
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel B(Ar′)2(Ar′′) hetero-tri(aryl)boranes: a systematic study of Lewis acidity†
Robin J.
Blagg
 *,
Trevor R.
Simmons
,
Georgina R.
Hatton‡
,
James M.
Courtney
,
Elliot L.
Bennett
,
Elliot J.
Lawrence
and
Gregory G.
Wildgoose
*
*,
Trevor R.
Simmons
,
Georgina R.
Hatton‡
,
James M.
Courtney
,
Elliot L.
Bennett
,
Elliot J.
Lawrence
and
Gregory G.
Wildgoose
*
School of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK. E-mail: g.wildgoose@uea.ac.uk; r.blagg@uea.ac.uk
First published on 30th October 2015
Abstract
A series of homo- and hetero-tri(aryl)boranes incorporating pentafluorophenyl, 3,5-bis(trifluoromethyl)phenyl, and pentachlorophenyl groups, four of which are novel species, have been studied as the acidic component of frustrated Lewis pairs for the heterolytic cleavage of H2. Under mild conditions eight of these will cleave H2; the rate of cleavage depending on both the electrophilicity of the borane and the steric bulk around the boron atom. Electrochemical studies allow comparisons of the electrophilicity with spectroscopic measurements of Lewis acidity for different series of boranes. Discrepancies in the correlation between these two types of measurements, combined with structural characterisation of each borane, reveal that the twist of the aryl rings with respect to the boron-centred trigonal plane is significant from both a steric and electronic perspective, and is an important consideration in the design of tri(aryl)boranes as Lewis acids.
Introduction
Since the initial report by Welch and Stephan1 there has been rapid growth in studies of frustrated Lewis pairs (FLPs).2–7 In the archetypal system the Lewis acid, B(C6F5)3, and Lewis base, P(tBu)3, are combined and are restricted from forming a classical Lewis acid–base adduct due to their steric bulk. Upon the addition of H2, however, this FLP heterolytic cleaves H2 to give protic and hydridic products.FLPs in conjunction with H2 have found applications as mediators or catalysts for metal-free hydrogenation of numerous functional groups including; aldehydes and ketones,8–10 N-heterocyclic aromatics,11 imines and nitriles,12 and silyl enol ethers.13 FLPs have also been shown to react with other small molecules such as oxides of carbon,14–16 nitrogen,17 and sulfur,18 along with alkenes and alkynes.19,20
Tris(pentafluorophenyl)borane, B(C6F5)3, is the most commonly used Lewis acidic component of FLPs; although other electrophilic boranes have been used in FLPs (or suggested for use in FLPs), such as other halogenated tri(aryl)boranes9,15,21–23 including the stepwise-substitution series B(C6Cl5)n(C6F5)3−n (n = 1–3),24,25 and B{2,4,6-(CH3)3C6H2}n(C6F5)3−n (n = 1–3);26 and borenium cations (commonly stabilised by N-heterocycle carbenes).27,28 Not all Lewis acids used in FLPs have been boron based, with other examples including; Ingleson and co-workers’ carbon-based, water tolerant N-methylacredinium salts,29 tri(aryl)aluminium analogues of classical boron based species,30 both phosphorus(III) and phosphorus(V)-based species,31 and silicon based species.32,33
We have previously introduced the concept of “combined electrochemical-frustrated Lewis pairs”,28,34–37 where the heterolytic cleavage of H2 by an FLP is coupled with in situ electrochemical oxidation of the resulting Lewis acid-hydride, liberating two electrons, a proton, and regenerating the parent Lewis acid. Hence, we have shown these systems to be electrocatalytic for the oxidation of H2 to yield two protons and two-electrons overall – a key process for many hydrogen-based energy technologies.
To further develop our “combined electrochemical-frustrated Lewis pair” concept, we sought to expand the range of tri(aryl)borane Lewis acids allowing us to probe the effects of further controlled modification of the boranes. Our existing studies having focused on the archetypal B(C6F5)3,34,35 its perchlorinated analogue,36 three isomers of B{C6H3(CF3)2}3,37 along with the borenium cation [(iPrN)2H2C3BC9H14]+.28 The stepwise substitution of the aryl rings would result in a range of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hetero-tri(aryl)boranes, in addition to the existing homo-tri(aryl)boranes. It was envisaged that by altering the electronic and steric nature of successive aryl groups, that the Lewis acidic boranes would exhibit varying properties (e.g. reactivates towards H2 activation, chemical tolerances, redox properties). Such a systematic study could also allow for insights into the chemical origin of these properties and provide further information toward a predictive model for reactivities of tri(aryl)boranes.
:1 hetero-tri(aryl)boranes, in addition to the existing homo-tri(aryl)boranes. It was envisaged that by altering the electronic and steric nature of successive aryl groups, that the Lewis acidic boranes would exhibit varying properties (e.g. reactivates towards H2 activation, chemical tolerances, redox properties). Such a systematic study could also allow for insights into the chemical origin of these properties and provide further information toward a predictive model for reactivities of tri(aryl)boranes.
