
A concise synthesis and biological study of evodiamine and its analogues†‡
Jie-Dan
Deng§
ab,
Shuai
Lei§
a,
Yi
Jiang
 a,
Hong-Hua
Zhang
a,
Xiao-Ling
Hu
a,
Huai-Xiu
Wen
c,
Wen
Tan
*a and
Zhen
Wang
*ab
a,
Hong-Hua
Zhang
a,
Xiao-Ling
Hu
a,
Huai-Xiu
Wen
c,
Wen
Tan
*a and
Zhen
Wang
*ab
aSchool of Pharmacy, Lanzhou University, West Donggang Road, No. 199, Lanzhou 730000, China. E-mail: tanwen@lzu.edu.cn; zhenw@lzu.edu.cn
bInstitution State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China
cKey Laboratory of Tibetan Medicine Research, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, No. 23 Xining Road, Xining, P. R. China
First published on 1st February 2019
Abstract
Efficient access to evodiamine and its analogues is presented via Lewis acid catalysis. In this reaction, three chemical bonds and two heterocyclic-fused rings are constructed in one step. The reaction shows good functional group tolerance and atom economy, and various heteroatom-containing evodiamine analogues are obtained in moderate to excellent yields even on a gram scale. An anti-tumor study in vitro demonstrates compound 2b possesses potent efficacy against hepatoma cell line (IC50 = 5.7 μM).
Polycyclic heterocycles are crucial motifs that widely exist in drugs and alkaloids,1 and many of them have shown superior bioactivity in anti-bacterial (roquefortine C), anti-tumor (camptothecin), and cerebral vasodilatory (vincoline) agents etc. according to the known reports (Fig. 1).2 Thus, the rapid construction of polycyclic heterocycle skeletons is always a hotspot in synthetic chemistry.3 Among them, evodiamine is one of the most typical polycyclic heterocycles, the synthesis and modification of which have aroused broad interest in both synthetic and pharmaceutical industries because of its diverse biological activities.4 In the last decades, several groups have done elegant work in this domain. In 1997, Chen firstly employed evodiamine for anti-inflammatory purposes.5 Subsequently, Kamiya demonstrated evodiamine's efficacy in anti-obesity in 2001.6 After that, Zhang successfully applied evodiamine in a transgenic mouse model for Alzheimer's disease (AD) treatment.7 Through chemical decorations, Sheng et al. discovered several evodiamine analogues as potent anti-cancer drug candidates in recent years.8 The structure–activity relationships of evodiamine derivatives have been reported previously, in which R1 and X follow the listed orders respectively. For R1: OH > I > F > OMe > Br > Cl or Me; for X: O, S > N-Me > CH > C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N. Meanwhile, when different positions of Ar2 are substituted by different groups, the activity also varies.8b,d Despite evodiamine and its analogues having shown great potential benefits for human health, the approaches to synthesise these compounds are still limited.9 Conventional routes to such kind of chemicals usually suffer from multi-step preparation and an imine intermediate needing to be synthesized stepwise, along with this process employing some toxic reagents as well as transition metals (Scheme 1).10 In this context, methodologies to prepare evodiamine and its analogues concisely are highly desired in synthetic chemistry. Recently, Wu and co-workers developed an elegant work on the one-pot synthesis of evodiamine and its analogues; unfortunately, the substrates were merely constrained to N-methylisatoic anhydride accordingly.11 Therefore, related works on one-step preparation of diversified heteroatom-containing evodiamine analogues still remain scarce. Herein, we report concise access to evodiamine and various heteroatom-containing evodiamine analogues via Lewis acid catalysis. In this protocol, different fused heterocycles are efficiently constructed within one step.
N. Meanwhile, when different positions of Ar2 are substituted by different groups, the activity also varies.8b,d Despite evodiamine and its analogues having shown great potential benefits for human health, the approaches to synthesise these compounds are still limited.9 Conventional routes to such kind of chemicals usually suffer from multi-step preparation and an imine intermediate needing to be synthesized stepwise, along with this process employing some toxic reagents as well as transition metals (Scheme 1).10 In this context, methodologies to prepare evodiamine and its analogues concisely are highly desired in synthetic chemistry. Recently, Wu and co-workers developed an elegant work on the one-pot synthesis of evodiamine and its analogues; unfortunately, the substrates were merely constrained to N-methylisatoic anhydride accordingly.11 Therefore, related works on one-step preparation of diversified heteroatom-containing evodiamine analogues still remain scarce. Herein, we report concise access to evodiamine and various heteroatom-containing evodiamine analogues via Lewis acid catalysis. In this protocol, different fused heterocycles are efficiently constructed within one step.
