
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmyloid-β25–35 peptides aggregate into cross-β sheets in unsaturated anionic lipid membranes at high peptide concentrations
Jennifer
Tang
,
Richard J.
Alsop
,
Matilda
Backholm
,
Hannah
Dies
,
An-Chang
Shi
and
Maikel C.
Rheinstädter
*
Department of Physics and Astronomy, McMaster University, ABB-241, 1280 Main Street West, Hamilton, Ontario L8S 4M1, Canada. E-mail: rheinstadter@mcmaster.ca; Fax: +1-(905)-546-1252; Tel: +1-(905)-525-9140-23134
First published on 8th February 2016
Abstract
One of the hallmarks of Alzheimer's disease is the formation of protein plaques in the brain, which mainly consist of amyloid-β peptides of different lengths. While the role of these plaques in the pathology of the disease is not clear, the mechanism behind peptide aggregation is a topic of intense research and discussion. Because of their simplicity, synthetic membranes are promising model systems to identify the elementary processes involved. We prepared unsaturated zwitterionic/anionic lipid membranes made of 1-palmitoyl-2-oleoyl-sn-glycero-phosphocholine (POPC) and 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine (DMPS) at concentrations of POPC/3 mol% DMPS containing 0 mol%, 3 mol%, 10 mol%, and 20 mol% amyloid-β25–35 peptides. Membrane-embedded peptide clusters were observed at peptide concentrations of 10 and 20 mol% with a typical cluster size of ∼11 μm. Cluster density increased with peptide concentration from 59 (±3) clusters per mm2 to 920 (±64) clusters per mm2, respectively. While monomeric peptides take an α-helical state when embedded in lipid bilayers at low peptide concentrations, the peptides in peptide clusters were found to form cross-β sheets and showed the characteristic pattern in X-ray experiments. The presence of the peptides was accompanied by an elastic distortion of the bilayers, which can induce a long range interaction between the peptides. The experimentally observed cluster patterns agree well with Monte Carlo simulations of long-range interacting peptides. This interaction may be the fundamental process behind cross-β sheet formation in membranes and these sheets may serve as seeds for further growth into amyloid fibrils.
1 Introduction
A primary feature in the pathogenesis of Alzheimer's disease is the deposition of insoluble fibrillar plaques in the extracellular space of brain tissue.1 The major component of these plaques is the amyloid-β peptide (Aβ). Misfolding and aggregation of Aβ peptides is involved in the development of Alzheimer's disease, although the exact relationship between the protein structure and the pathology of Alzheimer's is still unclear.2 One stream of anti-Alzheimer's drugs directly addresses amyloid fibres to prevent their formation3 and growth,4 or reduce their toxicity.5 While the aggregation of proteins appears to be to some extent an inherent part of aging,6 increasing evidence suggests a link between the neurodegenerative disease and changes in the composition of brain tissue.The Aβ25–35 peptide comprises the transmembrane segment of the amyloid precursor protein (APP) and also comprises part of the full length Aβ1–42 polypeptide with 42 amino acids. Amyloid fibres are elongated protein structures, consisting of arrays of β-sheets running parallel to the long axis of the fibrils, the so-called cross-β motif,7 which is connected through steric zippers.1 It is believed that these fibres need a nucleus to form.8 While Aβ peptides are frequently reported in an extracellular location, Aβ1–40 and Aβ1–42 molecules were found to strongly interact with negatively charged lipids and to bind to anionic, negatively charged membranes,9–16 orienting parallel to the membrane surface. Through X-ray and neutron diffraction, Mason et al.,17 Dies et al.18 and Dante, Hauss and Dencher19–21 observed embedded states for Aβ1–42 and the Aβ25–35 segment in anionic lipid membranes. Both peptides were found to embed as α-helical monomers at low peptide concentrations of 3 mol%.12,18
Van der Waals and electrostatic interactions over long distances alone are typically too weak to overcome thermal fluctuations and cause peptides to aggregate. However, when embedded in a membrane, the physical properties of the lipid bilayer contribute to how peptides interact. The inclusion of peptides often leads to local distortions in membranes, which can interact with each other and lead to repulsive or attractive forces.22–26 As these forces are mediated by the membrane, they strongly depend on the membrane environment.
The membrane active segment amyloid-β25–35 was included in anionic synthetic bilayers at molar concentrations of 3 mol%, 10 mol%, and 20 mol% and the resulting structures were studied using optical microscopy and X-ray diffraction. Micrometer sized membrane-embedded peptide aggregates were observed at concentrations of more than 10 mol%, showing the characteristic diffraction pattern of cross-β sheets. Cluster formation inside the bilayers was accompanied by structural changes in the bilayers and local distortions. These distortions lead to long-ranged peptide–peptide interactions and Monte Carlo simulations including long-range interactions reproduce the experimentally observed peptide cluster patterns. These aggregates may serve as nuclei for extracellular plaque growth and we suggest that the corresponding distortion driven aggregation process is one of the fundamental mechanisms for nuclei formation.
2 Materials and methods
2.1 Preparation of the highly-oriented multi-lamellar membrane samples
Highly oriented multi-lamellar membranes were prepared on single-side polished silicon wafers. 100 mm diameter, 300 μm thick silicon [100] wafers were pre-cut into 1 × 1 cm2 chips. The wafers were first pretreated by sonication in dichloromethane (DCM) at 310 K for 30 minutes to remove all organic contamination and leave the substrates in a hydrophobic state. Each wafer was thoroughly rinsed three times by alternating with ∼50 mL of ultrapure water and methanol.Solutions of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) at a concentration of 20 mg of lipid per mL of solvent and 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine (DMPS) at a concentration of 20 mg of lipid per mL of solvent were each dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 chloroform:2,2,2-trifluoroethanol (TFE) solution. The amyloid-β peptides were prepared by pretreatment with trifluoroacetic acid (TFA) to disaggregate the peptide, as described by.27 This pretreatment included dissolving the peptide in a 1 mg mL−1 solution of TFA, sonicating with a tip sonicator for four three second intervals, and then removing the solvent through evaporation for 12 hours in a vacuum at 298 K. The peptide was then redissolved in a 20 mg ml−1 solution of 1:1 TFE:chloroform. Each solution underwent several centrifugations in a Vortex mixer until the solution was homogeneous. The POPC, DMPS, and peptide solutions were then mixed in appropriate ratios to produce the desired membrane samples for the experiment. Schematics of the POPC, DMPS and Aβ25–35 molecules are shown in Fig. 1.
:1 chloroform:2,2,2-trifluoroethanol (TFE) solution. The amyloid-β peptides were prepared by pretreatment with trifluoroacetic acid (TFA) to disaggregate the peptide, as described by.27 This pretreatment included dissolving the peptide in a 1 mg mL−1 solution of TFA, sonicating with a tip sonicator for four three second intervals, and then removing the solvent through evaporation for 12 hours in a vacuum at 298 K. The peptide was then redissolved in a 20 mg ml−1 solution of 1:1 TFE:chloroform. Each solution underwent several centrifugations in a Vortex mixer until the solution was homogeneous. The POPC, DMPS, and peptide solutions were then mixed in appropriate ratios to produce the desired membrane samples for the experiment. Schematics of the POPC, DMPS and Aβ25–35 molecules are shown in Fig. 1.
 | ||
| Fig. 1 Schematic representations of POPC, DMPS and amyloid-β25–35 molecules. | ||
The tilting incubator (VWR Incubating Rocker/3-D Rotator Waver) was heated to 313 K and the lipid solutions were placed inside to equilibrate. 65 μL of lipid solution was applied on each wafer, and the solvent was then allowed to slowly evaporate for 10 minutes at a speed of 15, tilt of 1, such that the lipid solution spread evenly on the wafers. After drying, the samples were placed in vacuum at 313 K for 12 hours to remove all traces of the solvent. The bilayers were annealed and rehydrated before use in a saturated K2SO4 solution, which provides ∼97% relative humidity (RH). The hydration container was allowed to equilibrate at 293 K in an incubator. The temperature of the incubator was then increased gradually from 293 K to 303 K over a period of ∼5 hours to slowly anneal the multi-lamellar structure. This procedure results in highly oriented multi-lamellar membrane stacks and a uniform coverage of the silicon substrates. About 3000 highly oriented stacked membranes with a thickness of ∼10 μm are produced using this protocol.
The high sample quality and high degree of order is a prerequisite to determine in-plane and out-of-plane structure of the membranes separately, but simultaneously.
2.2 Inverted light microscope
Optical microscopy was performed using an Olympus BX51 microscope. Samples were imaged in dark field reflection mode with a CCD camera (QIClick, QImaging), which provided high resolution images (1392 × 1040 pixels) for subsequent image analysis. In dark field microscopy, the unscattered beam is excluded from the image by illuminating the sample with light that when reflected will not be collected by the objective lens. A 50× magnification objective (UMPlanFI, Olympus) was used to obtain images with a resolution of 130 nm per pixel. Additional images with lower resolution were taken using a Nikon P520 digital camera in high-resolution macro setting mode.2.3 X-ray scattering experiment
X-ray diffraction data was obtained using the Biological Large Angle Diffraction Experiment (BLADE) in the Laboratory for Membrane and Protein Dynamics at McMaster University. BLADE uses a 9 kW (45 kV, 200 mA) CuK-α Rigaku Smartlab rotating anode at a wavelength of 1.5418 Å. Both source and detector are mounted on movable arms such that the membranes stay horizontal during the measurements. Focussing multi-layer optics provides a high intensity parallel beam with monochromatic X-ray intensities up to 1010 counts s−1 × mm−2. This beam geometry provides optimal illumination of the solid supported membrane samples to maximize the scattering signal. By using highly oriented membrane stacks, the in-plane (q||) and out-of-plane (qz) structure of the membranes can be determined separately, but simultaneously.The result of such an X-ray experiment is a 2-dimensional intensity map of a large area (0.03 Å−1 < qz < 1.1 Å−1 and 0 Å−1 < q|| < 3.1 Å−1) of the reciprocal space. The corresponding real-space length scales are determined by d = 2π/|Q| and cover length scales from about 2.5 to 60 Å, incorporating typical molecular dimensions and distances. These 2-dimensional data are essential to detect and identify signals from bilayers and peptides and determine orientation of the molecules. All scans were carried out at 28 °C and 97% RH. The membrane samples were mounted in a temperature and humidity controlled chamber, a so-called humidity chamber, during the measurements. The membranes were hydrated by water vapour and allowed to equilibrate for 6 hours before the measurements to ensure full re-hydration of the membrane stacks.