Herein we report studies of four novel hetero-tri(aryl)boranes 2, 3, 5 and 6, incorporating combinations of the pentafluorophenyl (C6F5, ArF5), 3,5-bis(trifluoromethyl)phenyl {3,5-(CF3)2C6H3, ArF6}, and pentachlorophenyl (C6Cl5, ArCl5) halogenated-aryl rings; together with further studies of the previously reported24 tris(pentachlorophenyl)borane, 7, and the hetero-(pentachlorophenyl)(pentafluorophenyl)boranes 8 and 9. Comparing these with the archetypal Lewis acidic borane B(C6F5)3, 1, and B{3,5-(CF3)2C6H3}3, 4, allows for a comprehensive study of the effects of stepwise substitution of the aryl rings (Fig. 1). Both electrochemical and conventional NMR methodologies are employed to quantify the Lewis acidity/electrophilicity of the boranes, and the results of these methodologies compared. Finally, their potential to act as the Lewis acidic component of an FLP {in combination with the Lewis base tri-tert-butylphosphine (tBu3P)} for the heterolytic cleavage of H2 under mild conditions is studied.
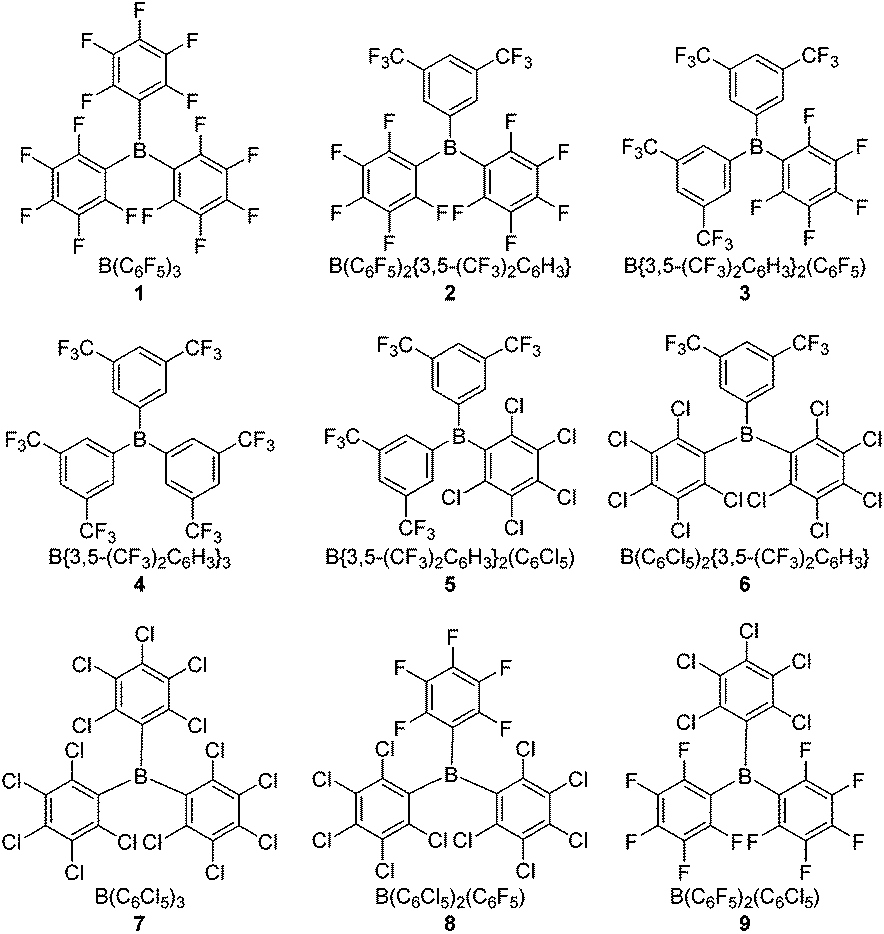 | ||
| Fig. 1 Homo- and hetero-tri(aryl)boranes, B(Ar′)2(Ar′′)/BArHalx. | ||
Results & discussion
The novel hetero-tri(aryl)boranes 3, 5 and 6, were all synthesised by broadly similar methods (Scheme 1), involving the reaction of a bis(aryl)haloborane with a suitable metal-aryl transfer agent. Reaction of B{3,5-(CF3)2C6H3}2Br with Cu(C6F5) in CH2Cl2 at room temperature, or Zn(C6Cl5)2 in toluene at +75 °C, led to synthesis of 3 and 5 respectively. In certain cases however, the impurity B{3,5-(CF3)2C6H3}2(OH) was obtained (characterisation data in ESI†), due to reaction of the bis(aryl)bromoborane precursor with trace water or hydroxide, fortunately 3 and 5 can be isolated by sublimation or recrystallization, respectively. 6 is synthesised by the reaction of a toluene solution of B(C6Cl5)2Cl with in situ generated Li{3,5-(CF3)2C6H3} in Et2O solution at −78 °C followed by slow warming to room temperature.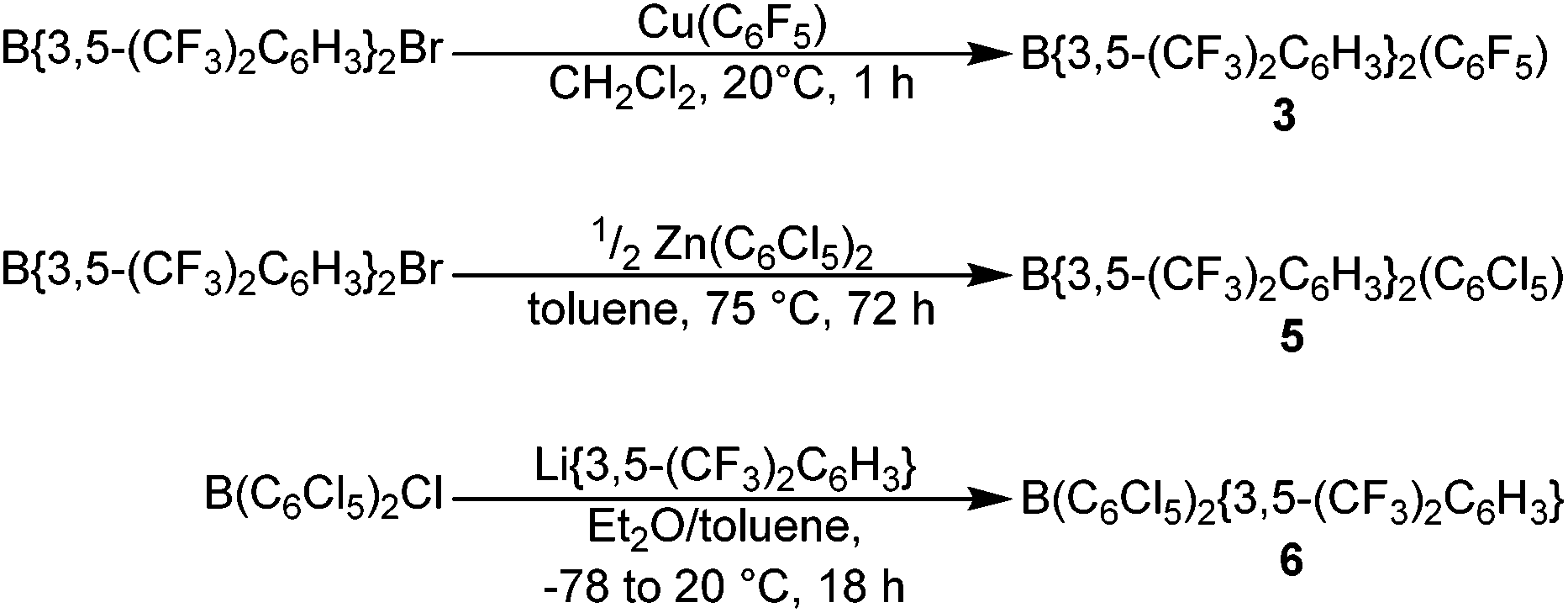 | ||
| Scheme 1 Synthetic routes for the synthesis of 3, 5 and 6. | ||
Applying this methodology for the synthesis of 2, by reaction of B(C6F5)2X (X = F, Cl) with M{3,5-(CF3)2C6H3} (M = Li, Cu) under various conditions, we were unable to obtain 2 as anything other than a minor component in a mixture of species. Therefore an alternative synthetic route was developed (Scheme 2). Following on from the publication by Samigullin et al.38 of a high yielding stepwise route for the synthesis of B{3,5-(CF3)2C6H3}2Br, we were able to successfully adapt their methodology for the synthesis of B{3,5-(CF3)2C6H3}Br2. This required the generation of Li{3,5-(CF3)2C6H3} at −78 °C, and it's reaction in Et2O with BH3·SMe2 to give [Li(OEt2)n][H3B{3,5-(CF3)2C6H3}]. Hydride abstraction using one equivalent Me3SiCl gave BH2{3,5-(CF3)2C6H3}, and reaction with excess methanol converts the bis-hydride to the bis-methoxide B{3,5-(CF3)2C6H3}(OMe)2 (characterisation data in ESI†). Conversion of the bis-methoxide to the bis-bromide was readily achieved by reaction with excess BBr3, leading to the isolation of highly reactive B{3,5-(CF3)2C6H3}Br2 as a pale yellow oil. Further reaction with two equivalents of Cu(C6F5) in CH2Cl2, results in the rapid generation of 2, which may be purified via sublimation.
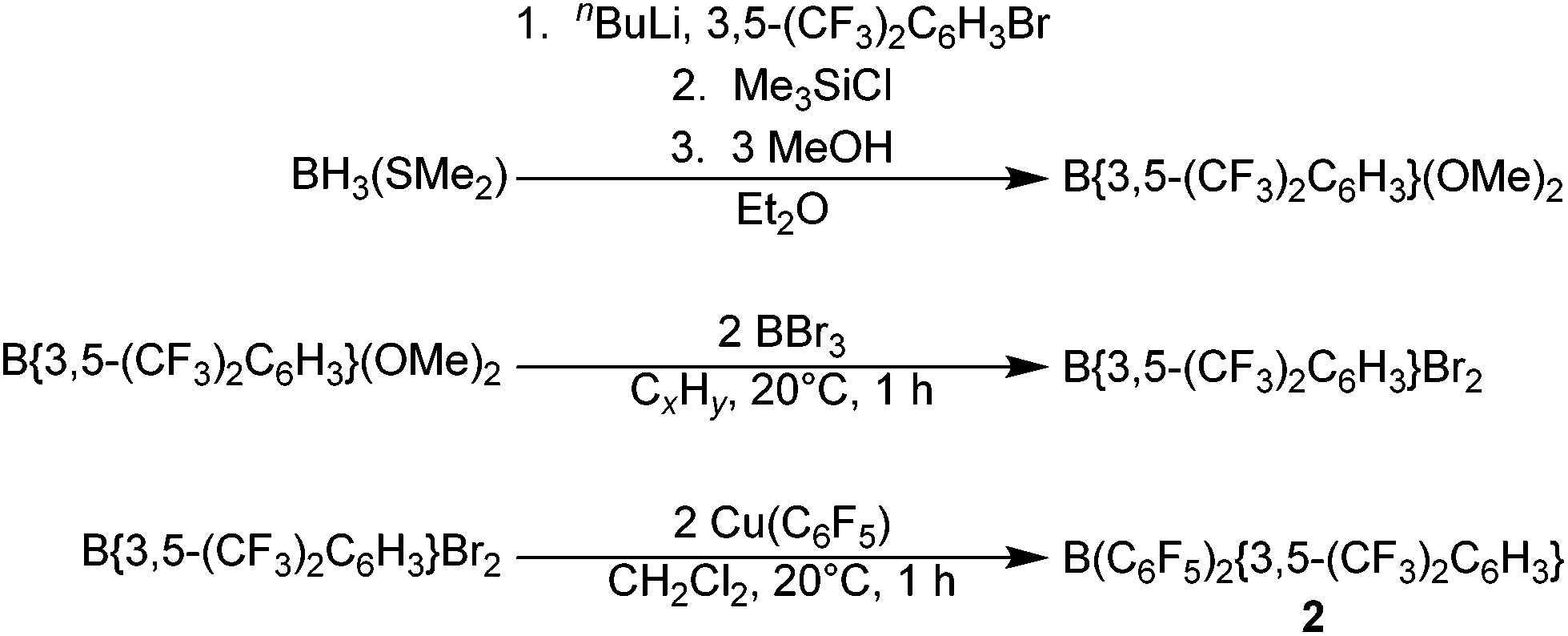 | ||
| Scheme 2 Synthetic route for the synthesis of 2. | ||
The syntheses of 7–9 have previously been reported by Ashley et al.,24 however we found a number of minor changes to the published methodologies were necessary to synthesise these compounds. For the lithium–halogen exchange of C6Cl6 and nBuLi to generate LiC6Cl5, we found control of temperature was critical with any temperature increase above −78 °C leading to an unacceptably high generation of decomposition products. Additionally we found that due to the sparing solubility of C6Cl6 in Et2O using as large a volume of solvent as practical was advantageous for initial lithiation. Of equal importance was the addition of an anti-solvent (n-pentane) in approximately a 3:2 ratio to precipitate out the aryl-lithium compound, so as to avoid the risk of undesirable Et2O cleavage products, as previously reported by Ashley et al.24 These modifications allowed for the synthesis of 7 (by reaction of three equivalents of LiC6Cl5 with BCl3), and B(C6Cl5)2Cl (by reaction of two equivalents of LiC6Cl5 with BCl3, precursor for 8), and B(C6Cl5)Br2 (by reaction of half an equivalent of Zn(C6Cl5)2 with BBr3, precursor for 9), to be readily achievable. Compound 9 was obtained by reaction of B(C6Cl5)Br2 with two equivalents of Cu(C6F5) and purification by sublimation as previously reported. Rather than using the published synthesis of 8, by reaction of B(C6Cl5)2Cl with Cu(C6F5) in toluene at +80 °C, We successfully synthesised 8 by reaction of B(C6Cl5)2Cl with freshly generated LiC6F5 in a toluene solution at −78 °C followed by slow warming to room temperature, and extraction into n-hexane.