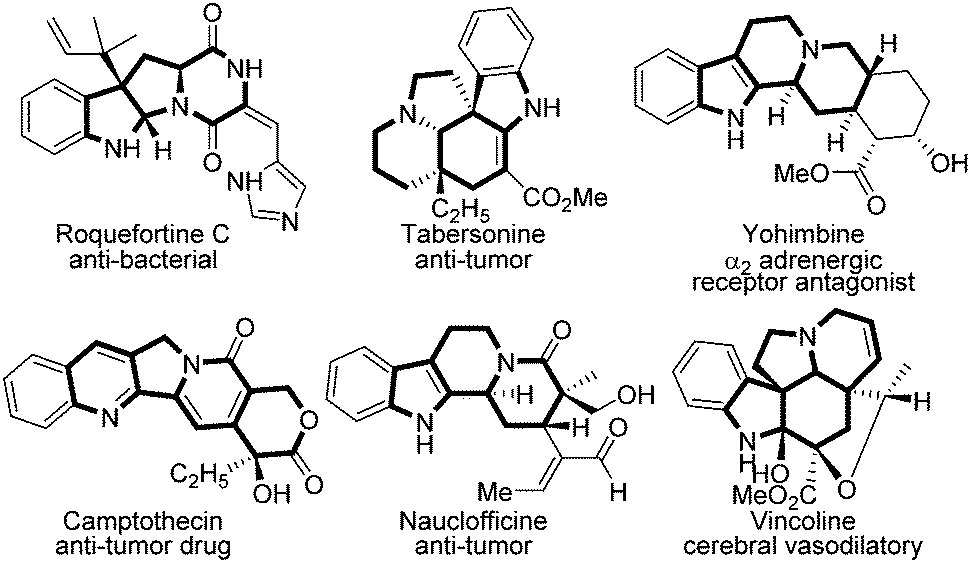 | ||
| Fig. 1 Polycyclic heterocycle skeletons in drugs and natural products. | ||
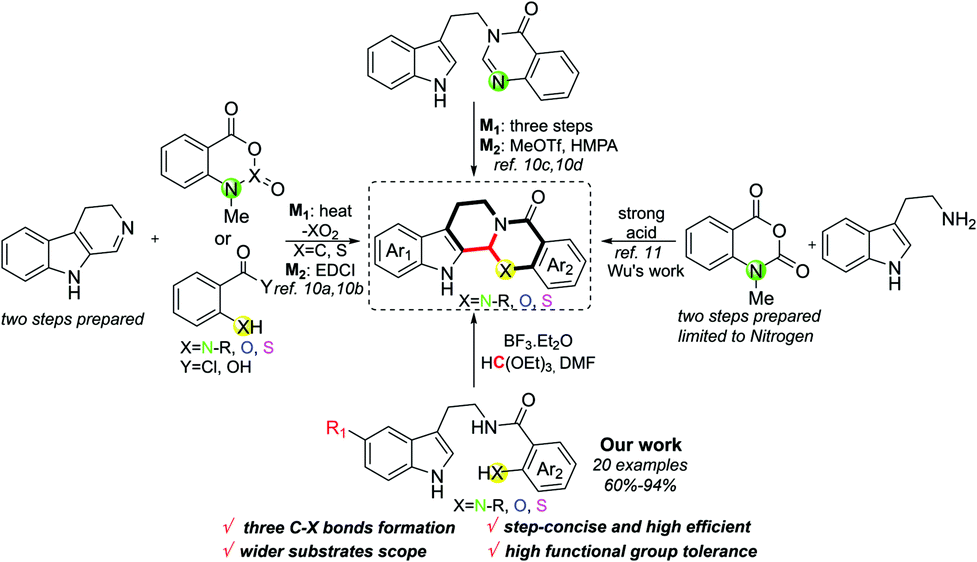 | ||
| Scheme 1 Approaches to evodiamine and its analogues. | ||
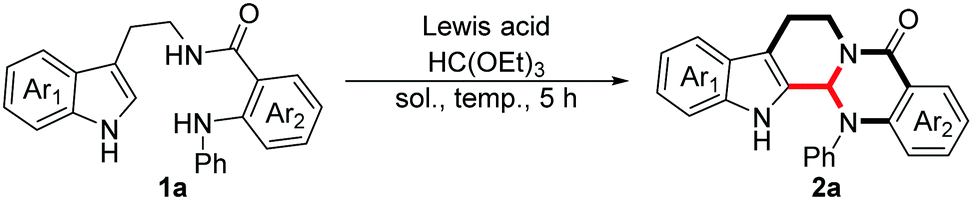
We initiated this study with 1a as the model substrate, and chose HC(OEt)3 as the carbon source with Lewis acid AlCl3 as the catalyst (Table 1). After screening various solvents, DMF was confirmed to be the optimal solvent and the desired product was obtained in 73% yield (entry 1). Considering the crucial role of Lewis acid in this transformation, different catalysts were checked subsequently (entries 5–9), and pleasingly, 2a was generated in 93% yield when 1 equivalent BF3·Et2O was employed (entry 9). Inspired by this pleasing result, we tried to lower the amount of BF3·Et2O, and no obvious decrease in efficiency was observed when 0.5 equivalent BF3·Et2O was used (91%, entry 10). However, the yield reduced sharply on reducing the amount of BF3·Et2O further (75%, entry 11), and in the absence of Lewis acid merely trace 2a was detected (entry 12). Meanwhile, a similar result was provided when we regulated the amount of HC(OEt)3 (48%, entry 13). Lastly, we performed this reaction in different temperature gradients, and an excellent yield was provided at 100 °C (92%, entry 14), and no obvious effect was observed even at 70 °C (entry 15). However, this reaction was suppressed thoroughly at room temperature (entry 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Lewis acid | HC(OEt)3 (eq.) | Temp. (°C) | Yieldb (%) |
| N.R. = no result.a Reaction conditions: 1a (1 eq., 0.10 mmol), HC(OEt)3 (3 eq., 0.30 mmol), Lewis acid (1 eq., 0.10 mmol), solvent (1 mL) under an Ar atmosphere for 5 h.b Isolated yields.c BF3·Et2O was used in 0.5 eq.d BF3·Et2O was used in 0.3 eq. | |||||
| 1 | DMF | AlCl3 | 3 | 135 | 73 |
| 2 | CH3CN | AlCl3 | 3 | 135 | 46 |
| 3 | Toluene | AlCl3 | 3 | 135 | 28 |
| 4 | DMSO | AlCl3 | 3 | 135 | N.R. |
| 5 | DMF | ZnCl2 | 3 | 135 | 81 |
| 6 | DMF | ZnBr2 | 3 | 135 | 70 |
| 7 | DMF | SnCl4 | 3 | 135 | 45 |