In order to unambiguously determine the small peptide signals, the experimental background was measured and it shown in Fig. 2. The 2-dimensional data show contributions of the silicon wafer, the [111] reflection normal to the wafer, and the aluminium[111] reflection in the plane as contribution from the sample can. The wafers used were cut such that the strong [111] silicon reflection is not perfectly perpendicular to the surface of the wafers to avoid overlap with the membrane and peptide signals.
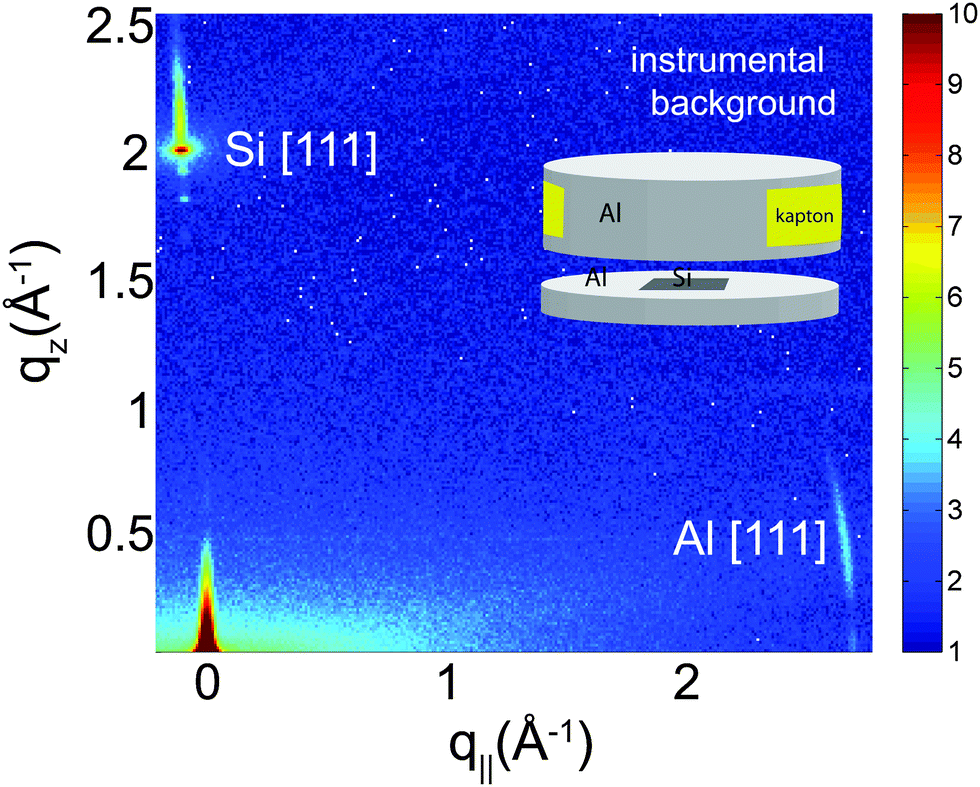 | ||
| Fig. 2 Background signals of the pure silicon wafer show the [111] reflection normal to the wafer. The aluminum[111] reflection is observed in the plane of the wafers as contribution from the sample can. | ||
The degree of orientation of the bilayers and lipid tails within the membrane samples was determined from the 2-dimensional X-ray maps. The intensity as a function of Q and angle γ from the q|| axis was used to determine orientation of lipid tail signals. Pixels within a wedge of the reciprocal space map, defined by γ and γstep (where γstep = 2°), were integrated as a function of Q = (qz2 + q||2)1/2 and normalized by pixel count at each Q. γ varied from 10° to 35° for samples with 0 mol%, 3 mol%, and 10 mol% peptide and from 10° to 85° for the sample with 20 mol% peptide. γ < 10° was not included due to high absorption at low angles.28 The integrated I(Q,γ) could be fit with Lorentzian functions. By calculating the area under the Lorentzian fits, I(γ) was determined and fit with a Gaussian distribution.
To determine the degree of orientation of membranes in the stack, the intensity as a function of the meridional angle δ was determined. The intensity was integrated around the second Bragg peak, at Q ≈ 0.22 Å−1, from 18° < δ < 40°. δ < 18° was not used in order to avoid contributions from incoherent scattering.29 The second Bragg peak was chosen, as incoherent contributions were weaker than the first Bragg peak. Pixel density at low-Q was too low to calculate I(Q,δ), as with the peptide samples, so I(δ) was calculated by direct summation of pixels within δ and δstep (where δstep = 2°), and within Q and Qstep, where the Q-range was chosen to include only scattering from the second Bragg peak. I(δ) was fit with a Gaussian distribution centred at δ = 0, which was then used to calculate the degree of orientation using Hermans orientation function:
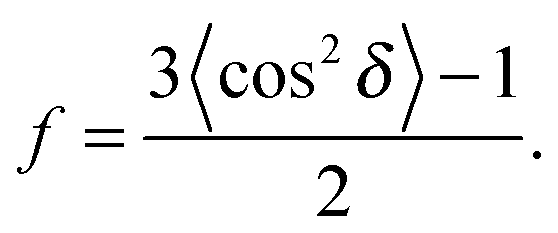 | (1) |
2.4 Monte Carlo simulations
The simulation was implemented in the multiagent NetLogo environment.30,31 The simulation box contained 2000 lipid molecules and peptides at concentrations of 0%, 3%, 10% and 20%, in agreement with the experiment. The lipid bilayer was modelled by an attractive lipid–lipid force of −0.5kBT. An attractive force between peptides of −1.5kBT was implemented, whose interaction distance could be varied from a direct peptide–peptide interaction to a long-range interaction over 20 nearest neighbour distances. The parameters of the simulation are listed in Table 1. All simulations started at a random configuration and were run for 20000 time steps to ensure that they reach equilibrium.
| Simulation parameter | Value |
|---|---|
| Lipid–lipid | −0.5kBT |
| Peptide–peptide | −1.5kBT |
| Lipid–peptide | — |
| Too-close-distance | 1.5 |
| Interaction distance | 1.5–20 |
3 Results
Anionic lipid membranes were prepared with 97 mol% 1-palmitoyl-2-oleoyl-sn-glycero-phosphocholine (POPC), a 16:0–18:1 zwitterionic phospholipid with one saturated and one unsaturated tail, and 3 mol% 1,2-dimyristol-sn-glycero-3-phospho-L-serine (DMPS), a 14 chain fully saturated anionic phospholipid. A small percentage of charged phospholipids is found in cell membranes in the brain and is essential for proper cell signalling, protein sorting, and cell adhesion.32 Four different membrane complexes were prepared for this study, as detailed in the Materials and methods section and listed in Table 2.
| Aβ25–35 | d z (mol%) | d HH (Å) | d water (Å) | a T (Å) | ΔaT (Å) | A T (Å2) | V T (Å3) | f Membranes | Lipid tail tilt (°) |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 59.0 ± 0.1 | 39.4 ± 1.0 | 19.6 ± 0.5 | 5.20 ± 0.05 | 0.59 ± 0.02 | 23.4 ± 0.1 | 922 ± 1 | 0.96 ± 0.02 | 19.2 ± 5 |
| 3 | 54.9 ± 0.1 | 39.1 ± 1.0 | 15.8 ± 0.4 | 5.21 ± 0.05 | 0.61 ± 0.02 | 23.5 ± 0.1 | 919 ± 1 | 0.92 ± 0.03 | 21.4 ± 2.3 |
| 10 | 61.6 ± 0.4 | 39.2 ± 1.0 | 22.4 ± 0.6 | 5.21 ± 0.05 | 0.67 ± 0.02 | 23.5 ± 0.1 | 921 ± 1 | 0.90 ± 0.03 | 20.5 ± 2.3 |
| 20 | 58.0 ± 0.2 | 39.3 ± 1.0 | 18.7 ± 0.5 | 5.06 ± 0.05 | 0.74 ± 0.02 | 22.6 ± 0.1 | 886 ± 1 | 0.86 ± 0.03 | 25.4 ± 3 |
The Results section is organized as follows: size distribution and density of the peptide aggregates were determined from optical microscopy. From intensity and distribution of membrane and peptide signals in high resolution X-ray diffraction experiments, molecular structure of the peptide aggregates and effect of the peptides on bilayer structure was determined and a model for cluster structure was developed. The effect of local bilayer distortions on peptide–peptide interactions was studied through long-ranged peptide interactions in Monte Carlo computer simulations.
3.1 Optical microscopy
Microscope images of POPC/DMPS bilayers at (a) 0 mol% Aβ25–35 and (b) 20 mol% Aβ25–35 are shown in Fig. 3. The pure lipid bilayers in part (a) show a smooth and aggregate free surface. No peptide aggregates were observed at 3 mol% peptides. At 10 mol% Aβ25–35, almost circular structures become visible whose density increases at 20 mol% peptide, as shown in part (b). Number and size of these structures were determined by analyzing several images at each concentration taken at different spots of the solid supported membrane stacks. | ||
| Fig. 3 Optical microcopy images of a (a) pure POPC/DMPS membrane and (b) a POPC/DMPS + 20 mol% Aβ25–35. While the pure lipid matrix shows a smooth surface, inclusions were observed at peptide concentrations of 10 and 20 mol%. | ||
The corresponding histograms are shown in Fig. 4. A normal distribution was fit and size of the clusters was determined to be 11.0 ± 1.1 μm for 10 mol% and 10.9 ± 2.5 μm for 20 mol%. While the size of clusters stayed approximately constant between 10 and 20 mol%, the density increased from 59 ± 3 mm−2 to 920 ± 64 mm−2.