Structural studies
X-ray crystal structures were obtained from single crystals of 5 (grown by slow diffusion of a saturated CH2Cl2 solution into n-hexane at −25 °C), and 6 (grown from a saturated n-hexane solution at −25 °C) (Fig. 2a and b and S1a and b,†Table 5).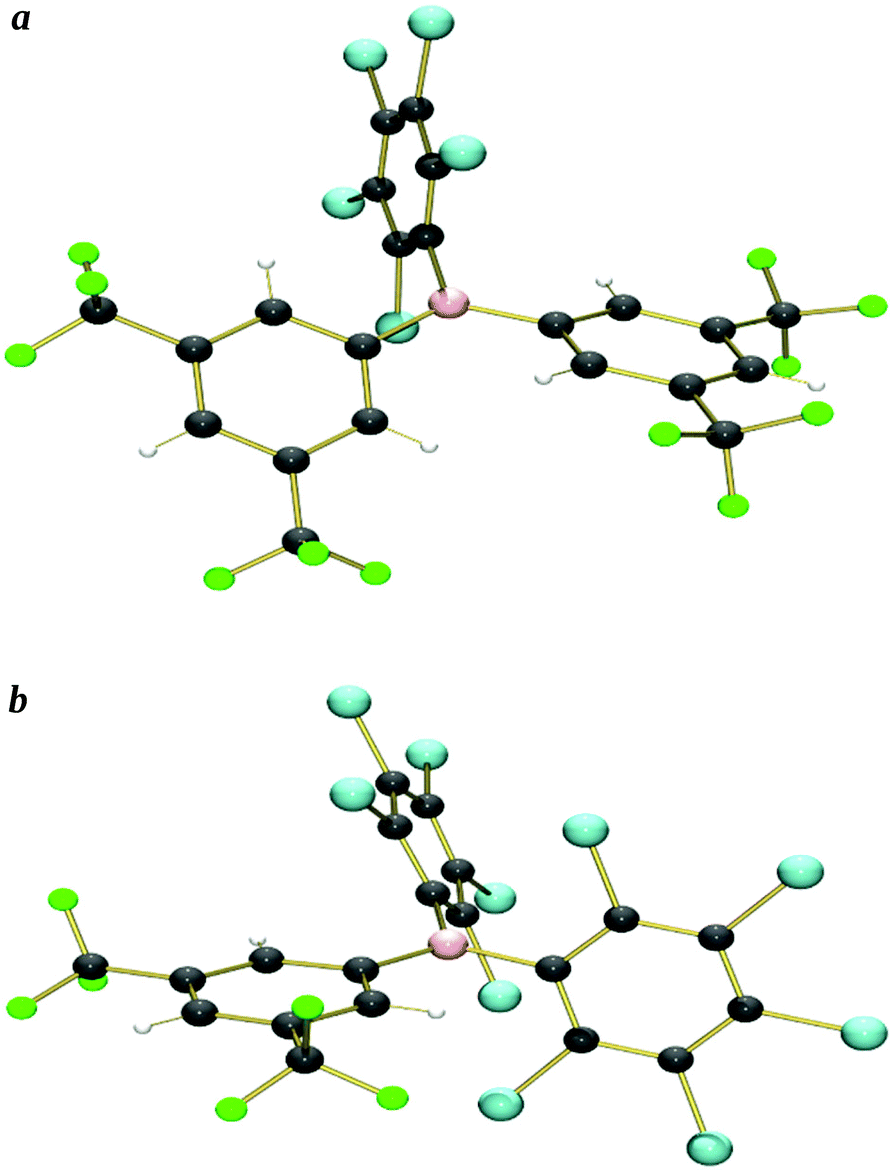 | ||
| Fig. 2 (a) X-ray crystallographic structure of B{3,5-(CF3)2C6H3}2(C6Cl5) 5. (b) X-ray crystallographic structure of B(C6Cl5)2{3,5-(CF3)2C6H3} 6 (minor component of crystallographic disorder removed for clarity). | ||
We have been unable to grow single crystals of 1–3, so instead, geometry optimised structures have been calculated computationally {at the B3LYP/6-311+G(d,p) level of theory, see ESI† for further details} (we previously reported the calculated structure of 134) (Fig. S2a–c†); calculated structures of 4–8 do not show significant differences when compared to their X-ray crystal structures, thereby validating this approach. {9 is an exception, due to the two ArF5 rings having significantly different twist-angles (assumed to be due to crystal packing effects) in the crystal structure, a feature that is not reproduced in the calculated structure.}
The crystallographic and calculated structures of 1–3, 5 and 6, along with the previously published crystal structures of 439 and 7–9,24 all show similar features: a trigonal-planar boron centre and the three aryl rings twisted with respect to the BC3 plane, leading to a propeller type conformation, thereby minimising steric interactions between the aryl rings. The ortho-substituents would dominate such steric interactions leading to the smallest (13–36°) twist occurring for ArF6 rings (ortho-H) the largest (56–80°) for the ArCl5 rings (ortho-Cl) with the ArF5 rings (ortho-F) in the middle (22–52°), as shown in Table 1.
| ArF5 | ArF6 | ArCl5 | |
|---|---|---|---|
| a Crystal structure not known, angles from optimised {B3LYP/6-311+G(d,p)} structure. b Crystallographic symmetry leads to only one unique twist-angle. c Each aryl ring has significantly different twist-angle: 52(1), 24(2)°. | |||
| B(C6F5)3134 |
37.0°a |
— | — |
| B(C6F5)2{3,5-(CF3)2C6H3} 2 | 51.7°a |
25.1°a |
— |
| B{3,5-(CF3)2C6H3}2(C6F5) 3 | 51.1°a |
30.8°a |
— |
| B{3,5-(CF3)2C6H3}3439 |
— | 36(2)° | — |
| B{3,5-(CF3)2C6H3}2(C6Cl5) 5 | — | 26(1)° | 79.7°b |
| B(C6Cl5)2{3,5-(CF3)2C6H3} 6 | — | 13(0)° | 62(2)° |
| B(C6Cl5)3724 |
— | — | 56(3)° |
| B(C6Cl5)2(C6F5) 824 |
22(1)° | — | 59(3)° |
| B(C6F5)2(C6Cl5) 924 |
38(16)°c |
— | 70(1)° |
As the twist-angles of the aryl rings vary, changes in the electronic interactions of the ring and its substituents with the boron centre can be implied. Firstly, large twist-angles orientate the rings such that their ortho-substituents are above/below the boron centred trigonal plane, creating the potential for through-space donation of any lone pair electron density on the ortho-substituents into the formally vacant boron 2pz orbital, as we have shown to occur for the ortho-CF3 substituents of the 2,4- and 2,5-isomers of 4.37 Secondly, as the twist-angle decreases, symmetry considerations imply an increase in overlap between the boron 2pz orbital and the filled (2pz derived) π orbitals of the aromatic ring (while electronic effects via σ-bonding between the boron and aryl rings should be independent of the twist-angle of the aryl rings). In both these cases, donation of electron density into the boron 2pz orbital would be expected to attenuate the borane's Lewis acidity, influencing both its observed electrochemical properties and reactivity.
Electrochemical studies
Cyclic voltammograms were obtained at varying scan rates for 2, 3, 5–9 (Fig. 3), at a glassy carbon electrode, in the weakly-coordinating solvent CH2Cl2 using [nBu4N][B(C6F5)4] as the added electrolyte, and compared with those previously reported by this group under the same conditions for 1 and 4 (reproduced in Fig. S3a and b†).37 Note that whilst electrochemical studies of 7–9 have been previously reported,24 we have repeated the measurements herein to ensure they are comparable with our other results. This is especially true considering our use of the weakly-coordinating anion [B(C6F5)4]− in the electrolyte together with a glassy carbon macro-electrode, in contrast to the previously reported voltammograms using an electrolyte containing the potentially non-innocent [BF4]− anion and a platinum micro-electrode.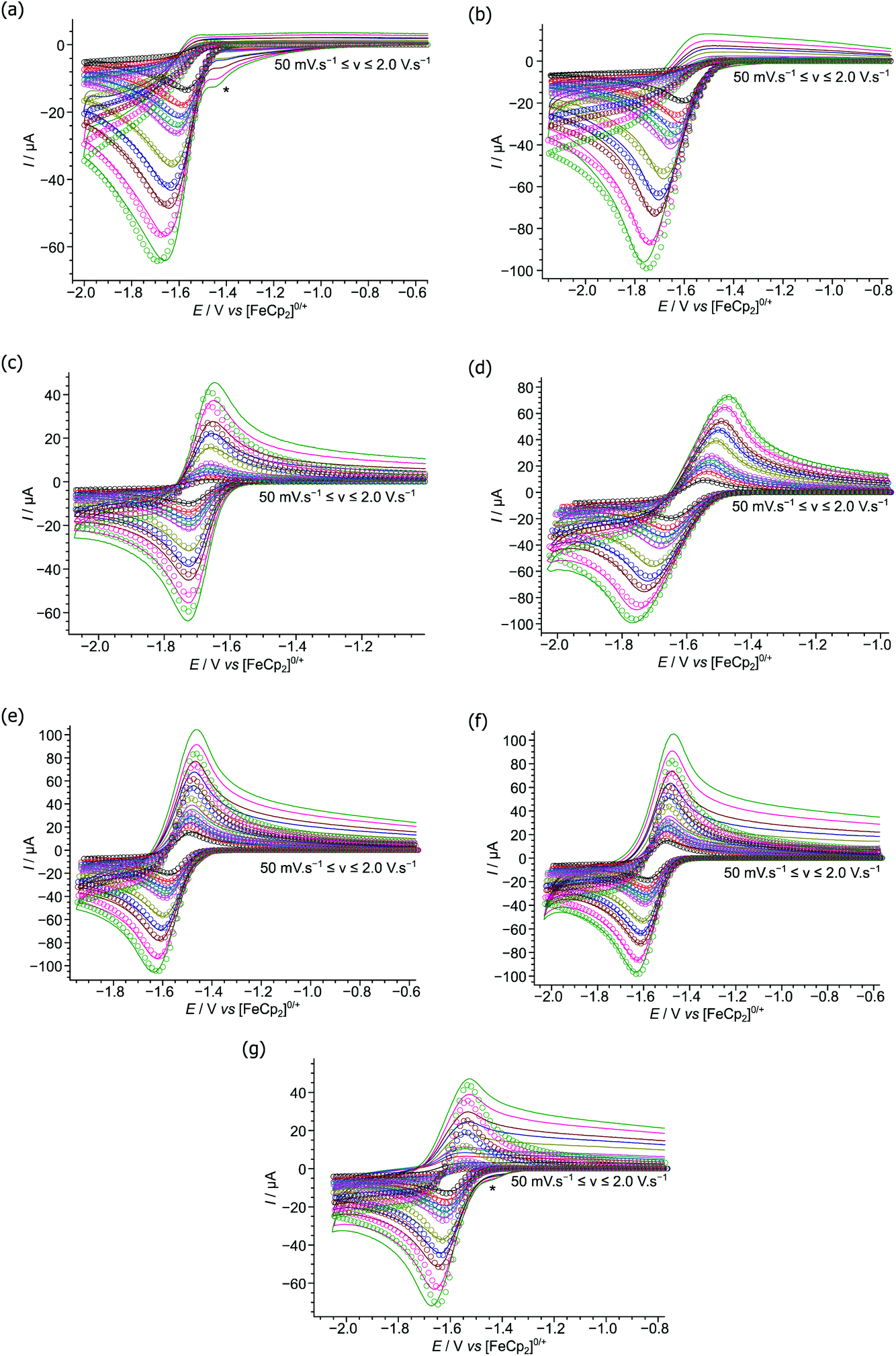 | ||
| Fig. 3 Experimental (line) and simulated (open circles) cyclic voltammograms for the reduction of: (a) B(C6F5)2{3,5-(CF3)2C6H3} 2; (b) B{3,5-(CF3)2C6H3}2(C6F5) 3; (c) B{3,5-(CF3)2C6H3}2(C6Cl5) 5; (d) B(C6Cl5)2{3,5-(CF3)2C6H3} 6; (e) B(C6Cl5)37; (f) B(C6Cl5)2(C6F5) 8; (g) B(C6F5)2(C6Cl5) 9. Shoulders (*) visible at the higher scan rates due to trace impurity in the solvent/electrolyte. | ||
There are three general behaviours observed in the cyclic voltammograms recorded for boranes 1–9. The first, for 2–4, showing completely irreversible reductions at all scan rates studied (50 mV s−1 to 2.0 V s−1), indicating vary rapid reaction/decomposition of the radical-anion intermediate generated upon reduction of the parent borane.34 The second, for 1, 5 & 9, appear irreversible at slower scan rates yet as the scan rates increase appear quasi-reversible as the kinetics of the homogeneous follow-on chemical decomposition step are outrun on the voltammetric timescale. Finally, 6–8, appear quasi-reversible over the entire range of scan rates.
To quantify the observed redox behaviours we performed digital simulations of the experimental voltammetric data, modelling the redox processes using an EC-mechanism34 (i.e. a reversible, heterogeneous electron transfer step followed by an irreversible, homogeneous chemical step generating electro-inactive products – other postulated mechanisms produce a poor fit to the data). These digital simulations allowed us to extract pertinent mechanistic parameters such as the formal redox potentials and charge transfer coefficients (E° and α respectively) and kinetic parameters for the electron transfer (k0) and follow-on chemical step (kf) as shown in Table 2 (together with our previously reported parameters for 1 and 4).
BArHalx + e− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) BArHalx˙− BArHalx˙− |
BArHalx˙− ⇒ decomposition |
D(BArHalx) = D(BArHalx˙−)/cm2 s−1a |
|||
|---|---|---|---|---|---|
| E°/V vs. [FeCp2]0/+ | α | k 0/cm s−1 |
k
f/s−1b |
||
| a With exception of 7, all diffusion constants (D) obtained via1H and/or 19F DOSY NMR spectroscopy. b k f values are modelled as a pseudo first-order process. | |||||
| B(C6F5)3137 |
−1.52 ± 0.01 | 0.379 | 1.45 × 10−2 | 9.2 | 0.85 × 10−5 |
| B(C6F5)2{3,5-(CF3)2C6H3} 2 | −1.56 ± 0.01 | 0.300 | 8.45 × 10−3 | ≥30 | 1.41 × 10−5 |
| B{3,5-(CF3)2C6H3}2(C6F5) 3 | −1.57 ± 0.01 | 0.387 | 4.85 × 10−3 | ≥25 | 1.18 × 10−5 |
| B{3,5-(CF3)2C6H3}3437 |
−1.61 ± 0.01 | 0.419 | 4.56 × 10−3 | ≥25 | 3.76 × 10−5 |
| B{3,5-(CF3)2C6H3}2(C6Cl5) 5 | −1.70 ± 0.01 | 0.512 | 2.69 × 10−1 | 0.36 | 2.54 × 10−5 |
| B(C6Cl5)2{3,5-(CF3)2C6H3} 6 | −1.60 ± 0.01 | 0.415 | 1.14 × 10−2 | 0.056 | 1.44 × 10−5 |
| B(C6Cl5)37 | −1.54 ± 0.01 | 0.425 | 1.20 × 10−2 | ≤10−5 | 1.22 × 10−5 |
| B(C6Cl5)2(C6F5) 8 | −1.54 ± 0.01 | 0.416 | 1.13 × 10−2 | ≤10−5 | 1.26 × 10−5 |
| B(C6F5)2(C6Cl5) 9 | −1.58 ± 0.01 | 0.445 | 5.59 × 10−2 | 0.88 | 1.68 × 10−5 |
These formal redox potentials are a measure of the electrophilicity of the LUMO (formally the vacant boron 2pz orbital), and one might expect them to correlate to the Lewis acidity of the free borane; the more negative the E° value, the less electrophilic the boron. When considering only the homo-tri(aryl)boranes 1, 4 & 7, the observed redox potential imply that the net electron withdrawing effect of the aryl rings increases (thereby making the boron more electrophilic) in the order ArF6 < ArCl5 ≈ ArF5. This is contrary to the description of ArCl5 as more electron withdrawing that ArF5 (due to back-donation of the filled fluorine 2p orbitals into the aromatic π* orbitals counteracting its high electronegativity, this effect is much reduced for chlorine due to the poorer overlap of 3p orbitals with the aromatic π orbitals; as shown by Hammett parameters: σparaCl = 0.227, σparaF = 0.062).24 We propose that this is due to through-space interaction between the ortho-Cl substituents with the boron centre in 7 (B⋯Cl ca. 3.1 Å), quenching the electrophilicity by donation of electron density from the chlorine lone pairs into the formally empty boron 2pz orbital. The orientation {large twist-angle, 56(3)°, orientating ortho-Cl above/below the BC3 trigonal-plane} and size of the chlorine 3p orbitals, make such interaction much more favourable than for 1; an effect which might also be expected to occur to a varying degree for all the other boranes incorporating ArCl5 substituents, 4, 5, 8 & 9.
Further discussion of the trends in E° is given below, but a linear trend can clearly be observed for the series 1–4: B(ArF5)x(ArF6)y (the stepwise substitution of ArF5 with ArF6). Similar experiments performed for the series B{2,4,6-(CH3)3C6H2}n(C6F5)3−n (n = 1–3)26 at a Pt disc in thf/[nBu4N][B(C6F5)4], displayed a pronounced linear change in E1/2 varying by ca. 500 mV as each mesityl ring was substituted with a C6F5 ring.
Additionally, since the reversibility of the voltammograms is dependent on the stability of the radical-anion intermediate generated upon reduction of the parent borane, and as we have previously reported,34,37 it is assumed that the decomposition pathway proceeds via interaction of the radical-anion with the solvent, it may be implied that the slower the rate of decomposition (quantified by the rate constant kf), the more steric shielding is present around the boron centre to stabilise the radical anion. Hence, the presence of ArCl5 ring(s) reduces kf values by up to five orders of magnitude due to its high steric bulk (due to the ortho-Cl substituents and the associated high twist-angles), compared to the ArF5 and ArF6 rings alone.
Measurements of Lewis acidity
To-date common methods for the quantification of Lewis acidity have been based on spectroscopic techniques. One such technique, the “Gutmann–Beckett method”,40,41 involves adduct formation between the Lewis acid of interest and the Lewis base triethylphosphine-oxide (Et3PO); and measurement of its 31P chemical shift (commonly reported as the “acceptor number” – a normalised proxy for the observed chemical shift relative to that of free Et3PO in hexane). With the rational that increased Lewis acidity, results in de-shielding (and thereby an increase in chemical shift) of the bound phosphorus atom. A comprehensive study of Lewis acidic boron compounds was recently published by Sivaev and Bregadze,42 identifying a number of electronic effects which may influence the measurement of Lewis acidity by this method. As a measure of Lewis acid–base adduct formation the “Gutmann–Beckett method” is also somewhat limited by steric effects, and is blind to any associated electronic influences present in the trigonal planar parent borane that are no longer present in the tetrahedral adduct (such as through-space donation from ortho-substituents into the boron 2pz orbital, as discussed above for the ArCl5 substituents). Considering that all our Lewis acids are boron based, we will also consider both the 11B chemical shifts of the free boranes and their Et3PO adducts as tools to measure Lewis acidity.Spectral data for Et3PO adducts of 1–6, 8, 9, are detailed in Tables 3 and S1.† (As previously reported,24 due to its steric bulk 7 will not form an adduct with Et3PO, clearly identifying a limit on such methodology.)
| δ P(adduct)/ppm | δ B(adduct)/ppm | |
|---|---|---|
| All values herein have been re-measured by the authors, internally referenced to δP (Et3PO) = +50.70 ppm.a Previously reported in ref. 42 over the range δP +77.0 to +78.1.b Previously reported in ref. 24 as δP +74.5 and δB +0.3.c Previously reported in ref. 24 as δP +75.8 and δB −1.1. | ||
| B(C6F5)31a |
+77.15 | −2.46 |
| B(C6F5)2{3,5-(CF3)2C6H3} 2 | +78.01 | +0.27 |
| B{3,5-(CF3)2C6H3}2(C6F5) 3 | +77.68 | +2.95 |
| B{3,5-(CF3)2C6H3}34 | +78.80 | +4.27 |
| B{3,5-(CF3)2C6H3}2(C6Cl5) 5 | +76.7 (br) | +4.37 |
| B(C6Cl5)2{3,5-(CF3)2C6H3} 6 | +76.9 (v.br) | +6.06 |
| B(C6Cl5)37 | No adduct formation | |
| B(C6Cl5)2(C6F5) 8b |
+74.70 | +2.27 |
| B(C6F5)2(C6Cl5) 9c |
+75.35 | −0.31 |
Thereby we have three measures to quantify the Lewis acidity of different boranes: the 11B chemical shift of the free borane, and the 31P and 11B chemical shifts of the Lewis acid–base adduct Et3POB(Ar′)2(Ar′′), together with the standard reduction potential (E°) of the free borane which measures the electrophilicity of the formally vacant 2pz orbital of the boron (Table 2). Comparing the variation in these measures across the stepwise substitution of aryl rings in the boranes 1–9 (Fig. 4), together with considering any correlation between all four measures (Fig. S5†), allows for general trends (and outliers) to be identified.