| 8 | DMF | TiCl4 | 3 | 135 | 33 |
| 9 | DMF | BF3·Et2O | 3 | 135 | 93 |
| 10c | DMF | BF3·Et2O | 3 | 135 | 91 |
| 11d | DMF | BF3·Et2O | 3 | 135 | 75 |
| 12 | DMF | — | 3 | 135 | Trace |
| 13c | DMF | BF3·Et2O | 1 | 135 | 48 |
| 14c | DMF | BF3·Et2O | 3 | 100 | 92 |
| 15c | DMF | BF3·Et2O | 3 | 70 | 80 |
| 16c | DMF | BF3·Et2O | 3 | 25 | N.R. |
With the optimized conditions in hand, we investigated the scope of the reaction with regard to various substituents on the indole rings as well as the ortho-position of benzamides, and moderate to excellent yields were obtained in all cases (Scheme 2) (2a–2t, 60–94%). Different electron-donating substituents on the indole rings were well tolerated in this reaction (2b, 91%; 2c, 93%). When an electron-withdrawing group was introduced the yield was reduced slightly (2d, 71%). Meanwhile, various aryl-substituted amidogens on the ortho-position of the benzamides provided the desired products in excellent yields (2e, 94%; 2f, 90%). It is noteworthy that evodiamine 2g was directly obtained within our protocol in 92% yield. Subsequently, various ortho-hydroxyl substituted benzamides were checked in this scenario (1h–1q), and electron-donating substituents showed no effects on the reaction while the electron-withdrawing ones gave a lower yield on the contrary (2i–2k, 60–75%), which is in accordance with the aforementioned results. Unexpectedly, diversified substituents on the para-position of ortho-hydroxyl substituted benzamides made no difference to the yields (2l–2n, 70–73%), no matter whether they were electron-donating or -withdrawing ones. Likewise, for the meta-substituted substrates, the same phenomenon was observed as the different electronic substituents on the indole rings (2o, 74%; 2p, 60%). Lastly, three ortho-sulfydryl benzamides were also tested in this reaction (1r–1t), since the stronger nucleophilicity of the sulphur atom compared with nitrogen and oxygen atoms enabled these reactions to be performed under milder conditions. Different electron-donating groups on the indole rings showed no effects on the yields (2s, 75%; 2t, 81%).