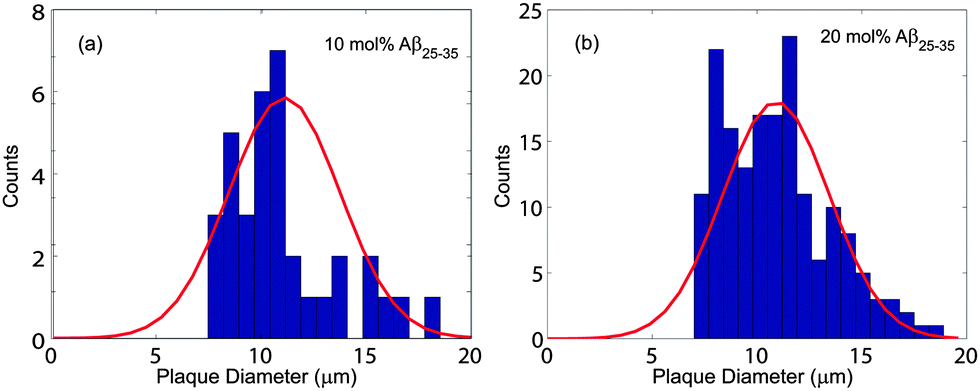 | ||
| Fig. 4 Histograms of the cluster size distribution in bilayers containing (a) 10 mol% Aβ25–35 and (b) 20 mol% Aβ25–35. The cluster sizes were determined from normal distributions to be 11.0 ± 1.1 μm and 10.9 ± 2.5 μm with cluster densities of 59 ± 3 and 920 ± 64 clusters per mm2, respectively. | ||
3.2 X-ray diffraction
The multi-lamellar membrane complexes were oriented in the diffractometer such that the q|| axis probed lateral membrane structure and the perpendicular axis, qz, probed out-of-plane structure. The samples were kept in a temperature and humidity controlled chamber during the measurements. Data were collected at T = 28 °C and in a 97% H2O atmosphere to ensure full hydration of the membranes to study structure in the fluid, physiologically relevant fluid state.Fig. 5(a)–(d) show 2-dimensional X-ray intensity maps for (a) POPC/DMPS, (b) POPC/DMPS + 3 mol% Aβ25–35 and, (c) POPC/DMPS + 10 mol% Aβ25–35 and, (d) POPC/DMPS + 20 mol% Aβ25–35. The out-of-plane scattering along qz shows pronounced and equally spaced Bragg intensities due to the multi-lamellar structure of the membranes, as reviewed for instance in.33,34
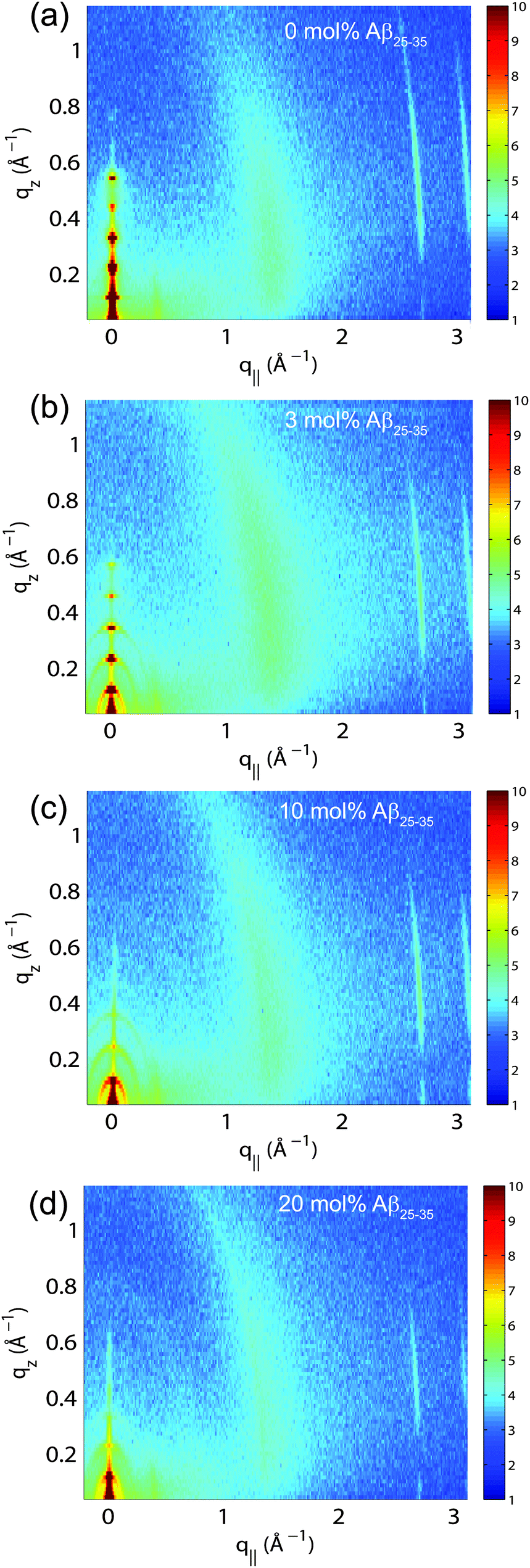 | ||
| Fig. 5 Two-dimensional X-ray data were collected for all membrane complexes to study molecular structure perpendicular to the solid supported membranes (out-of-plane) and parallel to the membranes (in-plane). Data for 0, 3, 10 and 20 mol% Aβ25–35 are shown in (a)–(d). Peptide signals are visible at 10 and 20 mol%. | ||
The diffracted intensity shows a well developed in-plane Bragg feature along the q||-axis at q|| ∼ 1.4 Å−1, related to the packing of the lipid tails in the hydrophobic membrane core. This peak is the result of a hexagonal packing of lipid tails35 (planar group p6). The distance between two acyl tails is determined to be 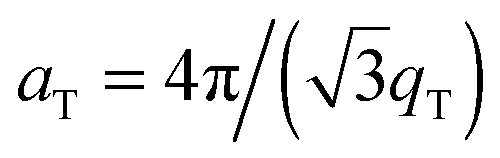 , where qT is the position of the tail correlation peak. The area per lipid acyl chain is obtained to
, where qT is the position of the tail correlation peak. The area per lipid acyl chain is obtained to 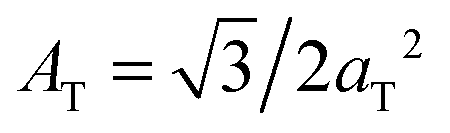 . Additional features appear at high peptide concentrations.
. Additional features appear at high peptide concentrations.
For a quantitative analysis of the diffracted intensities, the 2-dimensional data were cut along the out-of-plane and in-plane axes. Reflectivities for all samples are shown in Fig. 6. While 5 Bragg reflections were observed for the pure lipid matrix and at 3 mol% peptides, addition of 10 and 20 mol% reduces the number of peaks to 3, indicative of a less well-ordered lamellar structure. The lamellar spacings, dz, of the membrane complexes, i.e., the distance between two neighbouring bilayers in the membrane stack, was determined from the distance between the well developed Bragg reflections (dz = 2π/Δqz). The thickness of the membrane, defined by the head group to head group spacing (dHH), and the thickness of the hydration water layer, dwater, were determined from electron density distributions through Fourier transformation of the reflectivity data, the results of which are given in Table 2. While changes were observed in the lamellar spacing dz upon addition of Aβ25–35, the membrane thickness dHH stayed almost constant.
