
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast excited state dynamics of iridium(III) complexes and their changes upon immobilisation onto titanium dioxide layers†
Stefanie
Tschierlei
 *ab,
Antje
Neubauer
ac,
Nils
Rockstroh
d,
Michael
Karnahl
bd,
Patrick
Schwarzbach
a,
Henrik
Junge
d,
Matthias
Beller
d and
Stefan
Lochbrunner
*a
*ab,
Antje
Neubauer
ac,
Nils
Rockstroh
d,
Michael
Karnahl
bd,
Patrick
Schwarzbach
a,
Henrik
Junge
d,
Matthias
Beller
d and
Stefan
Lochbrunner
*a
aInstitute of Physics, University of Rostock, Albert-Einstein-Straße 23, 18059 Rostock, Germany. E-mail: stefan.lochbrunner@uni-rostock.de
bInstitute of Organic Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: stefanie.tschierlei@oc.uni-stuttgart.de
cBecker & Hickl GmbH, Nahmitzer Damm 30, 12277 Berlin, Germany
dLeibniz-Institute for Catalysis at the University of Rostock (LIKAT), Albert-Einstein-Str. 29a, 18059 Rostock, Germany
First published on 16th March 2016
Abstract
Time-resolved spectroscopy was applied to investigate the excited state dynamics of two heteroleptic Ir(III) complexes with the general formula [Ir(C^N)2(N^N)]+, where C^N and N^N represent different cyclometalating and diimine ligands, respectively. The excited state relaxation is influenced by the ligand substitution as well as the light polarisation. Vibrational relaxation occurs in the sub-ps timescale and interligand charge transfer results in polarisation dependent signal dynamics with a time constant of about 30 ps. Electron injection from the iridium dye to TiO2 is analysed with respect to potential applications in solar energy conversion.
Cyclometalated Ir(III) complexes1 have been known for more than 30 years and their photophysical properties as well as excited state processes have been extensively studied since then, especially by Watts and coworkers.2,3 Their absorption, emission and electrochemical behaviour can be controlled by the exact choice of the ligands, which render them as ideal candidates for applications with respect to solar energy conversion and in photo(electro)chemical devices. Especially during the last decade cyclometalated Ir(III) complexes gained considerable scientific attention due to their usage in organic light emitting diodes,1,4 in light emitting electrochemical cells,5,6 as oxygen sensors7,8 and as photosensitisers for photocatalytic water splitting.9–18 In particular, their ability to generate hydrogen by the light-driven reduction of protons is still superior compared to Ru(II) polypyridine or Cu(I) bisdiimine complexes.15,19 Therefore, several systematic series of Ir(III) complexes with the general formula [Ir(C^N)2(N^N)]+, where C^N is a monoanionic cyclometalating ligand, have been prepared to broaden the scope of available sensitisers and to gain insight into the relationship between the structure and the resulting photophysical properties.1,5,6,17 In this regard, modifications were made to meet the following properties: (i) strong absorption in the visible region, (ii) long excited state lifetime, (iii) intense luminescence, (iv) electrochemical reversibility and (v) high stability under ambient conditions. As a result, highly luminescent Ir(III) complexes with quantum yields above 67% and emission lifetimes in the μs time range were prepared,15 which increased the probability of an electron-transfer process occurring prior to radiative or non-radiative relaxation. In-depth investigations of the underlying intermolecular reaction pathways of the iridium photosensitiser in the presence of a sacrificial electron donor such as triethylamine and an iron water reduction catalyst within a photocatalytic hydrogen evolving system revealed the different catalytic intermediates and the rate-limiting steps of the electron transfer processes.20–23 However, the initial light-induced intramolecular excited state processes in such [Ir(C^N)2(N^N)]+ complexes and their respective time constants are still not fully clarified. It is generally accepted that upon photoexcitation with visible light a transition into a singlet metal-to-ligand charge transfer (MLCT) state occurs. Subsequently, as a result of the iridium-induced strong spin–orbit coupling an ultrafast intersystem crossing (ISC) to the 3MLCT state takes place within the first 100 fs,24–26 probably followed by vibrational relaxation to the respective lowest lying vibrational state in less than 700 fs.24,26–28 Furthermore, vibrational cooling involving energy transfer to the solvent25,26,29 or charge separation processes due to different types of surrounding ligands28,30 are found in the low picosecond time-range. However, the finally populated lowest lying triplet excited state, where the emission originates, is stable for several nano- or microseconds as required for photocatalytic applications.15,21,31,32