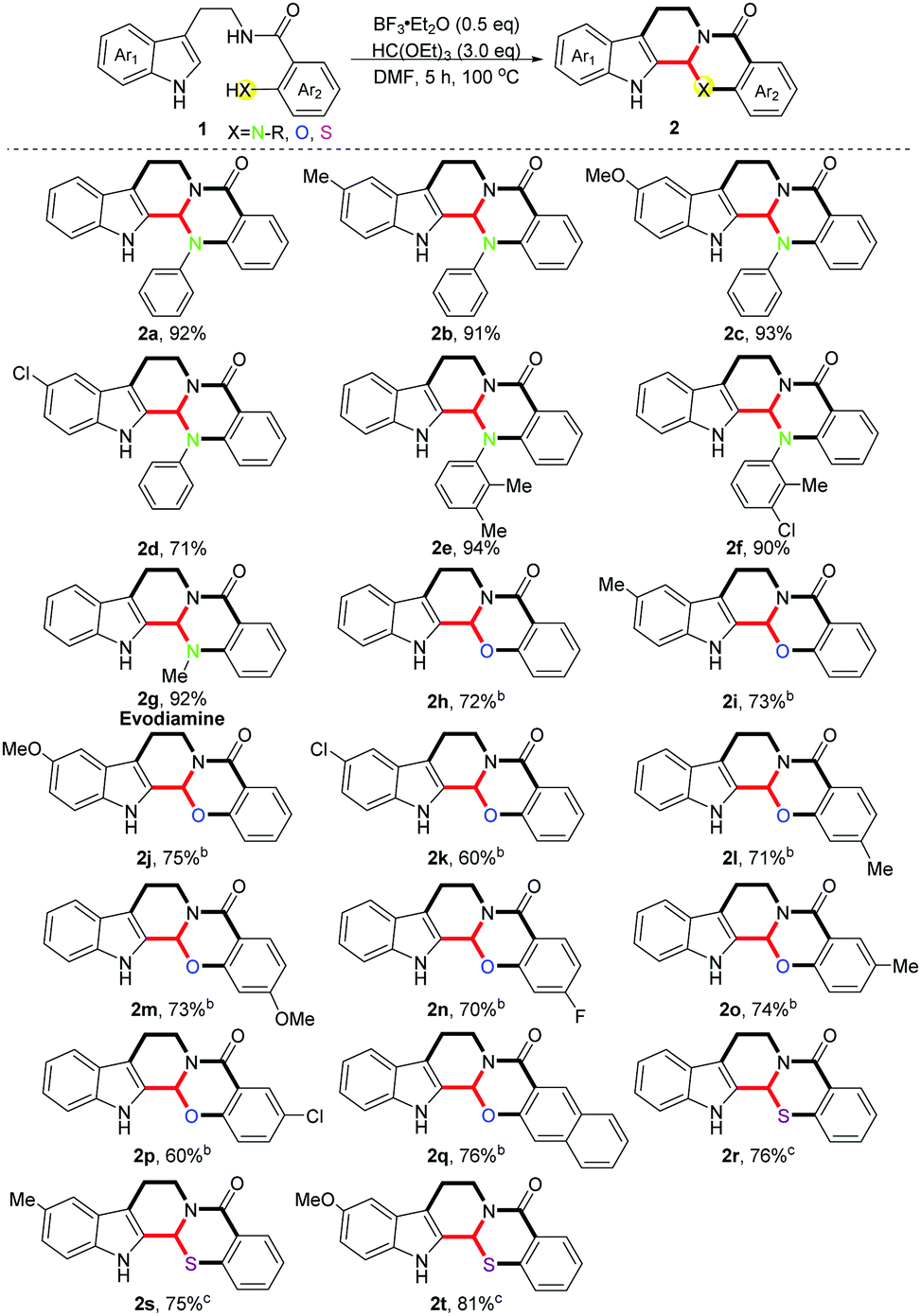 | ||
Scheme 2 Investigation of substrate scope.a a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Standard conditions. bReaction temperature was 135 °C. cCH2Cl2 was used as the solvent, room temperature. Standard conditions. bReaction temperature was 135 °C. cCH2Cl2 was used as the solvent, room temperature. | ||
According to previous studies,8 since evodiamine analogues usually possess anti-tumor activity, these unreported compounds synthesized by us were evaluated for their cytotoxicity using the MTT method (Table 2 and Fig. 2). The results data showed that the compound 2b exhibited higher activity than other compounds in the field of inhibiting proliferation of SMMC-7721 cells, and its IC50 value was 5.7 μM which was close to the IC50 of vorinostat. Moreover, the selectivity of 2b on both the tumor cell lines and normal cell line (WI-38) was also better than that of vorinostat.
| Compds | CAL-27 | A549 | SMMC-7721 | WI-38 |
|---|---|---|---|---|
| a All values are the mean ± SEM (n = 3). | ||||
| 2a | >200 | 14.6 ± 2.9 | 12.6 ± 5.7 | 64.5 ± 7.8 |
| 2b | 75.1 ± 2.3 | 13.4 ± 0.9 | 5.7 ± 4.5 | 43.5 ± 5.0 |
| 2c | 9.9 ± 1.0 | >200 | 12.9 ± 2.1 | >200 |
| 2d | >200 | 64.1 ± 1.8 | 16.8 ± 2.2 | 58.7 ± 2.0 |
| 2e | >200 | 56.7 ± 2.8 | 43.3 ± 2.3 | 42.8 ± 5.7 |
| 2f | >200 | 29.4 ± 3.2 | 9.2 ± 1.9 | 27.4 ± 2.1 |
| 2q | >200 | >200 | >200 | 84.0 ± 3.6 |
| Vorinostat | 1.3 ± 0.3 | 3.1 ± 0.5 | 3.6 ± 0.5 | 10.2 ± 2.2 |
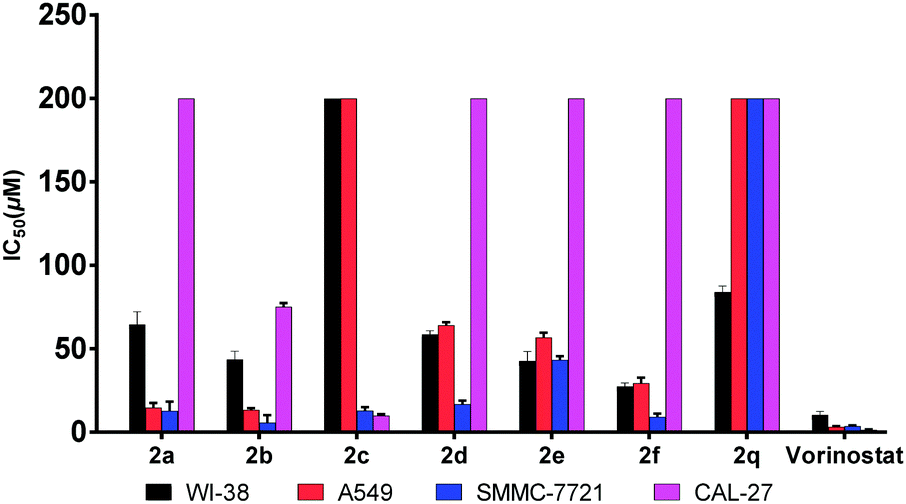 | ||
| Fig. 2 In vitro antitumor activity of the new compounds (IC50, μM). | ||
The induced apoptosis in SMMC-7721 cells of compound 2b was further confirmed by DAPI fluorescence after a 72 h treatment. Under a fluorescence microscope, the fluorescence of the cells in the control group was more diffuse and uniform, and the nucleus was obviously a regular ellipsoid shape (Fig. 3A). But when the cells were exposed to compound 2b for 72 h, the nuclei were apparently damaged. Most cells showed condensed and fragmented nuclei (arrows) (Fig. 3B and C). This indicated that compound 2b could induce significant cell apoptosis in SMMC-7721 cells. All data mentioned above suggests that these new compounds merit further research to identify promising features as antitumor candidates in the future.
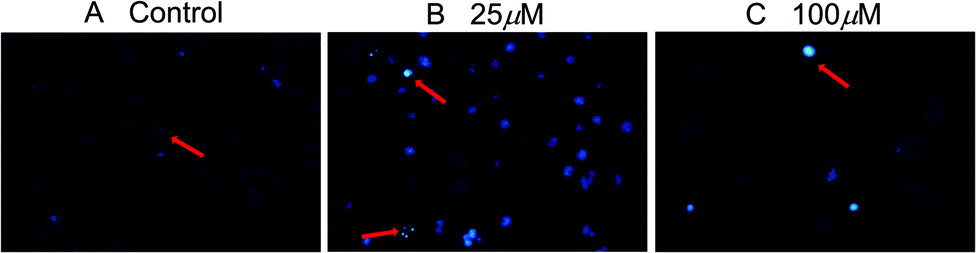 | ||
| Fig. 3 Apoptosis of SMMC-7721 cells after compound 2b treatment (200×). SMMC-7721 cells were incubated with compound 2b for 72 h, and the nuclei were stained by DAPI. | ||
To demonstrate the robust nature of this protocol, we performed a gram scale reaction using 1b as the typical substrate, and the corresponding product 2b was obtained in good yield (0.89 g, 79%), which could ensure a sufficient reagent supply in the following insightful bioactivity assessments (Scheme 3).
