, and An-PIC show three peaks at about 385 nm, 500 nm and 700 nm, while those of Py-PIC show four peaks at 400 nm, 480 nm, 600 nm and 720 nm. All the transient absorption spectra of PICRs have a broad absorption band at 700–750 nm. This characteristic absorption band is due to the radical–radical interaction between the photogenerated imidazolyl and phenoxyl radicals.27 Interestingly, while the absorption spectra of the closed ring isomers of PIC derivatives are largely shifted to a longer wavelength as compared to that of the PIC,27,29 the characteristic absorption bands at 500 nm and 720 nm to the biradical species of the PIC are not largely changed irrespective of the aryl substituents. The plausible reason for this phenomenon is due to the no changes in the spin-density distributions of the PICR as described below. This result indicates that the substitutions at the phenyl ring of the 2-position of the imidazole ring do not affect the absorption spectra of the open ring isomers.
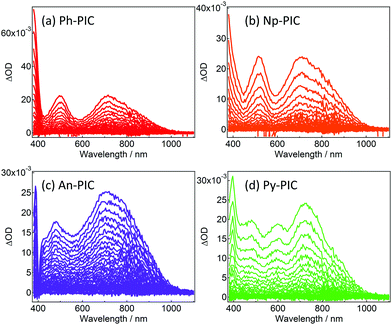 |
| | Fig. 2 Transient vis-NIR spectra of (a) Ph-PIC, (b) Np-PIC, (c) An-PIC and (d) Py-PIC in toluene (excitation wavelength: 355 nm, temp.: 183 K, power: 4.2 mJ, time interval: 4.0 ns). | |
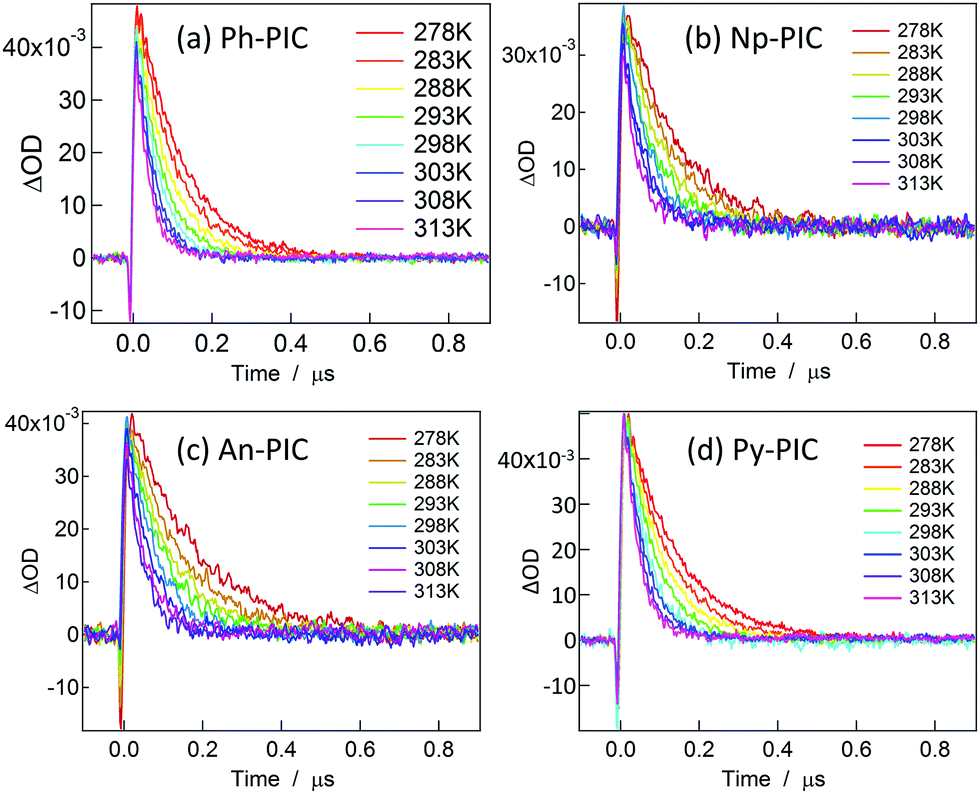
Fig. 3 shows the decay profiles of the transient absorbance of Ph-PIC, Np-PIC, An-PIC and Py-PIC (3.2 × 10−5, 3.5 × 10−5, 3.1 × 10−5, and 3.3 × 10−5 M, respectively) probed at 700 nm after irradiating with a 355 nm nanosecond laser pulse in benzene. Similar to the transient absorption spectra (Fig. 2), the rates of the thermal back reaction of the open ring isomers for all the PIC derivatives have more or less similar half-lives (around 40 ns at room temperature). In order to investigate the reason for the invariable thermal back reaction rates of the PIC derivatives, the Eyring analysis for the rate constants for the thermal back reaction was performed (Fig. S45, ESI†). The activation parameters of all the PIC derivatives for the thermal back reaction are summarized in Table 1.