In this work, time-resolved optical spectroscopy is used to study and compare the intramolecular relaxation dynamics of two different heteroleptic Ir(III) complexes, namely [Ir(ppy)2(bpy)]+1 and [Ir(ppy)2(bpy(COOH)2)]+2 (ppy = monodeprotonated 2-phenyl-pyridine and bpy = 2,2′-bipyridine) (Fig. 1). While compound 1 often serves as a reference complex for photophysical and photocatalytic studies, compound 2 bears two carboxyl anchor groups at the 4,4′-position in the bipyridine ligand for the immobilisation on semiconducting materials like TiO2 or NiO.33 This strategy of dye sensitisation is commonly used for the preparation of heterogeneous composite materials with the aim to broaden the light absorption of the semiconductor.33–35 Moreover, this immobilised system (abbreviated as 2T) can be considered as a simple model of an anode of a dye-sensitised solar cell, where it is important to study the electron injection into the TiO2 conduction band upon light irradiation.36,37
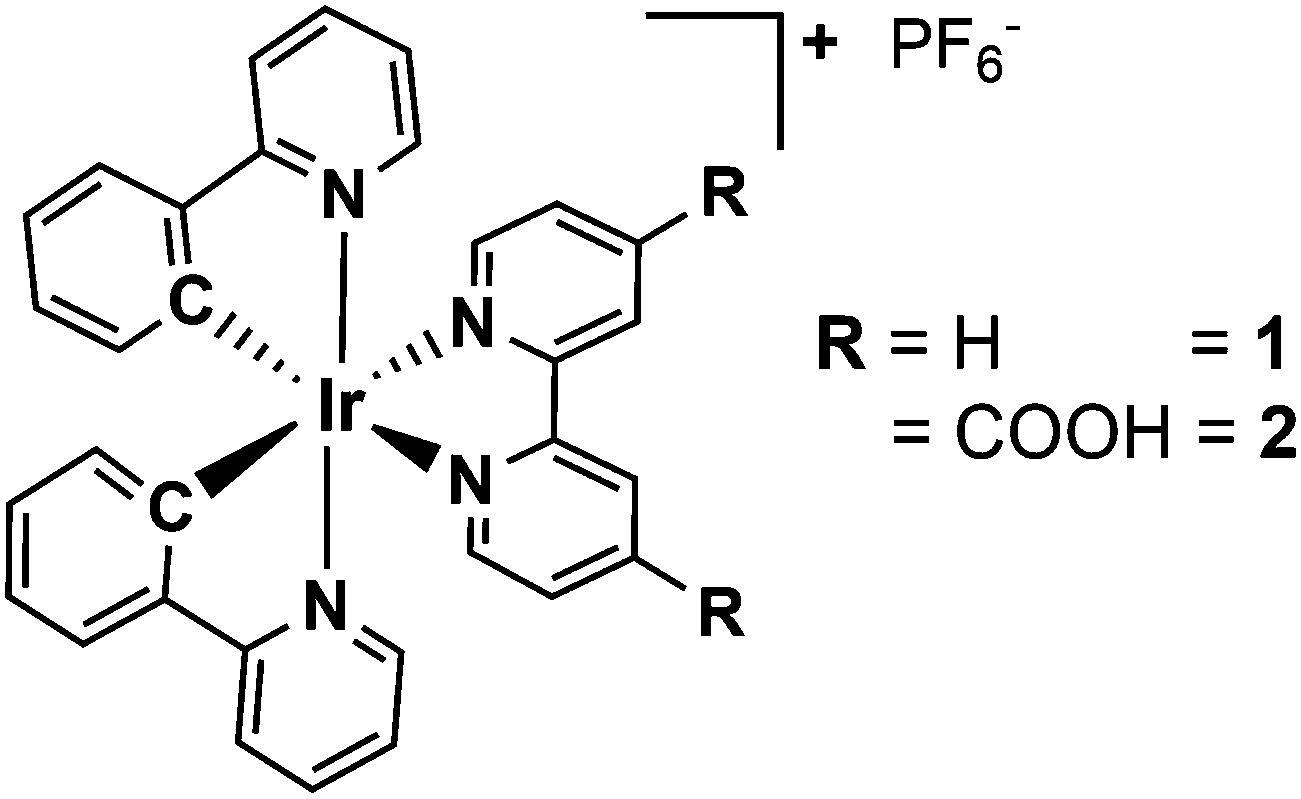 | ||
| Fig. 1 Structure of the cyclometalated Ir(III) complexes 1 and 2 applied in this study. | ||
Femtosecond time-resolved transient absorption spectroscopy addresses the intramolecular relaxation steps in 1 and 2 upon photoexcitation. Comparison of the behaviour of the two Ir(III) complexes discloses the influence of the carboxyl substituents and allows for conclusions on structure-dynamic-properties. Moreover, the effect of attaching 2 onto titania on the ultrafast excited state dynamics of 2 will be revealed and compared with the plain molecular system. Finally, this knowledge should contribute to the development of more efficient photosensitisers for applications in the field of solar energy conversion in the future.
Results and discussion
The Ir(III) complexes 1 and 2 were both synthesised and analysed according to literature procedures (Fig. 1).14,31,35,38 After purification 1 and 2 were obtained as yellow to orange solids with yields of 82% and 36%, respectively. The structures were confirmed by 1H NMR spectroscopy and showed the typical pattern of the aromatic signals (ESI†). For the synthesis of 1 a slightly modified procedure was applied using ethanol as the only solvent. The iridium dimer [(ppy)2IrCl]2 (ppy = monodeprotonated 2-phenyl-pyridine) and a slight excess of the bpy ligand were reacted in ethanol in a pressure tube at 100 °C for 2 days. Precipitation with excess of ammonium hexafluorophosphate and thoroughly washing with water and diethylether yielded the pure compound (ESI†). The complexes 1 and 2 possess a d6 electron configuration and an octahedral structure (Fig. 1). Two more isomers, where either the ppy carbon atoms are trans to each other or one ppy carbon is trans to a ppy nitrogen and the other is trans to a bpy nitrogen, are not likely due to the stronger trans effect of Ir–C compared to Ir–N.1The absorption spectra of 1 and 2 (Fig. 2a and Fig. SI1, ESI†), which are similar to related cyclometalated Ir(III) complexes like the homoleptic analogue fac-[Ir(ppy)3],32 exhibit the absorption bands of the spin-allowed π–π* transitions to the ppy and bpy ligand below 320 nm. In the visible region the absorption is assigned to spin-allowed metal-to-ligand charge transfer (MLCT) transitions (Ird–π*) centred at about 390 and 420 nm.26,27,31,39 Furthermore, the very weak absorption above 460 nm is a contribution of the spin-forbidden singlet-to-triplet MLCT transitions as a consequence of the heavy-atom effect resulting in a strong spin–orbit coupling,32 which is also supported by theoretical calculations.40,41 A comparison of 1 and 2 reveals only a small impact of the carboxyl groups on the absorption properties.