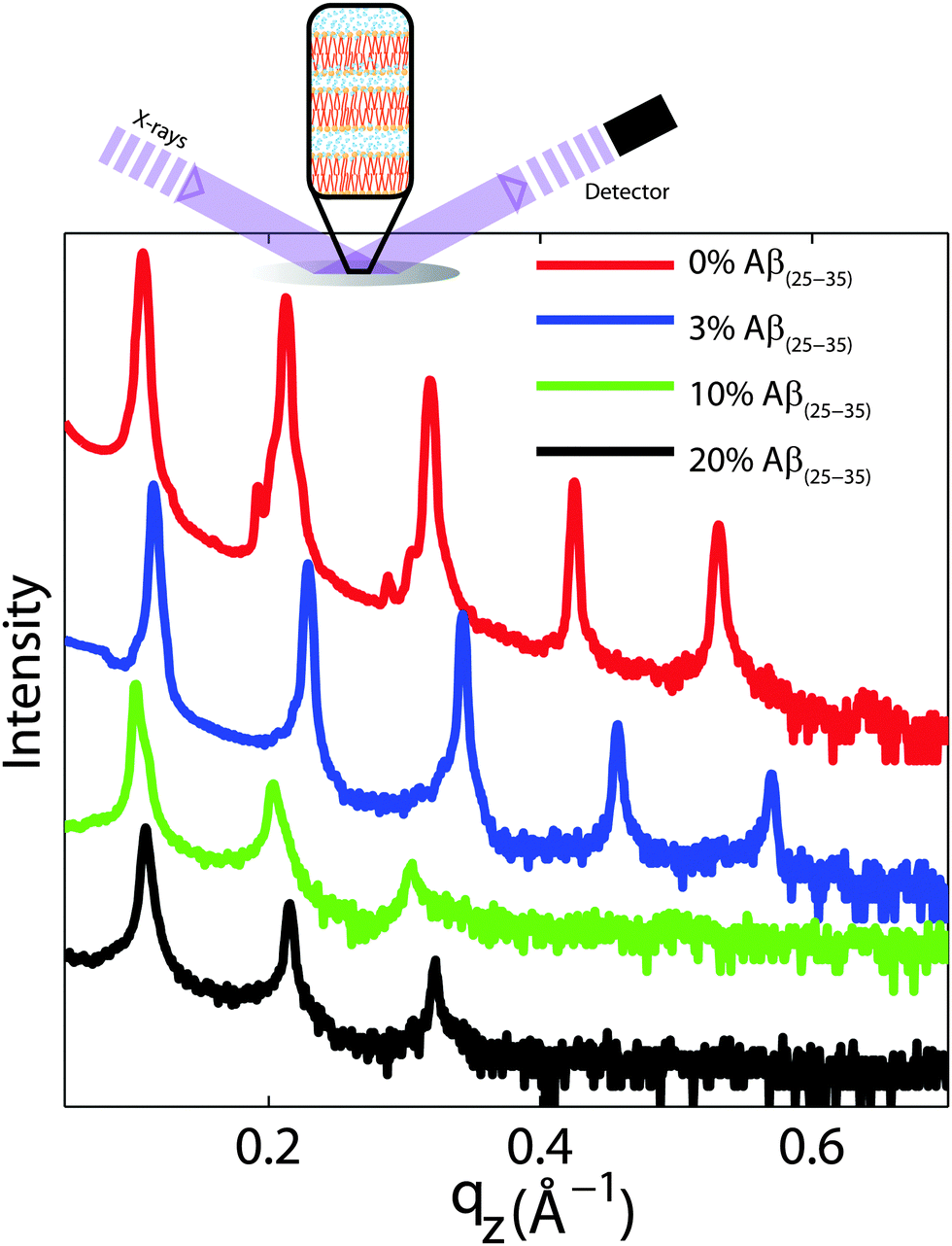 | ||
| Fig. 6 Out-of-plane diffraction for all membrane complexes. The lamellar structure of the membrane stack leads to a well defined series of Bragg peaks. Five diffraction orders were observed for 0 and 3 mol% Aβ25–35, while only 3 lamellar orders are visible for the cluster forming concentrations of 10 and 20 mol% peptide. Lamellar dz-spacings, head group-head group distances, dHH, and water layer thickness, dwater, were determined from this data and are listed in Table 2. | ||
In-plane diffraction data are shown in Fig. 7. Different signals are assigned to different molecular components. A signal from the membrane hydration water, i.e., water molecules in the water layer between stacked membranes at 3.4 Å (q|| = 1.85 Å−1), is observed for all concentrations.36,37 We note that the characteristic distance in bulk water is slightly smaller, namely ∼3.1 Å (2 Å−1). Due to interactions with the bilayer, membrane hydration water has distinct properties as compared to bulk water, such as structure, freezing temperature, and dynamics.38,39
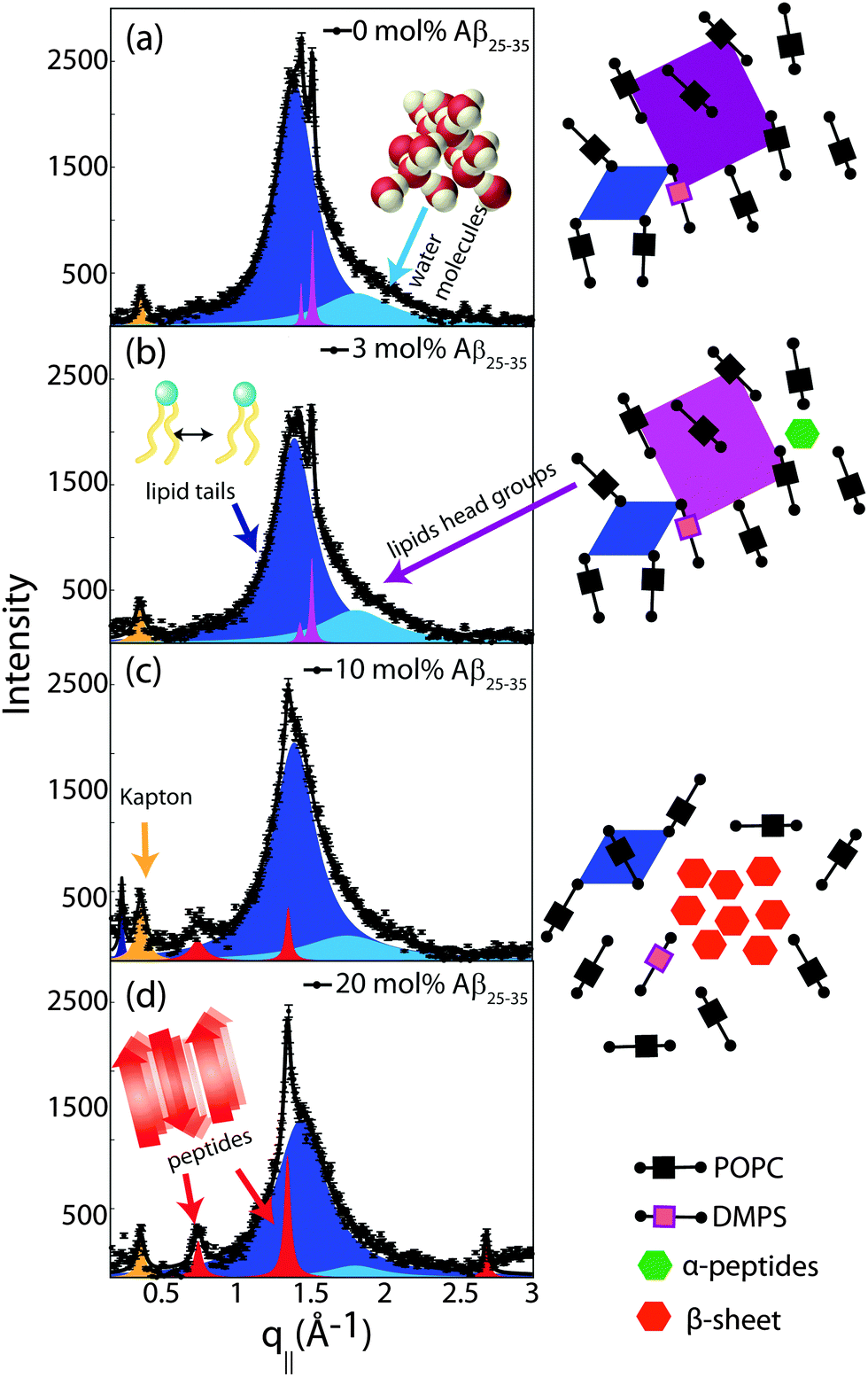 | ||
| Fig. 7 In-plane X-ray diffraction of the POPC/DMPS lipid membranes at different concentrations of amyloid-β25–35. The signals at 0 and 3 mol% Aβ can be assigned to organization of lipid tails, lipid head groups and hydration water molecules. The corresponding structures are depicted in the cartoons on the right. Peptides occur in monomeric α-helical states at these low peptide concentrations. Signals corresponding to formation of cross-β peptide sheets were observed at peptide concentrations of 10 and 20 mol%. The volume concentration of the different phases is proportional to the integrated intensities of the corresponding correlation peaks. The position, width, and intensity of the peaks are fit by a least-square algorithm. 15 free parameters are varied per fit. The signal at q|| = 0.4 Å−1 is a contribution of the kapton window of the X-ray chamber. | ||
The distance between lipid chains can be determined from the position of the acyl chain correlation peak while the width of the peak is related to the packing density in the hydrocarbon core. Values for aT, ΔaT, as well as the area and the volume per lipid acyl chain are given in Table 2. The chain distance and area continuously decrease with peptide concentration while packing order decreases. The volume occupied by the lipid tails decreases by ∼7% upon addition of 20 mol% peptide.
Additional narrow signals are observed at q||-values of 1.43 and 1.5 Å−1 in the 0 and 3 mol% membranes. These signals have been reported before40–42 and assigned to the organization of the lipid head groups within the lipid matrix. Peak positions are well described by a rectangular unit cell with dimensions a = 8.38 Å and b = 8.79 Å. The corresponding arrangements of lipid tails and head groups are pictured in the cartoon next to the diffraction patterns in Fig. 7.
Two signals appear with increasing peptide concentration. While it has been reported previously that amyloid-β25–35 peptides dissolve as monomeric α-helices in anionic membranes at a low peptide concentration of 3 mol%,12,18 the signals at 10 Å (q|| = 0.6 Å−1) and 4.7 Å (q|| = 1.35 Å−1) are the pattern of amyloid peptides forming cross-β amyloid sheets. These signals are observed at 10 mol%, when cluster formation was detected, and increase with increasing peptide concentration.
The structure of a cross-β sheet is depicted in Fig. 8(d). The two reflections observed in the X-ray pattern correspond to inter-strand and inter-sheet distances of peptide chains. The reflection at 1.35 Å−1 is indicative of extended protein chains running roughly perpendicular to the membrane plane and spaced 4.7 Å apart. The reflection at 0.6 Å−1 shows that the extended chains are organized into sheets spaced 10 Å apart.
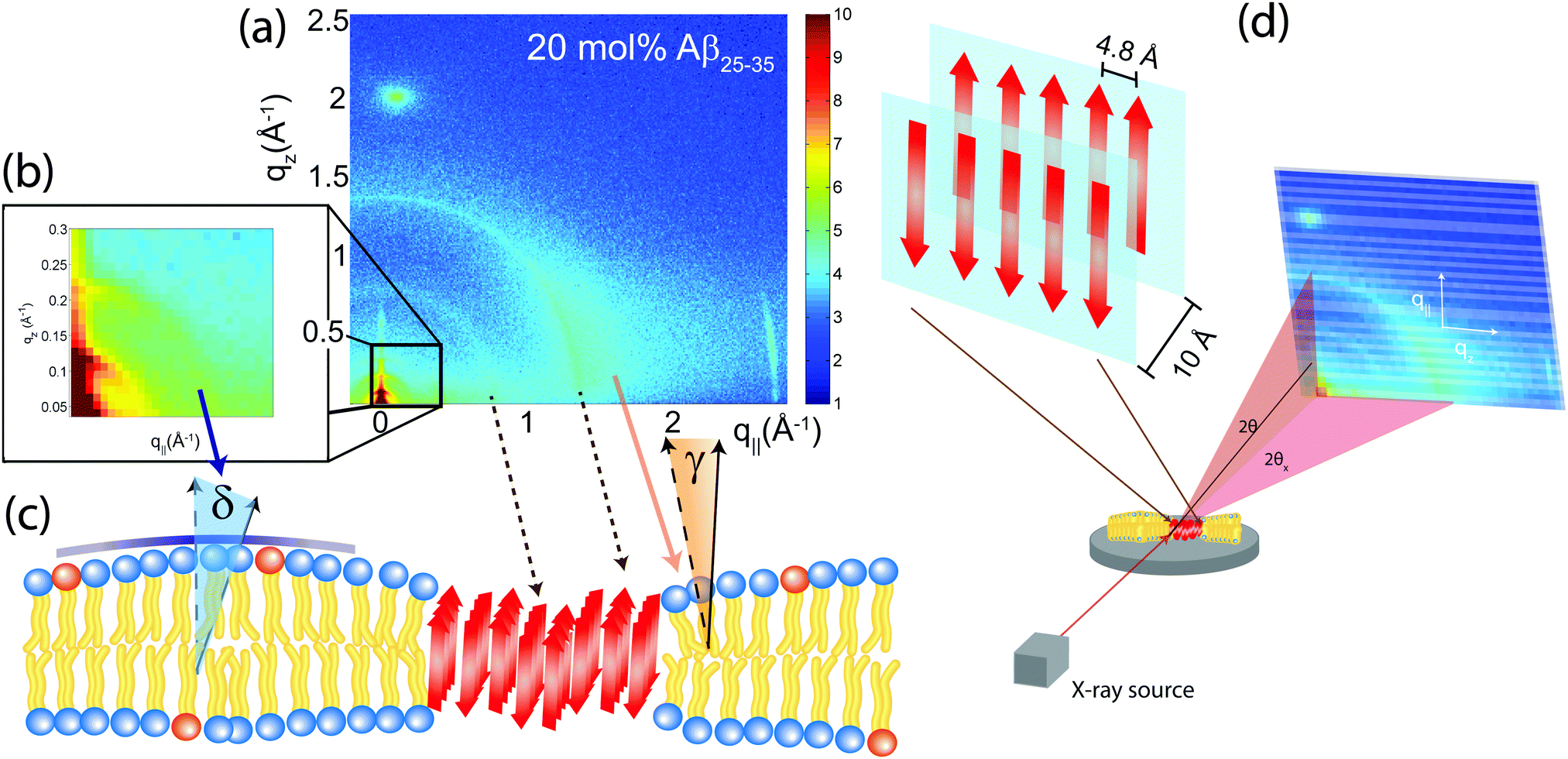 | ||
| Fig. 8 (a) Two-dimensional X-ray diffraction image of a membrane with 20 mol% Aβ peptides. The various features can be assigned to lipid and peptide structures in the membrane. (b) An inset of the image at low-Q, highlighting the presence of more isotropic scattering and elastic distortions. (c) A model of the peptide cluster in the membrane. Hydrophobic matching leads to distortions in the membrane structure. (d) Structure of a cross-β sheet. The 4.8 Å distance corresponds to chain distances within a sheet while the 10 Å distance is the distance between antiparallel sheets. | ||
These measurements indicate that amyloid-β peptides partition in anionic lipid bilayers at high peptide concentrations of 10 and 20 mol% and form cross-β sheets. Inclusion of the peptides has a profound effect on the structure of the bilayers: it leads to a reduction of distance between neighboring chains and a distortion in chain packing. The interaction between peptides and bilayers and the role of bilayer distortions will be investigated further in the next section.
3.3 Angular distribution of membrane and peptide signals
The average orientation of the lipid bilayers and the tilt of the lipid molecules can be determined by studying the angular dependence of the corresponding diffraction signals on the 2-dimensional X-ray intensity maps. Data for 20 mol% and the assignment of the scattering signals are depicted in Fig. 8(a). The intensity at the lipid tail position was integrated as function of the azimuth, γ, to determine the average tilt angle of the lipid acyl chains.Fig. 8(b) shows the small angle region around the reflectivity Bragg peaks in magnification. The corresponding intensity shows a circular pattern and was integrated over the meridian, δ, and analyzed using Hermans orientation function, as detailed in the Materials and methods section. Hermans function describes the degree or extent of orientation of the molecular axis relative to the membrane normal. Completely aligned would result in f = 1, randomly oriented in f = 0 and a perpendicular orientation would give f = −0.5.
For a perfectly ordered membrane stack, where all bilayers are planar and perfectly parallel, the intensity of the reflectivity Bragg peaks is located along the qz-axis. There are two origins for a smearing of the intensity: Lamellar diffuse scattering, caused by bilayer undulations, results in horizontal sheets of intensity along q||. By analyzing this intensity, the elastic constants of the bilayers can be determined.33,43,44 In addition, bilayers which are bent permanently lead to a ring of intensity on the 2-dimensional plots (a powder ring). This intensity is a measure of the static distortions of the bilayers caused by the presence of the peptides.
Signals of membrane curvature and lipid tails orientation are plotted in Fig. 9 and the results are listed in Table 2. Bending of the bilayers and the average tilt angles of the lipid tails increase with peptide concentration, indicative of increasing bilayer distortions in the presence of peptide aggregates.
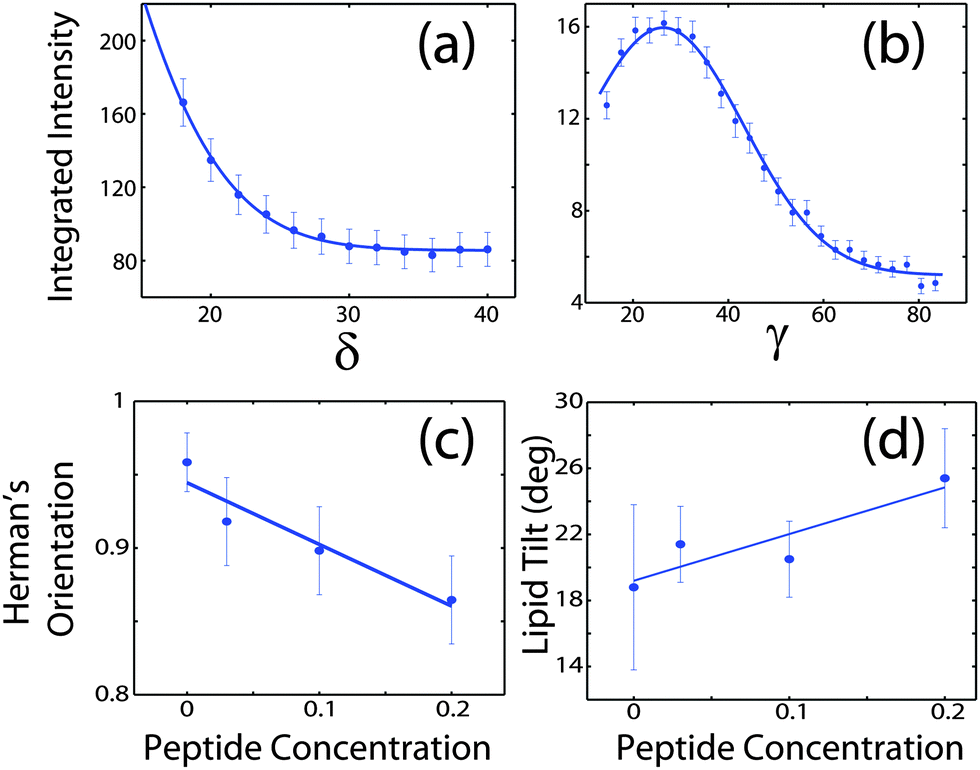 | ||
| Fig. 9 (a) Intensity of the second reflectivity Bragg peak as a function of angle δ from the qz axis. (b) Integrated intensity of the lipid correlation peak as a function of angle γ from the q|| axis. (c) Hermans orientation function, as defined in the Materials and methods section in eqn (1), for membrane bending for all peptide concentrations and (d) lipid tail tilt angle for all peptide concentrations. | ||
3.4 Monte Carlo modelling
In order to investigate aggregation of peptides and in particular the effect of the range of peptide interaction, Monte Carlo simulations were conducted. The computer modelling results are shown in Fig. 10. The system consisted of a planar system of 2000 lipid and peptide molecules, with peptide concentrations of 3, 10 and 20 mol% peptide. The lipid bilayer was modelled by an attractive lipid–lipid force of 0.5kBT. An attractive peptide–peptide force of 1.5kBT was included and simulation runs at different interaction distances were conducted for 3 mol%, 10 mol% and 20 mol% Aβ25–35 peptides. | ||
| Fig. 10 Monte Carlo simulations of a lipid bilayer containing different peptide concentrations and different ranges of the peptide–peptide interaction. A nearest-neighbour interaction between peptides was simulated in (a)–(c). A long-range interaction including up to 10 molecular distances in (g)–(i) was found to best mimic the experimental findings. | ||
A direct peptide–peptide interaction, as modelled at a range of 3 molecular distances, is shown in Fig. 10(a)–(c) and was found to lead to cluster formation at all peptide concentrations. Cluster size increases with peptide concentration, in conflict with the experimental findings. Truly long range interactions over 20 molecular distances (Fig. 10(m)–(o)) were found to suppress cluster formation at low peptide concentrations of 3 mol%, however, also led to an increasing cluster size at higher peptide concentrations.
A peptide–peptide interaction of 10 molecular distances in parts (g)–(i) was found to best reproduce the experimental patterns: (1) there is a threshold, i.e., no clusters form at low peptide concentrations and (2) cluster size is independent of the amount of peptides at higher concentrations.
