
High-strength hydrogel-based bioinks
Fei
Gao
a,
Changshun
Ruan
 *b and
Wenguang
Liu
*a
*b and
Wenguang
Liu
*a
aSchool of Materials Science and Engineering, Tianjin Key Laboratory of Composite and Functional Materials, Tianjin University, Tianjin 300350, China. E-mail: wgliu@tju.edu.cn
bResearch Center for Human Tissue and Organs Degeneration, Institute Biomedical and Biotechnology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China. E-mail: cs.ruan@siat.ac.cn
First published on 15th July 2019
Abstract
In the past few decades, the emerging three-dimensional printing (3D printing) techniques have revealed great potential for precise medicine and individualized therapy, thus attracting intensive attention in biomedical applications. 3D printing is heavily dependent on the properties of biomaterials, i.e., bioinks. High-strength hydrogels, resembling natural tissues with exceptional mechanics have demonstrated great potential as bioinks for 3D printing load-bearing tissue engineering scaffolds. However, achieving high-strength hydrogel inks and 3D printing them to construct a complex scaffold with the desired physicochemical and biofunctions for a definite biomedical application is still in its infancy. This review summarizes the progress of high-strength hydrogels for 3D printing from our team and other groups; their applications in biomedical areas are also presented. Meanwhile, the opportunities and challenges associated with 3D printing fabrication of high-strength hydrogels are discussed.
1. Introduction
3D printing is a layer-by-layer assembly technique to fabricate complex and customized structures precisely following a 3D digital model, and has developed rapidly since it was first proposed by Charles W. Hull in 1986.1–4 Due to its prominent advantages over the traditional manufacturing techniques,5–8 3D printing has been widely utilized in biomedical fields, including wearable devices,9 personalized implants,10 renewable artificial tissues,2etc. As a vital part of 3D printing, bio-printing that directly fabricates cell-laden tissue constructs and organs has also obtained increasing attention in the past decade and has great potential to solve the ever-increasing organ shortage crisis.9 The advancement of both 3D printing and bioprinting is heavily dependent on the development of biomaterials that can be used as inks/bioinks. For instance, different materials used as printed-inks require distinct printing methods or modes, which determined the performance of the printed-products. Therefore, exploring novel biomaterials with excellent printability, better biocompatibility and appropriate mechanical stability to satisfy the requirements of inks for creating complex biological constructs by 3D printing is highly desired in biomedical applications.Due to their unique compositional and structural similarities to the natural extracellular matrix (ECM), hydrogels have received soaring interest as leading candidates for biomedical applications.11–17 Since 2001, novel hydrogels have been designed with different toughening mechanisms to improve the mechanical strength, including slide-ring hydrogels,18 nanocomposite hydrogels,19 double-network hydrogels20 and ionic-covalent crosslinked hydrogels.21 In particular, supramolecular interactions have recently been introduced as the enhancing factors to construct a series of multifunctional high-strength hydrogels.22–25 High-strength hydrogels can overcome the weak mechanical properties of conventional hydrogels to closely match the exceptional mechanics of natural load-bearing tissues; thus they have become ideal candidates as inks/bioinks for 3D printing to accelerate the potential application of hydrogels in biomedical fields. To date, several printing methods have been proposed to fabricate 3D hydrogel scaffolds, like inkjet printing using microdrops,26,27 stereolithography using UV photopolymerization,28 laser printing29 and extrusion printing.30–32 Extrusion printing, a modified fused deposition modeling which extrudes continuous liquid inks to form a solid layered structure, is supposed to be the most commonly used strategy for 3D printing of high-strength hydrogels. Apart from the exciting features of quickly positioning selected multiple materials with distinct elasticity modulus to different regions within a single component, extrusion printing with layer-by-layer accumulation is also a logical method for homogenously and controllably distributing biological factors such as cells, growth factors and bioactive molecules throughout a biomaterial matrix, as well as designing biomimetic scaffolds with functionality and complexity of native tissues. Furthermore, 3D printing technology presents an opportunity to customize hydrogel implants based on medical images from X-ray, magnetic resonance imaging (MRI), computed tomography (CT) or ultrasound of a patient's own anatomy which fits the model perfectly with no trimming after printing.33,34 Consequently, combination of high-strength hydrogels with 3D printing techniques, will substantially broaden their potential application in biomedical fields.
The 3D printing technique breaks through the limitations of traditional methods to design biological functional hydrogel scaffolds. However, not all hydrogels can be printed, at least, not all 3D-printed hydrogel-based scaffolds can be used clinically. Therefore, in the present review, high-strength hydrogels for 3D printing are first and comprehensively reviewed with an emphasis on how to design high-strength hydrogels with printability and biological characters for biomedical applications. The opportunities and challenges associated with 3D printing fabrication of high-strength hydrogels are also discussed to point out the future directions of building the next generation of hydrogel scaffolds.
2. Hydrogel performances to meet the requirements of inks
In 3D printing, the ideal high-strength hydrogel-based inks for common extrusion printing should have the following characteristics (Fig. 1).35–37 Firstly, the ink itself must have a certain consistency and viscosity with a shear-thinning property, making sure that the ink could be smoothly squeezed out of the needle without frequent clogging of the nozzle, thus avoiding extrusion fracture of the printed filaments. Secondly, the viscosity and consistency of the as-printed filaments should restore or gel quickly enough once they are printed onto the receiver substrate to keep the outline of the filaments approximate to a circle from the cross-sectional view. Thirdly, the mechanical modulus of the as-printed filaments should be strong enough to self-support, realizing the fabrication of certain-scale constructs. In this way, the distortion of the scaffold induced by the self-gravity of deposited filaments can be retarded and the side voids can be maintained well as the printing layers add up. These three requirements are the basic guarantees for high-strength hydrogel-based inks to fabricate a 3D scaffold with a high shape-fidelity used in biomedical areas. Additionally, to obtain an ideal scaffold with excellent shape fidelity and pre-designed porosity, the interfacial bonding between the printed layers should be strong enough to prevent delamination within multilayer stacks especially for complex shaped scaffolds. Furthermore, the print parameters for getting an ideal three-dimensional scaffold, with smooth and uniform filaments as well as a regular macroporous structure, are also critical. Printing pressure, printing speed, feeding rate, and the distance between the needle and the printer substrate should be matched. More importantly, integration of the mechanical stability and swelling stability in physiological environments as well as multi-functional properties in a printed hydrogel-based scaffold will create new opportunities to solve practical problems in biomedical applications. Compared with conventional hydrogels, high-strength hydrogels can well meet the above mentioned requirements, especially in the performances of self-supporting and mechanical stability.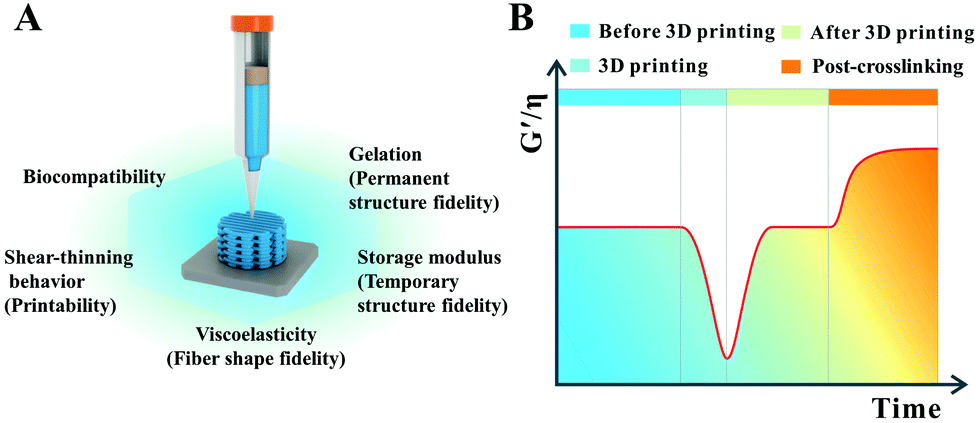 | ||