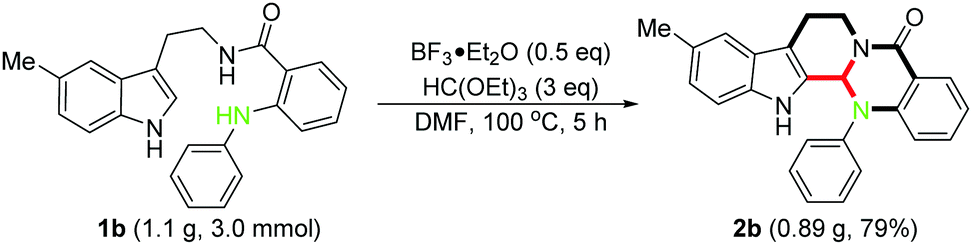 | ||
| Scheme 3 Gram scale experiment. | ||
In summary, we have developed an efficient route to evodiamine and various heteroatom-containing analogues. In this transformation three chemical bonds and two fused rings were constructed within one step. The reaction showed good functional group tolerance, and all the expanded examples were obtained in good to excellent yields even on gram scales. Notably, the unreported evodiamine analogue 2b synthesized in our protocol exhibited potent efficacy against SMMC-7721 (IC50 = 5.7 μM). The fluorescence photomicrography results showed compound 2b could particularly induce significant apoptosis in SMMC-7721 cells. More insightful bioactivity assays and applications of the reaction are in progress in our lab.
We thank Prof. Quan-Yi Zhao and Prof. Dian He for helpful discussions. Financial support was provided by the Recruitment Program of Global Experts (1000 Talents Plan).
Conflicts of interest
The authors declare no competing financial interest.Notes and references
- For reviews of polycyclic heterocycle skeletons in drugs and natural products see: (a) A. K. Mukherjee, S. Basu, N. Sarkar and A. C. Ghosh, Curr. Med. Chem., 2001, 8, 1467–1486 CrossRef CAS PubMed; (b) R. v. d. Heijden, D. I. Jacobs, W. Snoeijer, D. Hallard and R. Verpoorte, Curr. Med. Chem., 2004, 11, 607–628 CrossRef; (c) G. M. Cragg and D. J. Newman, J. Ethnopharmacol., 2005, 100, 72–79 CrossRef CAS PubMed; (d) Y. Pommier, Nat. Rev. Cancer, 2006, 6, 789–802 CrossRef CAS PubMed; (e) S. E. Kulling and H. M. Rawel, Planta Med., 2008, 74, 1625–1634 CrossRef CAS PubMed . For selected examples of polycyclic heterocycles' application in Medicinal Chemistry see: ; (f) D. E. Bierer, L. G. Dubenko, P. Zhang, Q. Lu, P. A. Imbach, A. W. Garofalo, P.-W. Phuan, D. M. Fort, J. Litvak, R. E. Gerber, B. Sloan, J. Luo, R. Cooper and G. M. Reaven, J. Med. Chem., 1998, 41, 2754–2764 CrossRef CAS PubMed; (g) J.-G. Delcros, S. Tomasi, S. Duhieu, M. Foucault, B. Martin, M. Le Roch, V. Eifler-Lima, J. Renault and P. Uriac, J. Med. Chem., 2006, 49, 232–245 CrossRef CAS PubMed; (h) J. C. Lukesh, D. W. Carney, H. Dong, R. M. Cross, V. Shukla, K. K. Duncan, S. Yang, D. M. Brody, M. M. Brütsch, A. Radakovic and D. L. Boger, J. Med. Chem., 2017, 60, 7591–7604 CrossRef CAS PubMed.
- (a) M. E. Wall, M. C. Wani, C. E. Cook, K. H. Palmer, A. T. McPhail and G. A. Sim, J. Am. Chem. Soc., 1966, 88, 3888–3890 CrossRef CAS; (b) L. Vereczkey, Eur. J. Drug Metab. Pharmacokinet., 1985, 10, 89–103 CrossRef CAS PubMed; (c) B. Kopp-Holtwiesche and H. J. Rehm, J. Environ. Pathol., Toxicol. Oncol., 1990, 10, 41–44 CAS; (d) Y.-P. Liu, Y. Li, X.-H. Cai, X.-Y. Li, L.-M. Kong, G.-G. Cheng and X.-D. Luo, J. Nat. Prod., 2012, 75, 220–224 CrossRef CAS PubMed; (e) J. T. Payne, C. B. Poor and J. C. Lewis, Angew. Chem., Int. Ed., 2015, 54, 4226–4230 CrossRef CAS PubMed; (f) Q.-L. Liu, A.-H. Chen, Z.-H. Jiang, Y.-L. Ma, J.-Y. Tang, W. Xu, Y.-P. Liu and Y.-H. Fu, Chin. J. Org. Chem., 2018, 38, 1833–1836 CrossRef.
- For reviews on construction of polycyclic heterocycles see: (a) D. M. D'Souza and T. J. J. Müller, Chem. Soc. Rev., 2007, 36, 1095–1108 RSC; (b) A. Padwa, Chem. Soc. Rev., 2009, 38, 3072–3081 RSC; (c) L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Acc. Chem. Res., 2012, 45, 1278–1293 CrossRef CAS PubMed; (d) K. C. Majumdar, S. Samanta and B. Sinha, Synthesis, 2012, 817–847 CrossRef CAS; (e) R. Lahiri, A. A. Ansari and Y. D. Vankar, Chem. Soc. Rev., 2013, 42, 5102–5118 RSC; (f) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2014, 43, 3106–3135 RSC; (g) P. Majumdar, A. Pati, M. Patra, R. K. Behera and A. K. Behera, Chem. Rev., 2014, 114, 2942–2977 CrossRef CAS PubMed; (h) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed; (i) U. K. Sharma, N. Sharma, D. D. Vachhani and E. V. Van der Eycken, Chem. Soc. Rev., 2015, 44, 1836–1860 RSC . For selected examples of construction of polycyclic heterocycles see: ; (j) Y.-P. Zhu, M.-C. Liu, Q. Cai, F.-C. Jia and A.-X. Wu, Chem. – Eur. J., 2013, 19, 10132–10137 CrossRef CAS PubMed; (k) H. Ikemoto, T. Yoshino, K. Sakata, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2014, 136, 5424–5431 CrossRef CAS PubMed; (l) Z. Meng, H. Yu, L. Li, W. Tao, H. Chen, M. Wan, P. Yang, D. J. Edmonds, J. Zhong and A. Li, Nat. Commun., 2015, 6, 6096 CrossRef CAS PubMed; (m) M. Feng, B. Tang, H.-X. Xu and X. Jiang, Org. Lett., 2016, 18, 4352–4355 CrossRef CAS PubMed; (n) H. Li, Q. Chen, Z. Lu and A. Li, J. Am. Chem. Soc., 2016, 138, 15555–15558 CrossRef CAS PubMed; (o) Y. Li, S. Zhu, J. Li and A. Li, J. Am. Chem. Soc., 2016, 138, 3982–3985 CrossRef CAS PubMed; (p) R. Kanno, S. Yokoshima, M. Kanai and T. Fukuyama, J. Antibiot., 2017, 71, 240 CrossRef PubMed; (q) J. Zhang, X. Wu, Q. Gao, X. Geng, P. Zhao, Y.-D. Wu and A. Wu, Org. Lett., 2017, 19, 408–411 CrossRef CAS PubMed; (r) N. Wang, J. Liu, C. Wang, L. Bai and X. Jiang, Org. Lett., 2018, 20, 292–295 CrossRef CAS PubMed.
- (a) C. L. King, Y. C. Kong, N. S. Wong, H. W. Yeung, H. H. S. Fong and U. Sankawa, J. Nat. Prod., 1980, 43, 577–582 CrossRef CAS PubMed; (b) W.-F. Chiou, J.-F. Liao and C.-F. Chen, J. Nat. Prod., 1996, 59, 374–378 CrossRef CAS PubMed; (c) L.-L. Yu, L.-K. Ho, J.-F. Liao and C.-F. Chen, J. Nat. Prod., 1997, 60, 1196–1198 CrossRef CAS; (d) L. V. Pearce, P. A. Petukhov, T. Szabo, N. Kedei, F. Bizik, A. P. Kozikowski and P. M. Blumberg, Org. Biomol. Chem., 2004, 2, 2281–2286 RSC; (e) L.-C. Lin, S.-H. Li, Y.-T. Wu, K.-L. Kuo and T.-H. Tsai, J. Agric. Food Chem., 2012, 60, 1595–1604 CrossRef CAS PubMed; (f) H. Yu, H. Jin, W. Gong, Z. Wang and H. Liang, Molecules, 2013, 18, 1826 CrossRef CAS PubMed; (g) X. Yuan, B. Zhang, Y. Wang, J. Ma and X. Hou, Anal. Methods, 2013, 5, 5767–5774 RSC; (h) Y. Niu, J. Bai, R. D. Kamm, Y. Wang and C. Wang, Mol. Pharm., 2014, 11, 2022–2029 CrossRef CAS PubMed; (i) S. Wang, S. Yamamoto, Y. Kogure, W. Zhang, K. Noguchi and Y. Dai, J. Nat. Prod., 2016, 79, 1225–1230 CrossRef CAS PubMed; (j) Y. Feng, N.-X. Li, H.-L. Yin, T.-Y. Chen, Q. Yang and M. Wu, Mol. Pharm., 2019, 16, 422–436 CrossRef CAS PubMed.
- W.-F. Chiou, Y.-J. Sung, J.-F. Liao, A. Y.-C. Shum and C.-F. Chen, J. Nat. Prod., 1997, 60, 708–711 CrossRef CAS PubMed.
- Y. Kobayashi, Y. Nakano, M. Kizaki, K. Hoshikuma, Y. Yokoo and T. Kamiya, Planta Med., 2001, 67, 628–633 CrossRef CAS PubMed.
- S.-M. Yuan, K. Gao, D.-M. Wang, X.-Z. Quan, J.-N. Liu, C.-M. Ma, C. Qin and L.-F. Zhang, Acta Pharmacol. Sin., 2011, 32, 295 CrossRef CAS PubMed.
- (a) G. Dong, C. Sheng, S. Wang, Z. Miao, J. Yao and W. Zhang, J. Med. Chem., 2010, 53, 7521–7531 CrossRef CAS PubMed; (b) G. Dong, S. Wang, Z. Miao, J. Yao, Y. Zhang, Z. Guo, W. Zhang and C. Sheng, J. Med. Chem., 2012, 55, 7593–7613 CrossRef CAS PubMed; (c) S. He, G. Dong, Z. Wang, W. Chen, Y. Huang, Z. Li, Y. Jiang, N. Liu, J. Yao, Z. Miao, W. Zhang and C. Sheng, ACS Med. Chem. Lett., 2015, 6, 239–243 CrossRef CAS PubMed; (d) S. Wang, K. Fang, G. Dong, S. Chen, N. Liu, Z. Miao, J. Yao, J. Li, W. Zhang and C. Sheng, J. Med. Chem., 2015, 58, 6678–6696 CrossRef CAS PubMed.
- (a) T. Kametani, T. Higa, L. Chu Van, M. Ihara, M. Koizumi and K. Fukumoto, J. Am. Chem. Soc., 1976, 98, 6186–6188 CrossRef CAS; (b) J. Bergman and S. Bergman, J. Org. Chem., 1985, 50, 1246–1255 CrossRef CAS; (c) W. P. Unsworth, C. Kitsiou and R. J. K. Taylor, Org. Lett., 2013, 15, 258–261 CrossRef CAS PubMed; (d) K. R. Rao, A. Raghunadh, R. Mekala, S. B. Meruva, T. V. Pratap, T. Krishna, D. Kalita, E. Laxminarayana, B. Prasad and M. Pal, Tetrahedron Lett., 2014, 55, 6004–6006 CrossRef CAS; (e) C. Kitsiou, W. P. Unsworth, G. Coulthard and R. J. K. Taylor, Tetrahedron, 2014, 70, 7172–7180 CrossRef CAS; (f) J. Ye, Y. Lin, Q. Liu, D. Xu, F. Wu, B. Liu, Y. Gao and H. Chen, Org. Lett., 2018, 20, 5457–5460 CrossRef CAS PubMed.
- (a) F. Pin, S. Comesse and A. Daïch, Tetrahedron, 2011, 67, 5564–5571 CrossRef CAS; (b) Y. Yang, C. Zhu, M. Zhang, S. Huang, J. Lin, X. Pan and W. Su, Chem. Commun., 2016, 52, 12869–12872 RSC; (c) W. P. Unsworth, G. Coulthard, C. Kitsiou and R. J. K. Taylor, J. Org. Chem., 2014, 79, 1368–1376 CrossRef CAS PubMed; (d) A. Clemenceau, Q. Wang and J. Zhu, Org. Lett., 2017, 19, 4872–4875 CrossRef CAS PubMed.
- Z.-X. Wang, J.-C. Xiang, M. Wang, J.-T. Ma, Y.-D. Wu and A.-X. Wu, Org. Lett., 2018, 20, 6380–6383 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Lanzhou University for its 110th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc00434c |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |