
Photodegradation of polymer-CNT nanocomposites: effect of CNT loading and CNT release characteristics†
Ronald S.
Lankone
a,
Jingjing
Wang
b,
James F.
Ranville
b and
D. Howard
Fairbrother
 *a
*a
aDepartment of Chemistry, Johns Hopkins University, Baltimore, MD 21218, USA. E-mail: howardf@jhu.edu
bDepartment of Chemistry & Geochemistry, Colorado School of Mines, Golden, CO 80401, USA
First published on 8th March 2017
Abstract
Information is currently lacking on the effect that carbon nanotubes (CNTs) have on the mechanism and extent of polymer photodegradation as well as the quantity, kinetics, and form of CNTs released. To address this, we followed in detail the photodegradation of a photolabile CNT-polymer nanocomposite (CNT-PNC), composed of single walled CNTs and polycaprolactone. Analysis of released CNT-containing fragments was accomplished with single particle inductively coupled plasma mass spectrometry (sp-ICP-MS). The mechanism of polymer photodegradation remained unchanged upon CNT inclusion, although polymer mass loss decreases systematically as CNT loading increases. This inhibitory effect is due to the light absorption and scattering properties of CNTs, which reduces the depth of photolysis and consequently the extent of CNT-PNC mass loss. Preferential CNT retention in the polymer during photodegradation results in CNT release at lower quantities than predicted based on their mass loading. The form of the released CNTs depends on CNT loading and evolves as the CNT-PNC photodegradation process proceeds. For CNT-PNCs with higher CNT loadings, multiple CNTs are initially released embedded within polymer fragments; as CNT-PNC degradation slows, released fragments contain predominantly isolated CNTs. For sufficiently long irradiation times, a dense CNT-mat forms at the surface, stabilizing the CNT-PNC towards further polymer or CNT loss. Extrapolating our findings to other CNT-PNC systems suggests that the quantity and form of released CNTs, as well as the extent of CNT-PNC photodegradation, will be influenced by the CNT loading and will evolve over the course of the CNT/PNC photodegradation process.
Environmental significanceAs carbon nanotube polymer nanocomposites (CNT-PNCs) enter the environment in increasing quantities, a more detailed understanding of the role CNTs play in regulating the photodegradation of CNT-PNCs, and the release of CNTs is needed. To address this issue we have conducted accelerated photodegradation studies and spectroscopic characterization of CNT-PNCs with a span of initial CNT concentrations. Our findings indicate that CNTs restrict the depth of photolysis and thereby reduce the extent of photodegradation. We also utilize single particle ICP-MS to characterize the released CNT-containing fragments as a function of both initial CNT concentration and the extent of photodegradation. These findings establish a systematic relationship between CNTs and CNT-PNC photodegradation and inform the nature of CNTs released into the environment. |
1 Introduction
Polymer nanocomposites (PNCs), particularly those containing carbon nanotubes (CNT-PNCs), are being utilized in a growing variety of commercial products such as sporting equipment, shoe soles, bicycles, and wind turbine blades1 and applications in highly-engineered materials such as the wingtip fairings of Lockheed Martin's F-35 Joint Strike Fighter.2 With the increased use of CNT-PNCs, the potential for CNT release into the environment and exposure to different ecosystems (aquatic, terrestrial, human) will inevitably increase as well. Release is an important factor to consider because it is the precursor to exposure, which along with hazard determines the risk posed by a material.3 Extensive research has already been conducted to explore the toxicity (hazard) of CNTs.4–7 In contrast, far less information is available on nanoparticle (NP) release from consumer products, especially the factors that control release from products. For the case of CNT-PNCs, this includes the influence that CNTs have on the polymer photodegredation and release mechanism(s), the kinetics of CNT release, the quantity and overall fraction of incorporated CNTs released, and the form of the released CNTs (dispersed vs. embedded in polymer fragments). Without such information, well-informed risk assessments of PNC products are difficult to develop.The release of CNTs from PNC-CNTs as a result of photochemical weathering can occur during PNC-CNT use or end of life phases and as such represents an important release scenario.8 Previous studies have shown that nanomaterial release from polymer nanocomposites occurs as a consequence of the photodegradation of the surrounding polymer matrix.9,10 As highlighted in a review by Kingston et al., PNCs composed of a variety of polymer matrices, such as epoxy, polyamide, polyurethane, and polycarbonate, are found to be susceptible to nanomaterial release through polymer photodegradation.11 However, polymer photodegredation is slow and one of the difficulties in studying polymer photodegradation in the natural environment is that it may take months or more typically years for measureable degradation to occur. Consequently, CNT-PNC weathering is often examined using conditions that accelerate the degradation process, although specific methods vary. Such accelerated degradation approaches include the NIST SPHERE (simulated photodegradation via high energy radiant exposure)12,13 and standardized accelerated weathering tests, such as ISO 3892-2:2006 (ref. 14) or ISO 4892/06.15 Accelerated degradation is not an approach that is restricted to studying the weathering of CNT-PNCs weathering. For example, Pillai et al. submerged quantum dot (QD)-low density polyethylene PNCs in either water or 3% acetic acid, and heated the system to 75 °C for up to 15 days to accelerate release in a manner similar to the protocol developed by the FDA to study the migration of components from food contact materials.16 By using conventional ICP-MS to detect dissolved elements (Cd, Se, Zn, and S) indicative of released QDs, they were able to determine release mechanisms and identify that release in this system is driven largely by particle dissolution and impacted by QD size.16
The extent of CNT release during weathering of CNT/PNCs has been reported to vary considerably depending on the details of the release scenario, as defined by both the CNT-PNC and the environmental conditions to which it is exposed. Thus, Wohlleben et al. did not find any evidence for the spontaneous release of CNTs following the weathering of CNT-polyurethane nanocomposites.17 However, using radiolabelled CNTs (14C-CNT), Rhiem et al. estimated that following a UV dose equivalent to three years of natural exposure in Florida, 64 mg CNT m−2 released from a polycarbonate polymer nanocomposites and that photodegraded polymers were more likely to release CNTs at the end-of-life disposal scenarios.18 In a review of over a dozen studies on the weathering of CNT-PNCs,19 Schlagenhauf et al. found that the quantification of released CNTs was left largely unaddressed. However, four of these studies did attempt to monitor CNT release through characterization of released material with TEM imaging20 or analytical ultracentrifugation14,17,21 and in two studies, it was concluded that CNT release was unlikely to have occurred based on SEM imaging of the surrounding sample holder10 or AFM scratch lithography of the weathered PNC surface.13 Wohlleben et al. provided a more quantitative assessment in a review of 27 different nanocomposite materials weathered under the ISO 4892 protocol and found their release rates (mg MJ−1 of UV) to span five orders of magnitude.22 Regarding CNT-PNCs specifically, release rates spanned roughly 0.05 mg MJ−1 (CNT – cross-linked polyurethane) to 8 mg MJ−1 (CNT – epoxy).
A constant challenge that pervades CNT release studies is that CNTs are released from CNT-PNCs at concentrations at or below the CNT detection limit of most analytical techniques. As a result, CNT release is often expressed in qualitative terms without quantification of released CNTs or the form of the released CNTs (embedded within polymer fragments or purely as CNTs) identified. As highlighted in a recent review,23 while many techniques currently exist that are capable of detecting CNTs, each technique has its own unique set of limitations. For example, although UV-vis spectroscopy is an inexpensive and readily available analytical method to measure the concentration of suspended CNTs,24 it lacks both the sensitivity needed to detect the low concentrations of CNTs expected to be released from CNT-PNCs and the selectivity to distinguish CNTs from other naturally occurring environmental particles. In contrast, although transmission electron microscopy has the requisite sensitivity to identify release engineered nanomaterials and can directly image and confirm the presence of CNTs25 as well as resolve their form (individual tubes vs. embedded in polymer fragments), it is costly, time consuming, and not an intrinsically quantitative technique. Near infrared fluorescence is both quantitative and can detect CNTs in environmental matrices, but it requires CNTs that are both well dispersed and of a specific chirality (semi-conducting).26,27 Scintillation counting of 14C-labelled carbon nanotubes may also be used to quantify CNT release following weathering,18 however 14C-labelled-CNTs are inherently expensive, difficult to synthesize and require specialized apparatus to be detected. One approach that combines sensitivity with selectivity for CNT detection involves the use of ICP-MS to measure metal nanoparticles, intimately bound to the CNTs, as a means to quantify CNT release. For example, following the abrasion of 1% (w/w) CNT-epoxy samples, Schlagenhauf et al. collected submicron sized fragments (PM1), submerged them in dilute nitric acid, and measured the dissolved metal ion concentration with ICP-MS as a route to quantify protruding and free standing CNTs, under the assumption that only exposed CNTs would lead to their bound metal nanoparticles being available for dissolution. From their findings, they estimate that for every one gram of material abraded, PM1 particles produced will have 40 μg of MWCNTs protruding from epoxy fragments and just 0.4 μg of MWCNTs will be free standing.28
The present study focuses on developing a detailed, mechanistic understanding of CNT-PNC photodegradation. A key experimental component of the present study was the use of single particle inductively coupled plasma mass spectrometry (sp-ICP-MS) to analyze released CNTs through the use of metal nanoparticles, residual from CNT synthesis, as proxies for CNT detection.29,30 This novel approach to analysis of released CNT distinguishes itself from the characterization techniques discussed above due to the fact that it can simultaneously quantify the concentration and characterize the form of released CNTs from data collected during a single analysis of a given sample. In order to develop a more fundamental mechanistic understanding of how CNTs influence the photodegradation of CNT-PNCs and CNT release characteristics, we have selected to photodegrade CNT-PNCs composed of SWCNTs and poly-ε-caprolactone (PCL) with UVC light; 254 nm irradiation was used to accelerate the rate of photodegradation because it is close to the absorption maximum in the ester's π to π* band (Fig. SI 1†). SWCNTs were selected, as opposed to more commercially relevant MWCNTs, due to the presence of residual yttrium nanoparticle catalysts remaining intimately bound to the SWCNTs. These Y nanoparticles act as proxies for the SWCNTs and allow for their detection and quantification with sp-ICP-MS.30 Additionally, because SWCNTs and MWCNTs produced from a variety of different methods all share similar extinction coefficients,31 the light attenuating behavior of the SWCNTs utilized in this study will likely be similar to other CNT-PNCs. PCL was chosen because it is both photolabile and because CNT-PNCs can readily be prepared with well-defined properties via solution blending. PCL also possesses characteristic spectral features in both infrared (IR) and X-ray photoelectron (XP) spectroscopies that allow for the extent of its degradation to be accurately monitored. A particular point of emphasis in this study was to elucidate the role that CNT loading plays in regulating the extent of CNT release and polymer degradation during weathering. Indeed, although CNT-PNCs contain a range of different CNT loadings (typically in the range of 0.1–5% wt), there is a surprising lack of information on the role that CNT loading plays in determining the kinetics of CNT-PNC photodegradation as well as CNT release. To address this issue, we studied photodegradation on CNT-PNCs prepared with a range of CNT loadings (0–5 wt%). By monitoring changes to the polymer on both the macroscopic level (mass loss) and molecular level (IR and XPS), and acquiring information on released fragments during CNT-PNC photodegradation, we are able to compare the extent of polymer degradation to the magnitude and rate of CNT release. Collectively, this information on released material and the remaining photolyzed nanocomposite enables us to identify the mechanism by which CNTs influence the rate and extent of polymer photodegradation, as well the dependence of CNT release characteristics on initial CNT loading and irradiation time.
2 Experimental methods
Chemicals
Chloroform and tetrahydrofuran (Fisher Scientific) were used without further purification or drying. HPLC grade water was purchased from VWR International. Ethyl cellulose (EC), poly-ε-caprolactone (PCL) and sodium deoxycholate (SDC) was purchased from Sigma Aldrich; for PCL, the manufacturer reports the number-average molecular weight (Mn 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). Single-walled carbon nanotubes (SWCNTs) were purchased from Carbon Solutions, Inc (Product number: AP-SWNT). The AP-SWNT sample is described by the manufacturer to be “as prepared” and not purified. They were used as received and characterized with TEM imaging (Fig. SI 2†) and Raman spectroscopy. Raman spectra (Fig. SI 3†) of the SWCNTs is dominated by the G band, indicating a high level of SWCNT purity and a relative lack of damage to the sidewalls. Raman spectra also contain two well defined radial breathing modes at 155 cm−1 and 176 cm−1 (with a third mode potentially at 188 cm−1), corresponding to a SWNCT tube diameter of 1.4–1.6 nm.32
000). Single-walled carbon nanotubes (SWCNTs) were purchased from Carbon Solutions, Inc (Product number: AP-SWNT). The AP-SWNT sample is described by the manufacturer to be “as prepared” and not purified. They were used as received and characterized with TEM imaging (Fig. SI 2†) and Raman spectroscopy. Raman spectra (Fig. SI 3†) of the SWCNTs is dominated by the G band, indicating a high level of SWCNT purity and a relative lack of damage to the sidewalls. Raman spectra also contain two well defined radial breathing modes at 155 cm−1 and 176 cm−1 (with a third mode potentially at 188 cm−1), corresponding to a SWNCT tube diameter of 1.4–1.6 nm.32
Nanocomposite preparation
All CNT-PNCs were prepared via solution blending.33 This technique allows for both a high degree of SWCNT dispersion and batch to batch consistency between composites. Characterization using SEM found no signs of SWCNT clumping or aggregation at the surface (Fig. 3). In addition to CNT-free PCL, nanocomposite samples were prepared at three SWCNT mass loadings (% w/w): 0.25% SWCNT, 1.5% SWCNT, and 5.0% SWCNT. Nanocomposites were prepared by first weighing out the required mass of SWCNT powder and adding it to a 50 mL Erlenmeyer flask. Next, 21 mL of THF, 21 mL of chloroform, and 31.5 mg of EC were added. EC is required to create well dispersed SWCNTs in the nanocomposites; 31.5 mg was sufficient to disperse all SWCNT masses used in this study and prevented any visible SWCNT aggregation in the suspensions.To create a uniform SWCNT dispersion the flask containing the mixture of SWCNT, EC, chloroform and THF was sealed with a septa, partially submersed in a low power (70 W) bath sonicator (Branson 1800) and sonicated for 3 hours. PCL (420 mg) was then added to this solution and the PCL/SWNCT mixture sonicated for an additional hour. The final solution was poured, in 6 mL portions, into 7 separate Fisherbrand disposable aluminum dishes which were then covered with a cardboard box, and allowed to dry overnight at room temperature. Circular nanocomposite samples, referred to as coupons (approximately 30 μm thick and 40 mm in diameter), were formed as the solvent mixture evaporated. All nanocomposites were allowed to dry for at least one additional day in a desiccator prior to characterization and UV irradiation. Examples of CNT-PNCs with different CNT loadings are shown in Fig. 1. The average starting mass of the nanocomposite samples used in this study was 31.0 mg (±2.3 mg). Hereafter, PCL samples free of CNTs are referred to as PCL/EC, while PCL samples free of CNTs and EC are referred to as PCL.
 | ||
| Fig. 1 (Top) Polymer nanocomposites (PNC) prepared by solution blending with a poly-ε-caprolactone (PCL) polymer matrix and 0–5% (by weight) SWCNT nanoparticle filler. (Bottom) Samples are bound to a glass slide using Teflon tape, submerged in water, and irradiated in a quartz test tube. | ||
UV irradiation of polymer nanocomposites
Polymer nanocomposites were irradiated in a RPR-100 Rayonet photochemical reactor by 16 cylindrically oriented low pressure mercury lamps (purchased from The Southern New England Ultraviolet Co.). To accelerate photodegredation, irradiation was performed principally at 254 nm (UVC) with the fluence = 1.62 × 1017 photon per second, as determined by actinometry.34 Additionally, 300 nm (UVB) was used in some comparative control studies to assess photodegradation in more environmentally relevant conditions. During irradiation, the temperature in the reactor was maintained at ≈30 °C by an internal fan. To prepare CNT-PNC samples for irradiation, individual nanocomposite samples were weighed and bound to a glass slide by thin strips of Teflon tape. These samples were then placed upright in a 15 mL quartz test tube (Fig. 1). Each test tube was subsequently filled with 12 mL of HPLC grade water and sealed with septa to prevent supernatant evaporation; the septa was wrapped with aluminum to prevent its photodegradation. Samples were submersed in water for the entire period of irradiation to ensure that all released nanomaterial is captured for subsequent sp-ICP-MS analysis. Up to 12 samples, each in individual test tubes could be irradiated simultaneously in a rotating carousel. Samples used in this study were continuously irradiated for time periods ranging from 0 hours up through, and including 125 days.Supernatant collection
Two different methods were used to determine SWCNT release from irradiated nanocomposites.After 20 days of irradiation had elapsed, one nanocomposite from each SWCNT loading (PCL/EC, 0.25% CNT-PCL, and 5.0% CNT-PCL) was removed from the photochemical reactor, dried for one day in a desiccator, weighed, and then characterized by scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS). These remaining samples were exposed to a total of 40, 80, or 125 days of irradiation. For these longer periods of irradiation, the supernatant was not periodically collected and replenished, as it was for the first 20 days, but instead collected at the conclusion of sample exposure. At the end of 40, 80, or 125 days one nanocomposite from each SWCNT loading was removed from the photochemical reactor, dried, weighed and characterized. The release supernatant from each nanocomposite was analyzed by single particle inductively coupled mass spectrometry (sp-ICP-MS).
Determination of CNTs in “release supernatants” by single particle ICPMS (sp-ICPMS)
Prior to sp-ICP-MS analysis, 1 wt% of surfactant was added to each “release supernatant” along with a 30 min period of sonication to ensure that all released CNTs were suspended. Samples were then diluted to a concentration suitable for sp-ICP-MS analysis. After each dilution, solutions were placed in 15 mL polypropylene centrifuge tubes and sonicated (Fisher FS60H bath sonicator, Pittsburgh, PA) for 15–20 minutes.Single particle ICP-MS analysis was performed to determine the SWCNT content of the release supernatants using 89Y pulses as surrogates for SWCNT detection, as detailed in Reed et al.29 All sp-ICP-MS analyses were performed in triplicate using a NexION 300Q ICP-MS (Perkin Elmer, Waltham, MA). Data collection used a dwell time of 0.1 ms, with 60 second total acquisition time. The sample introduction flow rate was 0.3 mL min−1. To determine the background 89Y signal Nanopure water was analyzed by sp-ICP-MS at the beginning of each experiment. Data collection and sp-ICP-MS calculations were performed using the SyngistixTM (version 1.0) Nano Application Module, with additional data processing being performed using EXCEL. The Syngistix software was used to determine the background counts, and any readings that were greater than 3 sigma above this background were considered to be a possible CNT entering the plasma. To discriminate these readings from random noise they were further examined, and only those that were part of two or more consecutive readings greater than 3 sigma above the background were considered part of a pulse generated by a CNT. All consecutive readings that occurred above background intensity were combined to provide the total intensity of that CNT.
The ICP-MS was tuned daily with Perkin Elmer NexION Setup solution (1% HNO3, 10 μg L−1 Be, Ce, Fe, In, Li, Mg, Pb, U, Perkin Elmer, Waltham, MA). Dissolved Au calibration solutions (0, 2, 5, 10 μg L−1) were prepared using Claritas PPT (SPEX Certiprep, Metuchen, NJ) ICP-MS standards and 2% hydrochloric acid (Optima, Fisher Chemical, Fair Lawn, NJ). A gold NP standard (56 nm, National Institute of Standards and Technology (NIST) RM 8013, Gaithersburg, MD) was diluted to 500 ng L−1 from the stock 50 mg L−1 (∼2.98 ± 0.04 × 1013 particles per mL) solution with 18.2 MΩ cm−1 Nanopure water (Barnstead Nanopure Diamond, Lake Balboa, CA) and analyzed to determine transport efficiency.36 Similarly, dissolved yttrium ion calibration solutions (0, 1, 2, 5 μg L−1) were prepared using Claritas PPT ICP-MS standards and 2% nitric acid (Optima, Fisher Chemical, Fair Lawn, NJ) to establish instrument sensitivity to 89Y and to allow the total NP mass in each pulse to be determined.
To directly establish the relation of the sum of 89Y response determined by sp-ICPMS with CNT mass concentration, CNT solutions (0, 100, 500, 1000 ng L−1) were prepared by suspending a known mass of SWCNTs in a known volume of ultrapure water (HPLC grade) containing 1 wt% sodium deoxycholate (Sigma-Aldrich, Saint Louis, MO). A bath Branson 1510 ultrasonicator (Danbury, CT) was used to suspend CNTs overnight. Because this suspension was not immediately characterized following its preparation, the dilutions prepared from it were sonicated for 30 minutes immediately prior to sp-ICP-MS analysis to debundle any CNTs that may have aggregated. This ensures that particle number measurements from the sample are accurate. A calibration curve was constructed between the known CNT mass in suspension and the 89Y response (Fig. SI 4†) so that release supernatants containing an unknown CNT concentration could be measured and quantified.
Characterization of nanocomposites
i) X-ray photoelectron spectroscopy (XPS). Surface analysis of CNT-PNC samples was performed before and after periods of irradiation by XPS using a PHI 5600 with Mg Kα X-ray (1253.6 eV) radiation. Prior to analysis, samples were dried overnight in a desiccator and were cut to size and affixed to a sample stub by copper tape. Quantitative analysis of the carbon (C(1s)) and oxygen (O(1s)) regions was completed using a pass energy of 58.7 eV, 10 sweeps, and a step size of 0.125 eV. XPS data analysis was performed with CasaXPS. Survey scans were also collected to ensure samples were free of contamination.
ii) Attenuated total internal reflectance-infrared spectroscopy (ATR-FTIR). ATR-FTIR was performed on select nanocomposite samples before and after specific periods of irradiation. Spectra were collected with a Nicolet iS5 FT-IR spectrometer, equipped with a diamond window. Prior to analysis, all samples were dried overnight in a desiccator. The manufacturer reports a sampling depth of approximately two microns for the diamond window. Samples were analyzed at a resolution of 0.482 cm−1, with 32 scans. Each ATR-FTIR was referenced to the ambient atmosphere.
iii) Scanning electron microscopy (SEM). SEM was performed using a Jeol 6700F, FESEM. Nanocomposite samples were dried overnight in a desiccator and mounted on a conductive metal stub with copper tape prior to imaging. To minimize charging, all samples were sputter coated with platinum for 300 seconds, at a rate of 2 nm per minute. For each sample imaged, four locations on the surface were randomly selected and imaged at increasing magnifications.
3 Results
Effect of photolysis on CNT polymer nanocomposites
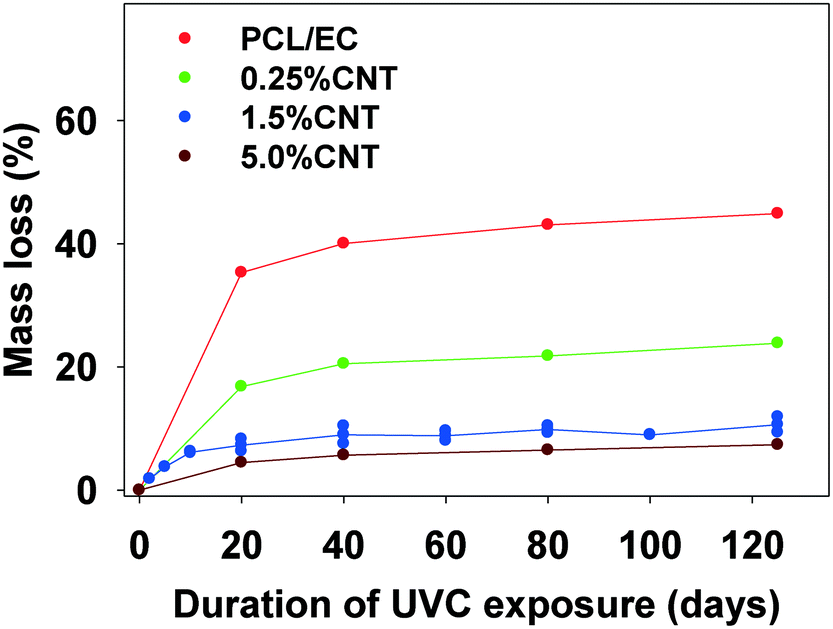 | ||
| Fig. 2 Mass loss of SWCNT-PCL nanocomposites with varying SWCNT loading (% w/w) following incremental periods of irradiation with 254 nm light. The average initial mass of all samples used in the study is 31.0 mg (±2.3 mg). | ||
In experiments using a longer wavelength of light more representative of sunlight, PCL/EC irradiated with UVB light also showed photodegradation and mass loss albeit at decreased magnitude and rate, as compared to PCL/EC irradiated with UVC. Specifically, UVB irradiation of PCL/EC following 20 and 40 days of exposure produces a mass loss of 3.18% and 6.44%, respectively. In contrast, following 20 and 40 days of UVC exposure, PCL/EC has lost 35.3% and 40.0% of its mass, respectively.
 | ||
| Fig. 3 SEM imaging shows CNT accumulation on the surface of 5.0% CNT sample following UVC exposure. Lower CNT loading samples do not show visible SWCNT accumulation. | ||
Following 125 days of irradiation, the dense and continuous CNT mat persists on the surface of the 5.0% CNT nanocomposite, with no signs of fragmentation or perforation. The PCL/EC and 0.25% CNT samples also retain a similar surface morphology to that observed following 20 days of irradiation. Additionally, the surface morphology of the 1.5% CNT sample, not shown, was found to be similar to the PCL/EC and 0.25% CNT samples following 0, 20, and 125 days of irradiation. An absence of observable CNTs at the surface following photolysis of CNT-PNCs has been previously documented by Schlagenhauf et al., in the study of 1% CNT-epoxy composites.41
:C ratio, as measured by XPS, of the 1.5% CNT nanocomposite samples as a function of irradiation time, which decreases by ≈50% through the first 40 days of irradiation before remaining constant thereafter.
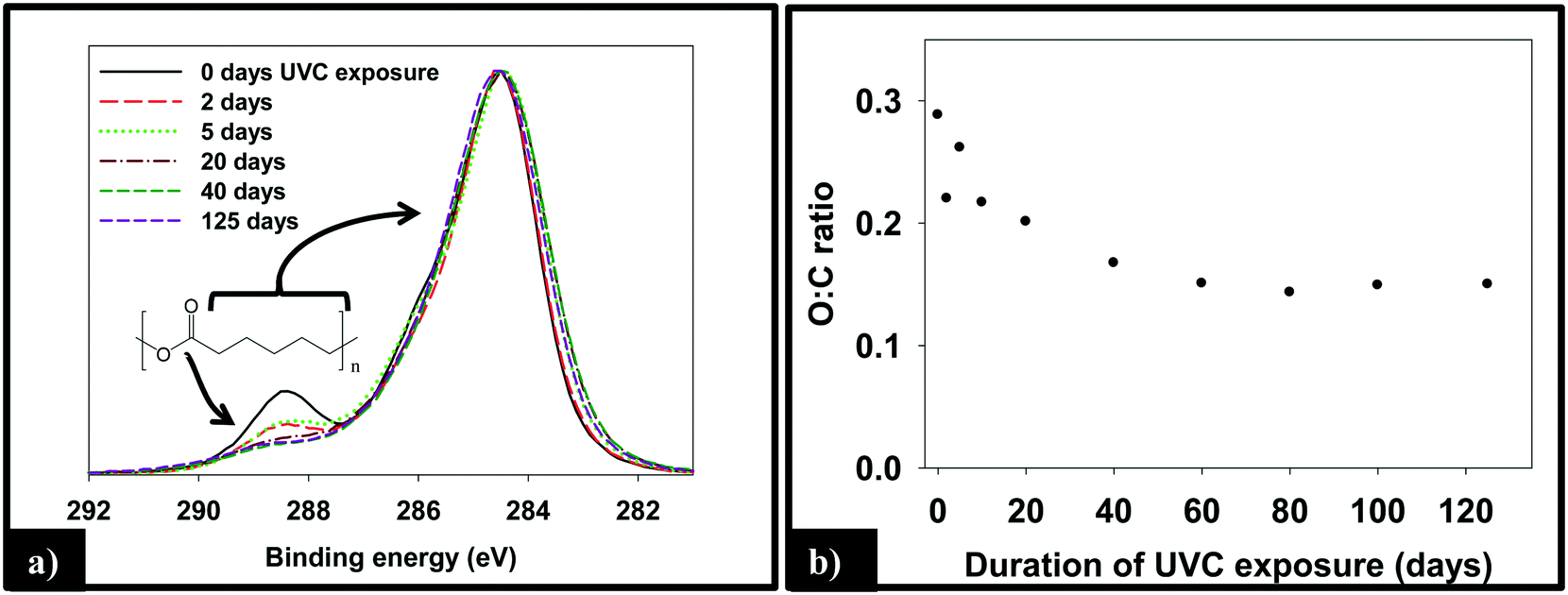 | ||
| Fig. 4 Transformations in (a) the C(1s) region and (b) O:C ratio of the 1.5% CNT composites as a function of UVC irradiation. The largest changes are seen to occur during the by first 20–40 days with little change in either the C(1s) region or the O:C ratio thereafter. | ||
ATR-FTIR was used to probe changes that occur to the chemical bonding within the near surface region (∼2 μm) of the PNCs as a result of UVC irradiation. Pure PCL, PCL/EC, and all nanocomposite samples were analyzed prior to UVC exposure and then again following 125 days of irradiation, as shown in Fig. 5a. Prior to irradiation, the ATR-FTIR of each sample was identical to literature spectra of PCL42 with absorption bands at 2866 cm−1 and 2942 cm−1 due to the symmetric and asymmetric C–H stretching modes of the alkyl carbon atoms. In addition to the characteristic fingerprint region of PCL <1500 cm−1, each sample also exhibits a sharp and intense ester stretch at 1720 cm−1 due to the ester group.42 No spectroscopic signature of CNTs was observed. Following 125 days of irradiation, the IR intensity in the C–H stretching region is relatively unchanged following irradiation regardless of CNT loading, with each sample still exhibiting an absorption band at 2866 cm−1, although subtle changes to the spectral envelope have occurred. Specifically, two small new bands appear at 2959 cm−1 and 2925 cm−1, replacing the band originally observed at 2942 cm−1. No other new features or absorption bands are observed as a consequence of photolysis. Although the C–H stretching region remains relatively unchanged after 125 days irradiation, inspection of Fig. 5a reveals that the intensity of the ester peak and signatures of PCL in the fingerprint region decrease for all samples. However, as the CNT loading increases the extent to which these spectral features decrease in intensity decreases significantly.
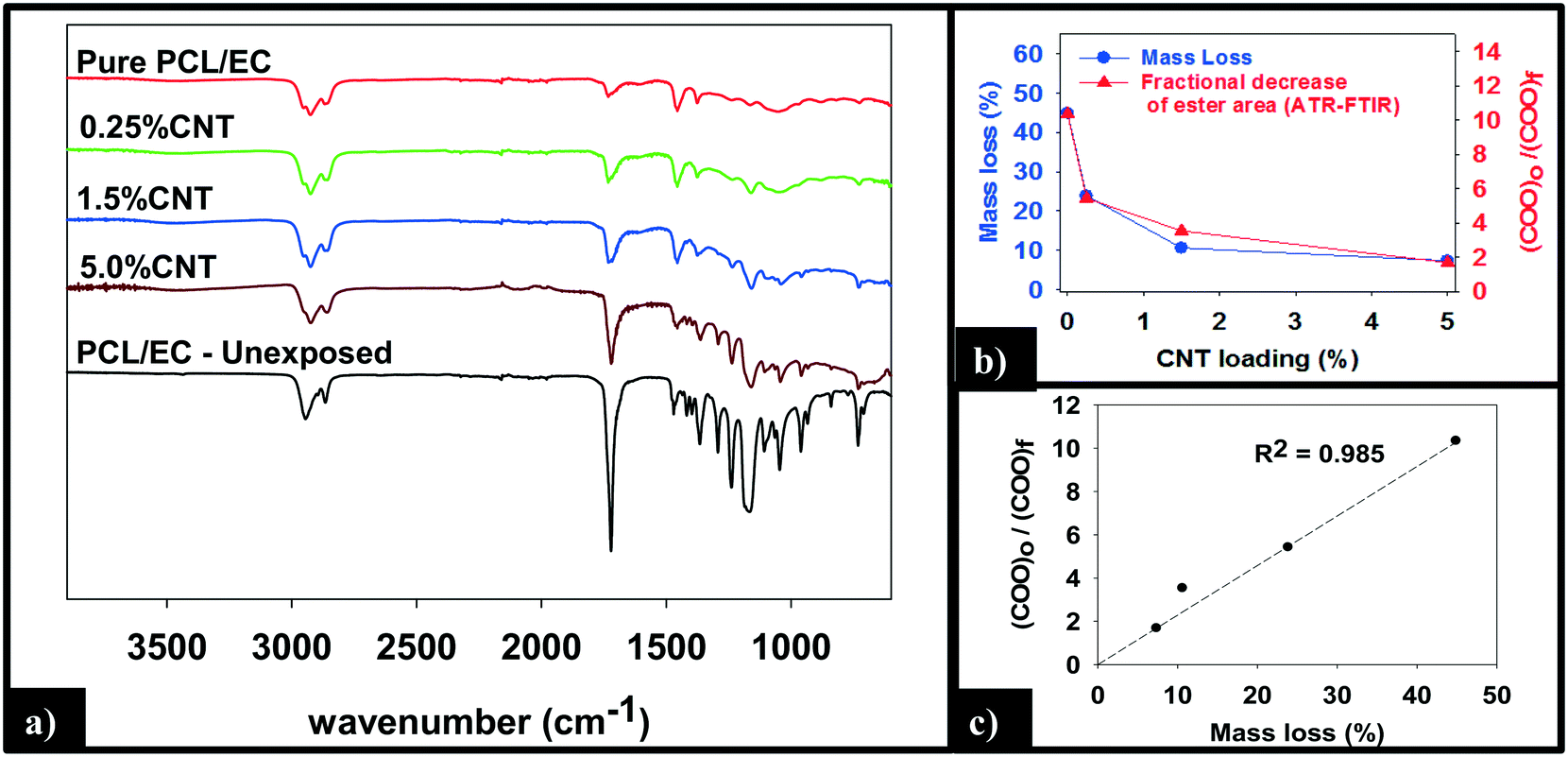 | ||
| Fig. 5 (a) ATR-FTIR analysis of the nanocomposites shows a reduction in intensity of the ester peak (1720 cm−1) following 125 days irradiation. (b) Following 125 days of exposure, the final mass loss and fractional decrease of ester band area show a similar dependence on CNT loading. (c) Mass loss increases linearly with the fractional decrease in ester band area. | ||
Fig. 5b and c quantify the relationships between CNT loading, mass loss, and the depletion of the ester groups in the near surface region, the latter measured by ATR-FTIR. We observe that the mass loss of PNCs and the fractional decrease in the ester group's concentration in the near surface region, following 125 days of UVC exposure, both decrease with increasing CNT loading43 (Fig. 5b). Moreover, Fig. 5c demonstrates that there is a linear relationship (R2 = 0.98) between the fractional decrease in the ester group's near surface concentration and the extent of polymer mass loss.
Carbon nanotube release and detection
Fig. 6a is a representative TEM image of the SWCNTs used in this study. Residual metal nanoparticles, left over from the fabrication process, are clearly observed attached to the SWCNTs, with most appearing to be fully encapsulated by carbonaceous material. The manufacturer reports a total metal content of <30%, by mass. In all images captured, no free metal nanoparticles were observed. The nanoparticles are roughly spherical with an average diameter of 15–20 nm, although the number of nanoparticles per nanotube is very heterogeneous.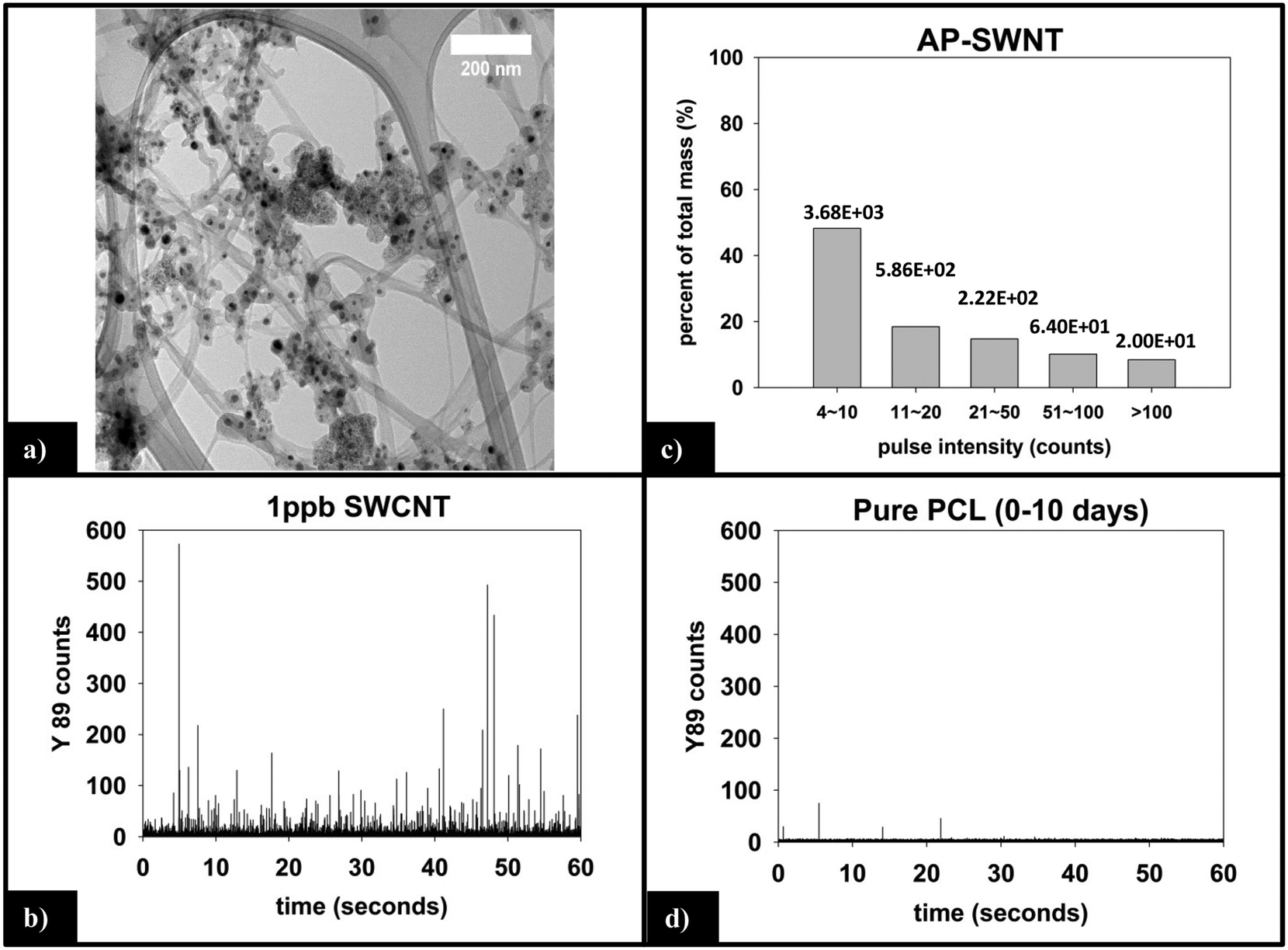 | ||
| Fig. 6 (a) TEM of SWCNTs shows embedded residual yttrium and nickel catalyst nanoparticles. (b) sp-ICP-MS analysis of 1 ppb SWCNT suspension prepared with 2% SDC. (c) Y mass distribution for SWCNT suspension, with the corresponding number of particles detected noted above each bin. (d) sp-ICP-MS of supernatant generated by 10 day irradiation of pure PCL is absent of 89Y response (i.e. no CNT release or interference from the polymer matrix). | ||
000 total dwell times recorded, 98.5% are 0–3 counts. Only 10 total pulses are measured to be greater than 10 counts (false positive detections), are not of statistical relevance, and attributed to trace amounts of yttrium found in the sample or solution.
(i) Measured vs. predicted SWCNT mass release. Integration of the 89Y signal allows the total mass of SWCNTs released to be determined using the calibration curve obtained with the SWCNT dispersions (Fig. SI 4†). Fig. 7a displays the cumulative SWCNT mass released from each nanocomposite into solution during different periods of photolysis. Fig. 7b depicts the predicted cumulative CNT release mass assuming that as polymer mass is lost, CNTs release proportionally with the polymer based on the initial CNT loading. A summary of the measured and predicted/proportional mass loss of SWCNTs from 0.5%, 1.5% and 5% CNTs during different stages of photolysis is provided in Table 1. We note that the error depicted in Fig. 7a and shown in Table 1 for the 0.25% CNT and 5.0% CNT samples was calculated by the summation of the samples' independent uncertainties.44 In separate control experiments the extent of CNT release from nanocomposite samples that were not irradiated but were submerged in water under the same conditions used for the photolysis experiments was assessed. For all nanocomposite samples, submersion resulted in <0.1 μg CNT release. Consequently, all of the SWCNT released in this study are a result of PNC photodegredation.
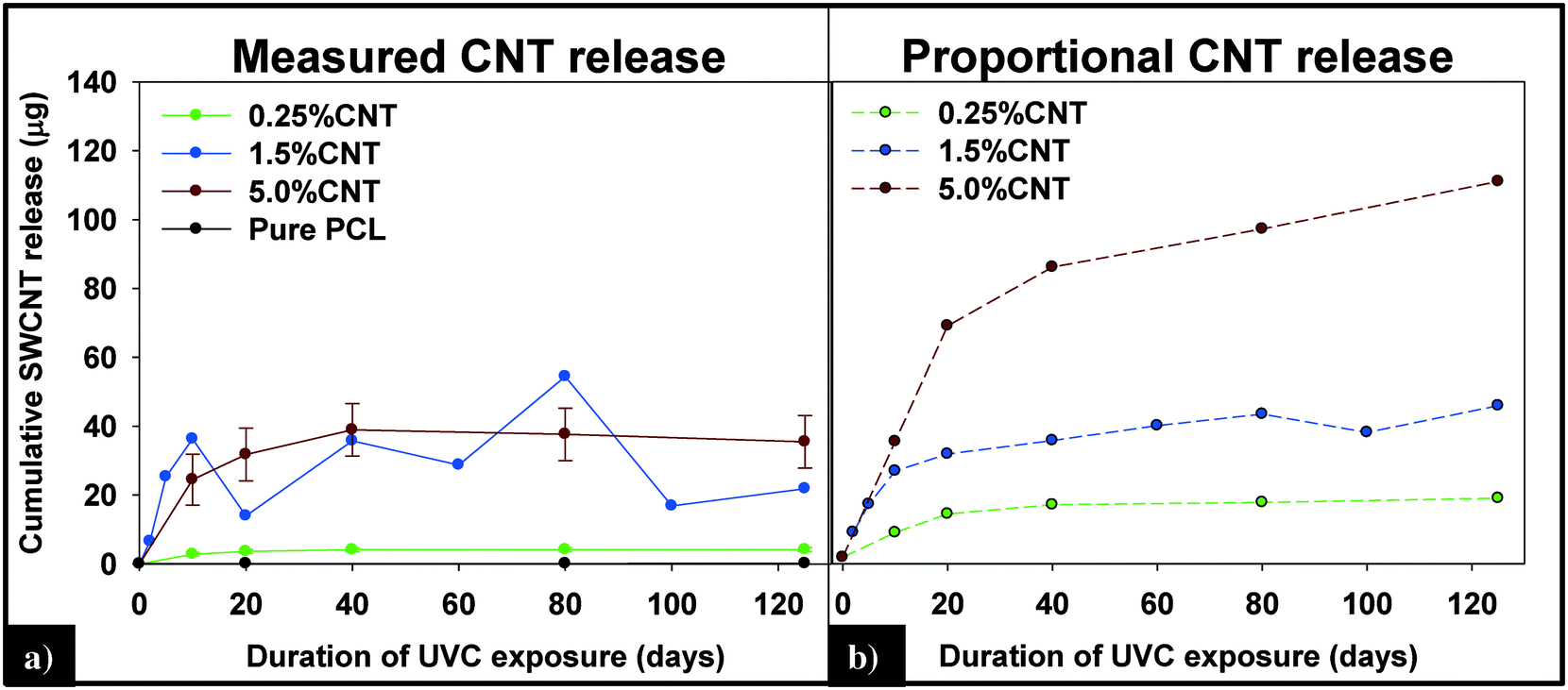 | ||
| Fig. 7 (A) Cumulative CNT release plateaus within the first 40 days of exposure and is less than the predicted release of CNTs (B), assuming CNTs are lost at the same rate as the polymer. | ||
| 0–20 days | 0–125 days | |||
|---|---|---|---|---|
| Measured | Predicted | Measured | Predicted | |
| 0.25% CNT | 3.66 (±0.56) | 13.76 | 4.19 (±0.56) | 18.63 |
| 1.5% CNT | 13.97 | 32.55 | 21.88 | 47.78 |
| 5.0% CNT | 31.79 (±7.63) | 73 | 35.46 (±7.63) | 118.5 |
From knowledge of the PNC mass, the CNT loading and the SWCNT mass released we can also determine the fraction of SWCNTs released at different stages of the photodegradation process. This analysis reveals that, for the same period of photolysis, the fraction of SWCNTs released decreases as the CNT loading increases. Thus, the 0.25% CNT sample lost 3.9% of its total CNTs during the first ten days of exposure, 1.1% from days 11–20, and 0.7% of initial CNTs during the last 21–125 days of exposure. In contrast, during the same intervals of irradiation, the 5.0% CNT sample lost 1.6%, 0.5%, and 0.4% of the CNTs initially present, respectively.
(ii) Functional form of SWNCT released. Fig. 8 displays the raw and processed sp-ICP-MS data collected for the 5.0% CNT nanocomposite during the initial (0–10 days), intermediate (11–20 days), and final (21–125 days) irradiation intervals. The data are binned by increasing 89Y pulse intensity (i.e. particle mass) and plotted as the fractional mass distribution of the SWCNTs detected in those intervals. During the first 10 days of irradiation (Fig. 8a), the largest number of yttrium pulses, as well as the most intense pulses, are detected. During the subsequent periods of irradiation, the pulse number and intensity steadily decrease, as shown in Fig. 8b and c. It should be noted that the lack of any increase in the background signal (0–4 counts) during PNC-CNT photolysis indicates that 89Y is not being released from the CNT-polymer matrix as ions or as individual 20 nm yttrium NPs (both of which would increase the background signal rather than be detected as a discrete pulse), but rather as multiple NPs still attached to the SWCNTs.
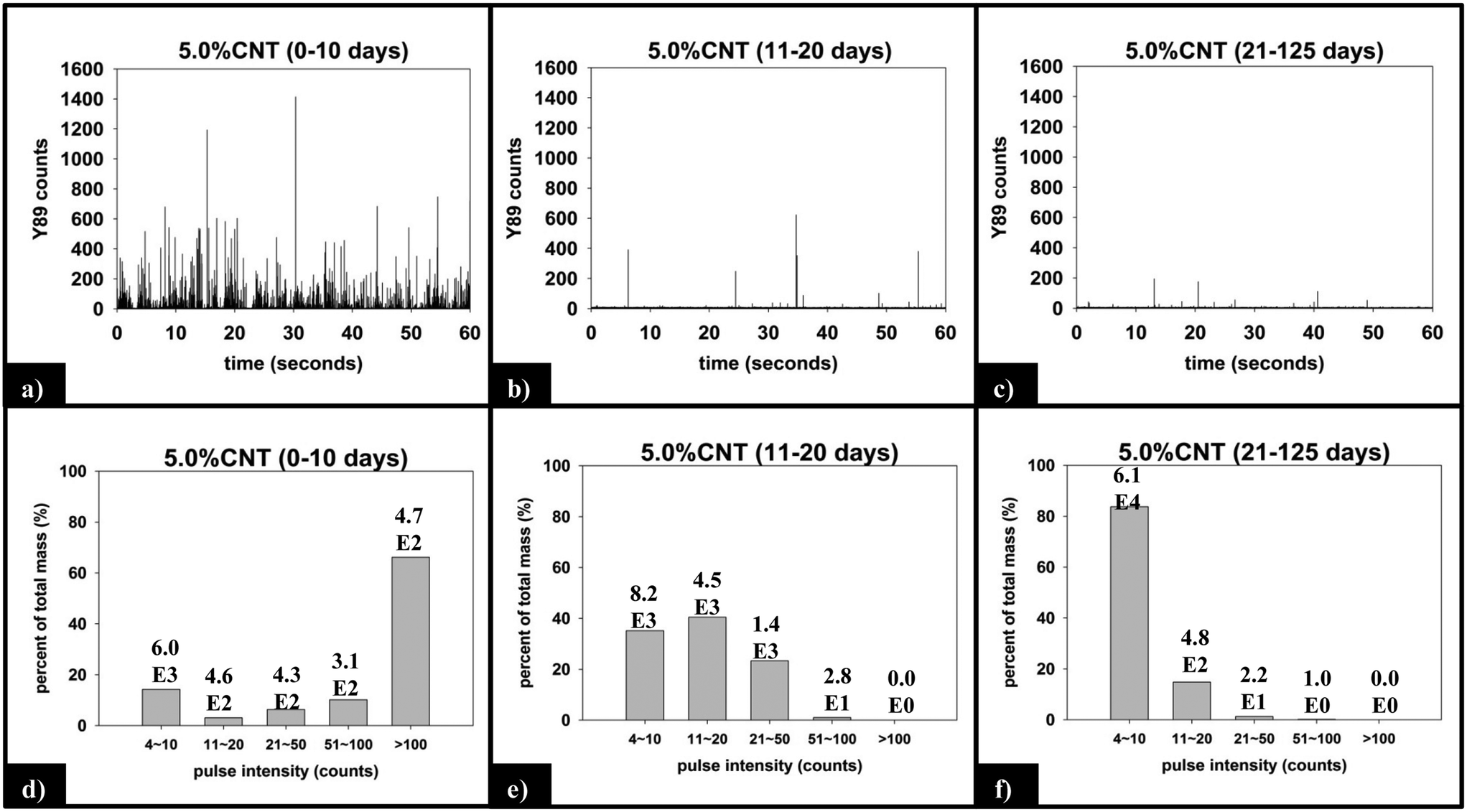 | ||
| Fig. 8 (a)–(c) Raw sp-ICP-MS data collected for 5.0% CNT nanocomposites with increasing duration of UVC exposure. (d)–(f) Y mass distribution for each period of exposure, with the corresponding number of particles detected noted above each bin. | ||
Fig. 9 displays the sp-ICP-MS data collected for the 0.25% CNT nanocomposite during the same initial (0–10 days), intermediate (11–20 days), and final (21–125 days) irradiation intervals described above for the 5.0% CNT samples, as well as the mass distribution of the SWCNTs detected from those intervals. Unlike the 5.0% CNT sample, in which a large number of particle detection events (pulses) are detected during each period of exposure, the 0.25% CNT samples released substantially less particles: during the initial, intermediate, final irradiation intervals; only 1021, 653, and 632 total particles are detected, respectively.
 | ||
| Fig. 9 (a)–(c) Raw sp-ICP-MS data collected for 0.25% CNT nanocomposites with increasing duration of UVC exposure. (d)–(f) Y mass distribution for each period of exposure, with the corresponding number of particles detected noted above. | ||
4 Discussion
Control studies
Comparing power data collected by the UV-B Monitoring and Research Program,35 we can estimate that at 300 nm our photochemical reactor accelerates photodegradation by a factor of approximately 100. Since our experimental data indicates that the use of 254 nm rather than 300 nm light accelerates photodegradation by an addition factor of 10, we can estimate an accelerated photodegradation rate on the order of 103 as compared to “natural” outdoor conditions.
PCL photodegradation in the absence of CNTs
Poly-ε-caprolactone (PCL) [–(CH2)5COO–]n was selected as the polymer matrix because of its ability to form well dispersed CNT composites and its relative ease of photodegradation, the latter a consequence of its molecular composition, specifically the presence of photolabile ester groups. Additionally, PCL's ester group provides characteristic spectral features in both ATR-FTIR and XPS that allow for the extent of photodegradation to be quantified spectroscopically.The depletion of the ester groups in the near surface region is even more convincingly demonstrated by the ATR-FTIR spectra shown in Fig. 5a. This shows that following 125 days of irradiation the carbonyl stretching band associated with the ester group in PCL, along with peaks in the fingerprint region below 1500 cm−1, are both greatly diminished in intensity. In contrast, the C–H stretching region stretching intensity remains nearly unchanged. Close inspection of C–H stretching region (Fig. SI 10†) suggests the formation of alkene bonds following UVC exposure, an expected product of the Norrish Type II reaction.46 However, the change in peak profile is subtle and definitive absorption band assignment is not possible. However, inspection of the difference spectra of each photodegraded sample (Fig. SI 10†), with respect to the unexposed PCL/EC spectrum makes the changes in the C–H stretching region more apparent and provides further evidence of alkene bond formation in the surface region following irradiation. In addition to mass loss and changes to the chemical bonding in the near surface region (<2 μm), SEM imaging shows that photodegradation transforms the initially smooth PCL surface (Fig. SI 6A†) into one with a high concentration of spherical structures (Fig. SI 6B†).
The combination of mass loss, XPS, and ATR-IR data supports the idea that the photodegradation of PCL/EC occurs through direct excitation and bond cleavage within the ester chromophore, leading initially to a rapid mass loss. As the photodegradation process proceeds, however, the ester group concentration in the near surface region decreases and a residual hydrocarbon rich layer builds up at the interface as the hydrophilic carboxylic acid capped fragments are released into solution; we believe that the spherical nodules observed by SEM are a manifestation of these residual hydrocarbon structures. The thickness of this hydrocarbon layer increases as the photolysis continues, leading to in a decrease in the concentration of ester groups within the near surface region that can be accessed/photolyzed by the incident irradiation. Ultimately, the hydrocarbon layer reaches a thickness that is comparable to the penetration depth of the light and at this stage, the photodegradation process and mass loss stop. This process of rapid initial polymer/PCL mass loss followed by the accumulation of a hydrocarbon layer is depicted pictorially in Scheme 1.
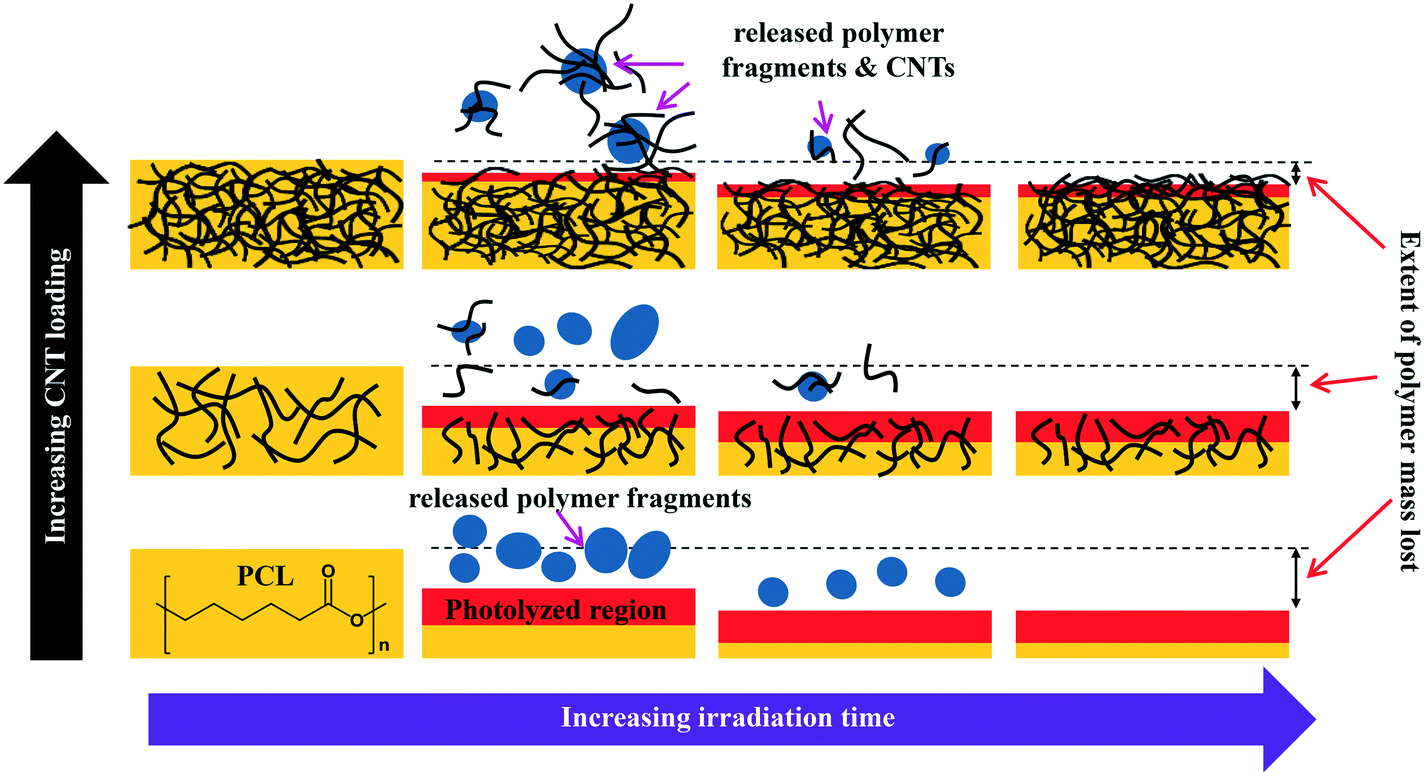 | ||
| Scheme 1 Depiction of how both the interface of polymer-CNT nanocomposites and the nature of release fragments evolve as photodegradation proceeds, shown as a function of CNT loading. | ||
Effect of CNT incorporation on photodegradation
To examine the impact of SWCNTs on the photodegradation process, SWCNT-PCL nanocomposites (PNCs) were produced with a range of SWCNT loadings (0.25%, 1.5%, and 5.0%, w/w). Analysis of the mass loss kinetics as well as the XPS and ATR-FTIR data (Fig. 4 and 5) reveals that at least qualitatively, similar changes occur to the SWCNT-PCL and PCL samples during photolysis. For example, XPS analysis of the 1.5% CNT sample (Fig. 4) reveals that there is a rapid reduction in the ester groups present at the surface during the first 20 days of exposure (Fig. 4a). Thereafter, the concentration of ester groups in the near surface region (<5 nm) has been depleted and there is little subsequent change in surface composition (Fig. 4b) and little to no additional mass loss (Fig. 2). A similar depletion of ester groups at the surface within the first 20 days was observed for all nanocomposite samples (Fig. SI 9†), although XPS data indicates that the 5.0% CNT nanocomposite retained ester groups in the near surface (topmost 5 nm or so) layers for at least 40 days of irradiation (Fig. SI 9D†). Thus, the experimental data points towards a photodegradation process that still proceeds through a Norrish Type II pathway, regardless of the presence of CNTs, mediated by the photolabile ester groups in the near surface region.Although the photodegradation mechanism is insensitive to the presence of CNTs, the extent of photodegradation is highly sensitive to the presence of CNTs. As seen in Fig. 2, the addition of just 0.25% CNT reduces the total mass loss of the nanocomposite to half that of the CNT-free polymer. Similarly, for higher CNT loadings of 1.5% CNT and 5.0% CNT, the extent of polymer mass loss following 125 days of exposure is limited to just 10.5% and 7.3% of the initial nanocomposite mass, respectively. The ATR-FTIR spectra in Fig. 5a show that after 125 days of photolysis, where photodegradation has ceased for all of the samples, the concentration of PCL in the near surface region (<2 μm), as observed by PCLs' characteristic ester stretch at 1720 cm−1, increases systematically as the CNT loading increases. This suggests that the reason for the reduction in mass loss observed as CNT loading increases is because CNTs reduce the effective depth of UV penetration and consequently the extent of polymer degradation. This is supported by the C(1s) data (Fig. SI 9D†), which shows that for the highest loading nanocomposite, 5.0% CNT, there is evidence of ester groups still being present in the uppermost surface layers (<5 nm depth) after 20 days of irradiation. In contrast, XPS analysis shows no evidence of ester groups being present for any of the other lower CNT loadings or the pure PCL after similar periods of irradiation.
Our experimental data also supports the idea that the metal nanoparticles associated with the CNTs do not alter the photodegradation mechanism (Norrish Type II reaction). Thus, the diminishment of spectral features in both XPS and ATR-FTIR indicate that the introduction of CNTs (and associated metal catalysts) reduce the depth of photolysis. However, there is no spectroscopic evidence of new features emerging as a result of a secondary degradation process driven by the presence of the metal catalysts. Additionally, TEM imaging reveals that the metal nanoparticles are encapsulated by carbonaceous material (Fig. 6a), preventing direct contact of the metal nanoparticles to either the polymer matrix or water/dissolved oxygen molecules that could generate reactive oxygen species. Thus, any impact the metal nanoparticles may have on photodegradation would seem to be limited to light attenuation and as such this would be spectroscopically indistinguishable from the impact of the CNTs.
A more detailed analysis of the relationship between mass loss and polymer degradation (Fig. 5b) reveals that after 125 days of UVC exposure, the measured mass loss and the fractional decrease in carbonyl peak area both exhibit a very similar decrease as a function of the CNT loading. Moreover, Fig. 5c reveals that a liner correlation exists between mass loss and the fractional decrease of carbonyl area, both measured after 125 days. The discovery of such a correlation between these two independently determined quantities provides strong support for the idea that the attenuation of mass loss observed upon CNT addition is a consequence of a “shading effect” caused by the CNTs absorbing UVC, reducing the effective penetration depth of the photolyzing radiation, and thus the extent of polymer mass loss. The idea that the CNTs are acting as photon absorbers/scatters is also supported by the observation that mass loss decreases exponentially with increased CNT loading/concentration, consistent with the Beer–Lambert law for light attenuation in the presence of a chromophore.
CNT release vs. retention
The detection and quantification of SWCNTs released during photolysis is accomplished with sp-ICP-MS, using embedded Y nanoparticles (Fig. SI 2A and B†), residual from their use as a catalyst during SWCNT production, as a proxy for CNTs.29,30 The integrated 89Y signal from well dispersed SWCNTs suspensions is proportional to the SWCNT mass concentration (Fig. SI 3†). This linear relationship between 89Y signal and SWCNT concentration allows us to determine mass of SWCNTs released from the nanocomposites at different stages of photolysis. In addition, analysis of the distribution of 89Y pulse intensities allows us to determine the form of released SWCNTs (individual SWCNTs vs. aggregates).Fig. 7a shows the total mass of CNT released from a CNT-PNC generally increases with the initial CNT loading, with the 5.0% CNT samples releasing over eight times the mass of CNTs as compared to the 0.25% CNT samples following 125 days of irradiation. However, although CNTs are released as the polymer nanocomposites photodegrade, a comparison of Fig. 7a and b indicates that for the 5.0% CNT and 0.25% CNT samples, CNTs are lost at a significantly lower rate than the polymer. This observation is also summarized in Table 1, where it is quantitatively clear that compared to mass of polymer lost the CNTs are releasing to lesser extent than predicted, based on their initial loading. For the 5.0% CNT this preferential CNT retention is clearly evinced by SEM images (Fig. 3), which reveal that a dense CNT mat has formed at the surface after 20 days of photolysis while no CNTs were observed at the surface prior to irradiation. This observation is similar to CNT surface aggregation observed in previous studies.10,21,38–40 The absence of visible CNTs on the surface of the 0.25% CNT and 1.5% CNT samples following photolysis is attributed to both their lower initial CNT concentration and the accumulation of hydrophobic capped polymer fragments, which occurs as a consequence of PCL photodegradation (Fig. SI 6B and C†).
The preferential retention of SWCNTs during photodegradation is attributed to the SWCNTs' high aspect ratio (1.4–1.6 nm in diameter as determined by Raman spectra shown in Fig. SI 3† and up to 3 μm long, as reported by the manufacturer). This facilitates an anchoring effect because many SWCNTs in the near surface region that experiences photolysis will still have some fraction embedded within the bulk of the nanocomposites. Additionally, CNT entanglement of anchored CNTs to surface confined CNTs will further restrain CNTs to the surface of the nanocomposite. The net result is a preferential retention of SWCNTs in the interfacial regions, as shown in Scheme 1.
The kinetics of CNT mass loss from the CNT-PNCs qualitatively mirrors the mass loss kinetics for the polymer (comparing Fig. 7a and 2, respectively). Thus, following the first 20 days of exposure, as polymer mass loss begins to plateau (Fig. 2), the rate of CNT release is abated as well. This similarity in behavior supports the idea that degradation of the surrounding polymer matrix is the principal means of CNT release from the PNCs as seen for other CNT-polymer nanocomposites.9 For the 5.0% CNT sample, Fig. 7a shows that once the CNT mat has formed there is almost no further measureable CNT mass loss, highlighting the photostability of this structure and also that the metal nanoparticles remain attached to the CNTs.
Form of released CNTs
The integrated 89Y signal in sp-ICP-MS reflects the mass loss of CNTs that occurs during polymer photodegradation. As a complement to this information, analysis of 89Y pulse distribution can provide insight into the form of released CNT (individual CNTs vs. aggregates of CNT contained in polymer fragments): thus, pulse binning of sp-ICP-MS data obtained from reference samples of 1 ppb SWCNT suspensions (Fig. 6c) shows that 48% of the measured mass is from CNTs yielding 4–10 89Y counts. The contribution to total measured mass quickly drops off with increasing pulse bin intensity, with no other pulse bin contributing more than 20% of total measured mass. Furthermore, only 20 CNTs (of 4572 total particles detected) were found to yield 89Y pulses with intensities greater than 100 89Y counts. This analysis of well dispersed SWCNTs provides a “baseline” for the 89Y pulse intensity distribution and establishes that individual SWCNTs are most likely to yield a signal of 4–10 89Y counts when detected with sp-ICP-MS. In contrast, large aggregates of CNTs will produce much larger pulse heights as they will contain many more Y nanoparticles.Examination of the measured 89Y pulses (Fig. 8a) and 89Y pulse distribution (Fig. 8d) from the 5.0% CNT during the initial 10 day period of photolysis and SWCNT release is in stark contrast to the pulse distribution found from individually suspended SWCNTs (Fig. 6c). Of 7668 total particles detected, 474 particles were detected with a 89Y pulse intensity greater than 100 89Y counts; of those particles, 206 particles yielded a pulse intensity greater than 200 89Y counts and 32 particles were detected with greater than 500 89Y counts (data not explicitly depicted within figure). Additionally, the 474 particles with pulse intensities greater than 100 89Y counts account for 66% of the total measured CNT mass released during the first 10 day period of exposure, the latter measured by the integrated 89Y signal. From the contrasting pulse intensity distributions, it is suggested that the 5.0% CNT sample released large CNT-containing fragments during the initial stage of photolysis, when mass loss (Fig. 2) and photolysis (Fig. 4 and SI 9†) are most rapid. Comparing the dominant pulse intensity of well dispersed SWCNTs (4–10 89Y counts) to the dominant pulse intensity of released material (100–500 89Y counts) suggests these released fragments contained anywhere from about 10–125 CNTs. We believe these CNTs are most likely being released embedded in polymer fragments rather than as CNT bundles because, prior to sp-ICP-MS analysis, all samples are spiked with dilute surfactant and sonicated for 30 minutes, in the same way that the suspensions of CNT powder are prepared. Such a treatment would therefore be expected to debundle CNT aggregates but would not loosen CNTs embedded in polymer fragments.
During the next 10 days of polymer mass loss and degradation, CNT release from the 5.0% CNT nanocomposite continues. However as seen in Fig. 7a the overall mass of CNTs released during this period of exposure (7.30 ± 1.79 μg) is significantly smaller than released initially (24.48 ± 7.42 μg). Additionally, we observe the form of released SWCNTs change during this intermediate period of irradiation. The overall pulse data in Fig. 8b shows particles with fewer CNTs/particle being released as compared to the first 10 days of irradiation (Fig. 8a). Quantitatively (Fig. 8e), this is reflected by the pulse intensity now being centered on particles of 11–20 89Y counts (contributing to 40% of the total measured mass), with very similar contributions to the total mass measured from particles of 4–10 89Y counts and 21–50 89Y counts. From this, we deduce that fewer CNTs are embedded in polymer fragments releasing from the surface than before. This may simply be a reflection of smaller polymer fragments being released. Alternatively, we note that the relative rate of CNT to polymer release decreases in this time interval (compare Fig. 7a and b) and this change in pulse intensity distribution may reflect a greater degree of CNT retention due to increased CNT concentration and connectivity in the near surface region. During the final period of exposure, from 21–125 days, where the CNT mat has formed, Fig. 7a reveals that there is very little CNT loss (3.7 μg), despite the continued loss of a small but measureable amount of polymer (Fig. 2). Additionally, we see the pulse distribution is largely made up of particles of 4–10 89Y counts, accounting for 92% of the total mass measured (Fig. 8f). This suggests that any released CNTs are in the form of individual CNT particles, although it is now not possible to determine if they are still embedded in polymeric fragments. The progression of CNT released-form as described above for PNCs with high initial CNT concentrations, as a function of duration of photolysis, is shown in Scheme 1.
For the 0.25% CNT sample, the 89Y pulse distribution during the first 10 days of photolysis (Fig. 9d) yields pulses with predominantly 4–10 89Y counts, in contrast to the 5.0% CNT sample. In fact, we observe that 4–10 89Y counts is the dominant pulse distribution of released material for all three periods of photolysis and release (Fig. 9d–f). Since the experimental evidence indicates that the nanocomposites degrade through the same mechanism, irrespective of CNT loading, we assert that the fragments of polymer released will likely be of comparable physical dimensions. From this, it follows that since the 0.25% CNT sample possesses 1/20th the initial CNT concentration as the 5.0% CNT sample, there would be on average 1/20th the number of CNTs released in each polymer fragment. As a result, during the first 10 days of photolysis, when SWCNT mass release is greatest, this would shift the pulse distribution to one where 89Y pulses in the 4–10 range dominate, as we observe experimentally in Fig. 9d. Thus, the 89Y pulse distribution and its invariance to irradiation time is simply a reflection of the fewer SWCNTs present in the 0.25% CNT sample when compared to the 5.0% CNT sample. The contrast in the behavior of the 0.25% CNT and 5.0% CNT sample does, however, demonstrate that nanocomposites with higher CNT loadings will likely exhibit greater variance in the form and number of released CNTs as they photodegrade, as shown in Scheme 1.
Although the detailed nature of polymer photodegradation processes are often complex and system specific, they are typically initiated by photon absorption.47 Consequently, our findings suggest that the presence of both SWCNTs and MWCNTs will decrease the extent of polymer photodegradation in other polymer CNT nanocomposites by virtue of their ability to absorb incoming light over a wide range of wavelengths.31 In addition to the decrease in polymer mass loss, another important consequence of this “shielding effect” is that the total number of CNTs released during PNC photodegradation will not vary in direct proportion to the CNT loading. Thus, although the absolute CNT mass loss will increase with CNT loading, the fraction of CNTs released from a CNT-PNC will decrease as the CNT loading increases as is seen Table 1. We postulate that similar “shading” effects will be operative for other nanoparticles/polymer additives such as fullerenes, graphene and carbon fibers.48 We expect that the mechanism by which CNTs mitigate polymer photodegradation (through their shading effect), including the functional dependence on CNT loading, to be conserved amongst different polymer/CNT systems. The absolute magnitude of the retardation effect exerted by CNTs, however will be system specific due to differences in both the wavelength of light responsible for polymer photodegradation and a given polymer matrix's overall susceptibility to both photodegradation and indirect effects such as increased temperature of black composites under irradiation lamps. We also expect that polymer fragments containing multiple CNTs will be prevalent during the initial stages of photodegradation, particularly for PNCs with higher CNT loadings. Additionally, photodegradation and release will also be regulated by exposed surface area. Consequently, when small PNC fragments, such as those released during an abrasion process are weathered, they will release more CNTs as compared to the original material of the same volume, by virtue of their increased surface area. However, CNTs contained within such fragments will still inhibit the extent of photodegradation by the same mechanism.
5 Conclusions
To better understand the effect that CNT inclusion has on polymer photodegradation, we photodegraded CNT-PCL polymer nanocomposites that contained a range of initial CNT loadings (0–5% w/w) under accelerated photodegradation conditions, monitoring changes to the polymer and correlating these changes to the extent, rate, and form of released CNTs. Through spectroscopic characterization (ATR-FTIR and XPS) and mass loss measurements of the CNT-PNCs, we determined that PCL's principle route of photolysis, cleavage of ester groups in the polymer backbone, occurs regardless of the presence or absence of CNTs. However, while the mechanism of photodegradation is invariant to the presence of CNTs, the extent of polymer photodegradation systematically decreases with increasing initial CNT loading. We have attributed this inhibition of polymer degradation to be a consequence of the CNTs' light absorbing/scatting properties. The kinetics of CNT release during polymer photodegradation mirrors polymer mass loss, although CNTs are preferentially retained in the CNT-PNC matrix. For CNT-PNCs of sufficiently high initial CNT loading (5%), sp-ICP-MS revealed that multiple CNTs are principally released embedded in photodegraded polymer fragments. As the rate of CNT-PNC mass loss decreases, the rate and magnitude of CNT release decreases as well, due to both increased attenuation of light by CNTs accumulating in the near surface region and the simultaneous accumulation of a hydrocarbon layer resulting from PCL photolysis. Our results indicate that the fractional release of CNTs as well as the extent of polymer photodegradation will decrease as the CNT loading increases.Acknowledgements
This work was supported by NSF grant number #1336168 and EPA grant no. RD83558001. The authors would also like to thank the Johns Hopkins Department of Chemistry and Department of Materials Science and Engineering for providing the necessary instruments for sample characterization – XPS, SEM, ATR-FTIR. We would also like to thank Ken Livi for providing TEM imaging.References
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535 CrossRef CAS PubMed.
- A. Kausar, I. Rafique and B. Muhammad, Polym.-Plast. Technol. Eng., 2016, 55, 1167–1191 CrossRef CAS.
- T. A. J. Kuhlbusch, C. Asbach, H. Fissan, D. Göhler and M. Stintz, Part. Fibre Toxicol., 2011, 8, 22 CrossRef PubMed.
- J. Muller, F. Huaux, N. Moreau, P. Misson, J.-F. Heilier, M. Delos, M. Arras, A. Fonseca, J. B. Nagy and D. Lison, Toxicol. Appl. Pharmacol., 2005, 207, 221–231 CrossRef CAS PubMed.
- K. Fent, in Nanoparticles in the Water Cycle: Properties, Analysis and Environmental Relevance, ed. H. F. Frimmel and R. Niessner, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010, pp. 183–205, DOI:10.1007/978-3-642-10318-6_11.
- K. Kostarelos, Nat. Biotechnol., 2008, 26, 774–776 CrossRef CAS PubMed.
- J. R. Peralta-Videa, L. Zhao, M. L. Lopez-Moreno, G. de la Rosa, J. Hong and J. L. Gardea-Torresdey, J. Hazard. Mater., 2011, 186, 1–15 CrossRef CAS PubMed.
- N. Tinh, W. Wendel and S. Lipiin, in Safety of Nanomaterials along Their Lifecycle, CRC Press, 2014, pp. 315–334, DOI:10.1201/b17774-18.
- T. V. Duncan, ACS Appl. Mater. Interfaces, 2015, 7, 20–39 CAS.
- T. Nguyen, B. Pellegrin, C. Bernard, X. Gu, J. M. Gorham, P. Stutzman, D. Stanley, A. Shapiro, E. Byrd, R. Hettenhouser and J. Chin, J. Phys.: Conf. Ser., 2011, 304, 012060 CrossRef.
- C. Kingston, R. Zepp, A. Andrady, D. Boverhof, R. Fehir, D. Hawkins, J. Roberts, P. Sayre, B. Shelton, Y. Sultan, V. Vejins and W. Wohlleben, Carbon, 2014, 68, 33–57 CrossRef CAS.
- J. E. Chin, N. Embree, J. Garver, B. Dickens, T. Finn and J. W. Martin, Rev. Sci. Instrum., 2004, 75, 8 CrossRef.
- E. J. Petersen, T. Lam, J. M. Gorham, K. C. Scott, C. J. Long, D. Stanley, R. Sharma, J. Alexander Liddle, B. Pellegrin and T. Nguyen, Carbon, 2014, 69, 194–205 CrossRef CAS.
- S. Hirth, L. Cena, G. Cox, Ž. Tomović, T. Peters and W. Wohlleben, J. Nanopart. Res., 2013, 15, 1–15 CrossRef PubMed.
- G. Vilar, E. Fernández-Rosas, V. Puntes, V. Jamier, L. Aubouy and S. Vázquez-Campos, J. Phys.: Conf. Ser., 2013, 429, 012044 CrossRef.
- K. V. Pillai, P. J. Gray, C.-C. Tien, R. Bleher, L.-P. Sung and T. V. Duncan, Environ. Sci.: Nano, 2016, 3, 657–669 RSC.
- W. Wohlleben, M. W. Meier, S. Vogel, R. Landsiedel, G. Cox, S. Hirth and Z. Tomovic, Nanoscale, 2013, 5, 369–380 RSC.
- S. Rhiem, A.-K. Barthel, A. Meyer-Plath, M. P. Hennig, V. Wachtendorf, H. Sturm, A. Schäffer and H. M. Maes, Environ. Pollut., 2016, 215, 356–365 CrossRef CAS PubMed.
- L. Schlagenhauf, F. Nüesch and J. Wang, Fibers, 2014, 2, 108–127 CrossRef CAS.
- G. Vilar, E. Fernández-Rosas, V. Puntes, V. Jamier, L. Aubouy and S. Vázquez-Campos, J. Phys.: Conf. Ser., 2013, 429, 012044 CrossRef.
- W. Wohlleben, S. Brill, M. W. Meier, M. Mertler, G. Cox, S. Hirth, B. von Vacano, V. Strauss, S. Treumann, K. Wiench, L. Ma-Hock and R. Landsiedel, Small, 2011, 7, 2384–2395 CrossRef CAS PubMed.
- W. Wohlleben and N. Neubauer, NanoImpact, 2016, 1, 39–45 CrossRef.
- E. J. Petersen, D. X. Flores-Cervantes, T. D. Bucheli, L. C. C. Elliott, J. A. Fagan, A. Gogos, S. Hanna, R. Kägi, E. Mansfield, A. R. M. Bustos, D. L. Plata, V. Reipa, P. Westerhoff and M. R. Winchester, Environ. Sci. Technol., 2016, 50, 4587–4605 CrossRef CAS PubMed.
- S. Attal, R. Thiruvengadathan and O. Regev, Anal. Chem., 2006, 78, 8098–8104 CrossRef CAS PubMed.
- A. E. Porter, M. Gass, K. Muller, J. N. Skepper, P. A. Midgley and M. Welland, Nat. Nanotechnol., 2007, 2, 713–717 CrossRef CAS PubMed.
- A. Schierz, A. N. Parks, K. M. Washburn, G. T. Chandler and P. L. Ferguson, Environ. Sci. Technol., 2012, 46, 12262–12271 CrossRef CAS PubMed.
- A. Schierz, B. Espinasse, M. R. Wiesner, J. H. Bisesi, T. Sabo-Attwood and P. L. Ferguson, Environ. Sci.: Nano, 2014, 1, 574–583 RSC.
- L. Schlagenhauf, T. Buerki-Thurnherr, Y.-Y. Kuo, A. Wichser, F. Nüesch, P. Wick and J. Wang, Environ. Sci. Technol., 2015, 49, 10616–10623 CrossRef CAS PubMed.
- R. B. Reed, D. G. Goodwin, K. L. Marsh, S. S. Capracotta, C. P. Higgins, D. H. Fairbrother and J. F. Ranville, Environ. Sci.: Processes Impacts, 2013, 15, 204–213 CAS.
- J. Wang, R. S. Lankone, R. B. Reed, D. H. Fairbrother and J. F. Ranville, NanoImpact, 2016, 1, 65–72 CrossRef.
- G. A. Rance, D. H. Marsh, R. J. Nicholas and A. N. Khlobystov, Chem. Phys. Lett., 2010, 493, 19–23 CrossRef CAS.
- S. Bandow, S. Asaka, Y. Saito, A. M. Rao, L. Grigorian, E. Richter and P. C. Eklund, Phys. Rev. Lett., 1998, 80, 3779–3782 CrossRef CAS.
- N. G. Sahoo, S. Rana, J. W. Cho, L. Li and S. H. Chan, Prog. Polym. Sci., 2010, 35, 837–867 CrossRef CAS.
- C. G. Hatchard and C. A. Parker, Proc. R. Soc. London, Ser. A, 1956, 235, 518 CrossRef CAS.
- UV-B Monitoring and Research Program, http://uvb.nrel.colostate.edu/UVB/index.jsf, (accessed Feb. 10th, 2017).
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- C. Bernard, T. Nguyen, B. Pellegrin, R. D. Holbrook, M. Zhao and J. Chin, J. Phys.: Conf. Ser., 2011, 304, 012063 CrossRef.
- T. Nguyen, B. Pellegrin, L. Mermet, A. Shapiro, X. Gu and J. Chin, Nanotechnology, 2009, 1, 90–93 CAS.
- J. Ging, R. Tejerina-Anton, G. Ramakrishnan, M. Nielsen, K. Murphy, J. M. Gorham, T. Nguyen and A. Orlov, Sci. Total Environ., 2014, 473–474, 9–19 CrossRef CAS PubMed.
- S. Hirth, L. Cena, G. Cox, Z. Tomovic, T. Peters and W. Wohlleben, J. Nanopart. Res., 2013, 15, 1504 CrossRef PubMed.
- L. Schlagenhauf, B. Kianfar, T. Buerki-Thurnherr, Y.-Y. Kuo, A. Wichser, F. Nuesch, P. Wick and J. Wang, Nanoscale, 2015, 7, 18524–18536 RSC.
- T. Elzein, M. Nasser-Eddine, C. Delaite, S. Bistac and P. Dumas, J. Colloid Interface Sci., 2004, 273, 381–387 CrossRef CAS PubMed.
- Fractional decrease in ester band area of a given sample is determined by the quotient of the ester band area of unexposed PCL/EC over the ester band area of the sample (COOi/COOf). Following this convention, an unphotolyzed sample will yield a value of 1 and with increasing loss of ester band area, the value will increase.
- J. R. Taylor, An Introduction to Error Analysis, The Study of Uncertainties in Physical Measurements, University Science Books Sausalito, CA, 2nd edn, 1997 Search PubMed.
- M. Iqbal, in An Introduction to Solar Radiation, Academic Press, 1983, pp. 43–58, DOI:10.1016/B978-0-12-373750-2.50008-2.
- E. Ikada, J. Photopolym. Sci. Technol., 1998, 11, 23–27 CrossRef CAS.
- J. F. Rabek, Photodegradation of Polymers: Physical Characteristics and Applications, Springer, Germany, 1996 Search PubMed.
- W. Wohlleben, J. Meyer, J. Muller, P. Muller, K. Vilsmeier, B. Stahlmecke and T. A. J. Kuhlbusch, Environ. Sci.: Nano, 2016, 3, 1036–1051 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption spectra of PCL is included in SI 1. Additional TEM and EDX characterization of the as received SWCNTs are shown in SI 2. Raman spectra of a 1.5% CNT sample is shown in SI 3. SWCNT calibration curve used for calculating CNT concentration of 89Y signal with sp-ICP-MS is shown in SI 4. Absolute mass loss measurements for nanocomposite samples are provided in SI 5. Additional SEM imaging of the CNT free polymers and exposed 0.25% CNT sample is shown in SI 6 and 7, respectively. Additional XPS spectra of the CNT free polymer and nanocomposite samples are included in SI 8 & SI 9, respectively. ATR-FTIR of C–H stretching region, as well as delta spectra of all exposed samples is shown in SI 10. See DOI: 10.1039/c6en00669h |
| This journal is © The Royal Society of Chemistry 2017 |