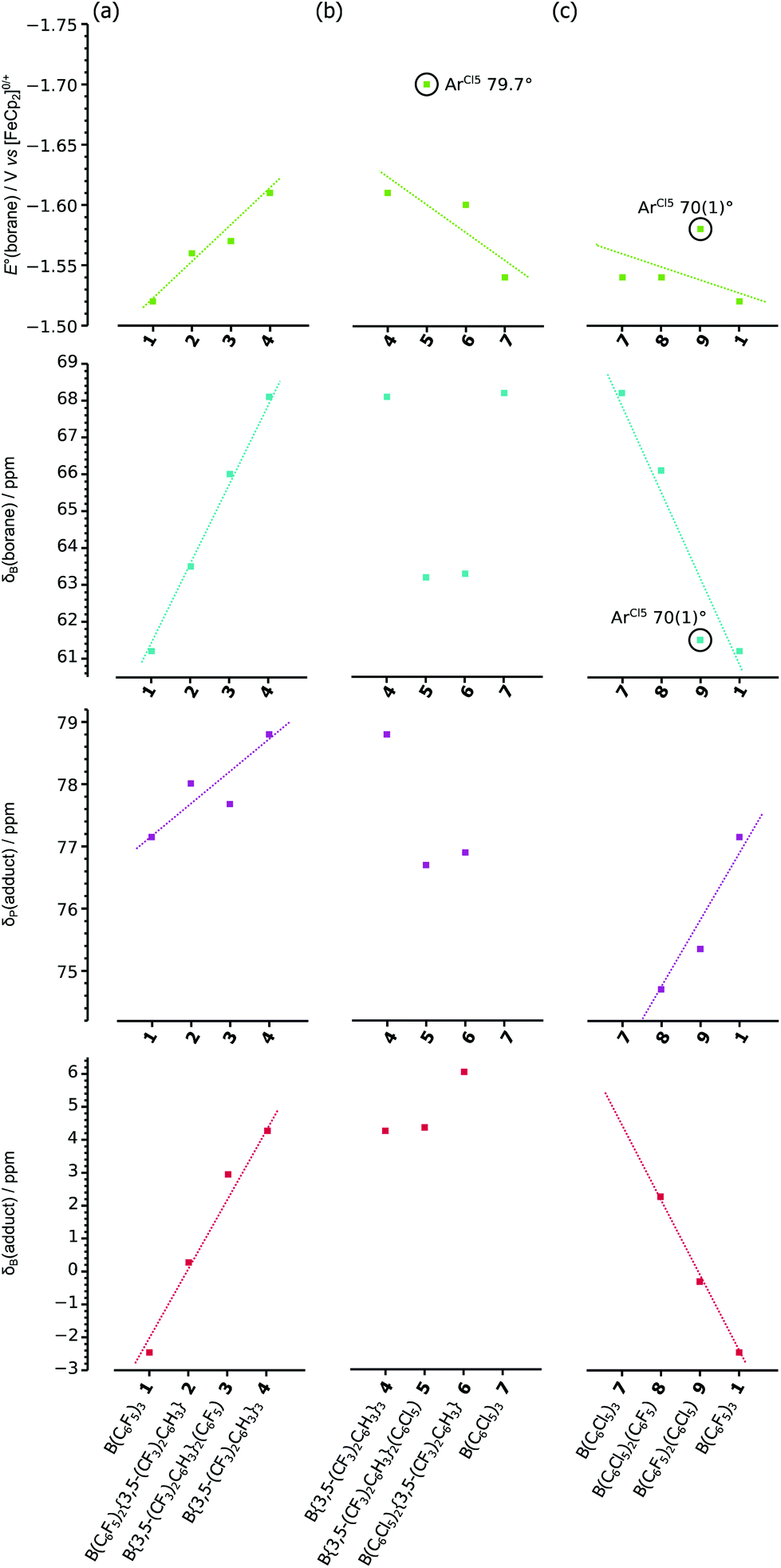 | ||
| Fig. 4 Variation of E°(borane), δB(borane), δP(adduct) and δB(adduct); for (a) B(ArF5)x(ArF6)y1–4, (b) B(ArF6)y(ArCl5)z4–7, and (c) B(ArF5)x(ArCl5)z7–9 & 1. | ||
The series B(ArF5)x(ArF6)y, 1–4, shows linear variations in all four measures across the series, with −E°, δB(borane), δP(adduct) and δB(adduct) all increasing as ArF5 rings are substituted with ArF6 rings.
The series B(ArF6)y(ArCl5)z, 4–7, shows clear trends only for −E° showing a general decrease as the ArF6 rings are substituted with ArCl5 rings (with exception of 5, which appears higher than the trend otherwise implies). For δB(borane), δP(adduct) and δB(adduct), any trends are relatively poor (further emphasised by the fact that an adduct of 7 cannot be formed).
Finally, the series B(ArF5)x(ArCl5)z, 7–9, 1, shows generally linear variations with δP(adduct) increasing, while −E°, δB(borane) and δB(adduct) all decrease as ArCl5 rings are substituted with ArF5 rings. However, compound 9, while consistent with the general trends, is clearly an outlier for −E° and δB(borane) in which cases its higher and lower (respectively) than the trends would otherwise imply.
The two most prominent outliers, in both cases where −E° is higher than the trends otherwise imply, 5 & 9 (circled in Fig. 4), incorporate a single ArCl5 ring with a large (>70°) twist-angle, leading to the ortho-Cl substituents being located above/below the boron centre. As previously suggested (vide supra) this orientation allows for significant through-space donation of electron density into the formally vacant boron 2pz orbital,37 thereby reducing the electrophilicity (increasing −E°) of the boron from that predicted otherwise by the trend {the observation of 9 as an outlier in the general trend of δB(borane), could also be rationalised similarly}. It should be noted that 9 does not appear as an outlier in plots of δP(adduct) and δB(adduct) (considering the poor trends of the 4–7 series, 5 cannot be identified as an outlier or not), since the geometry changes at the boron centre and the explicit filling of the boron 2pz orbital, eliminates the possibility for electronic effects due to ortho-substituents. Similar interactions, leading to the “Gutmann–Beckett method” failing to give adequate measures of Lewis acidity, have been previously noted for ferrocenyl substituted silicon cations; where the cationic silicon interacts with the ferrocenyl backbone, an interaction that is quenched upon adduct formation.43
Considering the correlations between all six combinations of the four measurements (see Fig. S5† for correlation plots), for the entire data set (1–9), there is little evidence of correlation between any of the potential measures of Lewis acidity with themselves nor with the measure of electrophilicity, −E°. However, there are generally linear correlations between all six combinations of measures for the B(ArF5)x(ArF6)y, 1–4, series (which it should be noted, are the only compounds which do not incorporate ArCl5 substituents).
This lack of clear correlation between any pair of measures of Lewis acidity, particularly that of E° (i.e. electrophilicity of the boron centre) and δP(adduct) (i.e. the “Gutmann–Beckett” acceptor number) (Fig. S5b†), is not surprising given that the “Gutmann–Beckett method” is highly dependent on steric constraints and adduct formation that will not occur for the most sterically bulky Lewis acids, such as 7, and the 2,4- and 2,5-isomers of 4;37 whereas, electron transfer is not influenced to any appreciable extent by the sterics surrounding the boron centre in these tri(aryl)boranes, where the pendant conjugated aryl systems ensure electron transfer to even the most crowded boron centres.37 What these correlations do reveal is that the electrochemical measurements clearly and easily identify outliers for the trends, and that where these occur there is an obvious, but often unconsidered electronic effect due to the twist-angle of the aryl rings affecting either the donation of electron density into the vacant 2pz orbital on boron from ortho-substituents on the rings, or π-electron density from the aryl rings or both.
H2 cleavage by FLPs
The ability of these boranes to act as the Lewis acidic component of an FLP for the cleavage of H2 was screened, in combination with the Lewis base P(tBu)3 in dichloromethane at room temperature, with the reactions monitored by 1H and 11B NMR spectroscopy (see Fig. S6–S14†).In addition to the now well studied 1, under these mild conditions, 2, 3, 5, 6, 8 & 9, irreversibly heterolytically cleave H2 to generate terminal-tri(aryl)borohydrides; as shown by the observation of a 1:1:1:1 quartet in the 1H NMR spectra and a doublet in the 11B NMR spectra with 1JHB coupling of 85–95 Hz (Table 4). (H2 cleavage has also been previously reported for the Lewis acids 8 & 9, with the Lewis bases tmp & lutidine at elevated temperatures,25,44 and as an equilibrium process with thf as the Lewis base.23) As previously reported,21,37 while 4 rapidly cleaves H2 it does not lead to generation of a terminal-hydride, but instead to the bridging-hydride species [μ-H(4)2]−. Resonances unequivocally associated with [μ-H(4)2]− could not be distinguished in either the 1H or 11B NMR spectra (despite [(tBu)3PH][μ-H(4)2] remaining soluble in CD2Cl2). Finally, while 7/P(tBu)3 and 7/thf FLPs have previously been shown to cleave H2, albeit at elevated temperatures,23,45 under these mild conditions there was no evidence for reaction of H2 with the FLP 7/P(tBu)3 over a period of five days.
| δ H(−HBArHalx)/ppm | δ B(−HBArHalx)/ppm | 1 J HB/Hz | |
|---|---|---|---|
| All values herein have been re-measured by the authors. | |||
| B(C6F5)31 | +3.61 | −25.3 | 93.9 |
| B(C6F5)2{3,5-(CF3)2C6H3} 2 | +3.69 | −19.9 | 86.1 |
| B{3,5-(CF3)2C6H3}2(C6F5) 3 | +3.71 | −14.7 | 86.1 |
| B{3,5-(CF3)2C6H3}34 | No terminal-hydride formation | ||
| B{3,5-(CF3)2C6H3}2(C6Cl5) 5 | +4.22 | −10.0 | 86.0 |
| B(C6Cl5)2{3,5-(CF3)2C6H3} 6 | +4.24 | −8.6 | 88.0 |
| B(C6Cl5)37 | No reaction | ||
| B(C6Cl5)2(C6F5) 8 | +4.11 | −14.3 | 86.1 |
| B(C6F5)2(C6Cl5) 9 | +3.94 | −19.6 | 90.0 |
| B{3,5-(CF3)2C6H3}2(C6Cl5) 5 | B(C6Cl5)2{3,5-(CF3)2C6H3} 6 | |
|---|---|---|
| Empirical formula | C22H6BCl5F12 | C20H3BCl10F6 |
| Formula weight | 686.33 | 722.53 |
| Temperature/K | 140(1) | 140(1) |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | P21/n |
| a/Å | 11.5422(8) | 8.5532(17) |
| b/Å | 13.1500(8) | 9.742(2) |
| c/Å | 16.6630(10) | 30.812(8) |
| α/° | 90.0 | 90.0 |
| β/° | 103.799(7) | 90.871(18) |
| γ/° | 90.0 | 90.0 |
| Volume/Å3 | 2456.1(3) | 2567.1(10) |
| Z | 4 | 4 |
| ρ calc/mg mm−3 | 1.856 | 1.870 |
| μ/mm−1 | 0.696 | 1.142 |
| F(000) | 1344.0 | 1408.0 |
| Crystal size/mm3 | 0.1 × 0.05 × 0.05 | 0.1 × 0.05 × 0.05 |
| Radiation | Mo Kα (λ = 0.71073 Å) | Mo Kα (λ = 0.71073 Å) |
| 2Θ range for data collection | 6.688 to 52.724° | 5.764 to 52.0° |
| Index ranges | −11 ≤ h ≤ 14, −16 ≤ k ≤ 16, −20 ≤ l ≤ 20 | −10 ≤ h ≤ 9, −12 ≤ k ≤ 12, −38 ≤ l ≤ 36 |
| Reflections collected | 9849 | 20074 |
| Independent reflections | 2506 [Rint = 0.0534, Rsigma = 0.0572] | 5027 [Rint = 0.2721, Rsigma = 0.3383] |
| Data/restraints/parameters | 2506/0/183 | 5027/75/357 |
| Goodness-of-fit on F2 | 1.009 | 1.013 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0417, wR2 = 0.0808 | R 1 = 0.0891, wR2 = 0.1457 |
| Final R indexes [all data] | R 1 = 0.0755, wR2 = 0.0925 | R 1 = 0.2819, wR2 = 0.2313 |
| Largest diff. peak/hole/e A−3 | 0.38/−0.27 | 0.54/−0.69 |
Since all our reactions were performed under the same conditions and monitored throughout, it is possible to make qualitative descriptions as to the kinetics and mechanism(s) of H2 cleavage by these boranes. We observed the reaction with 1 as the Lewis acid reaching completion fastest (ca. 5 hours following H2 addition), with the reaction time increasing in the order, 1 < 2 ≈ 9 < 3 < 8 < 5 ≈ 6 (ca. 23% conversion, after ca. 96 hours). Cleavage with 1–3 was near quantitative, while the Lewis acids 5, 6, 8 & 9 led to a mixture of products by 11B NMR spectroscopy (the by-products giving signals in the range δB +6 to −3 ppm, corresponding to four-coordinate boron centres – we speculate that some of these could be due to trace moisture and the products of its reaction with the FLP). One feature of note in the 1H NMR spectra recorded for 1–3 is initially the borohydride resonances are observed as sharp signals before broadening (for 1 and 2, without changing δH; for 3, initially observed at ca. +3.3 ppm and gradually shifting to ca. +3.7 ppm before the signal broadens) to eventually resolve into the characteristic broad 1:1:1:1 quartet. Further, the dissolved H2 is not observed until the signal has stopped shifting (while it is clearly observed as the reaction progresses for 5, 6, 8 & 9). These observations may provide further clues as to the mechanism of FLP H2 cleavage, and we intend to investigate them further in later work.
The reactivity of these boranes towards heterolytic H2 cleavage unsurprisingly depends on both the Lewis acidity and electrophilicity of the boron centre and the steric bulk of the substituents; hence the lack of reactivity under these conditions of the bulky 7 despite its apparently favourable electrophilicity. However, as we have discovered herein, the correlation between the various aryl substituents and the Lewis acidity – and even the steric buttressing around the boron centre – is by no means simple. There are subtle steric and electronic influences at work (e.g. through space interactions of ortho-substituents with the vacant 2pz orbital affecting both steric orientation of these rings and the electronics of the borane) that this report has highlighted, that require careful consideration when designing potential new Lewis acidic boranes for FLP reactions.
Conclusions
We have synthesised four new hetero-tri(aryl)boranes, 2, 3, 5 & 6. Which, together with three known homo-tri(aryl)boranes, 1, 4 & 7 and two known hetero-tri(aryl)boranes, 8 & 9, give a series of nine compounds linked by stepwise substitution of their aryl rings. Their redox chemistry has been investigated electrochemically, along with their suitability as Lewis acid components of FLP systems for the heterolytic cleavage of H2 under mild conditions.Rapid electrochemical techniques and analysis of the position and shape of the resulting voltammograms allow for the electrophilicity to be quantified along with a qualitative description of the steric shielding around the boron centre. While the correlations both between different spectroscopic measures of Lewis acidity, and between measures of Lewis acidity and electrochemical measures of electrophilicity are generally poor, the electrochemical measurements easily allow us to identify outliers from trends within specific series of substituted boranes. These arise due to electronic effects (such as, interactions of ortho-substituents and aryl π orbitals, with the formally vacant boron 2pz orbital) are not always obvious from spectroscopic measurements of Lewis acidity. Where these electronic interactions are identified, we have found them to correspond to a higher than average twist-angle between the boron trigonal-plane. Whilst steric buttressing and inductive/mesomeric through-bond effects are often considered in the design of new boranes for FLP studies, the degree of twist angle of each aryl ring and the resulting potential for through-space electronic effects is shown to be an important but often unconsidered factor in determining the Lewis acidity and reactivity of these compounds.
While all but one of these compounds cleaves H2 under mild conditions as part of an FLP, the rate of cleavage depends on both the Lewis acidity/electrophilicity of the boron centre and its surrounding steric bulk. Establishing that while increasing the formal Lewis acidity should increases the rate of reaction, it must be balanced by preventing too much steric bulk around the active site that might inhibit the rate of reaction. However, while these new boranes do not appear to cleave H2 as rapidly as the archetype 1, these slower reactions afford the opportunity to observe signals arising from potential intermediates formed during H2 cleavage by NMR spectroscopy. A comprehensive kinetic investigation of these reactions as part of our future work could possibly lead to a greater understanding of the mechanism of H2 cleavage by FLPs.
Experimental
All reactions and manipulations were performed under an atmosphere of dry oxygen-free N2, using either standard Schlenk techniques or in an MBraun UNIlab glovebox. All solvents were dried prior to use by refluxing over an appropriate drying agent {Na/benzophenone for n-pentane, n-hexane, petroleum ether (b.p 40–60 °C) and diethyl ether; Na for toluene; CaH2 for dichloromethane}, collected by distillation under a dry oxygen-free N2 atmosphere and stored over 4 Å molecular sieves prior to use.NMR Spectra were obtained on a Bruker Avance DPX-500 spectrometer; for 1H spectra residual protio-solvent was used as an internal standard; for 13C the solvent resonance(s) were used as an internal standard;46 for 19F spectra CFCl3 was used as an external standard; for 11B spectra BF3·Et2O was used as an external standard; for 31P spectra 85% H3PO4 was used as an external standard.
High resolution mass spectrometry was performed by the EPSRC Mass Spectrometry Service at the University of Swansea. Elemental analyses were performed by the Elemental Analysis Service at London Metropolitan University.
Single crystals of 5 were grown by slow diffusion of a saturated CH2Cl2 solution into n-hexane at −25 °C, single crystals of 6 were grown from a saturated n-hexane solution at −25 °C. For 5 and 6, suitable crystals were selected, encapsulated in a viscous perfluoropolyether and mounted on an Agilent Technologies Xcaliber-3 single crystal X-ray diffractometer using Mo Kα radiation where the crystals were cooled to 140 K during data collection and a full sphere of data collected. The data was reduced and an absorption correction performed using Agilent Technologies CrysAlisPro version 171.37.35.47 Using Olex2,48 the structures were solved and space group assigned with SuperFlip/EDMA using charge flipping,49 and then refined with the ShelXL version 2014/7 refinement program using least squares minimisation.50
CCDC 1418145 (5), and 1418144 (6) contains the supplementary crystallographic data for this paper.
Electrochemical studies were carried out using a Metrohm Autolab μ-II, PGSTAT30 or PGSTAT302N potentiostat linked to a computer running Metrohm Autolab NOVA version 1.11 software, in conjunction with a three electrode cell comprising: a glassy carbon disc working electrode (Bioanalytical Systems, Inc., ca. 7.0 mm2 area calibrated using the [FeCp2]0/+ redox couple), a platinum wire (99.99% purity) counter electrode, and a silver wire (99.99% purity) pseudo-reference electrode. All working electrodes were polished with 0.3 μm α-alumina and dried prior to use. All electrochemical measurements were performed at ambient temperature under a dry N2 atmosphere (using either a custom-built electrochemical cell or within a glovebox), in CH2Cl2 containing 50 mM [nBu4N][B(C6F5)4] as the supporting electrolyte and between 1.5 and 2.5 mM of the analyte species of interest. Cyclic voltammetric measurements were, where possible, iR-compensated using positive-feedback to within 85 ± 5% of the uncompensated solution resistance (ca. 666 Ω). [nBu4N][B(C6F5)4] was synthesised according to published methods.51 All potentials were referenced to the [FeCp2]0/+ redox couple, which was added as an internal standard. Simulations of electrochemical processes were performed using ElchSoft DigiElch version 7.096 software.52
B{3,5-(CF3)2C6H3}2Br,38 Cu(C6F5),53 Zn(C6Cl5)2, B(C6Cl5)37, and B(C6F5)2(C6Cl5) 9,24 were synthesised as previously reported. Characterisation data for the intermediate B{3,5-(CF3)2C6H3}(OMe)2, and the impurity B{3,5-(CF3)2C6H3}2(OH) is detailed in the ESI.† All other reagents were obtained from commercial suppliers and used as supplied.
B{3,5-(CF3)2C6H3}Br2
n BuLi (6.25 cm3, 10 mmol, 1.6 M in hexanes) was slowly added to a cooled (−78 °C) solution of 3,5-bis(trifluoromethyl)bromobenzene (1.72 cm3, 10 mmol) in Et2O (20 cm3) and stirred for 10 min. BH3·SMe2 (0.95 cm3, 10 mmol) is added, and after 10 min the reaction mixture warmed to room temperature and stirred for a further 30 min. Me3SiCl (1.27 cm3, 10 mmol) is added to the clear orange solution, resulting in the rapid formation of a white precipitate, the mixture is stirred for 30 min. Methanol (1.2 cm3, 30 mmol) is slowly added, resulting in rapid evolution of H2, and the mixture stirred for an hour. Volatiles are removed in vacuo to give a cloudy white oil; the intermediate B{3,5-(CF3)2C6H3}(OMe)2 is extracted into 25 cm3 petroleum ether and isolated as a clear pale yellow solution by filtration (via cannula). BBr3 (2.0 cm3, 20.8 mmol) is added and the mixture stirred for 30 min, the mixture is once again filtered (via cannula) isolating a clear pale yellow solution, all volatiles are removed in vacuo to give the product as a highly reactive yellow oil. Yield 1.63 g (4.2 mmol, 42%).1H NMR (500.21 MHz, CD2Cl2, 25 °C, δ): +8.66 (s, 2H, ArF6 2,6-H), +8.20 (s, 1H, ArF6 4-H); 11B NMR (160.49 MHz, CD2Cl2, 25 °C, δ): +57.3 (br.s); 19F NMR (470.67 MHz, CD2Cl2, 25 °C, δ): −63.3 (s, 6F, ArF6 3,5-CF3).
B(C6F5)2{3,5-(CF3)2C6H3} 2
Solutions of freshly prepared B{3,5-(CF3)2C6H3}Br2 (1.62 g, 4.2 mmol) in 10 cm3 CH2Cl2 and Cu(C6F5) (2.30 g, 10 mmol) in 40 cm3 CH2Cl2 are combined, and stirred for ca. 1 hour. The reaction mixture is filtered (via cannula) to remove the blue-grey precipitate, isolating the clear pale yellow solution, volatiles are removed in vacuo to give the off-white solid product. Yield: 1.78 g (3.2 mmol, 77%). The product can be further purified by sublimation at 10−1 mbar / 90 °C.1H NMR (500.21 MHz, CD2Cl2, 25 °C, δ): +8.22 (s, 1H, ArF6 4-H), +8.15 (s, 2H, ArF6 2,6-H); 11B NMR (160.49 MHz, CD2Cl2, 25 °C, δ): +63.5 (br.s); 13C{1H} NMR (125.78 MHz, CD2Cl2, 25 °C, δ): +148.3 (br.d, 1JCF = 248 Hz, ArF5 2,6-C), +145.2 (br.d, 1JCF = 260 Hz, ArF5 4-C), +141.3 (br.s, ArF6 1-C), +138.4 (br.d, 1JCF = 255 Hz, ArF5 3,5-C), +137.6 (br.q, 3JCF = 1.8 Hz, ArF6 2,6-C), +132.2 (q, 2JCF = 33.5 Hz, ArF6 3,5-C), +128.9 (sept., 3JCF = 3.7 Hz, ArF6 4-C), +123.7 (q, 1JCF = 273 Hz, ArF6 3,5-CF3); 19F NMR (470.67 MHz, CD2Cl2, 25 °C, δ): −63.3 (s, 6F, ArF6 3,5-CF3), −127.3 (m, 4F, ArF5 2,6-F), −145.2 (t, 3JFF = 18.7 Hz, 2F, ArF5 4-F), −160.4 (m, 4F, ArF5 3,5-F). HRMS-APCI (m/z): [M]+ calc. for C20H3BF16, 558.0070; found, 558.0063. Elemental analysis (calc. for C20H3BF16): C 43.17 (43.03), H 0.57 (0.54).
B{3,5-(CF3)2C6H3}2(C6F5) 3
Solutions of B{3,5-(CF3)2C6H3}2Br (0.72 g, 1.0 mmol) in 10 cm3 CH2Cl2 and Cu(C6F5) (0.23 g, 1.0 mmol) in 10 cm3 CH2Cl2 are combined, and stirred for ca. 1 hour. The reaction mixture is filtered (via cannula) to remove the precipitate, isolating the clear pale yellow solution, volatiles are removed in vacuo to give the off-white solid product. Yield: 0.46 g (0.75 mmol, 75%). The product can be further purified by sublimation at 10−1 mbar / 110 °C.1H NMR (500.21 MHz, CD2Cl2, 25 °C, δ): +8.22 (s, 2H, ArF6 4-H), +8.07 (s, 4H, ArF6 2,6-H); 11B NMR (160.49 MHz, CD2Cl2, 25 °C, δ): +66.0 (br.s); 13C{1H} NMR (125.78 MHz, CD2Cl2, 25 °C, δ): +148.3 (br.d, 1JCF = 250 Hz, ArF5 2,6-C), +144.9 (br.d, 1JCF = 260 Hz, ArF5 4-C), +142.3 (br.s, ArF6 1-C), +138.5 (br.d, 1JCF = 255 Hz, ArF5 3,5-C), +137.7 (br.q, 3JCF = 2.3 Hz, ArF6 2,6-C), +132.3 (q, 2JCF = 33.0 Hz, ArF6 3,5-C), +127.9 (sept., 3JCF = 3.7 Hz, ArF6 4-C), +123.8 (q, 1JCF = 273 Hz, ArF6 3,5-CF3), +113.2 (br.s, ArF5 1-C); 19F NMR (470.67 MHz, CD2Cl2, 25 °C, δ): −63.4 (s, 12F, ArF6 3,5-CF3), −126.0 (m, 2F, ArF5 2,6-F), −146.0 (tt, 3JFF = 19.9 Hz, 4JFF = 4.2 Hz, 1F, ArF5 4-F), −160.1 (m, 2F, ArF5 3,5-F). HRMS-APCI (m/z): [M − H]+ calc. for C22H5BF17, 603.0211; found, 603.0201. Elemental analysis (calc. for C22H6BF17): C 43.86 (43.73), H 1.08 (1.00).
B(C6Cl5)2Cl
C6Cl6 (14.24 g, 50 mmol) was suspended in Et2O (≥300 cm3) and the slurry cooled to −78 °C. nBuLi (35.5 cm3, 50 mmol, 1.41 M in hexanes) was added, and the cooled reaction mixture stirred for 4 hours to give a clear golden solution. Cooled (−78 °C) n-pentane (≥300 cm3) was added to give a fine white precipitate of LiC6Cl5; after 30 min, BCl3 (25 cm3, 25 mmol, 1.0 M in heptane) was slowly added, and the reaction mixture slowly warmed to room temperature and stirred for 12 hours. Removal of volatiles in vacuo, gave a pale orange solid; the product was extracted with toluene (3 × 100 cm3) and isolated by filtration to give an amber solution. Removal of volatiles in vacuo, washing with n-hexane, and drying in vacuo, gave B(C6Cl5)2Cl as a pale yellow solid. Yield: 8.80 g (16.1 mmol, 64.4%).Characterisation data as previously reported in ref. 24.
B{3,5-(CF3)2C6H3}2(C6Cl5) 5
B{3,5-(CF3)2C6H3}2Br (1.00 g, 1.93 mmol) and Zn(C6Cl5)2 (0.55 g, 0.97 mmol) are combined and suspended in 10 cm3 toluene. The reaction vessel is sealed and heated at ca. 75 °C for ca. 72 hours. Once cooled volatiles are removed in vacuo, and the product extracted into CH2Cl2 giving a pale green solution. The solid is precipitated by, addition of n-hexane, concentration in vacuo, and cooling at −25 °C. The micro-crystalline pale green solid was isolated and dried in vacuo. Yield: 0.46 g (0.64 mmol, 33%).1H NMR (500.21 MHz, CD2Cl2, 25 °C, δ): +8.20 (s, 2H, ArF6 4-H), +8.13 (s, 4H, ArF6 2,6-H); 11B NMR (160.49 MHz, CD2Cl2, 25 °C, δ): +65.7 (br.s); 13C{1H} NMR (125.78 MHz, CD2Cl2, 25 °C, δ): +137.4 (br.q, 3JCF = 3.7 Hz, ArF6 2,6-C) +135.4 (s, ArCl5 4-C), +132.9 (s, ArCl5 2,6/3,5-C), +132.3 (q, 2JCF = 33.0 Hz, ArF6 3,5-C), +131.4 (s, ArCl5 2,6/3,5-C), +127.7 (sept., 3JCF = 3.7 Hz, ArF6 4-C), +123.6 (q, 1JCF = 273 Hz, ArF6 3,5-CF3); 19F NMR (470.67 MHz, CD2Cl2, 25 °C, δ): −63.2 (s, 12F, ArF6 3,5-CF3). HRMS-APCI (m/z): [M]+ calc. for C22H6BCl5F12, 685.8784; found, 685.8773. Elemental analysis (calc. for C22H6BCl5F12): C 38.62 (38.49), H 0.81 (0.88).
B(C6Cl5)2{3,5-(CF3)2C6H3} 6
n BuLi (1.56 cm3, 2.2 mmol, 1.41 M in hexanes) was slowly added to a cooled (−78 °C) solution of 3,5-bis(trifluoromethyl)bromobenzene (0.38 cm3, 2.2 mmol) in Et2O (50 cm3) and left to stir for 1 hour. B(C6Cl5)2Cl (1.23 g, 2.3 mmol) was dissolved in toluene (20 cm3), cooled (−78 °C), and slowly added to the reaction mixture, which was left to stir at −78 °C for 3 hours and then slowly warmed to room temperature over 18 hours. Removal of volatiles in vacuo, gave a pale yellow solid. This was extracted with n-hexane (2 × 50 cm3), which after removal of volatiles in vacuo, gave a light yellow powder. Yield 0.78 g (1.1 mmol, 50%).1H NMR (500.21 MHz, CD2Cl2, 25 °C, δ): +8.15 (s, 1H, ArF6 4-H), +8.03 (s, 2H, ArF6 2,6-H); 11B NMR (160.49 MHz, CD2Cl2, 25 °C, δ): +66.0 (br.s); 13C{1H} NMR (125.78 MHz, CD2Cl2, 25 °C, δ): +137.3 (br.q, 3JCF = 2.3 Hz, ArF6 2,6-C) +137.0 (s, ArCl5 4-C), +133.5 (s, ArCl5 2,3,5,6-C), +132.5 (q, 2JCF = 33.4 Hz, ArF6 3,5-C), +128.4 (sept., 3JCF = 3.8 Hz, ArF6 4-C), +123.7 (q, 1JCF = 273 Hz, ArF6 3,5-CF3); 19F NMR (470.67 MHz, CD2Cl2, 25 °C, δ): −63.3 (s, 6F, ArF6 3,5-CF3). HRMS-APCI (m/z): [M]+ calc. for C20H3BCl10F6, 721.7058; found, 721.7054. Elemental analysis (calc. for C20H3BCl10F6): C 33.36 (33.24), H 0.50 (0.42).
B(C6Cl5)2(C6F5) 8
n BuLi (1.40 cm3, 1.97 mmol, 1.41 M in hexanes) was slowly added to a cooled (−78 °C) solution of bromopentafluorobenzene (0.25 cm3, 2.0 mmol) in toluene (50 cm3) and stirred for 15 minutes. A solution of B(C6Cl5)2Cl (1.09 g, 2.0 mmol) in toluene (20 cm3) was then slowly added to the reaction mixture, which was subsequently left to warm to room temperature over 18 hours. The solution was filtered through celite (via cannula), and the volatiles removed in vacuo to give a sticky amber solid. This was then extracted with n-pentane (5 cm3), which when cooled (−78 °C) precipitated the product as a pale yellow powder. Yield: 0.28 g (0.41 mmol, 21%).Characterisation data as previously reported in ref. 24.
Acknowledgements
G. G. W. thanks the Royal Society for financial support by a University Research Fellowship. The research leading to these results has received funding from the European Research Council under the ERC Grant Agreements no. 307061 (PiHOMER) and 640988 (FLPower). We acknowledge the use of the EPSRC funded National Chemical Database Service hosted by the Royal Society of Chemistry, and the EPSRC UK National Mass Spectrometry Facility (NMSF) at the University of Swansea. We thank the Research Computing Service at the University of East Anglia for access to the high performance computing cluster.References
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed.
- D. W. Stephan, Dalton Trans., 2009, 3129–3136 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, Comp. Inorg. Chem. II, 2013, 1, 1069–1103 CAS.
- D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2012, 136, 15809–15812 CrossRef PubMed.
- Á. Gyömöre, M. Bakos, T. Földes, I. Pápai, A. Domján and T. Soós, ACS Catal., 2015, 5, 5366–5372 CrossRef.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed; D. J. Scott, T. R. Simmons, E. J. Lawrence, G. G. Wildgoose, M. J. Fuchter and A. E. Ashley, ACS Catal., 2015, 5, 5540–5544 CrossRef PubMed.
- T. Mahdi, J. N. del Castillo and D. W. Stephan, Organometallics, 2013, 32, 1971–1978 CrossRef CAS.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed.
- H. Wang, R. Fröhlich, G. Kehra and G. Erker, Chem. Commun., 2008, 5966–5968 RSC.
- G. Ménard, T. M. Gilbert, J. A. Hatnean, A. Kraft, I. Krossing and D. W. Stephan, Organometallics, 2013, 32, 4416–4422 CrossRef.
- Z. Lu, Y. Wang, J. Liu, Y. jian Lin, Z. H. Li and H. Wang, Organometallics, 2013, 32, 6753–6758 CrossRef CAS.
- A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS PubMed.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918–9919 CrossRef CAS PubMed.
- M. Sajid, A. Klose, B. Birkmann, L. Liang, B. Schirmer, T. Wiegand, H. Eckert, A. J. Lough, R. Fröhlich, C. G. Daniliuc, S. Grimme, D. W. Stephan, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 213–219 RSC.
- J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968–4971 CrossRef CAS PubMed.
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396–8397 CrossRef CAS PubMed.
- T. J. Herrington, A. J. W. Thom, A. J. P. White and A. E. Ashley, Dalton Trans., 2012, 41, 9019–9022 RSC.
- S. C. Binding, H. Zaher, F. M. Chadwick and D. O'Hare, Dalton Trans., 2012, 41, 9061–9066 RSC.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2014, 53, 10218–10222 CrossRef CAS PubMed.
- A. E. Ashley, T. J. Herrington, G. G. Wildgoose, H. Zaher, A. L. Thompson, N. H. Rees, T. Krämer and D. O'Hare, J. Am. Chem. Soc., 2011, 133, 14727–14740 CrossRef CAS PubMed.
- D. O'Hare and A. Ashley, US Pat, 2012/0283340A1, 2012 Search PubMed.
- S. A. Cummings, M. Iimura, C. J. Harlan, R. J. Kwaan, I. V. Trieu, J. R. Norton, B. M. Bridgewater, F. Jäkle, A. Sundararaman and M. Tilset, Organometallics, 2006, 25, 1565–1568 CrossRef CAS.
- J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728–15731 CrossRef CAS PubMed.
- E. J. Lawrence, T. J. Herrington, A. E. Ashley and G. G. Wildgoose, Angew. Chem., Int. Ed., 2014, 53, 9922–9925 CrossRef CAS PubMed.
- E. R. Clark and M. J. Ingleson, Angew. Chem., Int. Ed., 2014, 53, 11306–11309 CrossRef CAS PubMed.
- G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 53, 8272–8275 CrossRef PubMed.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2015 10.1039/c5cs00516g.
- A. Schäfer, M. Reißmann, A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2011, 50, 12636–12638 CrossRef PubMed.
- B. Waerder, M. Pieper, L. A. Körte, T. A. Kinder, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201504171.
- E. J. Lawrence, V. S. Oganesyan, G. G. Wildgoose and A. E. Ashley, Dalton Trans., 2013, 42, 782–789 RSC.
- E. J. Lawrence, V. S. Oganesyan, D. L. Hughes, A. E. Ashley and G. G. Wildgoose, J. Am. Chem. Soc., 2014, 136, 6031–6036 CrossRef CAS PubMed.
- E. J. Lawrence, R. J. Blagg, D. L. Hughes, A. E. Ashley and G. G. Wildgoose, Chem. – Eur. J., 2015, 21, 900–906 CrossRef PubMed.
- R. J. Blagg, E. J. Lawrence, K. Resner, V. S. Oganesyan, T. J. Herrington, A. E. Ashley and G. G. Wildgoose, Dalton Trans., 2015 10.1039/c5dt01918d.
- K. Samigullin, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2014, 33, 3564–3569 CrossRef CAS.
- W. V. Konze, B. L. Scott and G. J. Kubas, Chem. Commun., 1999, 1807–1808 RSC.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. S. Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS.
- A. R. Nödling, K. Müther, V. H. G. Rohde, G. Hilt and M. Oestreich, Organometallics, 2014, 33, 302–308 CrossRef.
- H. Zaher, A. E. Ashley, M. Irwin, A. L. Thompson, M. J. Gutmann, T. Krämera and D. O'Hare, Chem. Commun., 2013, 49, 9755–9757 RSC.
- A. L. Travis, S. C. Binding, H. Zaher, T. A. Q. Arnold, J.-C. Buffet and D. O'Hare, Dalton Trans., 2013, 42, 2431–2437 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- CrysAlisPro, Agilent Technologies, Yarnton, UK Search PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 59–75 CrossRef CAS PubMed; O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS; L. Palatinus and A. van der Lee, J. Crystallogr. Cryst., 2008, 41, 975–984 CrossRef; L. Palatinus, S. J. Prathapab and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575–580 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef CAS PubMed; G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
- S. Lancaster, ChemSpider Synthetic Pages, 2003, 10.1039/SP215, http://cssp.chemspider.com/215; T. E. Krafft, US Pat, 5,679,289, Boulder Scientific Company, 1997 CrossRef CAS PubMed; R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS PubMed.
- DigiElch-Professional, ElechSoft, Kleinromstedt, Germany Search PubMed.
- A. Cairncross, W. A. Sheppard and E. Wonchoba, Org. Synth., 1979, 59, 122 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterisation data for the impurity B{3,5-(CF3)2C6H3}2(OH), characterisation data for the intermediate B{3,5-(CF3)2C6H3}(OMe)2; further details on the X-ray crystallographic studies of 5, 6; further details on the DFT calculations; reproductions of the previously published cyclic voltammograms of 1 and 4, cyclic voltammograms of B{3,5-(CF3)2C6H3}2(OH); further details for the NMR characterisation of the Et3PO adducts of 1–9, and correlation plots between the four different potential measures of Lewis acidity; further details for the H2 cleavage by 1–9/P(tBu)3, including the time resolved 1H and 11B NMR spectra monitoring the reactions. CCDC 1418145 (5) and 1418144 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt03854e |
| ‡ Current address: School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. |
| This journal is © The Royal Society of Chemistry 2016 |