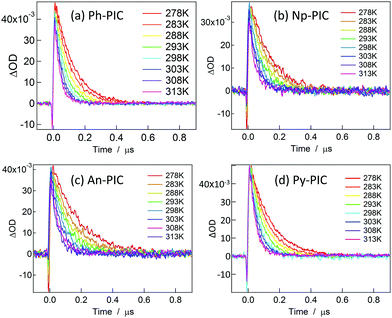 |
| | Fig. 3 Decay profiles of the transient biradical species of Ph-PIC, Np-PIC, An-PIC and Py-PIC (3.2 × 10−5, 3.5 × 10−5, 3.1 × 10−5, and 3.3 × 10−5 M, respectively) probed at 700 nm in benzene. | |
Table 1 Activation parameters of PIC, Ph-PIC, Np-PIC, An-PIC and Py-PIC
|
|
ΔH‡ [kJ mol−1] |
ΔS‡ [J mol−1 K−1] |
ΔG‡ [kJ mol−1] |
τ
1/2 [ns] |
|
PIC
|
26.3 |
−15.1 |
30.7 |
26 |
|
Ph-PIC
|
23.6 |
−28.8 |
32.2 |
35 |
|
Np-PIC
|
22.9 |
−30.3 |
32.0 |
46 |
|
An-PIC
|
25.1 |
−24.3 |
32.4 |
49 |
|
Py-PIC
|
22.8 |
−30.6 |
31.9 |
39 |
Except for An-PIC, the absolute values of activation enthalpy (ΔH‡) for the thermal back reactions gradually decrease by extending the π-conjugation of the introduced aromatic rings. The reason for the deviation of An-PIC is probably due to the larger dihedral angle between the anthracene and the imidazole ring of An-PIC (θ = 69.3°) than those of the other PIC derivatives (θ = 37–55°) (Fig. S46 and Table S17, ESI†). This large dihedral angle causes the inefficient π-electron conjugation between the anthracene moiety and the imidazole ring. On the other hand, according to the Eyring equation, the absolute values of the activation entropy (ΔS‡) for the thermal back reactions increase with decreasing ΔH‡, leading to the little differences between the activation free energy (ΔG‡) in the photochromic reactions of the PIC derivatives.
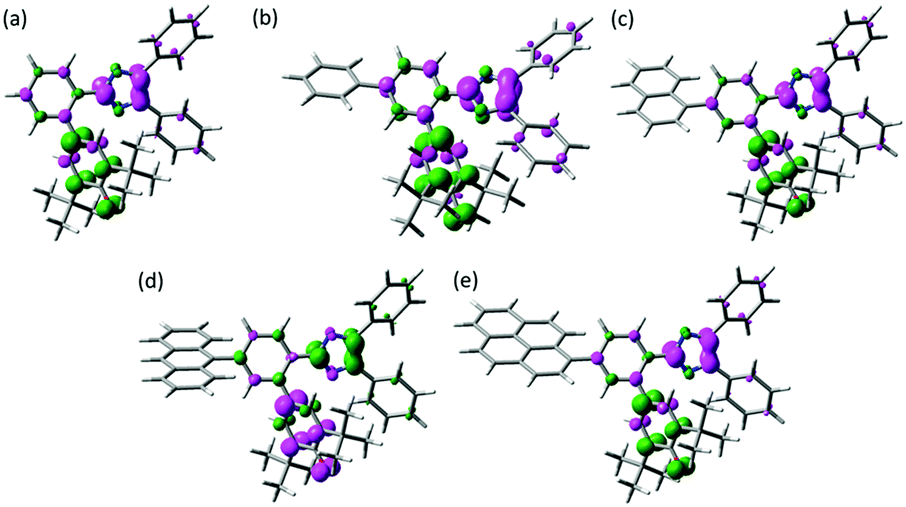
The spin density of the biradical is one of the measures to discuss about the similar transient absorption spectra and the decay profiles of the transient biradical species of Ph-PIC, Np-PIC, An-PIC and Py-PIC (Fig. 4). As discussed above, the broad absorption band at around 700 nm of the biradical species is attributable to the strong interaction between the imidazolyl and phenoxyl radicals at the o-position of the phenyl ring. Moreover, since the thermal back reaction of the biradical species is the intramolecular recombination reaction, the spin density on the imidazole and the phenoxyl rings could critically affect the rate of the thermal recombination reaction. The calculated spin density distributions of the singlet biradical species by DFT calculation are shown in Fig. 4. The introduction of the aromatic rings to the bridging phenyl ring induces almost no changes in the spin density distributions on both the phenoxyl and diphenyl imidazole rings. Therefore, the extension of the π-electron conjugation length is insensitive to the transient absorption spectrum and the speed of the thermal back reaction.
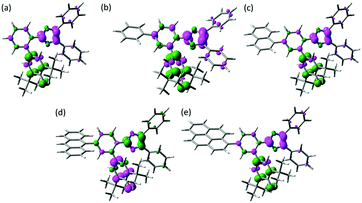 |
| | Fig. 4 Spin density distributions of (a) PICR, (b) Ph-PICR, (c) Np-PICR, (d) An-PICR, and (e) Py-PICR. (UMPW1PW91/6-31+G(d,p)//UM052X/6-31G(d)). | |
Conclusions
In summary, we developed PIC derivatives possessing high photosensitivity to UVA light. The introduction of the aromatic ring to the bridging part of the PIC is efficient to increase the photosensitivity and the molar extinction coefficient of the PIC derivatives at 365 nm. On the other hand, because of the similar activation free energy barrier and the almost invariable spin density distribution of the biradical species, the absorption spectrum of the transient biradical and the thermal back reaction rate are insensitive to the π-electron conjugation length. This result could give us a perspective for designing fast photochromic molecules with high photosensitivity and molar extinction coefficient without changing the absorption spectrum of the transient biradical and the thermal back reaction rate in the future.
Experimental
Materials and methods
All of the reactions were monitored via thin-layer chromatography carried out on 0.2 mm E. Merck silica gel plates (60F-254). Column chromatography was performed on silica gel (silica gel 60N, Kanto Chemical Co., Inc.). NMR spectra were recorded at 400 MHz on a Bruker AVANCE III 400 NanoBay spectrometer. DMSO-d6 and CDCl3 were used as deuterated solvents. ESI-TOF-MS spectra were recorded on a Bruker micrOTOF II-AGA1 spectrometer. Unless otherwise noted, all of the reagents and reaction solvents were purchased from Tokyo Chemical Industry Co., Ltd, Wako Pure Chemical Industries, Ltd, Sigma-Aldrich Inc., and Kanto Chemical Co., Inc. and used without further purification.
Synthetic procedures
Synthesis of compound 2-bromo-4-iodobenzaldehyde (3).
2-Bromo-1-(dibromomethyl)-4-iodobenzene (2) (5 g, 11 mmol) and sodium hydrogen carbonate (3.8 g, 45 mmol) in DMSO (55 mL) were refluxed for 1.1 h. After cooling to room temperature, the mixture was extracted with CH2Cl2 and the organic layer was washed with water, and dried over Na2SO4. After the removal of the solvents, the residue was purified via silica gel column chromatography (hexane/CH2Cl2 = 2/1) to give 3 (1.0 g, 28%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 10.28 (s, 1H), 8.06 (s, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.66 (d, J = 8.1 Hz, 1H).
Synthesis of compound 2-(2-bromo-4-iodophenyl)-1,3-dioxolane (4).
2-Bromo-4-iodobenzaldehyde (3) (80 mg, 0.29 mmol), ethylene glycol (27 mg, 0.44 mmol) and p-toluene sulfonic acid (2.2 mg, 0.017 mmol) in benzene (4 mL) were refluxed for 18 h with Dean–Stark apparatus. After cooling to room temperature, the mixture was extracted with AcOEt and the organic layer was washed with water, and dried over Na2SO4. After the removal of the solvents, the residue was purified via silica gel column chromatography (hexane/AcOEt = 10/1) to give 4 (38 mg, 42%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.93 (s, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.31 (d, J = 8.2 Hz, 1H), 6.02 (s, 1H), 4.16–4.04 (m, 4H).
Synthesis of compound 2-(3-bromo-[1,1′-biphenyl]-4-yl)-1,3-dioxolane (5a).
A THF (2 mL) and water (2 mL) solution of potassium carbonate (43 mg, 0.31 mmol) was added with 2-(2-bromo-4-iodophenyl)-1,3-dioxolane (4) (39 mg, 0.11 mmol) and 4,4,5,5-tetramethyl-2-phenyl-1,3,2-dioxaborolane (45 mg, 0.22 mmol), and the mixture was purged with N2 gas. The resulting solution was added with Pd(PPh3)4 (3.8 mg, 3.3 × 10−3 mmol), and refluxed for 25 h. The mixture was extracted with AcOEt, and the organic extract was washed with water, and dried over Na2SO4. After the removal of the solvents, the residue was purified via silica gel column chromatography (hexane/ethyl acetate = 10/1) to give 5a (32 mg, 95%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.79 (s, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.57–7.55 (m, 3H), 7.45 (t, J = 7.6 Hz, 2H), 7.38 (t, J = 7.2 Hz, 1H), 6.14 (s, 1H), 4.21–4.08 (m, 4H). HRMS (ESI-TOF) calcd for C15H16BrO2 [M + H]+, 326.9991; found, 326.9944.
Synthesis of compound 3,5-di-tert-butyl-6′-(1,3-dioxolan-2-yl)-[1,1′:3′,1′′-terphenyl]-4-ol (6a).
A THF (1 mL) and water (0.5 mL) solution of Na2CO3 (70 mg, 0.36 mmol) was added with 2-(3-bromo-[1,1′-biphenyl]-4-yl)-1,3-dioxolane (5a) (30 mg, 0.13 mmol) and 2,6-di-tert-butyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol (53 mg, 0.16 mmol), and the mixture was purged with N2 gas. The resulting solution was added with Pd(PPh3)4 (4.5 mg, 3.9 × 10−3 mmol), and refluxed for 24 h. The mixture was extracted with AcOEt, and the organic extract was washed with water, and dried over Na2SO4. After the removal of the solvents, the residue was purified via silica gel column chromatography (hexane/ethyl acetate = 10/1) to give 6a (27 mg, 65%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.79 (d, J = 7.8 Hz, 1H), 7.64–7.59 (m, 3H), 7.56 (s, 1H), 7.44 (t, J = 7.8 Hz, 2H), 7.36 (d, J = 7.4 Hz, 1H), 7.34 (s, 2H), 5.69 (s, 1H), 5.27 (s, 1H), 4.25–3.96 (m, 2H), 1.48 (s, 18H). HRMS (ESI-TOF) calcd for C29H34O3 [M + H]+, 431.2580; found, 431.2518.
Synthesis of compound 3,5-di-tert-butyl-6′-(4,5-diphenyl-1H-imidazol-2-yl)-[1,1′:3′,1′′-terphenyl]-4-ol (7a).
3,5-Di-tert-butyl-6′-(1,3-dioxolan-2-yl)-[1,1′:3′,1′′-terphenyl]-4-ol (6a) (18 mg, 0.042 mmol), benzil (10.6 mg, 0.050 mmol), and ammonium acetate (64.8 mg, 0.84 mmol) were stirred at 110 °C in AcOH (0.5 mL) in a sealed tube for 12 h. The reaction mixture was cooled to room temperature and neutralized by NH3 aq. The organic extraction was washed with water and dichloromethane. The crude product was purified via silica gel column chromatography (hexane/AcOEt = 10/1 → 6/1) to give 7a (63 mg, 63%) as a colorless solid. 1H NMR (400 MHz, DMSO-d6): δ 11.76 (s, 1H), 7.80 (d, J = 7.7 Hz, 2H), 7.74 (d, J = 6.4 Hz, 2H), 7.52 (t, J = 7.7 Hz, 2H), 7.43 (d, J = 7.3 Hz, 3H), 7.33–7.25 (m, 6H), 7.21–7.19 (m, 3H), 7.06 (s, 2H), 6.95 (s, 1H), 1.30 (s, 18H).
Synthesis of compound 3,5-di-tert-butyl-2′,3′,7′-triphenylspiro-[cyclohexane-1,5′-imidazo[2,1-a]isoindole]-2,5-dien-4-one (Ph-PIC, Scheme 2).
All manipulations were carried out with exclusion of light. A solution of potassium ferricyanide (267 mg, 0.81 mmol), and KOH (210 mg, 0.90 mmol) in water (2.0 mL) was added to a suspension of 3,5-di-tert-butyl-6′-(4,5-diphenyl-1H-imidazol-2-yl)-[1,1′:3′,1′′-ter-phenyl]-4-ol (7a) (15 mg, 0.26 mmol) in benzene (1.5 mL). After stirring for 6 h at room temperature, the resultant mixture was then extracted with benzene and the organic extract was washed with water and dried over Na2SO4. After the removal of the solvents, the crude product was purified via silica gel column chromatography (hexane/AcOEt = 6/1), to give Ph-PIC (11 mg, 72%) as a pale yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 7.99 (d, J = 7.7 Hz, 1H), 7.84 (d, J = 7.7 Hz, 1H), 7.57 (d, J = 7.3 Hz, 2H), 7.53 (d, J = 7.3 Hz, 2H), 7.49 (d, J = 7.3 Hz, 2H),7.41–7.33 (m, 6H), 7.25 (t, J = 7.7 Hz, 2H), 7.17 (t, J = 7.3 Hz, 1H), 7.11 (s, 1H), 6.52 (s, 1H), 1.06 (s, 18H). HRMS (ESI-TOF) calcd for C41H38N2O [M + H]+, 575.3056; found, 575.3005.
Synthesis of compound 2-(2-bromo-4-(naphthalen-1-yl)phenyl)-1,3-dioxolane (5b).
A THF (1 mL) and water (0.5 mL) solution of sodium carbonate (47 mg, 0.45 mmol) was added with 4 (56.7 mg, 0.160 mmol) and naphthalen-1-ylboronic acid (33 mg, 0.19 mmol), and the mixture was purged with N2 gas. The resulting solution was added with Pd(PPh3)4 (5.6 mg, 4.8 × 10−3 mmol), and refluxed for 25 h. The mixture was extracted with CH2Cl2, and the organic extract was washed with water, and dried over Na2SO4. After the removal of the solvents, the residue was purified via silica gel column chromatography (hexane/ethyl acetate = 10/1) to give 5b (53 mg, 93%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 8.5 Hz, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 8.5 Hz, 1H), 7.73–7.71 (m, 2H), 7.54–7.43 (m, 4H), 7.39 (d, J = 6.9 Hz, 1H), 6.20 (s, 1H), 4.25–4.14 (m, 4H).
Synthesis of compound 3,5-di-tert-butyl-2′-(1,3-dioxolan-2-yl)-5′-(naphthalen-1-yl)-[1,1′-biphenyl]-4-ol (6b).
A THF (1 mL) and water (0.5 mL) solution of sodium carbonate (66 mg, 0.62 mmol) was added with 2-(2-bromo-4-(naphthalen-1-yl)phenyl)-1,3-dioxolane (5b) (40 mg, 0.11 mmol) and 2,6-di-tert-butyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol (44 mg, 0.13 mmol), and the mixture was purged with N2 gas. The resulting solution was added with Pd(PPh3)4 (3.8 mg, 3.3 × 10−3 mmol), and refluxed for 24 h. The mixture was extracted with AcOEt, and the organic extract was washed with water, and dried over Na2SO4. After the removal of the solvents, the residue was purified via silica gel column chromatography (hexane/ethyl acetate = 10/1) to give 6b (37 mg, 68%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.50 (s, 1H), 8.04 (d, J = 8.5 Hz, 2H), 7.95 (d, J = 7.7 Hz, 1H), 7.79 (d, J = 8.9 Hz, 2H), 7.45–7.44 (m, 4H), 7.38–7.33 (m, 4H), 5.84 (s, 1H), 5.25 (s, 1H), 4.34–4.04 (m, 4H), 1.47 (s, 18H). HRMS (ESI-TOF) calcd for C33H36O3 [M + Na]+, 503.2556; found, 503.2523.
Synthesis of compound 3,5-di-tert-butyl-2′-(4,5-diphenyl-1H-imidazol-2-yl)-5′-(naphthalen-1-yl)-[1,1′-biphenyl]-4-ol (7b).
3,5-Di-tert-butyl-2′-(1,3-dioxolan-2-yl)-5′-(naphthalen-1-yl)-[1,1′-biphenyl]-4-ol (6b) (33 mg, 0.069 mmol), benzil (21 mg, 0.10 mmol), and ammonium acetate (107 mg, 1.38 mmol) were stirred at 110 °C in AcOH (0.5 mL) in a sealed tube for 17.5 h. The reaction mixture was cooled to room temperature and neutralized by NH3 aq. The organic extract was washed with water and dichloromethane. The crude product was purified via silica gel column chromatography (hexane/AcOEt = 10/1 → 6/1) to give 7b (20 mg, 46%) as a pale yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 12.07 (s, 1H), 8.74 (s, 1H), 8.20 (d, J = 8.3 Hz, 2H), 7.89 (d, J = 7.6 Hz, 1H), 7.73 (d, J = 8.7 Hz, 2H), 7.59–7.54 (m, 3H), 7.52–7.48 (m, 5H), 7.34–7.29 (m, 7H), 7.22 (t, J = 7.2 Hz, 1H), 7.09 (s, 2H), 6.93 (m, 1H), 1.25 (s, 18H). HRMS (ESI-TOF) calcd for C45H42N2O [M + H]+, 627.3369; found, 627.3378.
Synthesis of compound 3,5-di-tert-butyl-7′-(naphthalen-1-yl)-2′,3′-diphenylspiro[cyclohexane-1,5′-imidazo[2,1-a]isoindole]-2,5-dien-4-one (Np-PIC, Scheme 3).
All manipulations were carried out with the exclusion of light. A solution of potassium ferricyanide (207 mg, 0.63 mmol), and KOH (212 mg, 3.78 mmol) in water (2 mL) was added to a suspension of 3,5-di-tert-butyl-2′-(4,5-diphenyl-1H-imidazol-2-yl)-5′-(naphthalen-1-yl)-[1,1′-biphen yl]-4-ol (7b) (13 mg, 0.021 mmol) in benzene (1 mL). After stirring for 3 h at room temperature, the resultant mixture was extracted with benzene and the organic extract was washed with water and dried over Na2SO4. After the removal of the solvents, the crude product was purified via silica gel column chromatography (hexane/AcOEt = 6/1), to give Np-PIC (8 mg, 65%) as a pale yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 8.07 (d, J = 7.9 Hz, 1H), 8.02 (d, J = 8.3 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 8.6 Hz, 1H), 7.69 (d, J = 8.6 Hz, 1H), 7.62–7.53 (m, 4H), 7.50 (d, J = 6.9 Hz, 1H), 7.44–7.41 (m, 4H), 7.37–7.34 (m, 2H), 7.27 (t, J = 7.4 Hz, 2H), 7.18 (d, J = 7.2 Hz, 1H), 7.01 (s, 1H), 6.60 (s, 2H), 1.03 (s, 18H). HRMS (ESI-TOF) calcd for C45H40N2O [M + H]+, 625.3213; found, 625.3200.
Synthesis of compound 2-(4-(anthracen-9-yl)-2-bromophenyl)-1,3-dioxolane (5c).
A THF (1.0 mL) and water (0.5 mL) solution of potassium carbonate (116 mg, 0.84 mmol) was added with 4 (52.9 mg, 0.149 mmol) and anthracen-9-ylboronic acid (39.7 mg, 0.179 mmol), and the mixture was purged with N2 gas. The resulting solution was added with Pd(PPh3)4 (5.2 mg, 4.4 × 10−3 mmol), and refluxed for 23 h. The mixture was extracted with CH2Cl2, and the organic extract was washed with water, and dried over Na2SO4. After the removal of the solvents, the residue was purified via silica gel column chromatography (hexane/ethyl acetate = 10/1) to give 5c (37.9 mg, 53%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.52 (s, 1H), 8.05 (d, J = 8.5 Hz, 2H), 7.83 (d, J = 7.7 Hz, 1H), 7.68 (s, 1H), 7.63 (d, J = 8.9 Hz, 2H), 7.49–7.42 (m, 3H), 7.39–7.35 (m, 2H), 6.27 (s, 1H), 4.30–4.16 (m, 4H).
Synthesis of compound 5′-(anthracen-9-yl)-3,5-di-tert-butyl-2′-(1,3-dioxolan-2-yl)-[1,1′-biphenyl]-4-ol (6c).
A THF (1 mL) and water (0.5 mL) solution of potassium carbonate (66 mg, 0.48 mmol) was added with 2-(4-(anthracen-9-yl)-2-bromophenyl)-1,3-dioxolane (5c) (35 mg, 0.086 mmol) and 2,6-di-tert-butyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol (33 mg, 0.10 mmol), and the mixture was purged with N2 gas. The resulting solution was added with Pd(PPh3)4 (3.0 mg, 2.6 × 10−3 mmol), and refluxed for 25 h. The mixture was extracted with AcOEt, and the organic extract was washed with water, and dried over Na2SO4. After the removal of the solvents, the residue was purified via silica gel column chromatography (hexane/ethyl acetate = 10/1) to give 6c (30 mg, 65%) as a yellow amorphism. 1H NMR (400 MHz, CDCl3): δ 8.01 (d, J = 8.6 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.55–7.41 (m, 6H), 7.37 (s, 2H), 5.76 (s, 1H), 5.26 (s, 1H), 4.29–3.99 (m, 4H), 1.47 (s, 18H). HRMS (ESI-TOF) calcd for C37H38O3 [M + Na]+, 553.2713; found, 553.2703.
Synthesis of compound 5′-(anthracen-9-yl)-3,5-di-tert-butyl-2′-(4,5-diphenyl-1H-imidazol-2-yl)-[1,1′-biphenyl]-4-ol (7c).
5′-(Anthracen-9-yl)-3,5-di-tert-butyl-2′-(1,3-dioxolan-2-yl)-[1,1′-biphenyl]-4-ol (6c) (50 mg, 0.094 mmol), benzil (29 mg, 0.14 mmol), and ammonium acetate (145 mg, 1.88 mmol) were stirred at 110 °C in AcOH (0.7 mL) in a sealed tube for 13 h. The reaction mixture was cooled to room temperature and neutralized by NH3 aq. The organic extract was washed with water and dichloromethane. The crude product was purified via silica gel column chromatography (hexane/AcOEt = 10/1) to give 7c (15.1 mg, 24%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 12.02 (s, 1H), 11.82 (s, 1H), 11.72 (s, 2H), 7.52 (d, J = 8.4 Hz, 1H), 7.43–7.41 (m, 5H), 7.35–7.08 (m, 34H), 6.91 (d, J = 8.3 Hz, 1H), 6.78 (d, J = 7.7 Hz, 4H), 6.69 (d, J = 7.7 Hz, 4H), 4.63–4.58 (m, 4H), 3.18–3.12 (m, 12H). HRMS (ESI-TOF) calcd for C49H44N2O [M + H]+, 677.3526; found, 677.3514.
Synthesis of compound 7′-(anthracen-9-yl)-3,5-di-tert-butyl-2′,3′-diphenylspiro[cyclohexane-1,5′-imidazo[2,1-a]isoindole]-2,5-dien-4-one (An-PIC, Scheme 4).
All manipulations were carried out with the exclusion of light. A solution of potassium ferricyanide (148 mg, 0.45 mmol) and KOH (152 mg, 2.7 mmol) in water (1.5 mL) was added to a suspension of compound (7c) (10 mg, 0.015 mmol) in benzene (0.5 mL). After stirring for 2 h at room temperature, the resultant mixture was then extracted with benzene and the organic extract was washed with water and dried over Na2SO4. After the removal of the solvents, the crude product was purified via silica gel column chromatography (hexane/AcOEt = 6/1), to give An-PIC (7.5 mg, 75%) as a yellow powder. 1H NMR (400 MHz, DMSO-d6): δ 8.72 (s, 1H), 8.18–8.15 (m, 3H), 7.65–7.63 (m, 3H), 7.60 (d, J = 7.5 Hz, 2H), 7.54 (d, J = 7.24 Hz, 2H), 7.45–7.40 (m, 5H), 7.38–7.34 (m, 2H), 7.29 (t, J = 7.6 Hz, 2H), 6.96 (s, 1H), 6.66 (s, 2H), 1.00 (s, 18H). HRMS (ESI-TOF) calcd for C49H42N2O [M + H]+, 675.3369; found, 675.3341.
Synthesis of compound 2-(2-bromo-4-(1,9-dihydropyren-1-yl)phenyl)-1,3-dioxolane (5d).
A THF (1.0 mL) and water (0.5 mL) solution of sodium carbonate (47 mg, 0.45 mmol) was added with 4 (58 mg, 0.16 mmol) and pyren-1-ylboronic acid (44 mg, 0.18 mmol), and the mixture was purged with N2 gas. The resulting solution was added with Pd(PPh3)4 (5.5 mg, 4.8 × 10−3 mmol), and refluxed for 28 h. The mixture was extracted with AcOEt, and the organic extract was washed with water, and dried over Na2SO4. After the removal of the solvents, the residue was purified via silica gel column chromatography (hexane/ethyl acetate = 10/1) to give 5d (31 mg, 44%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.22 (d, J = 7.7 Hz, 2H), 8.19 (d, J = 7.4 Hz, 1H), 8.13–8.09 (m, 3H), 8.06–8.01 (m, 2H), 7.94 (d, J = 7.9 Hz, 1H), 7.86 (s, 1H), 7.80 (d, J = 7.9 Hz, 1H), 7.62 (d, J = 7.9 Hz, 1H), 6.24 (s, 1H), 4.28–4.16 (m, 4H).
Synthesis of compound 3,5-di-tert-butyl-5′-(1,9-dihydropyren-1-yl)-2′-(1,3-dioxolan-2-yl)-[1,1′-biphenyl]-4-ol (6d).
A THF (1 mL) and water (0.5 mL) solution of sodium carbonate (28 mg, 0.26 mmol) was added with 2-(2-bromo-4-(3a,3a1-dihydropyren-1-yl)phenyl)-1,3-dioxolane (5d) (40 mg, 0.093 mmol) and 2,6-di-tert-butyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol (37 mg, 0.11 mmol), and the mixture was purged with N2 gas. The resulting solution was added with Pd(PPh3)4 (3.2 mg, 2.8 × 10−3 mmol), and refluxed for 22 h. The mixture was extracted with AcOEt, and the organic extract was washed with water, and dried over Na2SO4. After the removal of the solvents, the residue was purified via silica gel column chromatography (hexane/ethyl acetate = 10/1) to give 6d (31 g, 62%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 9.3 Hz, 1H), 8.24–8.16 (m, 3H), 8.10 (s, 2H), 8.04–7.99 (m, 3H), 7.92 (d, J = 7.8 Hz, 1H), 7.67–7.63 (m, 2H), 7.42 (s, 2H), 5.81 (s, 1H), 5.27 (s, 1H), 4.32–4.03 (m, 4H), 1.48 (s, 18H).
Synthesis of compound 3,5-di-tert-butyl-2′-(4,5-diphenyl-1H-imidazol-2-yl)-5′-(pyren-1-yl)-[1,1′-biphenyl]-4-ol (7d).
Compound 6d (57 mg, 0.10 mmol), benzil (32 mg, 0.15 mmol), and ammonium acetate (154 mg, 2 mmol) were stirred at 110 °C in AcOH (0.6 mL) in a sealed tube for 12 h. The reaction mixture was cooled to room temperature and neutralized by NH3 aq. The organic extract was washed with water and dichloromethane. The crude product was purified via silica gel column chromatography (hexane/AcOEt = 10/1 → 6/1) to give 7d (15.4 mg, 22%) as a pale yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 11.96 (s, 1H), 8.43 (d, J = 7.8 Hz, 1H), 8.36 (d, J = 7.8 Hz, 1H), 8.32 (d, J = 7.8 Hz, 1H), 8.29–8.25 (m, 4H), 8.19 (d, J = 7.9 Hz, 1H), 8.12 (t, J = 7.5 Hz, 1H), 7.88 (d, J = 7.5 Hz, 1H), 7.76 (s, 1H), 7.71 (d, J = 7.9 Hz, 1H), 7.49–7.47 (m, 2H), 7.36–7.12 (m, 9H), 7.13 (s, 1H), 6.96 (s, 1H), 4.19 (s, 1H), 1.28 (s, 18H). HRMS (ESI-TOF) calcd for C51H42N2O [M + H]+, 701.3526; found, 701.3521.
Synthesis of compound 3,5-di-tert-butyl-2′,3′-diphenyl-7′-(pyren-1-yl)spiro[cyclohexane-1,5′-imidazo[2,1-a]isoindole]-2,5-dien-4-one (Py-PIC, Scheme 5).
All manipulations were carried out with the exclusion of light. A solution of potassium ferricyanide (207 mg, 0.063 mmol) and KOH (212 mg, 3.78 mmol) in water (1.5 mL) was added to a suspension of 7d (15 mg, 0.021 mmol) in benzene (0.8 mL). After stirring for 2 h at room temperature, the resultant mixture was then extracted with benzene and the organic extract was washed with water and dried over Na2SO4. After the removal of the solvents, the crude product was purified via silica gel column chromatography (hexane/AcOEt = 6/1), to give Py-PIC (10 mg, 73%) as a pale yellow powder. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (d, J = 8.1 Hz, 1H), 8.41 (d, J = 7.8 Hz, 1H), 8.38 (d, J = 7.8 Hz, 1H), 8.30 (s, 2H), 8.24 (d, J = 7.8 Hz, 2H), 8.19–8.10 (m, 3H), 7.93 (d, J = 7.8 Hz, 1H), 7.65 (d, J = 7.4 Hz, 2H), 7.51–7.41 (m, 5H), 7.29–7.22 (m, 2H), 6.71 (s, 2H), 1.11 (s, 18H). HRMS (ESI-TOF) calcd for C51H42N2O [M + H]+, 669.3369; found, 669.3373.
Laser flash photolysis
The setup for the laser photolysis measurements of the PIC derivatives was described in our previous paper.31,32 The laser flash photolysis experiments were carried out using a TSP-1000 time-resolved spectrophotometer (UNISOKU). A 10 Hz Q-switched Nd:YAG laser (Continuum Minilite II) with the third harmonic at 355 nm (pulse width = 5 ns) was employed for the excitation light. The probe beam from a halogen lamp (OSRAM HLX64623) was guided with an optical fiber scope to be arranged in an orientation perpendicular to the exciting laser beam. The probe beam was monitored using a photomultiplier tube (Hamamatsu R2949) using a spectrometer (UNISOKU MD200) for transient absorption spectra and decay profiles of the colored species. The excitation intensity of one pulse was estimated by an energy detector (Gentec Electro-Optics QE12LP-S-MB) using an energy monitor (Gentec Electro-Optics MAESTRO). Optical-grade solvents were used for all measurements.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported partly by the JSPS KAKENHI Grant Numbers JP18H05263 and JP26107010 in the Scientific Research on Innovative Areas “Photosynergetics”, and JSPS KAKENHI Grant Number JP17K14475 for K. M.
Notes and references
-
Organic Photochromic and Thermochromic Compounds, ed. J. C. Crano and R. J. Guglielmetti, Plenum, New York, 1999 Search PubMed.
-
M. Irie, Y. Yokoyama and T. Seki, New Frontiers in Photochromism, Springer, Tokyo, 2013 Search PubMed.
-
B. L. Feringa and W. R. Browne, Molecular Switches, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed.
-
H. Tian and J. Zhang, Photochromic Materials, Wiley-VCH, Weinheim, Germany, 2016 Search PubMed.
- E. Hadjoudis, Mol. Eng., 1995, 5, 301–337 CrossRef CAS.
- E. Hadjoudis and I. M. Mavridis, Chem. Soc. Rev., 2004, 33, 579–588 CAS.
- E. Hadjoudis, S. Chatziefthimiou and I. Mavridis, Curr. Org. Chem., 2009, 13, 269–286 CrossRef CAS.
-
H. Rau, Photochemistry and Photophysics, ed. J. F. Rabek, CRC Press, Inc., Boca Raton, Florida, 1990, ch. 4, vol. II, pp. 119–141 Search PubMed.
- H. M. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- T. Hayashi and K. Maeda, Bull. Chem. Soc. Jpn., 1960, 33, 565–566 CrossRef CAS.
- T. Hayashi and K. Maeda, Bull. Chem. Soc. Jpn., 1962, 35, 2057–2058 CrossRef CAS.
- K. Maeda and T. Hayashi, Bull. Chem. Soc. Jpn., 1971, 44, 533–536 CrossRef CAS.
-
J. C. Crano and R. J. Guglielmetti, Organic Photochromic and Thermochromic Compounds, Springer, US, New York, 2001, pp. 11–83 Search PubMed.
- R. J. Hart, H. G. Heller and K. Salisbur, Chem. Commun., 1968, 1627–1628 RSC.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- K. Fujita, S. Hatano, D. Kato and J. Abe, Org. Lett., 2008, 10, 3105–3108 CrossRef CAS PubMed.
- K. Shima, K. Mutoh, Y. Kobayashi and J. Abe, J. Am. Chem. Soc., 2014, 136, 3796–3799 CrossRef CAS PubMed.
-
J. D. Hepworth and B. M. Heron, Functional Dyes, ed. S.-H. Kim, Elsevier Science, 2006, ch. 3, pp. 85–135 Search PubMed.
- H. Kuroiwa, Y. Inagaki, K. Mutoh and J. Abe, Adv. Mater., 2019, 31, 1805661 CrossRef.
- J. C. Crano, T. Flood, D. Knowles, A. Kumar and B. Van Gemert, Pure Appl. Chem., 1996, 68, 1395–1398 CAS.
- N. Ishii, T. Kato and J. Abe, Sci. Rep., 2012, 2, 819 CrossRef PubMed.
- F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327–336 RSC.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859–1867 RSC.
- K. Mutoh, N. Miyashita, K. Arai and J. Abe, J. Am. Chem. Soc., 2019, 141, 5650–5654 CrossRef CAS PubMed.
- A. Abdollahi, K. Sahandi-Zangabad and H. Roghani-Mamagani, ACS Appl. Mater. Interfaces, 2018, 10, 39279–39292 CrossRef CAS PubMed.
- H. Yamashita, T. Ikezawa, Y. Kobayashi and J. Abe, J. Am. Chem. Soc., 2015, 137, 4952–4955 CrossRef CAS PubMed.
- T. Ikezawa, K. Mutoh, Y. Kobayashi and J. Abe, Chem. Commun., 2016, 52, 2465–2468 RSC.
- Y. Kobayashi, Y. Mishima, K. Mutoh and J. Abe, Chem. Commun., 2017, 53, 4315–4318 RSC.
- K. Yamamoto, K. Mutoh and J. Abe, J. Phys. Chem. A, 2019, 123, 1945–1952 CrossRef CAS PubMed.
- A. Tokunaga, K. Mutoh, T. Hasegawa and J. Abe, J. Phys. Chem. Lett., 2018, 9, 1833–1837 CrossRef CAS PubMed.
- S. Toshimitsu, K. Shima, K. Mutoh, Y. Kobayashi and J. Abe, ChemPhotoChem, 2019, 3, 487–494 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00468h |
|
| This journal is © the Partner Organisations 2019 |

 b,
Katsuya
Mutoh
b,
Katsuya
Mutoh