4 Discussion
4.1 Effects of peptide insertion and cluster formation on bilayer properties
The structures that we observe in the optical microscopy images in Fig. 3 only appear at high levels of the amyloid peptides and go hand-in-hand with the occurrence of β-peptide signals in the X-ray diffraction experiment in Fig. 5 and 7. The intensity of these signals increases with peptide concentration, in agreement with the increase in cluster density in the microscopy images. From the structural parameters of the bilayers in Table 2, there is no significant increase in lamellar spacing in the presence of the peptides indicating that the peptide clusters do not form outside of the bilayers. We note that the constant amount of 3 mol% DMPS in the POPC/DMPS mixture is too small to explain the occurrence of the observed heterogeneities based on a lipid de-mixing or change in lipid structure. We, therefore, conclude that the observed structures are embedded in the bilayers and consist of a high concentration of peptides. The structural parameters give a detailed picture of organization and interaction between peptides and peptide clusters and anionic lipid membranes.With the addition of Aβ25–35 peptides to the anionic lipid bilayers (1) the average membrane thickness, as measured by the head group-head group distance dHH, does not change; (2) the distance between acyl chains, the area and volume per lipid tail continuously decrease while the distribution of tail distances increases. In order to accommodate the peptides the (3) average lipid acyl chain tilt increases, lipid tail disorder increases, and membrane orientation parameter decreases, indicative of increasing local distortions. The structure of the bilayer at high peptide concentrations is sketched in Fig. 8(c).
It is expected that membrane-mediated interactions between the inserted proteins play a major role in the aggregation behaviour of the Aβ25–35 peptides. In general, a number of mechanisms could generate membrane-mediated interactions between the peptides. First of all, the hydrophobic thickness of the transmembrane proteins does not necessarily match the equilibrium bilayer thickness, thus inducing hydrophobic mismatch in the system. Secondly, the inserted proteins may lead to localized lateral displacements of the lipids and/or curvature change of the membranes, resulting in local packing interactions. Finally the insertion of proteins could affect the fluctuation modes of the membranes, resulting in Casimir forces between the proteins. In the current system the inserted Aβ25–35 peptides could be influenced by one or more of these mechanisms, resulting in the clustering of the peptides at high concentrations.
One possible mechanism involves the local deformation of the lipid bilayers. The bending of a monolayer will arise, to some extent, due to thermal fluctuations in the membrane, but the most dominant energy cost associated with bending arises when there is an inclusion, such as a peptide, in the membrane. Hydrophobic mismatch occurs when the hydrophobic region of the peptide is larger, or smaller, than the bilayer thickness, which causes each monolayer leaflet to distort in order to ensure that the entire hydrophobic region of the peptide is contained within the hydrophobic core of the membrane. These local membrane distortions can result in a long-range interaction between the peptides.
The free energy per amphiphile of a monolayer can be written as:24,45–47
| f(u,aL) = γaL + G(u) + K(aL)(∇2u − κ(aL))2, | (2) |
Using eqn (2), the membrane perturbation profile and the membrane-induced interactions between an array of inclusions embedded in a two-dimensional membrane have been calculated,45,48,49 and are sketched in Fig. 11. In the case of small or vanishing spontaneous curvature, the global energy minimum is obtained at r = 0, which favours aggregation. A metastable, dispersed state exists, separated from the aggregated state by an energy barrier.
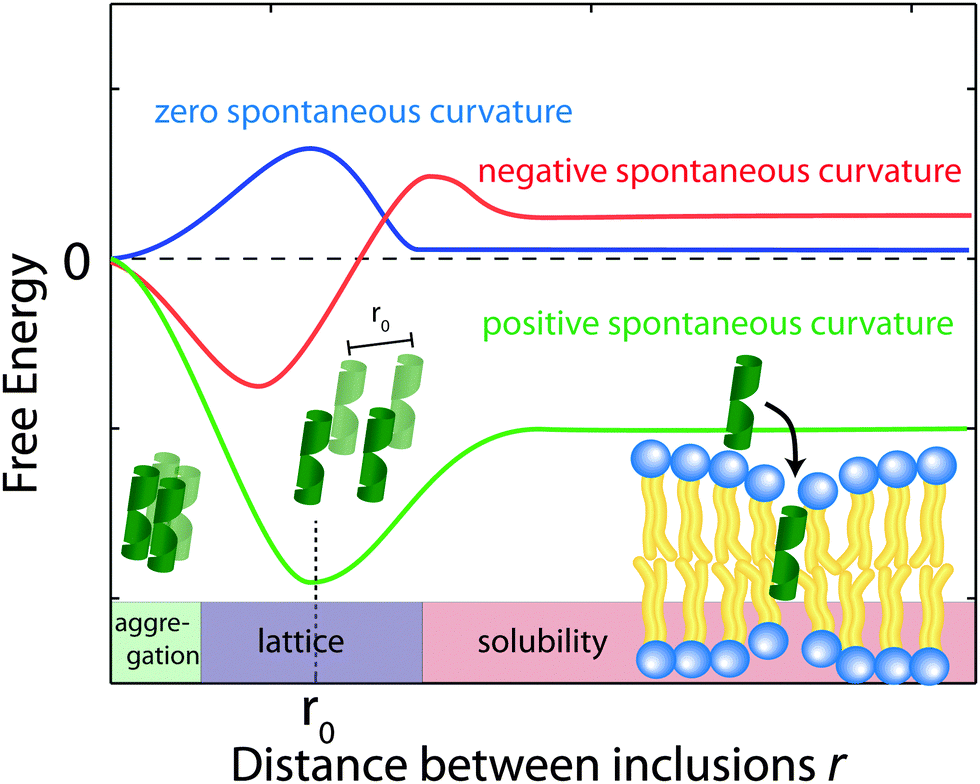 | ||
| Fig. 11 Schematics of the free energy profiles for lipids with zero, positive and negative spontaneous curvature as function of the distance between inclusions. | ||
Aggregation becomes unfavourable for nonzero spontaneous curvature (positive or negative) and the energy becomes minimal at a finite spacing (r0) between inclusions.24 In this state the peptides are expected to arrange on a regular lattice, as for instance observed in the case of purple membrane.22 The energy at r → ∞ is a measure of the energy related to insertion of the peptide into the bilayer. If this energy is negative peptides spontaneously embed in the bilayers. In the case of Aβ25–35, the peptide is shorter than the bilayer thickness, such that a positive spontaneous curvature favours peptide insertion.
However, POPC was reported to have a small, negative50 spontaneous curvature, while DMPS has a small positive spontaneous curvature. Using literature values, the total spontaneous curvature of the 0.97:0.03 mix can be calculated to 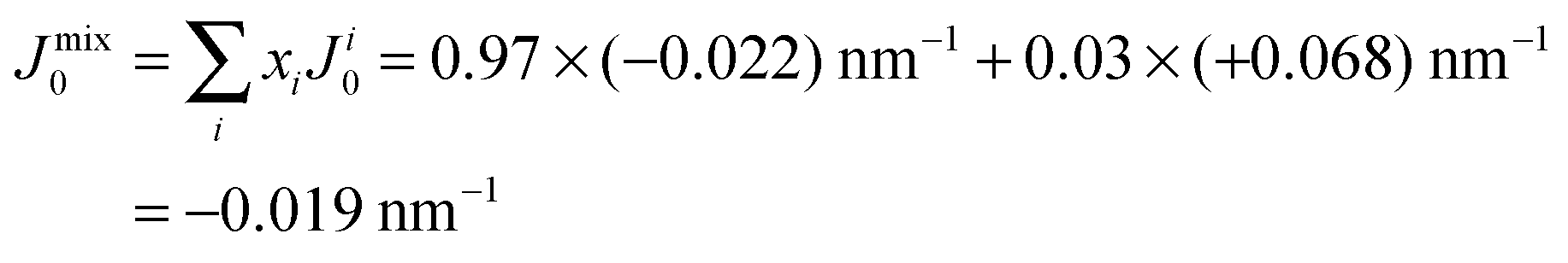 .50 The system is, thus, best described by the energy profile of a small spontaneous curvature in Fig. 11 and the global energy minimum in this case is the aggregated state at r = 0. There is an energy barrier to the inclusion of the peptides into the bilayer. Once embedded, the peptides first stay in the metastable, dispersed state. It seems that the energy barrier between local and global minimum can be overcome at higher peptide concentrations: the total energy cost increases with the number of inclusions such that the free energy is minimized by minimizing the number of membrane distortions. While the dispersed state is stable at low concentrations, peptides start to aggregate beyond a threshold concentration. From the experiments, the critical Aβ25–35 peptide concentration in POPC is between 3 and 10 mol%.
.50 The system is, thus, best described by the energy profile of a small spontaneous curvature in Fig. 11 and the global energy minimum in this case is the aggregated state at r = 0. There is an energy barrier to the inclusion of the peptides into the bilayer. Once embedded, the peptides first stay in the metastable, dispersed state. It seems that the energy barrier between local and global minimum can be overcome at higher peptide concentrations: the total energy cost increases with the number of inclusions such that the free energy is minimized by minimizing the number of membrane distortions. While the dispersed state is stable at low concentrations, peptides start to aggregate beyond a threshold concentration. From the experiments, the critical Aβ25–35 peptide concentration in POPC is between 3 and 10 mol%.
All-atom molecular dynamics (MD) simulations51–53 provide microscopic details of the corresponding structure: the lipid density next to the edge of a peptide is lower than its bulk value. This depletion layer is followed by a crowded region with high lipid density. The spontaneous curvature of the monolayer determines the shape of the membrane deformation profile. The perturbation length, i.e., the distance at which the membrane returns to its undistorted equilibrium thickness, is determined by compressibility and bending energy. In the computer simulations, this lateral perturbation of the lipid density was found to extend up to 25 Å from the edge of a small (cylindrical) inclusion with a radius of 5 Å, comparable to the size of the Aβ peptide. The corresponding distance between inclusions of 60 Å corresponds to about 6 molecular distances, in reasonable agreement with our Monte Carlo simulations.
The Monte Carlo simulations confirm that in order to reproduce the experimental findings (no aggregates formed at low peptide concentrations and cluster size is independent of peptide concentration) a long-ranged attractive interaction between peptides is necessary. A range of 10 molecular distances provided a good agreement with the experimental observations. While a direct, short-ranged peptide–peptide interaction led to cluster formation at all peptide concentrations, truly long-range interactions would lead to an increasing cluster size, in contradiction to the experiments.
4.2 Peptide conformations and properties of Aβ25–35-aggregates
Monomeric, single peptides were found in α-helical states when embedded in anionic membranes at low peptide concentrations.12,18 There is no sign of aggregates in the microscopy images and no additional signals in the X-ray diffraction pattern at 3 mol% peptide. We note that peptides often organize in bundles, whose structure is dominated by α-helical coiled-coils.54–58 Coiled coils consist of α-helices wound together to form a ropelike structure stabilized by hydrophobic interactions. The main features of this motif are a ∼9.5 Å (corresponding to q|| ∼ 0.6 Å−1) equatorial reflection corresponding to the spacing between adjacent coiled-coils and a ∼5.0 Å meridional reflection (corresponding to qz ∼ 1.25 Å−1) corresponding to the superhelical structure of α-helices twisting around each other within coiled-coils.59–61 The absence of those signals is indicative that peptides embed as monomers at low concentrations.Aggregates were observed to form at peptide concentrations of 10 and 20 mol% and signals of cross-β-sheets were observed in the X-ray diffraction patterns. This cross-β motif is found in the spine of amyloid fibrils,1,7 where the elementary β-strand building blocks assemble into larger structures through cross-β steric zippers. Cross-β sheets have been reported in short residue peptides,7 comparable to the length of Aβ25–35.
It has been observed previously that amyloid-β1–42 and amyloid-β25–35 peptides undergo a conformational change from α-helical to β-sheet structures in the presence of lipid membranes.62 The process behind β-sheet formation likely takes place in several steps. In a first step, the peptide makes contact with the membrane and aligns parallel to the membrane, before stronger bonds form and the peptide is embedded into the hydrocarbon core.63–66 From molecular dynamics computer simulations, α-helical peptides start to form small dynamical clusters of 4–6 peptides at low peptide concentrations.25 We note that as these cluster are small and transient structures, they would be difficult to observe in X-ray experiments. The transition into β-sheet structures inside of the hydrophobic core is then the result of (1) the hydrophobic effect and (2) a reduced conformational entropy of the peptide chains.67 Hydrophobic side chains orient towards the hydrophobic lipid acyl chains and reduction of the conformational freedom eventually promotes formation of β-sheets at higher peptide concentrations.
The amyloid literature typically distinguishes between the formation of a nucleus and eventually the growth of amyloid fibres.1 While the growth of the fibres appears to be somewhat understood, the formation of nuclei seems to be less well understood. While fibres are typically found in the extracellular space, our experiments provide plausible evidence that bilayers provide a site for peptide aggregation and nuclei formation inside of the bilayers. In this particular mechanism growth of the nuclei would occur by lateral attachment of further peptides.
5 Conclusion
Synthetic anionic membranes were fabricated and aggregation of amyloid-β25–35 peptides into cross-β sheets was observed and investigated using optical microscopy, X-ray diffraction, and computer modelling. Unsaturated zwitterionic/anionic lipid membranes made of POPC and DMPS at concentrations of POPC/3 mol% DMPS containing 0 mol%, 3 mol%, 10 mol% and 20 mol% amyloid-β25–35 peptides were prepared. Small, ∼11 μm sized peptide clusters were observed at peptide concentrations of 10 and 20 mol%. While cluster size was found to be independent of peptide concentration, cluster density increases with peptide concentration. Peptides in peptide clusters show the cross-β sheet motif.Inclusion of peptides and formation of peptide aggregates was found to lead to local distortions of the bilayers, which induces a long range interaction between the peptides. The experimentally observed cluster patterns agree well with Monte Carlo simulations of long-ranged interacting peptides. This elastic interaction may be the driving force behind peptide aggregation and cross-β sheet formation, which may serve as seeds for further growth into amyloid fibrils.
Acknowledgements
This research was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC), the National Research Council Canada (NRC), the Canada Foundation for Innovation (CFI) and the Ontario Ministry of Economic Development and Innovation. J. T. and H. D. are recipients of NSERC Undergraduate Research Awards (USRA), R. J. A. is the recipient of an NSERC PGSD scholarship, M. C. R. is the recipient of an Early Researcher Award of the Province of Ontario. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.References
- D. Eisenberg and M. Jucker, The amyloid state of proteins in human diseases, Cell, 2012, 148, 1188–1203 CrossRef CAS PubMed.
- B. J. Gilbert, The role of amyloid β in the pathogenesis of alzheimer's disease, J. Clin. Pathol., 2013, 66, 362–366 CrossRef CAS PubMed.
- A. J. Doig and P. Derreumaux, Inhibition of protein aggregation and amyloid formation by small molecules, Curr. Opin. Struct. Biol., 2015, 30, 50–56 CrossRef CAS PubMed.
- B. Bohrmann, K. Baumann, J. Benz, F. Gerber, W. Huber, F. Knoflach, J. Messer, K. Oroszlan, R. Rauchenberger and W. F. Richter, et al., Gantenerumab: a novel human anti-aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β, J. Alzheimer's Dis., 2011, 28, 49–69 Search PubMed.
- L. Jiang, C. Liu, D. Leibly, M. Landau, M. Zhao, M. P. Hughes and D. S. Eisenberg, Structure-based discovery of fiber-binding compounds that reduce the cytotoxicity of amyloid β, eLife, 2013, 2, e00857 Search PubMed.
- D. C. David, N. Ollikainen, J. C. Trinidad, M. P. Cary, A. L. Burlingame and C. Kenyon, Widespread protein aggregation as an inherent part of aging in c. elegans, PLoS Biol., 2010, 8, e1000450 Search PubMed.
- A. W. P. Fitzpatrick, G. T. Debelouchina, M. J. Bayro, D. K. Clare, M. A. Caporini, V. S. Bajaj, C. P. Jaroniec, L. Wang, V. Ladizhansky and S. A. Müller, et al., Atomic structure and hierarchical assembly of a cross-β amyloid fibril, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5468–5473 CrossRef CAS PubMed.
- J. T. Jarrett and P. T. Lansbury, Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in alzheimer's disease and scrapie?, Cell, 1993, 73, 1055–1058 CrossRef CAS PubMed.
- M. del Mar Martnez-Senac, J. Villalan and J. C. Gómez-Fernández, Structure of the alzheimer β-amyloid peptide (25–35) and its interaction with negatively charged phospholipid vesicles, Eur. J. Biochem., 1999, 265, 744–753 CrossRef.
- E. Maltseva and G. Brezesinski, Adsorption of amyloid β (1–40) peptide to phosphatidylethanolamine monolayers, ChemPhysChem, 2004, 5, 1185–1190 CrossRef CAS PubMed.
- G. Thakur, M. Micic and R. M. Leblanc, Surface chemistry of alzheimer's disease: a langmuir monolayer approach, Colloids Surf., B, 2009, 74, 436–456 CrossRef CAS PubMed.
- H.-H. G. Tsai, J.-B. Lee, S.-S. Tseng, X.-A. Pan and Y.-C. Shih, Folding and membrane insertion of amyloid-β (25–35) peptide and its mutants: Implications for aggregation and neurotoxicity, Proteins: Struct., Funct., Bioinf., 2010, 78, 1909–1925 CAS.
- M.-A. Sani, J. D. Gehman and F. Separovic, Lipid matrix plays a role in aβ fibril kinetics and morphology, FEBS Lett., 2011, 585, 749–754 CrossRef CAS PubMed.
- F. Hane, E. Drolle, R. Gaikwad, E. Faught and Z. Leonenko, Amyloid-β aggregation on model lipid membranes: An atomic force microscopy study, J. Alzheimer's Dis., 2011, 26, 485–494 CAS.
- H. Ding, J. A. Schauerte, D. G. Steel and A. Gafni, β-amyloid (1–40) peptide interactions with supported phospholipid membranes: A single-molecule study, Biophys. J., 2012, 103, 1500–1509 CrossRef CAS PubMed.
- H. Ahyayauch, M. Raab, J. V. Busto, N. Andraka, J.-L. R. Arrondo, M. Masserini, I. Tvaroska and F. M. Goni, Binding of β-amyloid (1–42) peptide to negatively charged phospholipid membranes in the liquid-ordered state: Modeling and experimental studies, Biophys. J., 2012, 103, 453–463 CrossRef CAS PubMed.
- R. P. Mason, J. D. Estermyer, J. F. Kelly and P. E. Mason, Alzheimer's disease amyloid β peptide 25–35 is localized in the membrane hydrocarbon core: X-ray diffraction analysis, Biochem. Biophys. Res. Commun., 1996, 222, 78–82 CrossRef CAS PubMed.
- H. Dies, L. Toppozini and M. C. Rheinstädter, The interaction between amyloid-β peptides and anionic lipid membranes containing cholesterol and melatonin, PLoS One, 2014, 9, e99124 Search PubMed.
- S. Dante, T. Hauss and N. A. Dencher, β-amyloid 25 to 35 is intercalated in anionic and zwitterionic lipid membranes to different extents, Biophys. J., 2002, 83, 2610–2616 CrossRef CAS PubMed.
- S. Dante, T. Hauss and N. A. Dencher, Cholesterol inhibits the insertion of the alzheimer's peptide aβ (25–35) in lipid bilayers, Eur. Biophys. J., 2006, 35, 523–531 CrossRef CAS PubMed.
- S. Dante, T. Hauss, R. Steitz, C. Canale and N. A. Dencher, Nanoscale structural and mechanical effects of β-amyloid (1–42) on polymer cushioned membranes: A combined study by neutron reflectometry and AFM force spectroscopy, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2646–2655 CrossRef CAS PubMed.
- M. C. Rheinstädter, K. Schmalzl, K. Wood and D. Strauch, Protein–protein interaction in purple membrane, Phys. Rev. Lett., 2009, 103, 128104 CrossRef PubMed.
- I. Casuso, P. Sens, F. Rico and S. Scheuring, Experimental evidence for membrane-mediated protein–protein interaction, Biophys. J., 2010, 99, L47–L49 CrossRef CAS PubMed.