| Fig. 1 Window for general requirements of desired high-strength hydrogel scaffolds with high structure fidelity and cell-friendly biocompatibility (A); specific transition window of viscosity (η) and storage modulus (G′) of high-strength hydrogel inks at different stages of the printing process (B). | ||
3. 3D printing of high-strength hydrogels
To meet those above general requirements of an ideal hydrogel ink and thus obtain a 3D-printed precisely high-strength or tough hydrogel scaffold, rheology modifiers are needed to thicken the viscosity of the synthetic polymer hydrogel precursor solution to facilitate their printing process. In these cases, LAPONITE®/nanoclay,38 carrageenan,39 agar,40 alginate,41 gelatin,42 collagen,43 chitosan,44 hyaluronic acid,45 chondroitin sulphate,46 cellulose47 or a mixture of them,48–51 are most commonly used as rheological modifiers. In addition, the synthetic polymers are often mixed with modified natural polymers or directly modified by natural polymers to adjust printable inks to have appropriate viscosity and shear-thinning properties.52–59 These hydrogel systems can allow us to print hydrogel scaffolds with a desirable mechanical property and excellent cellular biocompatibility. However, rheology modifiers and post-treatments such as ionic crosslinking and UV crosslinking make 3D printing of these hydrogel systems more complicated. Remarkably, supramolecular polymer hydrogels can be directly written to build the desired architectures.60–62 Because of the reversible spontaneous molecular interactions such as hydrogen bonding and guest–host interaction, supramolecular polymer hydrogel can be deposited successfully in a three dimensional scaffold via 3D printing and directly form self-supporting structures without any post-crosslinking processes.3.1. 3D printing of synthetic high-strength polymer hydrogels modified with inorganic nanoparticles
High-strength hydrogels can be made with diverse methods, but their monomer aqueous solutions usually have low viscosity and cannot be directly printed into 3D scaffolds. In order to print a synthetic polymer hydrogel into a 3D scaffold, it is often necessary to add a rheology modifier or a viscosity thickener to adjust the viscoelastic properties and rheological behaviors of the monomer aqueous solutions. Nanoclay is a two-dimensional sheet material that can fully adsorb molecular chains and increase the number of effective segments between crosslinking points. Thus, by adding nanoclay, the purpose of toughening hydrogel can be obtained, and the obtained hydrogel scaffold belongs to a filler-enhanced hydrogel scaffold. Nanocellulose, alginate, hyaluronic acid and gelatin can be entangled with synthetic polymer chains to form an interpenetrating network. If ionic crosslinking is further introduced, the obtained hydrogel scaffold demonstrates high-strength and toughness. All of these hydrogels can be assigned to non-covalently enhanced chemically crosslinked hydrogels. Subjecting to an external load, reversible physical crosslinking within the network is firstly dissociated to dissipative energy before chemical cross-linking damage, thus acting as a toughening mechanism.25 Therefore, adding the rheology modifier not only serves as a supporting frame material to improve the printability, but also enhances the mechanical properties.In our previous study, we printed a high-strength and swelling stable supramolecular polymer/clay nanocomposite hydrogel scaffold (PNAGA–clay scaffold) based on a bioactive ink composed of a hydrogen bonding monomer (N-acryloyl glycinamide) (NAGA) and nanoclay (Fig. 2).38 The viscosity of the hybrid ink was conveniently tailored by the additive, nanoclay, thus making the aqueous solution of NAGA 3D-printable, which further prevented the printed layer-by-layer construct on the printing substrate from collapsing before UV crosslinking. The combined physical crosslinking from dual amide hydrogen bonding and nanoclay–polymer chain interactions contributed to the superior mechanical performances – a high tensile strength (up to 1.17 MPa), outstanding compressive strength (up to 3.50 MPa), high Young's modulus (up to 0.18 MPa), and large break stain (up to 1300%). The 3D printed PNAGA–clay bioscaffolds showed swelling stability, providing reliable loading support in vivo. In addition, the release of bioactive ions (Mg2+ and Si4+) from the PNAGA–clay scaffold proved to promote the osteogenic differentiation of rat osteoblast cells in vitro, and highly efficiently facilitate osteogenesis in vivo. 3D printing of the hydrogen bonding monomer with a variety of bioactive inorganic nanoparticles will open up a new avenue to construct load-bearing tissue engineering scaffolds for precision and individualized repair of bone defects and degeneration.
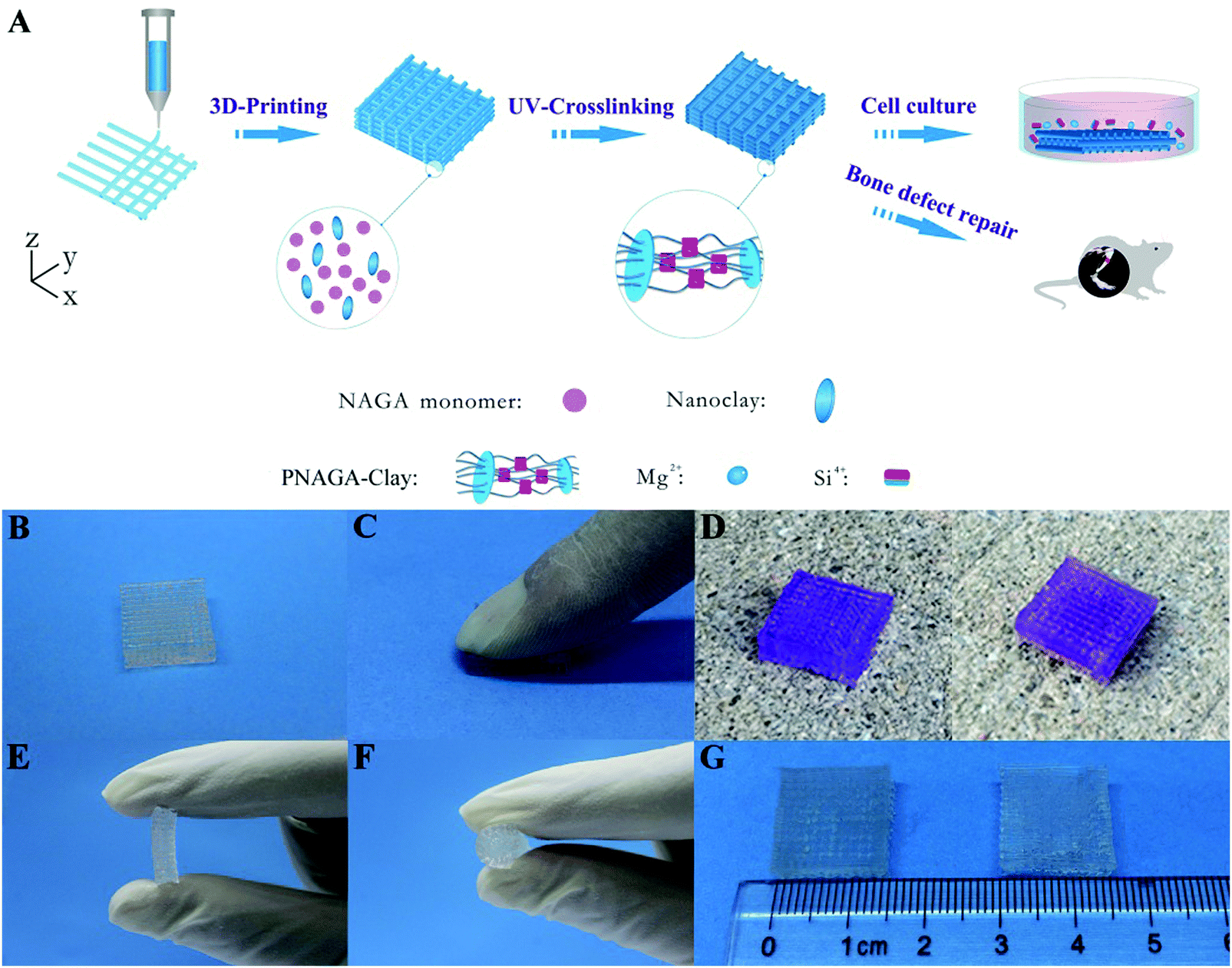 | ||
| Fig. 2 Procedure of 3D-printing PNAGA–clay scaffold (A) and photographs of PNAGA20%–clay scaffolds showing their ability to resist finger compression (B and C), car wheel pressing (D, left and right denote before and after pressing), and hand folding (E and F). The scaffold is very stable even after immersing in water for a long time (G, left and right denote before and after water immersion for 3 months). The scaffolds used for the car wheel pressing experiment were stained with gentian violet. Reproduced from ref. 38, with permission from American Chemical Society, Copyright 2017. | ||
Polyethylene glycol diacrylate (PEGDA), a common compound for the preparation of degradable synthetic high-strength hydrogels, has been used in many FDA-approved applications due to its well-known non-immunogenic biocompatible and highly hydrophilic characteristics.63–65 In our other previous study, PEGDA based hydrogel scaffolds were 3D printed after adding nanoclay (Fig. 3).66 The PEGDA–clay hydrogel scaffold could withstand nearly 1 MPa compression strength. Furthermore, a novel biodegradable rat osteoblast (ROB)–laden nanocomposite hydrogel construct was fabricated via a two-channel 3D-bioprinting method. In a simple way, the photocrosslinkable PEGDA and nanoclay pre-hydrogel solution was designed as bioink A, while the encapsulated cells-hyaluronic acid was used as bioink B. By optimizing the contents of nanoclay, bioink A could be printed smoothly and provided mechanical support before UV light-curing. Meanwhile, bioink B served to not only homogenously and precisely distribute cells into the 3D printed scaffolds, but also protect the encapsulated cells from UV-irradiation damage during the crosslinking process. In order to maintain a balance between the printability without incremental collapse of the printed scaffold and cell viability under lower printing pressure, the concentration of hyaluronic acid was chosen as 20%. We found that no obvious damage was observed even when the irradiation time was as long as 30 min; and furthermore, when the irradiation time was 10 minutes, the viability of the printed cells was higher than 95% after one-day of culture, and showed good adhesion, proliferation and differentiation within the 3D printed scaffolds. The novel biodegradable 3D printed PEG–nanoclay scaffolds also showed excellent osteogenic potential in rat tibia repair.
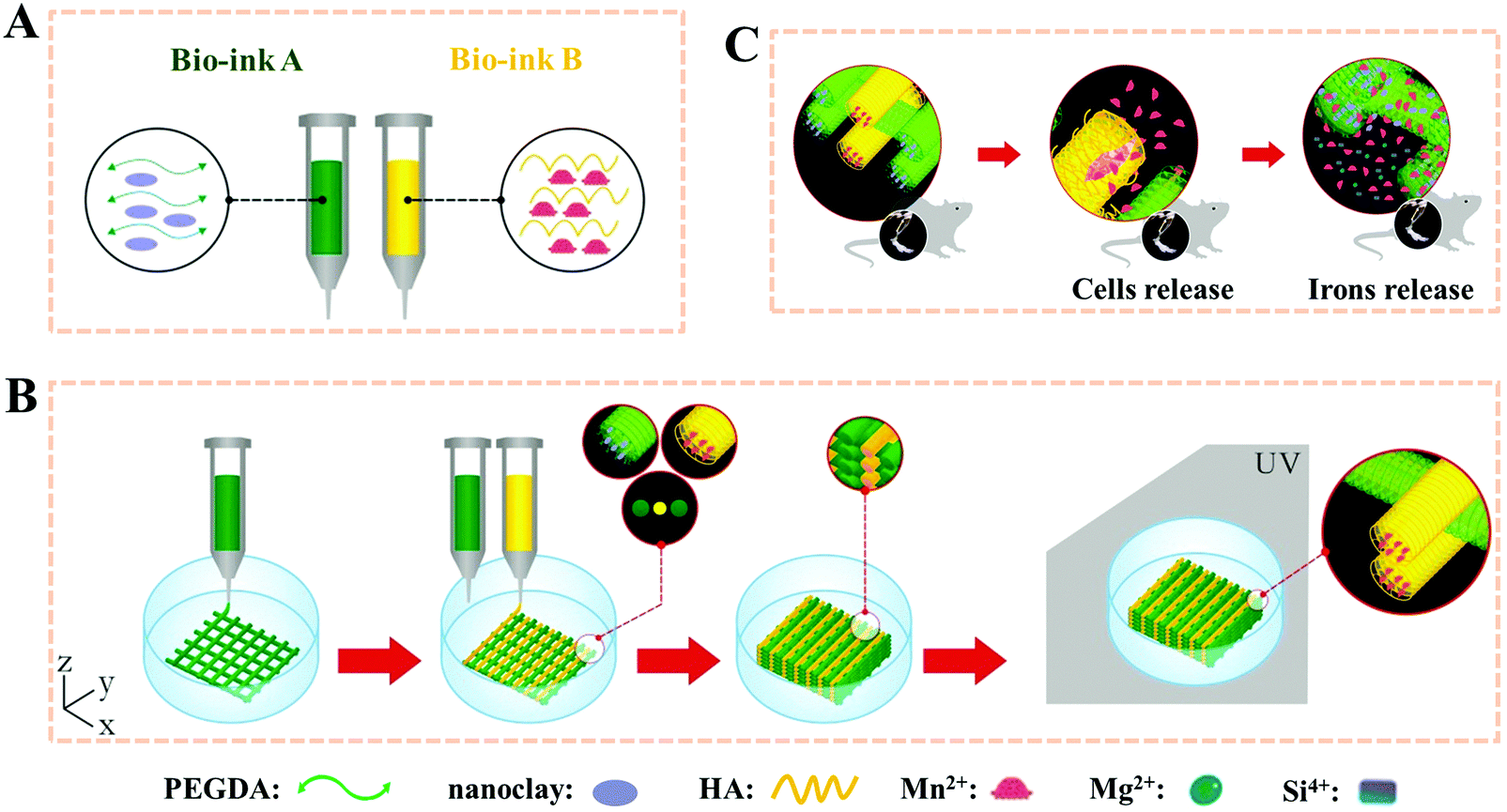 | ||
| Fig. 3 Stepwise illustration depicting the two-channel bioink system and method used for 3D-bioprinting and the in vivo experiments. The composition of bioink A and bioink B (A); 3D-bioprinting process of the ROB-laden constructs using a two-channel method (B) and the diagram for the in vivo experiments (C). Reproduced from ref. 66, with permission from John Wiley and Sons, Copyright 2018. | ||
Interpenetrating hydrogel is one of the most promising options because one fast curing network can be extruded to produce the shape of the structure while another network toughens and integrates the hydrogel. Inspired by this concept, Spinks et al. developed an ionic-covalent entanglement (ICE) hydrogel based ink consisting of alginate and acrylamide, which could be patterned into a defined shape due to the excellent viscosity and thixotropy of alginate and ionic crosslinking fixing as well as covalent crosslinking.67 These ICE hydrogels exhibited a remarkable mechanical performance because ionic crosslinking in the biopolymer network acted as sacrificial bonds that dissipated energy under stress. The 3D printed ICE hydrogels had a work of extension of 260 ± 3 kJ m−3. The mechanical properties were further improved by copolymerization of acrylated urethane based UV-curable adhesive material.68 Similarly, Li et al. incorporated amination and alginate noncovalently functionalized graphene oxide into this alginate and acrylamide based ICE hydrogels. Due to the dual energy dissipation mechanism and the hierarchy structure, the obtained hydrogels exhibited high mechanical properties (33.2 MPa compressive strength, 862.7 kPa tensile strength and 3324% break strain) with no influence on their printability.69 Kong et al. reported that the printed alginate and acrylamide based hydrogel microfibers could be stretched up to 21 times of their original length with a water content of 52%.70 Panhuis et al. demonstrated that another ICE hydrogel ink composed of kappa-carrageenan and poly(oxyalkylene amine), could be printed into a scaffold with a toughness as high as 1.4 ± 0.3 MJ m−3.39 Zhao et al. also created a 3D-printable and cell-friendly tough ICE hydrogel composed of PEG and alginate (Fig. 4).71 The toughening of this resultant hydrogel relied on combined mechanisms of mechanical energy dissipation caused by reversible Ca2+ crosslinking of alginate and high elasticity under large deformations originated from covalent crosslinking of PEG. The fracture toughness was as high as 1500 J m−2, which was higher than the value of articular cartilage. The tough PEG–alginate hydrogel was also cell-friendly and allowed cell encapsulation, which had a high cell viability (>75%) after being maintained over 7 days of culture. In addition, the actin-filament network and nuclei were highly deformed along with the stretching of the scaffold. Furthermore, incorporating nanoclay increased the viscosity of the prehydrogel and enhanced its shear-thinning properties, and thus achieved complex tough hydrogel 3D structures with a higher resolution.
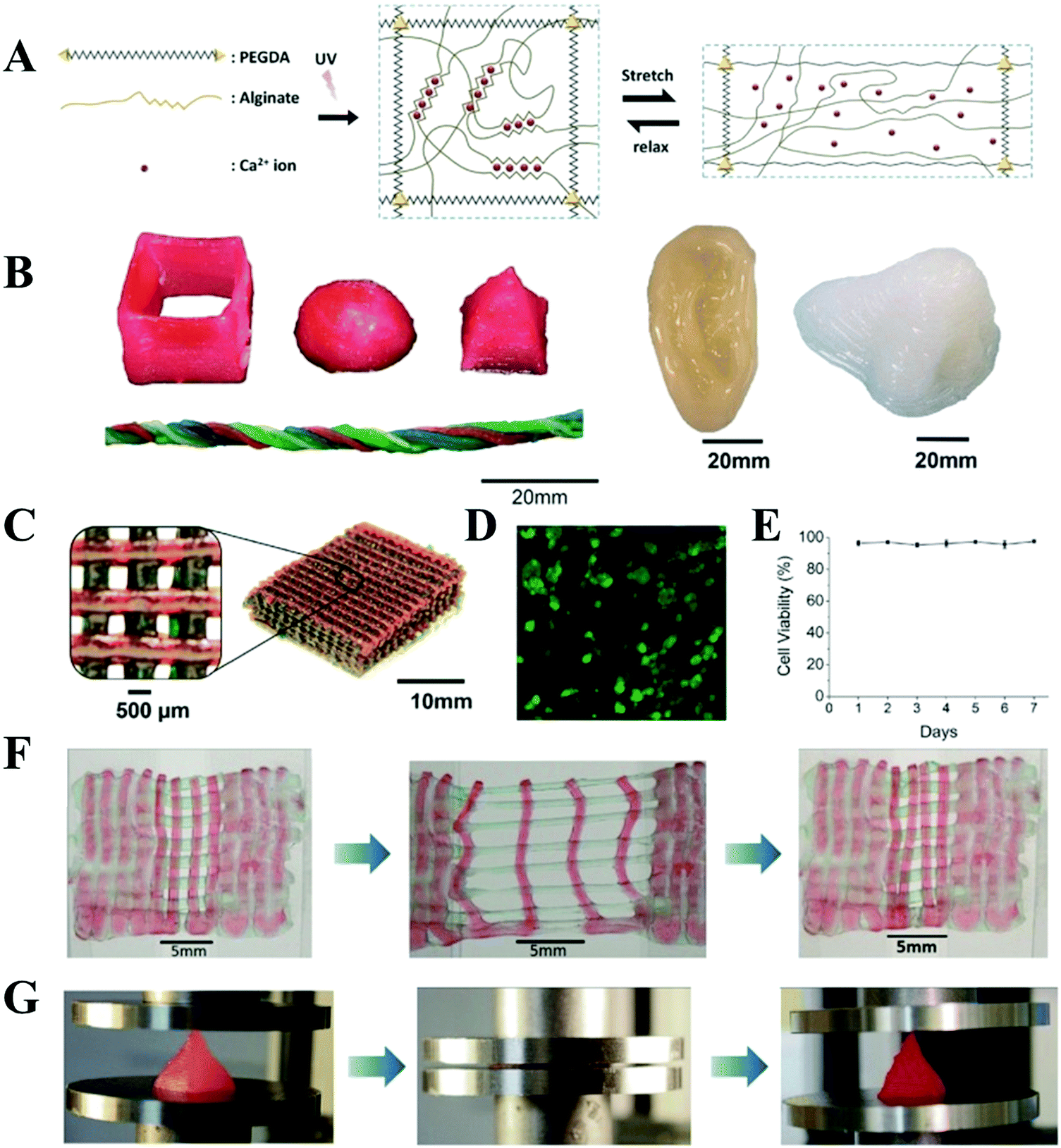 | ||
| Fig. 4 Schematic diagrams of the biocompatible and tough hydrogel. PEG and alginate polymers are covalently and ionically crosslinked through UV exposure and Ca2+, respectively. As the hydrogel is deformed, the alginate chains are detached from the reversible ionic crosslinks and mechanical energy is dissipated. Once the hydrogel is relaxed from deformation, it regains its original configuration since the covalently crosslinked PEG network maintains the elasticity of the hydrogel. Over time, some of the ionic crosslinks in the alginate network can reform in the deformed and relaxed hydrogel (A); 3D printing of tough and biocompatible PEG–alginate–nanoclay hydrogels. Various 3D constructs printed with the hydrogel (from left to right: hollow cube, hemisphere, pyramid, twisted bundle, the shape of an ear, and a nose. Nontoxic red food dye was added postprint on some samples for visibility) (B); a mesh printed with the tough and biocompatible hydrogel. The mesh was used to host Human Embryonic Kidney 293 cell (HEK-293) cells (C); live-dead assay of HEK-293 in a collagen hydrogel infused into the 3D printed mesh of the PEG–alginate–nanoclay hydrogel (D); viability of the HEK cells through 7 d (E); a printed bilayer mesh (top layer red, bottom layer green) is uniaxially stretched to three times of its initial length. Relaxation of the sample after stretching shows almost complete recovery of its original shape (F); a printed pyramid undergoes a compressive strain of 95% while returning to its original shape after relaxation (G). Reproduced from ref. 71, with permission from John Wiley and Sons, Copyright 2015. | ||
Recently, several groups have started to explore the double-network (DN) hydrogel based inks.40,71 Wiley et al. 3D-printed a DN hydrogel at room temperature with an interpenetrating network of poly(2-acrylamido-2-methylpropanesulfonate) and polyacrylamide, which achieved a compression strength of 93.5 MPa.72 LAPONITE® RDS, a layered silicate rheology modifier, was added to the first network precursor to thicken the viscosity of the ink and made it shear-thinning, and thus 3D printable via extrusion from a nozzle. After being printed and UV light-cured, the solidified bioink filaments were soaked in an acrylamide solution to form the second interpenetrating network, which was illuminated with UV light again. By tuning the ratio of the cross-linker in the first network to the acrylamide monomer in the second network, it was found that the formed tough DN hydrogels had a high compression strength, tensile strength, and elastic modulus surpassing those of most other 3D-printed hydrogel scaffolds. Cong et al. used a 3D-printer with a heated cartridge to print a super tough agar/PAAm DN hydrogel with a precise structure at 45–60 °C, which achieved a toughness of 3860 kJ m−3.40 After adding alginate, the high viscosity of the ink provided high surface tension, and the agar could be extruded in a semi-gelled state which ensured its quick gelling after extrusion. This method provides an innovative approach to fabricate excellent precise structures from a thermoreversible DN hydrogel, which is similar to the designed shape by regulating the alginate concentration. The entanglement of the alginate chains within the hydrogel networks restricts the agar helical chain bundles from pulling out under stress, which further toughens the printed DN hydrogel and makes the mechanical performance comparable to that of natural cartilage. In another study, Wu et al. developed a novel double-network SA/P(AAm-co-AAc)/Fe3+ hydrogel by taking advantage of dual Fe3+–COO– crosslinking.73 The optimal SA/P(AAm-co-AAc)/Fe3+ (SA 2 wt% and AAc 5 mol%) hydrogels exhibited outstanding mechanical properties (with 3.24 MPa tensile strength, 1228% strain 0.94 MPa elastic modulus, and 25.10 MJ m−3 toughness) and were highly swelling resistant. By properly adjusting the solution viscosity and gelation rate, the dual ionically crosslinked hydrogels were successfully 3D-printed into complicated 3D structures.
The above mentioned hydrogels were prepared with chemical crosslinking and physical crosslinking to achieve high mechanical properties. In a recent study, Xu et al. proposed a simple, nonpolymerization method to develop a novel type of dual physically crosslinked tough hydrogel, which consisted of poly(vinyl alcohol) (PVA) crystallite crosslinked network, and hyaluronic acid–Fe3+ physically crosslinked network.74 Nanosized PVA crystallites produced by a freeze–thaw method followed by annealing were chosen as major crosslinking sites for the primary network of the hydrogel. After following the crosslinking of the Fe3+-carboxylic group to construct the second network, their extraordinary mechanical performances including excellent tensile strength (up to 8 MPa), remarkable toughness (up to 19.6 MJ m−3) and high elastic modulus (up to 10 MPa) were successfully achieved. The PVA/HA–Fe3+ hydrogel performed better than most of the hydrogels in terms of the tensile strength and elastic modulus and showed a comparable performance to skin and cartilage in terms of elastic modulus and fracture energy. In particular, this PVA-assisted dual physically crosslinked tough hydrogel precursor solution with high molecular weight HA showed superior viscoelastic properties and could be printed into different shapes with no UV light curing.
3.2. 3D printing of synthetic high-strength polymer hydrogels mixed with modified natural polymers
When combining a synthetic polymer with a chemically modified natural polymer, such as methacrylated gelatin (GelMA) and methacrylated hyaluronic acid (MeHA), a chemically crosslinked stable hydrogel scaffold can be obtained.52–59 This strategy combines the advantages of natural polymers and synthetic polymers, resulting in a mechanically strong and long-term stable hydrogel scaffold, which is very important for their use in vivo; importantly, better cell adhesion and optimized degradation time benefit from the added natural polymers.GelMA, which is also frequently referred to as gelatin methacrylate, methacrylated gelatin, methacrylamide modified gelatin, or gelatin methacrylamide, is a photo-cross-linkable gelatin derivative. Note that the chemical modification of gelatin by methacrylic anhydride generally cannot significantly influence the arginine–glycine–aspartic acid motifs and matrix metalloproteinase motifs in the backbone chains of gelatin, which ensures the retention of excellent biocompatibility and enzymatic degradation properties of GelMA.75 Meanwhile, GelMA can also show good processability and is as operational as gelatin because of its retained viscosity especially at a low degree of methacrylation. At the same time, GelMA-based hydrogels overcome the temperature-dependent and reversible sol–gel transition of gelatin, thus precisely improving the stability and usability of gelatin. For these reasons, GelMA is receiving increasing interest in the biofabrication and tissue engineering fields. Very recently, investigations have shown that GelMA hydrogels are promising inks to build constructs with controlled architectures for various tissue engineering applications using 3D printing strategies and photopolymerization under UV light irradiation. However, their mechanical properties cannot meet the requirement of load-bearing scaffolds.76,77
To overcome this drawback, our group developed a novel supramolecular hydrogen bonding strengthened chemical crosslinked GelMA hydrogel (PACG–GelMA) by incorporating the reversible hydrogen bonds of N-acryloyl 2-glycine (ACG) into the GelMA hydrogel system (Fig. 5).53 The hydrogen bonds of the PACG side chain can reinforce and stabilize the GelMA network. The obtained PACG–GelMA hydrogel exhibited a high tensile strength (up to 1.1 MPa), outstanding compressive strength (up to 12.4 MPa), and high compression modulus (up to 837 kPa). Furthermore, owing to the thermoreversible gel–sol transition behavior and tunable viscosity of GelMA suited for 3D printing, biodegradable preprogrammed biohybrid gradient PACG–GelMA hydrogel scaffolds were 3D printed assisted with a low-temperature receiver, and stabilized under UV light irradiation. The obtained porous scaffolds demonstrated excellent compressive strengths and compressive modulus up to 2.51 MPa, 249 kPa, respectively. In addition, a biohybrid gradient scaffold consisting of a top layer of PACG–GelMA hydrogel–Mn2+, and bottom layer of PACG–GelMA hydrogel-bioactive glass was fabricated for the repair of osteochondral defects by a one-step 3D printing technique. In vitro biological experiments demonstrated that the biohybrid gradient hydrogel scaffold not only supported the cells’ attachment and spreading but also enhanced the gene expression of chondrogenic-related and osteogenic-related differentiation of human bone marrow stem cells. 12 weeks after in vivo implantation, the biohybrid gradient hydrogel scaffold significantly facilitated concurrent regeneration of cartilage and subchondral bone in a rat model.
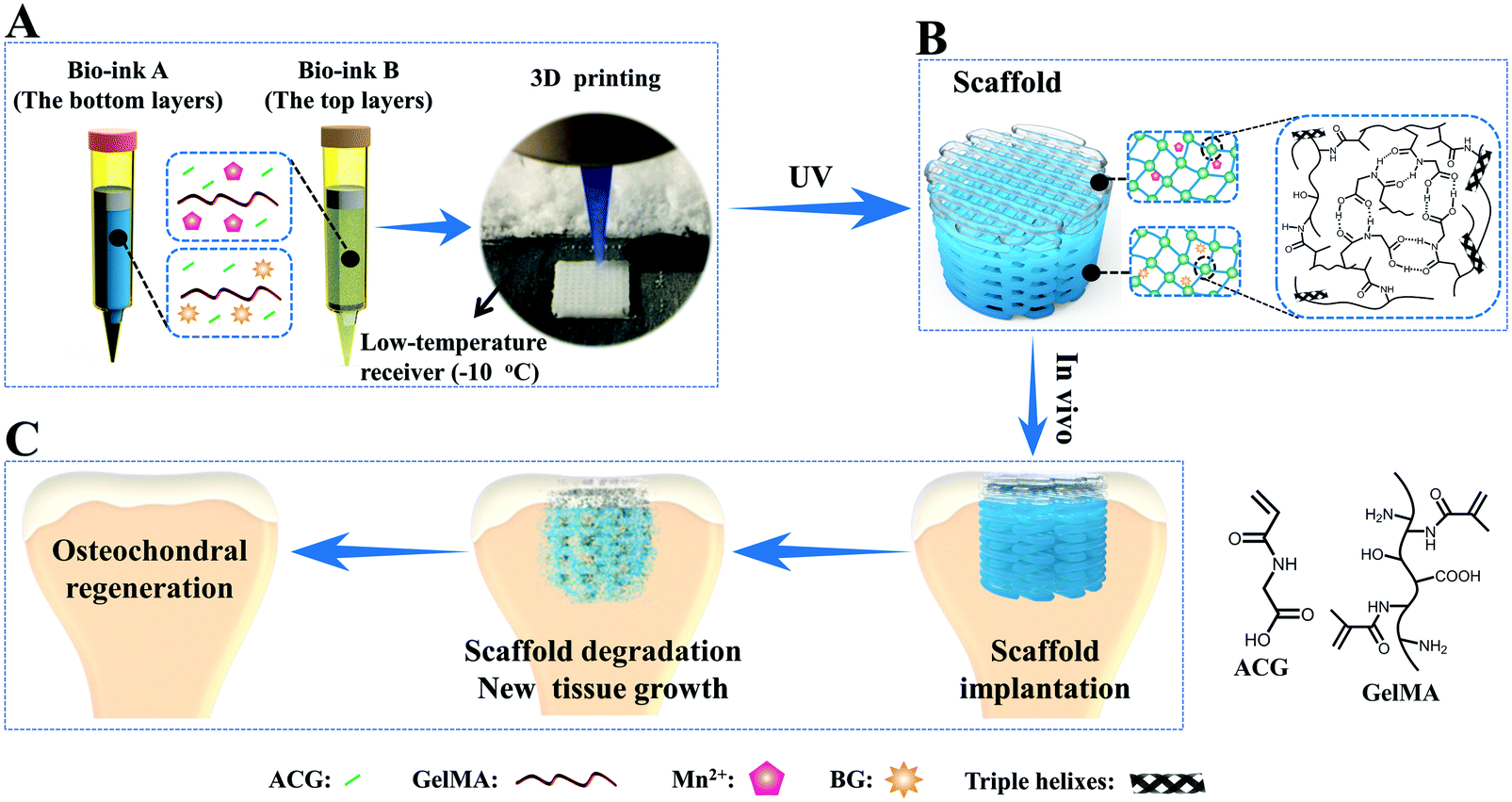 | ||
| Fig. 5 A schematic illustration of 3D printing of the biohybrid gradient scaffolds for repair of osteochondral defects. The compositions of bioink A and bioink B, and the 3D-bioprinting method of the biohybrid gradient scaffolds assisted with a low-temperature receiver (A); formation of a stable hydrogel scaffold after UV light-initiated polymerization and the main hydrogen bonding interactions in the PACG–GelMA network (B); the repair of osteochondral defects treated with the biohybrid gradient PACG–GelMA hydrogel scaffold with Mn2+ and BG being respectively loaded on the top layers and bottom layers in animal experiments (C). Reproduced from ref. 53, with permission from John Wiley and Sons, Copyright 2019. | ||
3.3. Direct 3D printing of high-strength supramolecular polymer hydrogels
With the great efforts of researchers, encouraging advances have been made in the 3D printing of synthetic high-strength hydrogels. However, it is still challenging to design a hydrogel ink that can be printed into a 3D scaffold in a direct writing mode that does not need the addition of a rheological modifier or post-crosslinking. In order to overcome this shortcoming, supramolecular polymer hydrogel inks are explored. Under shear stress, reversible non-covalent bonds in the network of supramolecular polymer hydrogels can be temporarily destroyed to lead to shear thinning properties, making it suitable for application in 3D printing. At the same time, the printed scaffolds do not need post-crosslinking due to the reversible reconstruction of the noncovalent bonds.Polyion complex (PIC) hydrogels are one type of tough supramolecular hydrogel obtained by polymerizing one charged monomer in the presence of oppositely charged polyelectrolyte with equivalent charge. These PIC hydrogels can form transparent viscous solutions when dissolved in a salt solution, which can form opaque hydrogels after being immersed into water to dialyze out the salts. These features of both reversible sol–gel transition and continuous acquisition of tough PIC gel fibers by directly extruding PIC solution into water encourage researchers to develop PIC hydrogel based scaffolds through extrusion-based 3D printing. Qian et al. reported various 3D PIC hydrogel structures based on poly(3-(methacryloylamino)propyl-trimethylammoniumchloride) and poly(sodium p-styrenesulfonate) (PMPTC/PNaSS) by exploiting the distinct strength of ionic bonding in PIC hydrogels at different stages of printing.61 They found that the dynamic nature of ionic bonds in PIC hydrogel favored the interfacial bonding between fibers and layers during the printing process, ensuring the integrity and toughness of printed hydrogel scaffolds. The printed PIC hydrogel structures showed excellent mechanical properties in terms of extensibility, strength, and toughness. The Young's modulus, tensile strength, and breaking strain of the printed fiber (with diameter of 0.2 mm) were 2.6 MPa, 1.2 MPa, and 530%, respectively, which are found to be comparable to those of bulk PIC hydrogels. Furthermore, inspired by molecular-level repeated modular domains of titin which could unfold and dissipate energy upon loading, they fabricated macroscopic ultratough hydrogel structures with different sizes of fibers by 3D printing with multiple nozzles.62 Under extension, the sacrificial bonds (thin fibers) were broken to release the “hidden length”, thereby relaxing the resultant force in the backbone chains. With consecutively releasing “hidden length”, the hydrogel could maintain the integrated structures and high stretchability. A spider-web-like construct was also demonstrated by incorporating multiple folded domains into the radial threads, revealing significantly enhanced extensibility and toughness. This work may provide a new avenue for the design of artificial materials with desired mechanical properties. Wu et al. 3D printed tough physical hydrogels based on highly viscous solutions of poly(acrylic acid-co-acrylamide) (P(AAc-co-AAm)) and poly(acrylic acid-co-N-isopropyl acrylamide) (P(AAc-co-NIPAm)) or their mixtures (Fig. 6).78 After being transferred into Fe3+ solution, the obtained P(AAc-co-AAm) hydrogel fiber showed a remarkable tensile breaking stress (2.38 MPa), breaking strain (802%), and elastic modulus (0.80 MPa). The integrated hydrogel scaffold demonstrated shape-morphing ability due to the volume contraction in concentrated saline solution of P(AAc-co-NIPAm) hydrogel fibers. The 3D-printed four-armed gripper could clamp plastic balls as large as 115 times its own weight.
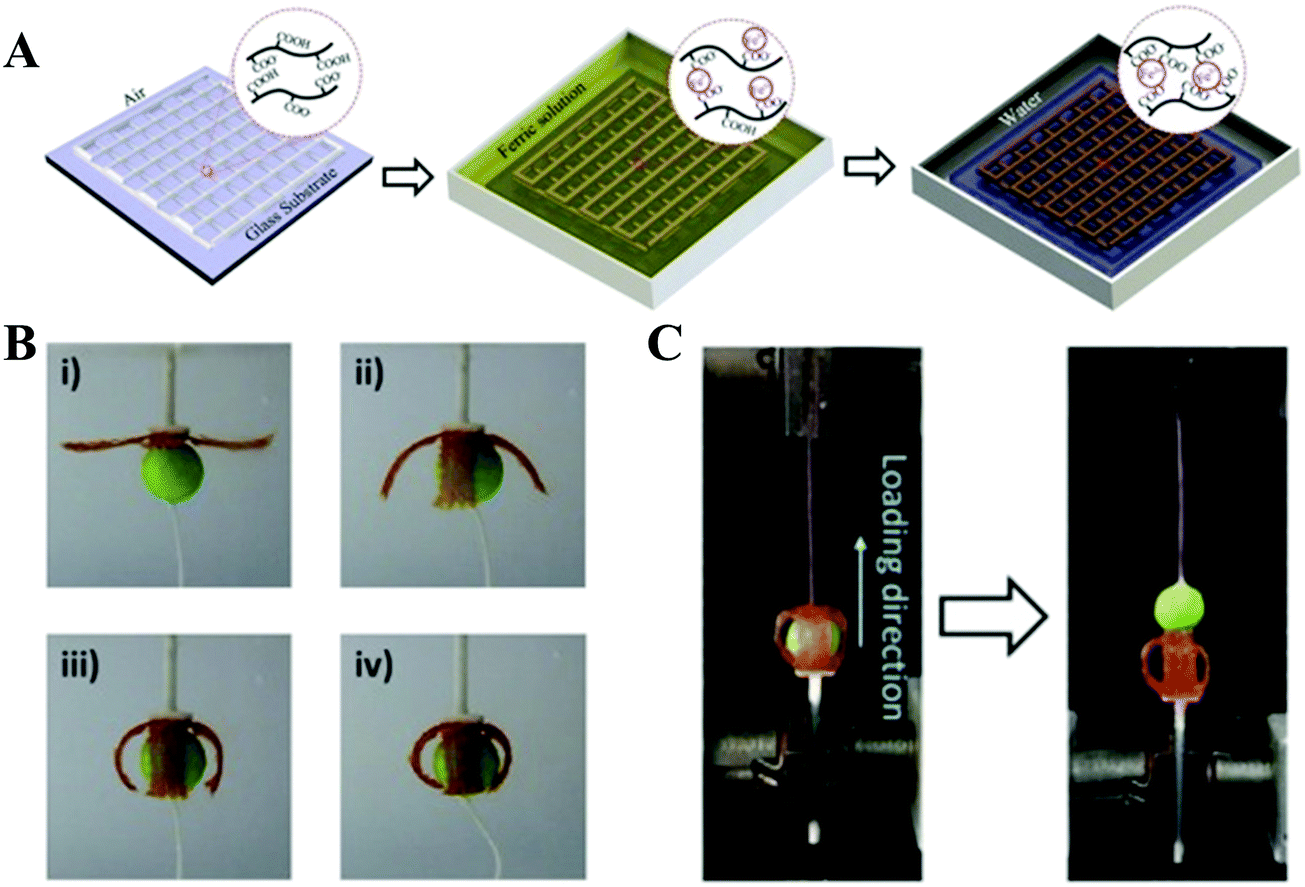 | ||
| Fig. 6 Schematic illustration of the fabrication of 3D-printed tough hydrogels. A highly viscous polymer solution was printed in air and incubated in FeCl3 solution, in which robust metal-coordination complexes are formed to crosslink the polymers. The as-prepared sample was then transformed into water to achieve the equilibrium state (A); 3D-printed four-armed gripper in 4 M saline solution to grip a plastic ball (diameter: 10 mm; weight: 13 mN) (B); measurement of the holding force by pulling the ball out of the gel gripper on a tensile tester (C). Reproduced from ref. 78, with permission from John Wiley and Sons, Copyright 2018. | ||
Thermo-reversible hydrogels may be an ideal material for extrusion printing due to their shear thinning behavior driven by temperature. Hydrogel inks with lower critical solution temperature (LCST) have been 3D printed in direct writing mode (without using rheology modifiers or post-crosslinking) into different structures.79 The resulting hydrogels exhibited a reversible and rapid modulus response to shear stress and shear strain. However, their mechanical properties are still not strong enough. Compared to the LCST hydrogels, only several polymer hydrogels with an upper critical solution temperature (UCST) have been reported. To our best knowledge, the UCST hydrogels were rarely printed into high-strength structure tissue engineering scaffolds. Our group reported on PNAGA supramolecular polymer hydrogels which demonstrated high mechanical properties resulting from multiple hydrogen bonding reinforcement (Fig. 7).80 However, the PNAGA hydrogel exhibited a high softening temperature and was difficult to be processed by melt-extrusion, which severely limited its direct printability. Copolymerizing N-acryloyl glycinamide with N-[tris(hydroxymethyl)methyl]acrylamide (THMMA), a hydrogen bonding monomer containing three hydroxyl groups in the side chain, could mitigate the hydrogen bonding interactions of PNAGA, thus resulting in a decrease of extrusion temperature, and simultaneously maintaining the swelling stability in aqueous environments. The synthesized P(NAGA-co-THMMA) copolymer hydrogel (PNT hydrogel) demonstrated excellent mechanical properties with robust tensile strength (up to 0.41 MPa), large stretchability (up to 860%), and high compressive strength (up to 8.4 MPa). The obtained copolymer hydrogels had a lower gel–sol transition temperature, which could be easily modulated by changing the monomer concentrations and NAGA/THMMA ratios. The rapid thermoreversible gel–sol transition behavior made this PNT copolymer hydrogel suitable for direct 3D printing. The resultant scaffold maintained high fidelity and resolution of architecture, featuring highly interconnected porosity and desirable mechanical properties. With large porosity (greater than 60%), the obtained scaffolds still had excellent compression strengths above 1 MPa. After 100 successive test cycles of continuous compression loading and unloading, the scaffold remained intact without any delamination due to the reversible H-bond as well as the additional physical crosslinkages from β-tricalciumphosphate (β-TCP). In mimicking articular cartilage-subchondral bone architecture, a bilayer biohybrid gradient PNT hydrogel scaffold with transforming growth factor (TGF-β1) loaded on the top layers and β-TCP incorporated on the bottom layers was fabricated by the thermal-assisted extrusion printing technique using three independently controlled print-heads in a predefined sequence. In vivo experiments showed that the 3D constructed biomimetic multiple scaffold could promote the regeneration of cartilage and subchondral bone simultaneously.
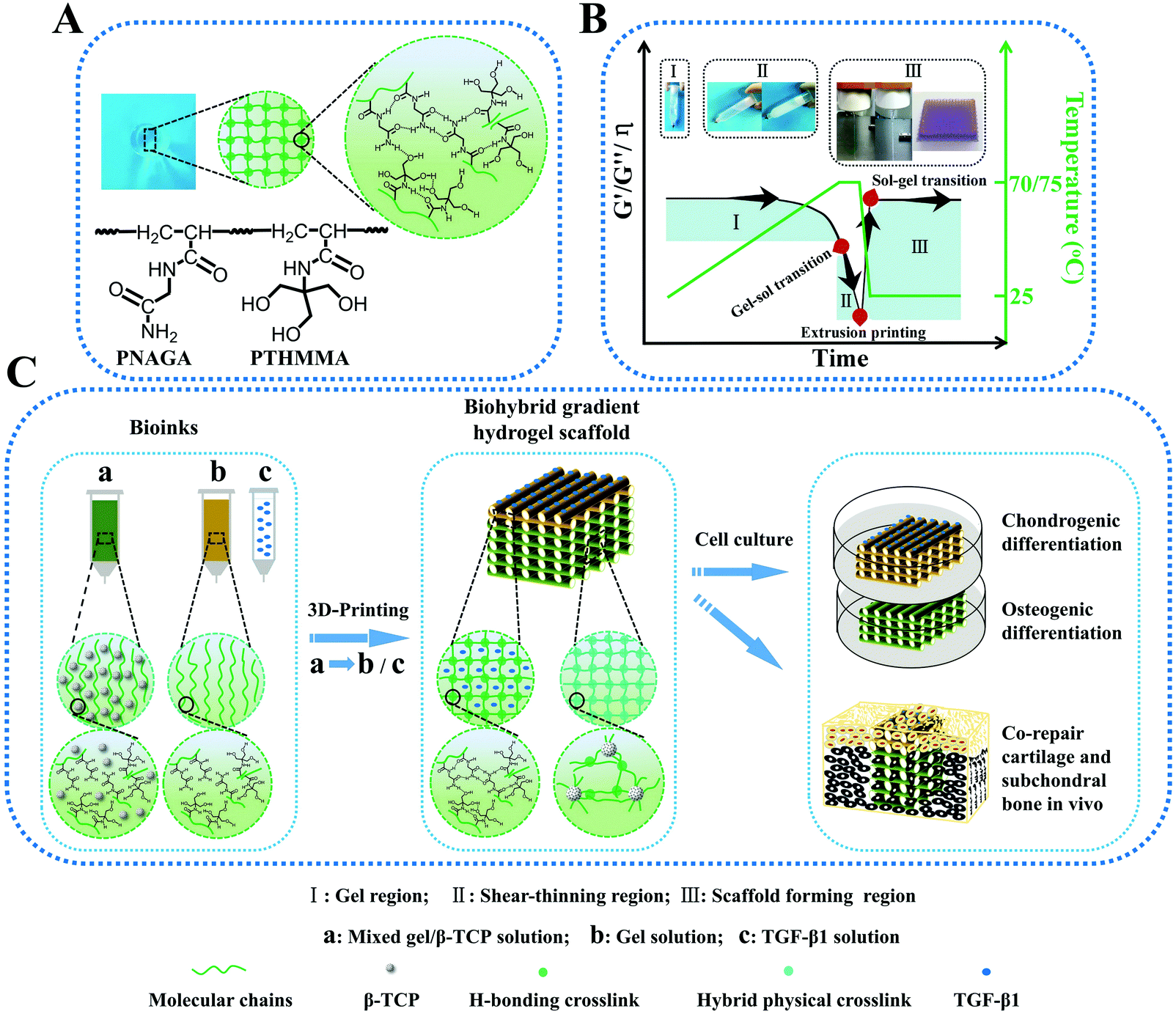 | ||
| Fig. 7 Schematic molecular structure and hydrogen bonding interactions in the PNT hydrogel (A); transition regions where PNT hydrogel underwent a reversible gel–sol transition during heating and cooling, by which melting printing was determined (B); procedure of thermal-assisted extrusion 3D printing of the biohybrid gradient scaffolds for repair of osteochondral defects (C). (I) Gel region: the PNT hydrogel maintained the gelling state in a lower temperature region, and underwent gel–sol transition with increasing temperature; (II) shear-thinning region: the viscosity of PNT sol decreased markedly with increasing shear rate so that it could be easily extruded out of the nozzles under certain pressure; (III) sol–gel transition region for scaffold formation: the extruded filaments gelled quickly on the printing substrate at room temperature to self-support the layer-by-layer construct, and the mechanical properties and viscosity recovered rapidly. The insets in panel B represent the procedure of I, II and III. Reproduced from ref. 80, with permission from John Wiley and Sons, Copyright 2018. | ||
4. Challenges and future outlook
Looking into the future, the general trends of biomedical areas, especially in tissue regeneration, should work towards high precision, multi-tissue integration and individualization, which will no doubt offer many opportunities for the development of high-strength hydrogel inks and 3D printing techniques. Of course, there are many challenges facing the design of high-strength hydrogel inks for a specific biomedical application. Firstly, besides biocompatibility, high-strength hydrogel inks with proper degradability, cell affinity, and the ability to integrate with tissues are extremely scarce in the literature. Secondly, the complex multi-level gradient structure of a native tissue or organ is highly required; thus a gradient high-strength hydrogel scaffold with continuous anisotropy is urgently needed. Thirdly, new printers or printing strategies such as extrusion printing combined with other printing technologies including inkjet printing, digital light processing-based printing and electrospinning with nanoscale resolution, to build 3D high-strength hydrogel-based scaffolds with micro-nano multi-scale structure, will greatly benefit the regeneration of a complex tissue or organ.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the support for this work from the National Key Research and Development Program (Grant No. 2016YFC1101301, 2018YFB1105600) and the National Natural Science Foundation of China (Grant No. 51733006, 31771041), the Youth Innovation Promotion Association of CAS (Grant No. 2019350), the Youth Talents of Guangdong Science and Technology Innovation (2015TQ01X076), and the Shenzhen Fundamental Research Foundation (Grant No. JCYJ20180507182237428).References
- A. A. Zadpoor and J. Malda, Ann. Biomed. Eng., 2017, 45, 1–11 CrossRef PubMed.
- S. V. Murphy and A. Atala, Nat. Biotechnol., 2014, 32, 773–785 CrossRef CAS PubMed.
- X. Li, J. He, W. Zhang, N. Jiang and D. Li, Materials, 2016, 9, 909–925 CrossRef PubMed.
- F. P. W. Melchels, M. A. N. Domingos, T. J. Klein, J. Malda, P. J. Bartolo and D. W. J. Hutmacher, Prog. Polym. Sci., 2012, 37, 1079–1104 CrossRef CAS.
- V. Mironov, N. Reis and B. Derby, Tissue Eng., 2006, 12, 631–634 CrossRef PubMed.
- E. Biazar, S. M. Najafi, K. S. Heidari, M. Yazdankhah, A. Rafiei and D. Biazar, J. Med. Eng. Technol., 2018, 42, 187–202 CrossRef PubMed.
- H. N. Chia and B. M. Wu, J. Biol. Eng., 2015, 9, 1–14 CrossRef CAS PubMed.
- U. Jammalamadaka and K. Tappa, J. Funct. Biomater., 2018, 9, 22–35 CrossRef PubMed.
- K. Tian, J. Bae, S. E. Bakarich, C. H. Yang, R. D. Gately, G. M. Spinks, M. Panhuis, Z. G. Suo and J. J. Vlassak, Adv. Mater., 2017, 29, 1604827 CrossRef PubMed.
- D. Radenkovic, A. Solouk and A. Seifalian, Med. Hypotheses, 2016, 87, 30–33 CrossRef PubMed.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- J. Thiele, Y. Ma, S. M. C. Bruekers, S. H. Ma and W. T. S. Huck, Adv. Mater., 2014, 26, 125–148 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS PubMed.
- J. L. Li, R. R. Xing, S. Bai and X. H. Yan, Soft Matter, 2019, 15, 1704–1715 RSC.
- I. Tomatsu, K. Peng and A. Kros, Adv. Drug Delivery Rev., 2011, 63, 1257–1266 CrossRef CAS PubMed.
- M. Burek and I. Wandzik, Polym. Rev., 2018, 54, 537–586 CrossRef.
- Y. Liu, W. He, Z. Zhang and B. P. Lee, Gels, 2018, 4, 46–75 CrossRef PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- M. J. Liu, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nature, 2015, 517, 68–72 CrossRef CAS PubMed.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- J. Y. Sun, X. H. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. G. Suo, Nature, 2011, 489, 133–136 CrossRef PubMed.
- M. J. Webber and P. Y. W. Dankers, Macromol. Biosci., 2019, 19, 1800452 CrossRef PubMed.
- Y. Wang, C. K. Adokoh and R. Narain, Expert Opin. Drug Delivery, 2018, 15, 77–91 CrossRef CAS PubMed.
- Z. Y. Xu and W. G. Liu, Chem. Commun., 2018, 54, 10540–10553 RSC.
- W. Wang, Y. Y. Zhang and W. G. Liu, Prog. Polym. Sci., 2017, 71, 1–25 CrossRef.
- R. Daly, T. S. Harrington, G. D. Martin and I. M. Hutchings, Int. J. Pharm., 2015, 494, 554–567 CrossRef CAS PubMed.
- L. D. Sarapuk, K. Kielbasinski, A. Arazna, K. Futera, A. Skalski, D. Janczak, M. Sloma and M. Jakubowska, Nanomaterials, 2018, 8, 602–612 CrossRef PubMed.
- H. Lin, D. N. Zhang, P. G. Alexander, G. Yang, J. Tan, A. W. M. Cheng and R. S. Tuan, Biomaterials, 2013, 34, 331–339 CrossRef CAS PubMed.
- Z. Zhang, C. Xu, R. Xiong, D. B. Chrisey and Y. Huang, Biomicrofluidics, 2017, 11, 034120 CrossRef PubMed.
- M. Hospodiuk, M. Dey, D. Sosnoski and I. T. Ozbolat, Biotechnol. Adv., 2017, 35, 217–239 CrossRef CAS PubMed.
- N. Annabi, A. Tamayol, J. A. Uquillas, M. Akbari, L. E. Bertassoni, C. Cha, G. Camci-Unal, M. R. Dokmeci, N. A. Peppas and A. Khademhosseini, Adv. Mater., 2014, 26, 85–124 CrossRef CAS.
- J. Malda, J. Visser, F. P. Melchels, T. Jüngst, W. E. Hennink, W. J. A. Dhert, J. Groll and D. W. Hutmacher, Adv. Mater., 2013, 25, 5011–5028 CrossRef CAS PubMed.
- S. Kyle, Z. M. Jessop, A. Al-Sabah and I. S. Whitaker, Adv. Healthcare Mater., 2017, 6, 1700264 CrossRef PubMed.
- D. Chimene, K. K. Lennox, R. R. Kaunas and A. K. Gaharwar, Ann. Biomed. Eng., 2016, 44, 2090–2102 CrossRef PubMed.
- Y. Yang, X. Song, X. Li, Z. Chen, C. Zhou, Q. Zhou and Y. Chen, Adv. Mater., 2018, 30, 1706539 CrossRef PubMed.
- D. Kirchmajer, R. Gorkin and M. Panhuis, J. Mater. Chem. B, 2015, 3, 4105–4117 RSC.
- P. S. Gungor-Ozkerim, I. Inci, Y. S. Zhang, A. Khademhosseini and M. R. Dokmeci, Biomater. Sci., 2018, 6, 915–946 RSC.
- X. Y. Zhai, Y. F. Ma, C. Y. Hou, F. Gao, Y. Y. Zhang, C. S. Ruan, H. B. Pan, W. W. Lu and W. G. Liu, ACS Biomater. Sci. Eng., 2017, 3, 1109–1118 CrossRef CAS.
- S. E. Bakarich, P. Balding, R. Gorkin III, G. M. Spinks and M. H. Panhuis, RSC Adv., 2014, 4, 38088–38092 RSC.
- J. H. Wei, J. L. Wang, S. H. Su, S. R. Wang, J. J. Qiu, Z. H. Zhang, G. Christopher, F. Ning and W. L. Cong, RSC Adv., 2015, 5, 81324–81329 RSC.
- J. H. Y. Chung, S. Naficy, Z. L. Yue, R. Kapsa, A. Quigley, S. E. Moulton and G. G. Wallace, Biomater. Sci., 2013, 1, 763–773 RSC.
- B. Duan, L. A. Hockaday, K. H. Kang and J. T. Butcher, J. Biomed. Mater. Res., Part A, 2013, 101, 1255–1264 CrossRef PubMed.
- S. Rhee, J. L. Puetzer, B. N. Mason, C. A. R. King and L. J. Bonassar, ACS Biomater. Sci. Eng., 2016, 2, 1800–1805 CrossRef CAS.
- Y. N. Yan, X. H. Wang, Y. Q. Pan, H. X. Liu, J. Cheng, Z. Xiong, F. Lin, R. D. Wu, R. J. Zhang and Q. P. Lu, Biomaterials, 2005, 26, 5864–5871 CrossRef CAS PubMed.
- I. L. Kim, R. L. Mauck and J. A. Burdick, Biomaterials, 2011, 32, 8771–8782 CrossRef CAS PubMed.
- D. A. Wang, S. Varghese, B. Sharma, I. Strehin, S. Fermanian, J. Gorham, D. H. Fairbrother, B. Cascio and J. H. Elisseeff, Nat. Mater., 2007, 6, 385–392 CrossRef CAS PubMed.
- J. Leppiniemi, P. Lahtinen, A. Paajanen, R. Mahlberg, S. Metsä-Kortelainen, T. Pinomaa, H. Pajari, I. Vikholm-Lundin, P. Pursula and V. P. Hytönen, ACS Appl. Mater. Interfaces, 2017, 9, 21959–21970 CrossRef CAS PubMed.
- H. J. Li, Y. J. Tan, K. F. Leong and L. Li, ACS Appl. Mater. Interfaces, 2017, 9, 20086–20097 CrossRef CAS PubMed.
- P. A. Levett, F. P. W. Melchels, K. Schrobback, D. W. Hutmacher, J. Malda and T. J. Klein, Acta Biomater., 2014, 10, 214–223 CrossRef CAS PubMed.
- S. Das, F. Pati, Y. Choi, G. Rijal, J. Shim, S. W. Kim, A. R. Ray, D. Cho and S. Ghosh, Acta Biomater., 2015, 11, 233–246 CrossRef CAS PubMed.
- K. Markstedt, A. Mantas, I. Tournier, H. M. Ávila, D. Hägg and P. Gatenholm, Biomacromolecules, 2015, 16, 1489–1496 CrossRef CAS PubMed.
- G. F. Gao, A. F. Schilling, K. Hubbell, T. Yonezawa, D. Truong, Y. Hong, G. H. Dai and X. F. Cui, Biotechnol. Lett., 2015, 37, 2349–2355 CrossRef CAS PubMed.
- F. Gao, Z. Y. Xu, Q. F. Liang, H. F. Li, L. Q. Peng, M. M. Wu, X. L. Zhao, X. Cui, C. S. Ruan and W. G. Liu, Adv. Sci., 2019, 1900867 CrossRef.
- D. Das, T. T. H. Pham and I. Noh, Colloids Surf., B, 2018, 170, 64–75 CrossRef CAS PubMed.
- I. Noh, N. Kim, H. N. Tran, J. Lee and C. Lee, Biomater. Res., 2019, 23, 3–11 CrossRef PubMed.
- D. Das, H. Cho, N. Kim, T. T. H. Pham, I. G. Kim, E. Chung and I. Noh, Carbohydr. Polym., 2019, 207, 628–639 CrossRef CAS PubMed.
- M. Costantini, S. Testa, P. Mozetic, A. Barbetta, C. Fuoco, E. Fornetti, F. Tamiro, S. Bernardini, J. Jaroszewicz, W. Swieszkowski, M. Trombetta, L. Castagnoli, D. Seliktar, P. Garstecki, G. Cesareni, S. Cannata, A. Rainer and C. Gargioli, Biomaterials, 2017, 13, 98–110 CrossRef PubMed.
- F. Maiullari, M. Costantini, M. Milan, V. Pace, M. Chirivì, S. Maiullari, A. Rainer, D. Baci, H. E. Marei, D. Seliktar, C. Gargioli, C. Bearzi and R. Rizzi, Sci. Rep., 2018, 8, 13532 CrossRef PubMed.
- L. Pescosolido, W. Schuurman, J. Malda, P. Matricardi, F. Alhaique, T. Coviello, P. R. Weeren, W. J. A. Dhert, W. E. Hennink and T. Vermonden, Biomacromolecules, 2011, 12, 1831–1838 CrossRef CAS PubMed.
- C. B. Highley, C. B. Rodell and J. A. Burdick, Adv. Mater., 2015, 27, 5075–5079 CrossRef CAS PubMed.
- F. B. Zhu, L. B. Cheng, J. Yin, Z. L. Wu, J. Qian, J. Z. Fu and Q. Zheng, ACS Appl. Mater. Interfaces, 2016, 8, 31304–31310 CrossRef CAS PubMed.
- F. B. Zhu, L. B. Cheng, Z. J. Wang, W. Hong, Z. L. Wu, J. Yin, J. Qian and Q. Zheng, ACS Appl. Mater. Interfaces, 2017, 9, 11363–11367 CrossRef CAS PubMed.
- M. B. Browning, S. N. Cereceres, P. T. Luong and E. M. Cosgriff-Hernandez, J. Biomed. Mater. Res., Part A, 2014, 102, 4244–4251 CAS.
- J. L. Zhang, N. Wang, W. G. Liu, X. L. Zhao and W. Lu, Soft Matter, 2013, 9, 6331–6337 RSC.
- Q. Wu, B. Xu, J. J. Wei, Q. Wang, Q. G. Wang and W. G. Liu, Chin. J. Polym. Sci., 2017, 35, 1222–1230 CrossRef CAS.
- X. Y. Zhai, C. S. Ruan, Y. F. Ma, D. L. Cheng, M. M. Wu, W. G. Liu, X. L. Zhao, H. B. Pan and W. W. Lu, Adv. Sci., 2017, 5, 1700550 CrossRef PubMed.
- S. E. Bakarich, M. Panhuis, S. Beirne, G. G. Wallace and G. M. Spinks, J. Mater. Chem. B, 2013, 1, 4939–4946 RSC.
- S. E. Bakarich, R. Gorkin III, R. Gately, S. Naficy, M. H. Panhuis and G. M. Spinks, Addit. Manuf., 2017, 14, 24–30 CrossRef CAS.
- S. J. Liu, A. K. Bastola and L. Li, ACS Appl. Mater. Interfaces, 2017, 9, 41473–41481 CrossRef CAS PubMed.
- S. S. Wei, G. Qu, G. Y. Luo, Y. X. Huang, H. S. Zhang, X. C. Zhou, L. Q. Wang, Z. Liu and T. T. Kong, ACS Appl. Mater. Interfaces, 2018, 10, 11204–11212 CrossRef CAS PubMed.
- S. Hong, D. Sycks, H. F. Chan, S. T. Lin, G. P. Lopez, F. Guilak, K. W. Leong and X. H. Zhao, Adv. Mater., 2015, 27, 4035–4040 CrossRef CAS PubMed.
- F. C. Yang, V. Tadepalli and B. J. Wiley, ACS Biomater. Sci. Eng., 2017, 3, 863–869 CrossRef CAS.
- X. F. Li, H. Wang, D. P. Li, S. J. Long, G. W. Zhang and Z. L. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 31198–31207 CrossRef CAS PubMed.
- A. Li, Y. Si, X. H. Wang, X. J. Jia, X. H. Guo and Y. H. Xu, ACS Appl. Nano Mater., 2019, 2, 707–715 CrossRef CAS.
- K. Yue, G. T. Santiago, M. M. Alvarez, A. Tamayol, N. Annabi and A. Khademhosseini, Biomaterials, 2015, 73, 254–271 CrossRef CAS PubMed.
- T. Billiet, E. Gevaert, T. D. Schryver, M. Cornelissen and P. Dubruel, Biomaterials, 2014, 35, 49–62 CrossRef CAS PubMed.
- W. J. Liu, M. A. Heinrich, Y. X. Zhou, A. Akpek, N. Hu, X. Liu, X. F. Guan, Z. Zhong, X. Y. Jin, A. Khademhosseini and Y. S. Zhang, Adv. Healthcare Mater., 2017, 6, 1601451 CrossRef PubMed.
- S. Y. Zheng, Y. Y. Shen, F. B. Zhu, J. Yin, J. Qian, J. Z. Fu, Z. L. Wu and Q. Zheng, Adv. Funct. Mater., 2018, 28, 1803366 CrossRef.
- M. Zhang, A. Vora, W. Han, R. J. Wojtecki, H. Maune, A. B. A. Le, L. E. Thompson, G. M. McClelland, F. Ribet, A. C. Engler and A. Nelson, Macromolecules, 2015, 48, 6482–6488 CrossRef CAS.
- F. Gao, Z. Y. Xu, Q. F. Liang, B. Liu, H. F. Li, Y. H. Wu, Y. Y. Zhang, Z. F. Lin, M. M. Wu, C. S. Ruan and W. G. Liu, Adv. Funct. Mater., 2018, 28, 1706644 CrossRef.
| This journal is © the Partner Organisations 2019 |