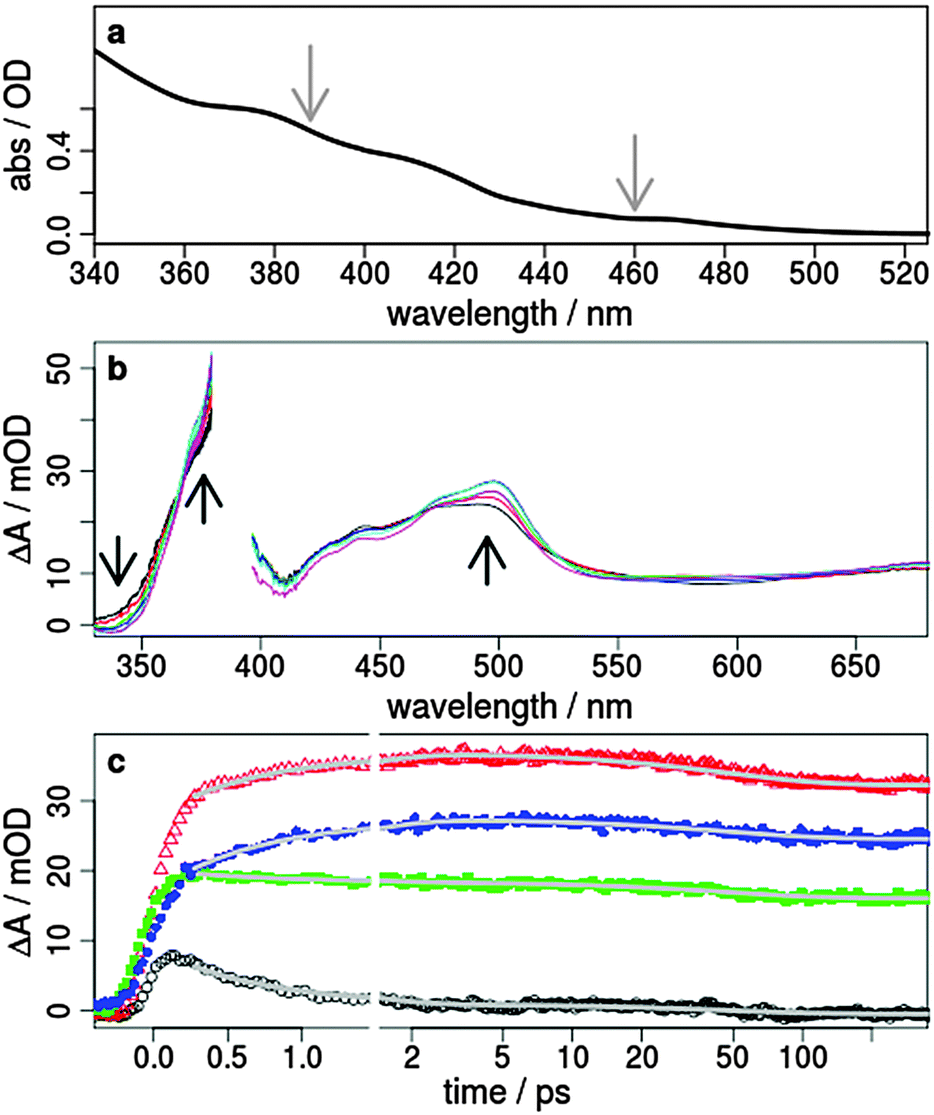 | ||
| Fig. 2 (a) Steady-state absorption spectrum of 1 in acetonitrile. The excitation wavelengths of 388 and 460 nm for the time-resolved transient absorption studies are marked with grey arrows, (b) transient absorption spectra after 0.5, 0.8, 1.25, 2, 5 and 50 ps excited at 388 nm with laser pulses polarised perpendicular to the probe (mOD = 10−3 optical density), and (c) transient signals as a function of the pump–probe delay time observed at 345 (black), 370 (red), 450 (green), and 500 nm (blue). The grey lines represent a double-exponential fit with the time constants τ1 = 0.7 ps and τ2 = 48 ps. The results of the decay associated spectra, comparison to other polarisations and transient spectra excited at 460 nm are summarised in the ESI.† | ||
In contrast, the emission (Fig. SI3, ESI†) is strongly influenced by the substituents resulting in different emission wavelengths and emission lifetimes. The emission maximum of 1 is located at 610 nm while it is red-shifted to 650 nm for 2 and the lifetime is decreased from 60.2 ± 0.4 ns (1) to 34.3 ± 0.2 ns (2). This behaviour is typical for the emission properties of similar Ir(III) or Ru(II) complexes with COOR substituents.31,42 Since the only structural difference between compound 2 and 1 is at the bpy ligand, it can be concluded that the lowest lying triplet excited state, from where the emission exclusively originates, corresponds to an MLCT state located at this ligand.
In order to analyse the relaxation dynamics of 1 and 2 in more detail, ultrafast time-resolved absorption measurements in acetonitrile solution were performed using 388 and 460 nm as pump wavelengths. These two wavelengths were selected, because they lie in the absorption band of the important MLCT transitions (Fig. 2a). Immediately after photoexcitation of 1 with 388 nm, a broad excited state absorption (ESA) appears in the visible region extending from 350 up to 700 nm, with two maxima at around 370 nm and 500 nm (Fig. 2b and ESI†). As apparent from the kinetics (Fig. 2c) the transient signals at 370 and 500 nm are increasing, while the bands at 340 and 440 nm decrease within the first 2 ps. For a quantitative evaluation the entire transient absorption change ΔA was globally analysed with the multi-exponential function 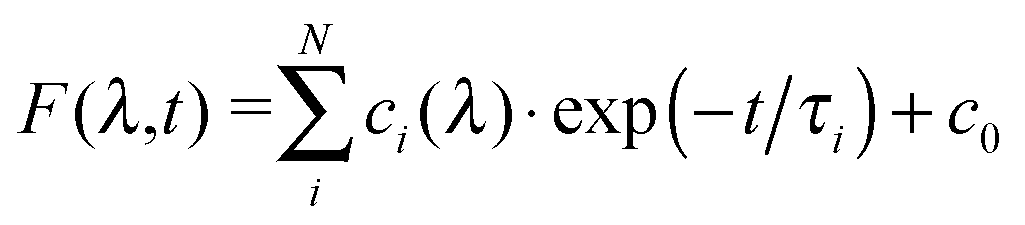 , with the probe wavelength λ, the delay time t and c0 as a long-lived component.43 It turns out that two exponential terms are needed for the description of the transient spectra. This global analysis of 1 resulted in a short time constant τ1 of 700 fs and a longer one τ2 of about 45 ps (Fig. 2 and Fig. SI4, SI5, ESI†; Table 1 and Table SI1, ESI†).
, with the probe wavelength λ, the delay time t and c0 as a long-lived component.43 It turns out that two exponential terms are needed for the description of the transient spectra. This global analysis of 1 resulted in a short time constant τ1 of 700 fs and a longer one τ2 of about 45 ps (Fig. 2 and Fig. SI4, SI5, ESI†; Table 1 and Table SI1, ESI†).
The fast component is associated with an absorption increase at around 370 and 500 nm and a decrease at 340 and 450 nm in the transient absorption spectra (Fig. 2). This is also confirmed by the negative and positive contributions of the fitted decay associated spectra (Fig. SI4, ESI†). The process is too slow for ISC in such Ir complexes and is more likely related to a subsequently occurring vibrational relaxation as also suggested by other groups.24,26–28 This assignment is further supported by the same observation for an excitation at 460 nm (Fig. SI5, ESI†), where the triplet MLCT states are directly populated.32 Moreover, such a vibrational relaxation does not change the dipole moment in 1, resulting in the independency of this process on the polarisation of the light pulses. Thus, no differences could be found in the decay associated spectra for perpendicular and parallel polarised pump light at 388 and 460 nm (Fig. SI6, ESI†).
With respect to the second time constant τ2 of about 45 ps (Table 1 and Table SI1, ESI†) the kinetics measured with parallel and perpendicular polarisations exhibit opposite trends (Fig. 3b and c, black rectangles). In the region between 350 and 530 nm the absorption increases for parallel polarised (Fig. 3c) and decreases somewhat with perpendicular polarised pump and probe pulses (Fig. 3b and Fig. SI4c, SI6, ESI†). This observation holds for both excitation wavelengths 388 nm and 460 nm. If the polarisations are set to magic angle, where both parallel and perpendicular parts of light are present, the contribution of the second exponential component is weak. These findings show that the second process must be associated with a change of orientation for the probed transition dipole. This cannot be explained by vibrational cooling processes or by interaction with the surrounding solvent molecules as suggested for other Ir(III) complexes.25,26,29 One possible explanation might be the occurrence of a light-induced interligand charge transfer (ILCT) process between ppy and bpy. This is in accordance with charge separation processes in slightly extended Ir(III) compounds28,30 and is comparable to interligand electron hopping in related coordination compounds such as Ru(II),44,45 Re(I)46 and Os(II)47 complexes. Therefore, we propose that after population of the two different triplet MLCT states of the ppy and bpy ligands an ILCT shifts the electron from the ppy onto the bpy ligand.
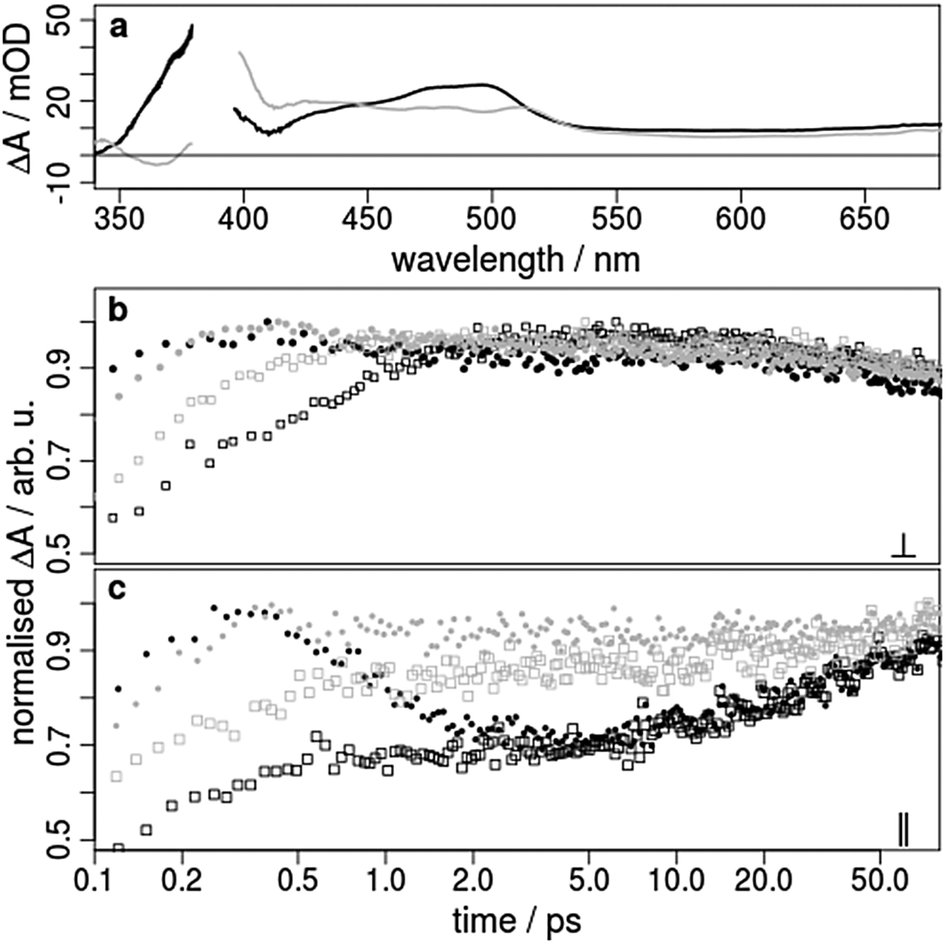 | ||
| Fig. 3 Transient absorption spectra and kinetics of 1 (black) and 2 (grey) excited with 388 nm laser pulses. (a) Transient absorption spectra after 1 ps excited with perpendicular polarised light. (b) Transient signals as a function of the pump–probe delay time observed at 440 nm (points) and 500 nm (rectangles) excited with perpendicular polarised light. (c) as (b) but excited with parallel polarised light. | ||
The observed transient absorption changes are dominated by dipole transitions of the reduced polypyridine ligands.23 This is concluded from the fact that the ESA has a very similar shape compared to the absorption spectrum of the photochemically reduced Ir complex.23 ILCT changes the transitions contributing to the ESA observed here. Since the ligands within the complex are almost perpendicularly oriented with respect to each other, the transition dipoles of the two interchanging ESA contributions are also more or less perpendicular. Accordingly, the electron transfer between the ligands changes the polarisation dependence of the ESA and causes the 30 ps dynamics. At the end, the final relaxation of the excited states to the ground state is independent from the polarisation (Fig. SI6 top, ESI†) and takes place from the lowest lying triplet state located at the bpy ligand (see above).
Another reason for the 45 ps dynamics can be seen in the potential rotation of the molecule.48 Calculation of the orientational relaxation time τor of 1 in acetonitrile results in approx. 52 ps (ESI†), which is on the same order of magnitude as that obtained in the experiment.
To evaluate the impact of substituents on the relaxation dynamics we studied complex 2 bearing two carboxyl anchor groups in 4,4′-position at the bpy ligand (Fig. 1). In the absorption spectrum of 2 the singlet MLCT band around 390 nm and the triplet MLCT absorption above 450 nm are somewhat more pronounced as in the spectrum of 1 (Fig. SI1, ESI†). This is probably an indication for an increased contribution of the substituted bpy ligand to the ground state absorption.42
Also the transient spectra of both complexes 1 and 2 excited at 388 nm are very similar and show broad ESA in the visible region (Fig. 3a). The global data analysis of 2 yields two time constants (τ1 = 0.3 ps and τ2 ≈ 25 ps), which are similar to those obtained for 1 (Table 1 and Table SI2, ESI†). Again τ1 is independent of the applied polarisation (Fig. SI7 and SI8, ESI†), but this time τ1 is slightly faster in 2 compared to 1, showing the influence of the carboxy groups in the bpy ligand. Further, the time traces of 2 are not substantially influenced by the polarisation of the pump light as apparent from the comparison of the quite equal slope of the grey points in Fig. 3b and c at 440 and 500 nm. For the global analysis the second time constant τ2 is solely necessary to fit kinetics below 420 nm as can be seen in the decay associated spectra (Fig. SI7, ESI†), which is in contrast to the analysis of 1. Thus, population and depopulation of the excited states of 2 in the time period after 2 ps is probably negligible. This finding supports our assignment of τ2 to an ILCT, because the triplet excited state of the substituted bpy ligand in 2 is lower in energy than the unsubstituted one. It seems that in 2 the electron is already promoted to the bpy ligand, and therefore, no ILCT is necessary to reach the lowest excited state (Fig. 5). Accordingly, the reorientation of the ESA dipole and the polarisation dependency is weak. These observations and conclusions also hold true for an excitation of 2 at 460 nm (Fig. SI8, ESI†).
Subsequently, the sensitisation of complex 2, bearing two carboxylate anchor groups, on the TiO2@glass layer was performed by dip coating resulting in 2T (ESI†). The successful immobilisation of 2 on the titania support was evidenced by UV/vis and Raman spectroscopy. It is found that the absorption of 2T is a combination of the absorption spectrum of TiO2 and of complex 2 (Fig. SI1, ESI†). Measurements of the non-resonant Raman spectra of 2 and 2T show that the Raman bands of 2, especially the modes of the ring stretching vibrations in the region between 1000 and 1650 cm−1,39 could be also detected for the 2T layer providing further evidence for the sensitisation of the TiO2 surface by the dye (Fig. SI2, ESI†).
This sensitisation enables a photoinduced electron transfer from the iridium dye into the conduction band of the semiconductor.33,36,37 To obtain efficient injection a strong coupling between the donor state of the dye and the acceptor state of titania is advantageous and the localisation of the excited MLCT state on the ligand with the anchoring substituents is beneficial. As shown for related Ru(II) complexes a transfer from singlet as well as triplet MLCT states can occur at sub-150 fs and tens of picoseconds, respectively.49,50 Further, time-resolved emission measurements for sensitised Ru(II) and Ir(III) complexes found multiexponential emission decays in about 500 ps.51,52 For 2T the transient absorption shows a multiexponential decay with time constants of 1.2, 13 and 150 ps (Fig. 4 and Fig. SI9, ESI†). In particular, the amplitudes of the short times are comparable to the results obtained for the lowest lying triplet excited state of 2 (Fig. SI7 and SI10, ESI†). This means that the triplet state is depopulated within 1.2 ps. The responsible process is most probably an electron transfer from the Ir complex to TiO2. In conclusion, the electron injection from the triplet MLCT localised at the bpy ligand to the conduction band of TiO2 seems to be very efficient, which is a strong hint for successful sensitisation (either physical or chemical) of TiO2 with 2. In the future, time-resolved infrared spectroscopy may help to reveal the respective transfer steps in the titania to elucidate the nature of the three time constants in more detail.
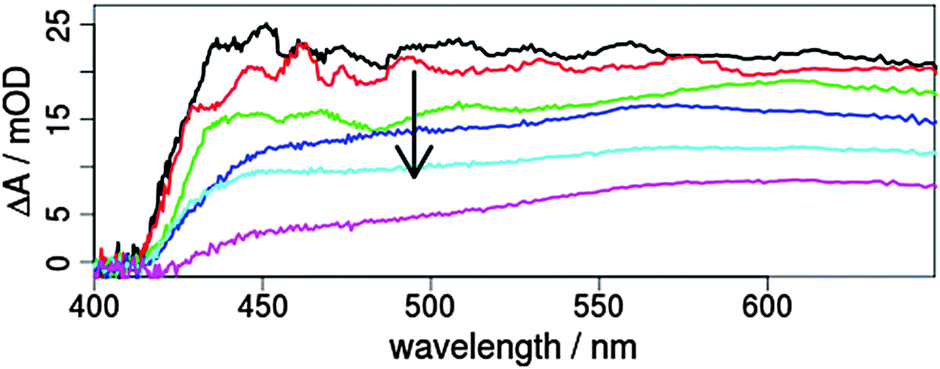 | ||
| Fig. 4 Transient absorption spectra of the dye-sensitised TiO2-layer 2T after 0.5 (black), 1 (red), 3 (green), 10 (blue), 50 (light blue) and 300 ps (magenta) excited at 388 nm with pulses perpendicular polarised to the probe. The observed changes are indicated with an arrow. | ||
Conclusions
In summary, we have prepared two iridium complexes 1 and 2 and successfully sensitised 2 onto a titania layer to form 2T. A combination of absorption, emission and ultrafast time-resolved spectroscopy was used to elucidate the photophysical properties of these dyes (Fig. 5). The absorption spectra of both compounds are fairly similar. The presence of carboxyl substituents in 2 leads to a pronounced bathochromical shift (40 nm) of the emission maximum and a shorter emission lifetime. The triplet excited state, from where the emission originates, is located at the bpy ligand and is lower in energy for 2 compared to 1.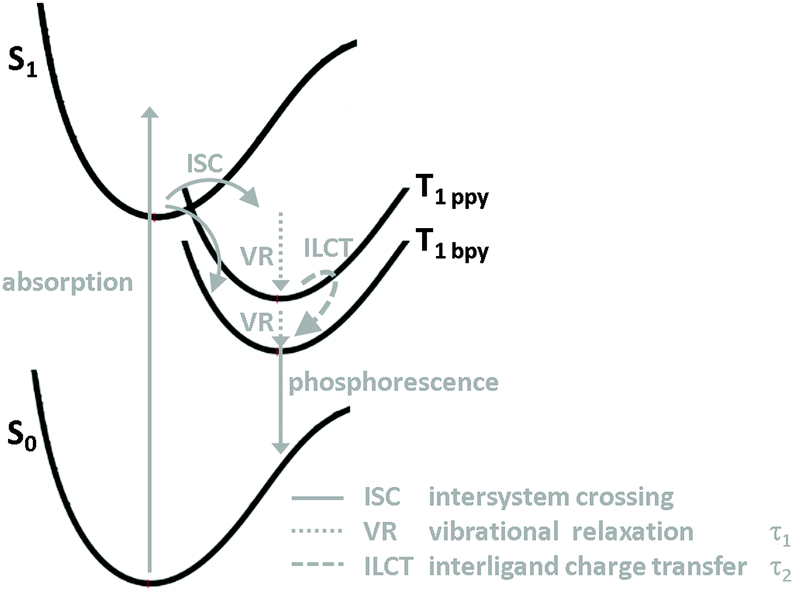 | ||
| Fig. 5 Schematic representation of the relaxation processes in 1 and 2. After light absorption and optical excitation of singlet metal-to-ligand charge transfer states involving the respective ppy and bpy ligands ISC to the triplet states occurs. This is followed by VR (τ1) and ILCT (τ2) between ppy and bpy, possibly accompanied by orientational relaxation of the molecule. Finally, phosphorescence (τem) from the triplet state takes place. | ||
After optical excitation and intersystem crossing vibrational relaxation occurs within 0.7 ps in 1 and is slightly faster in 2, most likely due to the decreased energy of the bpy triplet MLCT state in 2. In 1 an interligand charge transfer process from the ppy to the bpy ligand takes place within 45 ps and results in polarisation dependent transient spectra. The same process is inhibited in 2 due to the deeper level of the MLCT state involving the substituted bpy ligand. Investigation of 2T revealed three time constants associated with electron injection. The fastest one (1.2 ps) can be ascribed to a transfer from the triplet state located at the bpy ligand to the conduction band of TiO2. However, the other two processes could not be assigned so far and are subject of further investigations.
In general, this study contributes to a better understanding of photoinduced energy and electron transfer processes that are crucial for solar energy conversion.
Acknowledgements
We thank Wolfram Seidel (University of Rostock) for the permission to use the Raman spectrometer. We gratefully acknowledge the state of Mecklenburg-Western Pomerania, the Federal Ministry of Education and Research of Germany (BMBF, project “Light2Hydrogen”) as well as the European Social Funds (ESF, “PS4H”) for financial support. M. K. thanks the Fonds der Chemischen Industrie (FCI) for a fellowship.Notes and references
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS.
- K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1985, 107, 1431 CrossRef CAS.
- K. Dedeian, P. I. Djurovich, F. O. Garces, G. Carlson and R. J. Watts, Inorg. Chem., 1991, 30, 1685 CrossRef CAS.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750 CrossRef CAS PubMed.
- C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418 CrossRef CAS.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed.
- K. K.-W. Lo, S. P.-Y. Li and K. Y. Zhang, New J. Chem., 2011, 35, 265 RSC.
- X.-D. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666 RSC.
- E. D. Cline, S. E. Adamson and S. Bernhard, Inorg. Chem., 2008, 47, 10378 CrossRef CAS PubMed.
- S. Jasimuddin, T. Yamada, K. Fukuju, J. Otsuki and K. Sakai, Chem. Commun., 2010, 46, 8466 RSC.
- P. Zhang, P.-A. Jacques, M. Chavarot-Kerlidou, M. Wang, L. Sun, M. Fontecave and V. Artero, Inorg. Chem., 2012, 51, 2115 CrossRef CAS PubMed.
- H.-H. Cui, M.-Q. Hu, H.-M. Wen, G.-L. Chai, C.-B. Ma, H. Chen and C.-N. Chen, Dalton Trans., 2012, 41, 13899 RSC.
- F. Gärtner, B. Sundararaju, A.-E. Surkus, A. Boddien, B. Loges, H. Junge, P. Dixneuf and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 9962 CrossRef PubMed.
- F. Gärtner, D. Cozzula, S. Losse, A. Boddien, G. Anilkumar, H. Junge, T. Schulz, N. Marquet, A. Spannenberg, S. Gladiali and M. Beller, Chem. – Eur. J., 2011, 17, 6998 CrossRef PubMed.
- F. Gärtner, S. Denurra, S. Losse, A. Neubauer, A. Boddien, A. Gopinathan, A. Spannenberg, H. Junge, S. Lochbrunner, M. Blug, S. Hoch, J. Busse, S. Gladiali and M. Beller, Chem. – Eur. J., 2012, 18, 3220 CrossRef PubMed.
- D. R. Whang, K. Sakai and S. Y. Park, Angew. Chem., Int. Ed., 2013, 52, 11612 CrossRef CAS PubMed.
- A. Paul, N. Das, Y. Halpin, J. G. Vos and M. T. Pryce, Dalton Trans., 2015, 44, 10423 RSC.
- C.-H. Tsai, D. N. Chirdon, H. N. Kagalwala, A. B. Maurer, A. Kaur, T. Pintauer, S. Bernhard and K. J. T. Noonan, Chem. – Eur. J., 2015, 21, 11517 CrossRef CAS PubMed.
- S.-P. Luo, E. Mejía, A. Friedrich, A. Pazidis, H. Junge, A.-E. Surkus, R. Jackstell, S. Denurra, S. Gladiali, S. Lochbrunner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 419 CrossRef CAS PubMed.
- D. Hollmann, F. Gärtner, R. Ludwig, E. Barsch, H. Junge, M. Blug, S. Hoch, M. Beller and A. Brückner, Angew. Chem., Int. Ed., 2011, 50, 10246 CrossRef CAS PubMed.
- A. Neubauer, G. Grell, A. Friedrich, S. I. Bokarev, P. Schwarzbach, F. Gärtner, A.-E. Surkus, H. Junge, M. Beller, O. Kühn and S. Lochbrunner, J. Phys. Chem. Lett., 2014, 5, 1355 CrossRef CAS PubMed.
- S. Fischer, O. S. Bokareva, E. Barsch, S. I. Bokarev, O. Kühn and R. Ludwig, ChemCatChem, 2016, 8, 404 CrossRef CAS.
- S. I. Bokarev, D. Hollmann, A. Pazidis, A. Neubauer, J. Radnik, O. Kühn, S. Lochbrunner, H. Junge, M. Beller and A. Brückner, Phys. Chem. Chem. Phys., 2014, 16, 4789 RSC.
- H.-S. Duan, P.-T. Chou, C.-C. Hsu, J.-Y. Hung and Y. Chi, Inorg. Chem., 2009, 48, 6501 CrossRef CAS PubMed.
- G. J. Hedley, A. Ruseckas and I. D. W. Samuel, J. Phys. Chem. A, 2009, 113, 2 CrossRef CAS PubMed.
- G. J. Hedley, A. Ruseckas and I. D. W. Samuel, J. Phys. Chem. A, 2010, 114, 8961 CrossRef CAS PubMed.
- F. Spaenig, J.-H. Olivier, V. Prusakova, P. Retailleau, R. Ziessel and F. N. Castellano, Inorg. Chem., 2011, 50, 10859–10871 CrossRef CAS PubMed.
- J. H. Klein, T. L. Sunderland, C. Kaufmann, M. Holzapfel, A. Schmiedel and C. Lambert, Phys. Chem. Chem. Phys., 2013, 15, 16024 RSC.
- Y. You, K. S. Kim, T. K. Ahn, D. Kim and S. Y. Park, J. Phys. Chem. C, 2007, 111, 4052 CAS.
- C. Lambert, R. Wagener, J. H. Klein, G. Grelaud, M. Moos, A. Schmiedel, M. Holzapfel and T. Bruhn, Chem. Commun., 2014, 50, 11350 RSC.
- J. B. Waern, C. Desmarets, L.-M. Chamoreau, H. Amouri, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Inorg. Chem., 2008, 47, 3340 CrossRef CAS PubMed.
- T. Hofbeck and H. Yersin, Inorg. Chem., 2010, 49, 9290–9299 CrossRef CAS PubMed.
- R. Marschall, Top. Curr. Chem., 2016, 371, 143 CrossRef PubMed.
- M. Karnahl, E. Mejía, N. Rockstroh, S. Tschierlei, S.-P. Luo, K. Grabow, A. Kruth, V. Brüser, H. Junge, S. Lochbrunner and M. Beller, ChemCatChem, 2014, 6, 82 CrossRef CAS.
- A. Kruth, S. Peglow, N. Rockstroh, H. Junge, V. Brüser and K.-D. Weltmann, J. Photochem. Photobiol., A, 2014, 290, 31 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381 CrossRef CAS.
- M. S. Lowry, W. R. Hudson, R. A. Pascal and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129 CrossRef CAS PubMed.
- S.-H. Lai, J.-W. Ling, Y.-M. Huang, M.-J. Huang, C. H. Cheng and I.-C. Chen, J. Raman Spectrosc., 2011, 42, 332 CrossRef CAS.
- S. I. Bokarev, O. S. Bokareva and O. Kühn, J. Chem. Phys., 2012, 136, 214305 CrossRef PubMed.
- S. I. Bokarev, O. S. Bokareva and O. Kühn, Coord. Chem. Rev., 2015, 304–305, 133 CrossRef CAS.
- M. Schwalbe, M. Karnahl, S. Tschierlei, U. Uhlemann, M. Schmitt, B. Dietzek, J. Popp, R. Groake, J. G. Vos and S. Rau, Dalton Trans., 2010, 39, 2768 RSC.
- S. Tschierlei, M. Karnahl, N. Rockstroh, H. Junge, M. Beller and S. Lochbrunner, ChemPhysChem, 2014, 15, 3709 CrossRef CAS PubMed.
- S. Wallin, J. Davidsson, J. Modin and L. Hammarström, J. Phys. Chem. A, 2005, 109, 4697 CrossRef CAS PubMed.
- S. Tschierlei, M. Presselt, C. Kuhnt, A. Yartsev, T. Pascher, V. Sundström, M. Karnahl, M. Schwalbe, B. Schäfer, S. Rau, M. Schmitt, B. Dietzek and J. Popp, Chem. – Eur. J., 2009, 15, 7678 CrossRef CAS PubMed.
- D. J. Liard, M. Busby, I. R. Farrell, P. Matousek, M. Towrie and A. Vlcek, J. Phys. Chem. A, 2004, 108, 556 CrossRef CAS.
- G. B. Shaw, C. L. Brown and J. M. Papanikolas, J. Phys. Chem. A, 2002, 106, 1483 CrossRef CAS.
- A. Henseler and E. Vauthey, Chem. Phys. Lett., 1994, 228, 66 CrossRef CAS.
- G. Benkö, J. Kallioinen, J. E. I. Korppi-Tommola, A. P. Yartsev and V. Sundström, J. Am. Chem. Soc., 2002, 124, 489 CrossRef.
- J. Schindler, S. Kupfer, M. Wächtler, J. Guthmuller, S. Rau and B. Dietzek, ChemPhysChem, 2015, 16, 1061 CrossRef CAS PubMed.
- E. I. Mayo, K. Kils, T. Tirrell, P. I. Djurovich, A. Tamayo, M. E. Thompson, N. S. Lewis and H. B. Gray, Photochem. Photobiol. Sci., 2006, 5, 871 CAS.
- Y. Tachibana, I. V. Rubtsov, I. Montanari, K. Yoshihara, D. R. Klug and J. R. Durrant, J. Photochem. Photobiol., A, 2001, 142, 215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and synthetic details. UV/vis-, Raman- and time-resolved emission measurements. Transient absorption spectra of 1, 2 and 2T. See DOI: 10.1039/c6cp00343e |
| This journal is © the Owner Societies 2016 |