- C. L. Armstrong, E. Sandqvist and M. C. Rheinstädter, Protein–protein interactions in membranes, Protein Pept. Lett., 2011, 18, 344–353 CrossRef CAS PubMed.
- M. Pannuzzo, A. Raudino, D. Milardi, C. La Rosa and M. Karttunen, α-helical structures drive early stages of self-assembly of amyloidogenic amyloid polypeptide aggregate formation in membranes, Sci. Rep., 2013, 3, 2781 Search PubMed.
- S. Katira, K. K. Mandadapu, S. Vaikuntanathan, B. Smit, D. Chandler, The order–disorder phase transition in lipid bilayers mediates a force for assembly of transmembrane proteins, 2015, arXiv preprint arXiv:1506.04298.
- S.-C. Jao, K. Ma, J. Talafous, R. Orlando and M. G. Zagorski, Trifluoroacetic acid pretreatment reproducibly disaggregates the amyloid β-peptide, Amyloid, 1997, 4, 240–252 CrossRef CAS.
- T. T. Mills, G. E. S. Toombes, S. Tristram-Nagle, D.-M. Smilgies, G. W. Feigenson and J. F. Nagle, Order parameters and areas in fluid-phase oriented lipid membranes using wide angle x-ray scattering, Biophys. J., 2008, 95, 669–681 CrossRef CAS PubMed.
- M. S. Jablin, K. Akibori and J. F. Nagle, Experimental support for tilt-dependent theory of biomembrane mechanics, Phys. Rev. Lett., 2014, 113, 248102 CrossRef CAS PubMed.
- U. Wilensky, Netlogo, Center for Connected Learning and Computer-Based Modeling, Northwestern University, 1999, http://ccl.northwestern.edu/netlogo/.
- S. Tisue and U. Wilensky, Netlogo: A simple environment for modeling complexity, in International conference on complex systems, 2004, pp. 16–21.
- P. A. Leventis and S. Grinstein, The distribution and function of phosphatidylserine in cellular membranes, Annu. Rev. Biophys., 2010, 39, 407–427 CrossRef CAS PubMed.
- G. Pabst, N. Kučerka, M.-P. Nieh, M. C. Rheinstädter and J. Katsaras, Applications of neutron and X-ray scattering to the study of biologically relevant model membranes, Chem. Phys. Lipids, 2010, 163, 460–479 CrossRef CAS PubMed.
- G. Fragneto and M. Rheinstädter, Structural and dynamical studies from bio-mimetic systems: an overview, C. R. Phys., 2007, 8, 865–883 CrossRef CAS.
- C. L. Armstrong, D. Marquardt, H. Dies, N. Kučerka, Z. Yamani, T. A. Harroun, J. Katsaras, A.-C. Shi and M. C. Rheinstädter, The observation of highly ordered domains in membranes with cholesterol, PLoS One, 2013, 8, e66162 CAS.
- M. C. Rheinstädter, T. Seydel, F. Demmel and T. Salditt, Molecular motions in lipid bilayers studied by the neutron backscattering technique, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 061908 CrossRef PubMed.
- M. C. Rheinstädter, T. Seydel and T. Salditt, Nanosecond molecular relaxations in lipid bilayers studied by high energy resolution neutron scattering and in-situ diffraction, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 75, 011907 CrossRef PubMed.
- L. Toppozini, C. L. Armstrong, M. D. Kaye, M. Tyagi, T. Jenkins and M. Rheinstädter, Hydration water freezing in single supported lipid bilayers, ISRN Biophys., 2012, 2012, 520307 Search PubMed.
- L. Toppozini, F. Roosen-Runge, R. I. Bewley, R. M. Dalgliesh, T. Perring, T. Seydel, H. R. Glyde, V. G. Sakai and M. C. Rheinstädter, Anomalous and anisotropic nanoscale diffusion of hydration water molecules in fluid lipid membranes, Soft Matter, 2015, 11, 8354–8371 RSC.
- J. Katsaras, V. A. Raghunathan, E. J. Dufourc and J. Dufourcq, Evidence for a two-dimensional molecular lattice in subgel phase dppc bilayers, Biochemistry, 1995, 34, 4684–4688 CrossRef CAS PubMed.
- V. A. Raghunathan and J. Katsaras, Structure of the lc′ phase in a hydrated lipid multilamellar system, Phys. Rev. Lett., 1995, 74, 4456–4459 CrossRef CAS PubMed.
- M. A. Barrett, S. Zheng, G. Roshankar, R. J. Alsop, R. K. R. Belanger, C. Huynh, N. Kučerka and M. C. Rheinstädter, Interaction of aspirin (acetylsalicylic acid) with lipid membranes, PLoS One, 2012, 7, e34357 CAS.
- M. C. Rheinstädter, W. Häussler and T. Salditt, Dispersion relation of lipid membrane shape fluctuations by neutron spin-echo spectrometry, Phys. Rev. Lett., 2006, 97, 048103 CrossRef PubMed.
- C. L. Armstrong, W. Häußler, T. Seydel, J. Katsaras and M. C. Rheinstädter, Nanosecond lipid dynamics in membranes containing cholesterol, Soft Matter, 2014, 10, 2600–2611 RSC.
- H. Aranda-Espinoza, A. Berman, N. Dan, P. Pincus and S. Safran, Interaction between inclusions embedded in membranes, Biophys. J., 1996, 71, 648 CrossRef CAS PubMed.
- W. Helfrich, Elastic properties of lipid bilayers: theory and possible experiments, Z. Naturforsch., C: J. Biosci., 1973, 28, 693–703 CAS.
- R. Bruinsma and P. Pincus, Protein aggregation in membranes, Curr. Opin. Solid State Mater. Sci., 1996, 1, 401–406 CrossRef CAS.
- N. Dan, P. Pincus and S. A. Safran, Membrane-induced interactions between inclusions, Langmuir, 1993, 9, 2768–2771 CrossRef CAS.
- N. Dan, A. Berman, P. Pincus and S. A. Safran, Membrane-induced interactions between inclusions, J. Phys. II, 1994, 4, 1713–1725 CrossRef CAS.
- B. Kollmitzer, P. Heftberger, M. Rappolt and G. Pabst, Monolayer spontaneous curvature of raft-forming membrane lipids, Soft Matter, 2013, 9, 10877–10884 RSC.
- P. Lagüe, M. J. Zuckermann and B. Roux, Protein inclusion in lipid membranes: a theory based on the hypernetted chain integral equation, Faraday Discuss., 1999, 111, 165–172 RSC.
- P. Lagüe, M. J. Zuckermann and B. Roux, Lipid-mediated interactions between intrinsic membrane proteins: a theoretical study based on integral equations, Biophys. J., 2000, 79, 2867–2879 CrossRef.
- P. Lagüe, M. J. Zuckermann and B. Roux, Lipid-mediated interactions between intrinsic membrane proteins: dependence on protein size and lipid composition, Biophys. J., 2001, 81, 276–284 CrossRef.
- L. Pauling and R. B. Corey, Two hydrogen-bonded spiral configurations of the polypeptide chain, J. Am. Chem. Soc., 1950, 72, 5349 CrossRef CAS.
- N. Pinto, F.-C. Yang, A. Negishi, M. C. Rheinstädter, T. E. Gillis and D. S. Fudge, Self-assembly enhances the strength of fibers made from vimentin intermediate filament proteins, Biomacromolecules, 2014, 15, 574–581 CrossRef CAS PubMed.
- F.-C. Yang, R. D. Peters, H. Dies and M. C. Rheinstädter, Hierarchical, self-similar structure in native squid pen, Soft Matter, 2014, 10, 5541–5549 RSC.
- F.-C. Yang, Y. Zhang and M. C. Rheinstädter, The structure of peoples hair, PeerJ, 2014, 2, e619 Search PubMed.
- Y. Zhang, R. J. Alsop, A. Soomro, F.-C. Yang and M. C. Rheinstädter, Effect of shampoo, conditioner and permanent waving on the molecular structure of human hair, PeerJ, 2015, 3, e1296 Search PubMed.
- F. H. C. Crick, Is α-keratin a coiled coil?, Nature, 1952, 170, 882–883 CrossRef CAS PubMed.
- C. Cohen and D. A. Parry, α-helical coiled coils: more facts and better predictions, Science, 1994, 263, 488–489 CAS.
- A. N. Lupas and M. Gruber, The structure of α-helical coiled coils, Adv. Protein Chem., 2005, 70, 37–38 CrossRef CAS PubMed.
- A. Accardo, V. Shalabaeva, M. Cotte, M. Burghammer, R. Krahne, C. Riekel and S. Dante, Amyloid β peptide conformational changes in the presence of a lipid membrane system, Langmuir, 2014, 30, 3191–3198 CrossRef CAS PubMed.
- H. W. Huang, Action of antimicrobial peptides: two-state model, Biochemistry, 2000, 39, 8347–8352 CrossRef CAS PubMed.
- D. M. Engelman, Y. Chen, C.-N. Chin, A. R. Curran, A. M. Dixon, A. D. Dupuy, A. S. Lee, U. Lehnert, E. E. Matthews and Y. K. Reshetnyak, et al., Membrane protein folding: beyond the two stage model, FEBS Lett., 2003, 555, 122–125 CrossRef CAS PubMed.
- M. Heyden, J. A. Freites, M. B. Ulmschneider, S. H. White and D. J. Tobias, Assembly and stability of α-helical membrane proteins, Soft Matter, 2012, 8, 7742–7752 RSC.
- C.-C. Lee, Y. Sun and H. W. Huang, Membrane-mediated peptide conformation change from α-monomers to β-aggregates, Biophys. J., 2010, 98, 2236–2245 CrossRef CAS PubMed.
- A. Nikolic, S. Baud, S. Rauscher and R. Pomès, Molecular mechanism of β-sheet self-organization at water-hydrophobic interfaces, Proteins: Struct., Funct., Bioinf., 2011, 79, 1–22 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |