Self-healing systems based on disulfide–thiol exchange reactions†
Mark
Pepels
a,
Ivo
Filot
b,
Bert
Klumperman
*ac and
Han
Goossens
*a
aLaboratory of Polymer Materials, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: J.G.P.Goossens@tue.nl; Fax: +31 40 2436999; Tel: +31 40 2473899
bInorganic Materials Chemistry Group, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P. O. Box 513, 5600 MB Eindhoven, The Netherlands
cStellenbosch University, Department of Chemistry and Polymer Science, Private Bag X1, Matieland 7602, South Africa. E-mail: bklump@sun.ac.za
First published on 13th March 2013
Abstract
New thermoset systems based on disulfide bonds were synthesized with self-healing capabilities. The self-healing mechanism is not related to disulfide–disulfide exchange reactions, but to thiol–disulfide exchange reactions that are pH-dependent. Stress relaxation experiments showed large relaxation for systems having PTM2 as a curing agent, which indicates that the system can rearrange its molecular structure as a mechanism to release stress. However, relaxation rates decreased for samples tested longer after production. This indicates the disappearance of thiol-groups probably caused by thiol–thiol oxidation.
Introduction
In recent years, there has been increasing interest in the field of self-healing capabilities of polymer systems. The healing mechanism in self-healing materials should meet three conditions: the system should be mobile, to be able to bridge a crack. It should be temporary, to prevent creep, and local, to maintain the structural integrity of the material. Polymers of which the healing mechanism is triggered by crack initiation/propagation are referred to as autonomous self-healing polymers and polymers that heal by a different external trigger will be called self-healing polymers. Autonomous self-healing systems mostly comprise of systems containing encapsulated uncured resin.1–7 When a propagating crack encounters a capsule, the capsule ruptures and the uncured resin flows into the crack and subsequently cures, thereby restoring the crack. Self-healing systems can be split up into multiphase systems and systems with reversible chemistry. Multiphase systems, containing thermo-plastics and thermosets, use the ability of the thermoplastics to flow to heal cracks.8,9 Systems with reversible chemistry have a decreased viscosity when bonds are broken, giving them the ability to flow and heal cracks. This can be achieved using hydrogen bonding,10–12 metal–ligand bonding,13–16 and reversible covalent bonding.17–25Although systems using these techniques can have good self-healing properties, they all pose some problems for the application in composite systems. Encapsulated systems have the following problems: self-healing can only occur once at the same place and processability is more difficult than in the case of normal composite resins.26 Systems using hydrogen bonding and metal–ligand bonding are sensitive to creep and are hard to use in systems that need a high glass transition temperature. Thermosets having reversible covalent crosslinks that depolymerize above the ceiling temperature have the problem that high crosslink densities, which are often desired for composites, are hard to obtain due to the polymerization–depolymerization thermodynamics.
Sulfur chemistry has always been interesting but is still not fully understood. Especially in the field of rubber reclamation, the reduction and oxidation of the disulfide bond has been extensively investigated. Sastri and Tesoro reported on the reduction and reoxidation of epoxy systems cured with dithiodianiline.27 This chemistry is also used to make redox sensitive sol–gel systems, consisting of thiol-functionalized star polymers (sol), which upon addition of an oxidizing agent form a disulfide-bonded network (gel).28 Similar principles were used in thiol-functionalized dendrimers that upon oxidation form so-called megamers.29 The disulfide bond can also be opened forming sulfur radicals using heat or shear.30 This mechanism is also responsible for the exchange between disulfides, resulting in a mixture of products.31,32 It was also reported that the disulfide exchange reaction is catalyzed by thiols.31 The reaction, which has a radical mechanism, is not catalyzed by a thiol. However, disulfide bonds can react with thiols yielding again a disulfide and a thiol.30,31 In this reaction, a thiolate is formed due to the deprotonation of the thiol, which acts as a nucleophile and can attack the disulfide bond. The intermediate product can yield the starting products (R1–S–S–R1 + R2–SH) or yield the exchanged product (R1–S–S–R2 + R1–SH). Chemically crosslinked systems based on sulfur chemistry with the ability to reshuffle to potentially have self-healing properties were explored by a number of groups, such as the Bowman group33 and the Matyjaszewski group.34,35 In previous work we used a disulfide-based epoxy resin to produce a healable thermoset rubber that regained 100% of the original mechanical properties after healing for a short time (1 hour).20 The healing mechanism was ascribed to the ability of the disulfide to reversibly disconnect and connect. However, the timescale of disulfide exchange appears to be much longer36,37 in comparison to e.g. tetrasulfide exchange.38
The main objective of this study is to develop self-healing thermoset systems based on disulfides. In order to design a self-healing thermoset system based on disulfide bonds, first the mechanism behind the disulfide exchange has to be investigated, since other mechanisms have been postulated.39 Influences of the chemical structure, catalysts and other possible factors will be investigated. Secondly, thermoset systems based on this disulfide exchange will be synthesized and their ability to rearrange their network and healing capability will be investigated. We will also compare the behavior of the prepared systems with other systems with exchangeable bonds without interference with termination reactions, so-called vitrimers, as developed by Leibler, Tournilhac and co-workers.40–44
Experimental section
Materials
Polysulfide diglycidyl ether (Thioplast® EPS25, T25, 695 g equiv.−1), and polysulfides (Thioplast® G44, T44, Mn < 1100 g mol−1/Thioplast® G20, T20, Mn = 2000–3000 g mol−1/Thioplast® G130, T130, Mn = 5000–7000 g mol−1) were kindly provided by Akzo Nobel Sulfur Derivatives. The epoxy equivalent was determined by the Jay–Dijkstra–Dahmen method, variation Ciba.45 4,4′-Methylenedianiline (MDA, Aldrich, 97%), 4,4′-dithiodianiline (DTDA, Aldrich, 98%), pentaerythritol tetrakis(2-mercaptoacetate) (PTM1, Aldrich, technical grade), pentaerythritol tetrakis(2-mercaptopropionate) (PTM2, Aldrich, 97%), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, Fluka, >99%), 4-dimethylaminopyridine (DMAP, Aldrich 97%), dimethylaminopropylamine (DMAPA, Huntsman), dipropyl disulfide (DPDS, Aldrich, 98%), dibutyl disulfide (DBDS, Aldrich, 97%), and 1-penthanethiol (PT, Aldrich, 98%). The molecular structures are listed in Fig. 1. 2-Propanol (PrOH, Biosolve, AR) and ethylacetate (EtAc, Biosolve, AR) were used as received. 1,3-Dioxolane (DOx, Aldrich, 99%) was first distilled to remove the inhibitor. | ||
| Fig. 1 Chemical structures of the used model systems and thermoset systems. | ||
Instrumentation
Aliphatic disulfide model exchange experiments
A typical exchange experiment was performed by adding 1.000 g DPDS (6.65 mmol), 1.189 g DBDS (6.67 mmol), 0.690 g PT (6.62 mmol) and 0.068 g DBU (0.45 mmol) to a vial and stirring it for 1 hour at room temperature. A GC-MS sample was prepared and the reaction vial was subsequently placed in an oil bath and stirred at 60 °C for one hour. After this, again a GC-MS sample was prepared and both the samples were analyzed by GC-MS. The relative peak areas of the different product peaks were investigated. This was done for different combinations of starting materials and amount of additives.Model exchange experiments with structures resembling thermosets' molecular structure
A typical exchange experiment with structures resembling the thermosets' molecular structure was performed by adding 0.9 g T44 (5 mol eq. S–S per mol T44, 2 mol eq. S–H per mol T44), 3.31 g T130 (35 mol eq. S–S per mol T130, 2 mol eq. T130 S–H per mol T130), and 0.05 g DMAPA (4.9 mmol) in a vial and stirring it for 1 hour at 60 °C. A SEC sample was prepared and the vial was subsequently placed in an oven at 60 °C for 1 day. After this, again a SEC sample was prepared and both samples were analyzed by SEC. The shift in peaks was investigated for different combinations of starting materials and additives.Synthesis of thermoset rubbers
A typical synthesis of a thermoset system was performed by mixing 50.93 g T25 (6.5 mol eq. S–S per mol T25), and 9.07 g PTM2 (18.5 mmol, 4 mol eq. S–H per mol) in a 250 mL round-bottom flask. The mixture was heated to 60 °C during stirring and degassed by a vacuum–argon cycle for 1 hour. Next, the homogeneous mixture was cooled down to room temperature and 0.6 g DMAPA (5.8 mmol) was added. This was subsequently stirred for ½ hour under argon. After that, the mixture was cast in a PTFE mould, which was subsequently cured at 60 °C under nitrogen in an oven for 2 hours. The kinetics of the curing reaction were also monitored by FTIR spectroscopy.Stress relaxation
Stress relaxation experiments were performed using punched dumbbell-shaped tensile bars according to ISO 527-2 and using a grip-to-grip separation of 50 mm. The samples were stretched 10% at a speed of 1000 mm min−1 and kept at this position for 5 min, while measuring the stress at time intervals of 0.1 s.Healing experiments
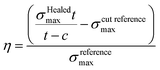
Healing experiments were performed by punching dumbbell-shaped tensile bars according to ISO 527-2, slicing these bars in half, leaving 150 μm attached, after which the bars were placed in the setup (see ESI S1†). The setup was subsequently heated in an oven for a given time. The healed bars were compared to virgin bars by performing a tensile test (grip-to-grip separation: 50 mm, testing speed: 10 or 20 mm min−1 for T3 samples) on both and comparing the stress at break. One bar was cut and not healed to be able to compensate for the 150 μm of material that was not cut. The healing efficiency was determined as follows:where σmax is the maximum strain of the healed, cut reference and reference sample respectively, t is the thickness of the sample, and c is the uncut thickness (150 μm).
Computational model
Density functional theory (DFT)46,47 electronic structure calculations were performed using the Gaussian 03 software package.48 The B3LYP functional was used in accordance with a 6-311++G(d,p) basis set.49,50 The vibrational frequencies were calculated using the conventional harmonic approximations employed in Gaussian. Some very small imaginary frequencies (>50 cm−1) were found. Several attempts were made to remove these imaginary frequencies by using slightly different geometries obtained by perturbation of the geometries along the eigenvectors corresponding to those vibrations. No reduction of imaginary frequencies was achieved indicating a very shallow potential energy surface. As the eigenvectors of the imaginary frequencies lie at a significant distance from the reactive center and moreover are caused by the large number of degrees of freedom in the aliphatic side chain, we chose to neglect them.Results and discussion
First, disulfide exchange experiments are reported with the objective to investigate the mechanism responsible for self-healing. Secondly, the influence of the molecular structure of the disulfides and possible additives on the ability to exchange is discussed followed by a theoretical analysis of the structures used, where the bond energy of the different disulfides and thiols was calculated by using electron density functional theory. Next, a comparison between the different disulfide exchange experiments is made and discussed with the use of the theoretical calculations.After this, the tensile properties of different thermoset systems containing disulfides are discussed with special attention to stress relaxation (and its mechanisms), since the ability of the system to rearrange can be determined by these results. Finally, the healing efficiency of the different thermoset systems containing disulfides is investigated and a discussion on the relationship between the molecular structure and the material properties is presented.
Aliphatic disulfide exchange
To investigate the disulfide exchange mechanism, experiments were performed with model compounds. The goal of these experiments was to investigate whether the disulfide exchange proceeds through a radical mechanism or a thiol–disulfide exchange mechanism. For this purpose, dipropyl disulfide (DPDS) and dibutyl disulfide (DBDS) were mixed under different conditions (temperature, presence of 1-pentanethiol (PT), presence of different bases and other additives) with the goal to investigate if propyl butyl disulfide (PBDS) was formed. (All exact amounts and results reported in this section can be found in ESI S2.†) Fig. 2 shows the resulting GC-MS-spectrum for the exchange reaction with equimolar (EM) amounts of DPDS, DBDS, and PT together with a small amount of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). In this experiment all the expected products for the exchange reaction were formed. | ||
| Fig. 2 GC-MS chromatogram for the exchange reaction of dipropyl disulfide (DPDS), dibutyl disulfide (DBDS) and 1-pentanethiol (PT) together with DBU at 20 °C. | ||
Similar experiments were performed with different amounts of DPDS, DBDS, PT, and DBU at 20 °C and at 60 °C (this temperature was chosen because it was the temperature at which known disulfide systems were healed20). The relative GC-MS peak intensities for DPDS, DBDS and PBDS (the exchange product) for the different experiments are shown in Fig. 3.
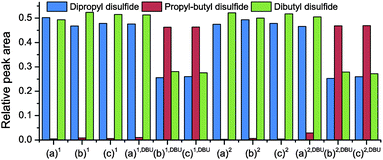 | ||
| Fig. 3 Relative GC-MS peak intensities for dipropyl disulfide (DPDS), dibutyl disulfide (DBDS) and propyl-butyl disulfide (PBDS) for different exchange experiments. All samples contain equimolar (EM) amounts of DPDS and DBDS. 1: one hour reaction at 20 °C, 2: one hour reaction at 20 °C and one hour at 60 °C of reaction time, DBU: 1 wt% of DBU (a) no additional components, (b) equimolar amount of PT, and (c) 0.13× 1-pentanethiol relative to DPDS. | ||
From the experimental data of Fig. 3 it can be seen that the presence of a thiol and a base is necessary to induce the exchange reaction. If only one of the two (thiol or base) is present, no significant exchange occurs. The addition of a relatively small amount of thiol (PT/DPDS = 0.13) leads to full exchange at 20 °C when also DBU is present in the mixture.
Next, the influence of the basicity of the tertiary amine on the thiol–disulfide exchange was investigated.51,52 To this extent, three different tertiary amines were investigated for their catalytic activity: 4-dimethylaminopyridine (DMAP, pKa = 9.8),53 dimethylaminopropyl-amine (DMAPA, pKa = 10.38),54 which is a commercially available curing accelerator and less toxic than DMAP, and DBU (pKa = 12),55 which was used for the initial exchange experiments. A concentration of 1 wt% was used in exchange experiments performed for 1 hour at 60 °C, containing equimolar amounts of DPDS, DBDS and PT. It can be seen that only for the experiment containing DBU, significant reaction occurs (Fig. 4).
 | ||
| Fig. 4 Relative GC-MS peak intensities for dipropyl disulfide (DPDS), dibutyl disulfide (DBDS) and propyl-butyl disulfide (PBDS) for exchange experiments containing equimolar amounts of DPDS, DBDS and 1-pentanethiol (PT), and (a) 1 wt% of DMAPA (pKa = 10.4), (b) 1 wt% of DMAP (pKa = 9.8), (c) DBU (pKa = 9.8) after one hour of reaction at 60 °C, (d) 1 wt% of DMAPA and 50 wt% 1,3-dioxalane before heating to 60 °C, (e) similar composition as for (d), after 1 hour of reaction at 60 °C, (f) similar composition as for (d), after 18 hours of reaction at 60 °C, (g) equimolar amount of ethylacetate, 1-propanol, 2 times equimolar amounts of 1,3-dioxalane and 1 wt% of DMAP after 1 hour of reaction at 60 °C. | ||
In the known disulfide-based self-healing system, DMAP was used as the base.20 Therefore, the possible influence of the system itself (e.g. polarity and other functional groups) was investigated. The repeating unit of the system consists of an acetal and disulfide bond (–O–CH2–O–(CH2)2–S–S–(CH2)2–). Therefore, exchange experiments were done using equimolar amounts of DPDS, DBDS and PT, 50 wt% of 1,3-dioxolane (DOx, which contains an acetal bond), and 1 wt% of DMAP. Samples were taken before heating to 60 °C, after 1 hour at 60 °C and after 18 hours at 60 °C, to be sure that the situation after 1 hour is not an equilibrium state. Next to this, an exchange experiment was performed using all functional groups present in the known self-healing thermoset. To this extent, a mixture was used containing equimolar amounts of DPDS, DBDS, PT, ethylacetate (EtAc), 1-propanol (PrOH), 2 equivalents (relative to DPDS) DOx, and 1 wt% DMAP.
From these results (Fig. 4), it can clearly be seen that the addition of DOx has no significant effect on the rate of exchange. Furthermore, it can be seen that the system is not in equilibrium after 1 hour, since the exchange reaction is still proceeding in the 17 hours that followed. The addition of all the functional groups that are present in the known self-healing thermoset slightly increases the exchange, but does not lead to enough exchange to promote self-healing on the time scale required.
Here, we showed that thiol–disulfide exchange under the right conditions is much faster than disulfide–disulfide exchange. However, it appears that thiol–disulfide exchange is highly dependent on the basicity of the system.
Disulfide exchange of structures resembling thermoset structure
The model exchange experiments using aliphatic disulfides did not show the expected exchange of disulfides, which is a requirement for a network to be able to rearrange. A reason for this could be that the model system did not have enough similarity with the molecular structure of the self-healing thermoset. However, because of the high molecular weight of the molecular structures resembling the backbone of the self-healing thermoset (T44, T20 and T130), these experiments could not be analyzed with GC-MS. Therefore, Size Exclusion Chromatography (SEC) was chosen as the technique, since it separates primarily based on the size of the polymer chain. It should be taken into account that the SEC-instrument was calibrated with PS standards, and that therefore the molecular weights cannot be taken as absolute. Although the experiments cannot be used in a quantitative manner, the results can be used qualitatively. This was done by investigating the product peaks of different starting mixtures, which were stirred at 60 °C for 1 hour. For some of these experiments the mixture was allowed to react for 24 hours (product after 24 hours of reaction in Fig. 5). In the chromatogram, this peak was compared with the product peak to see if the exchange reaction was already completed after 1 hour at 60 °C. The peaks of some of the starting products cannot be clearly distinguished in the chromatogram, since their molecular weight is too low for SEC. All results of the exchange experiments can be found in the ESI.†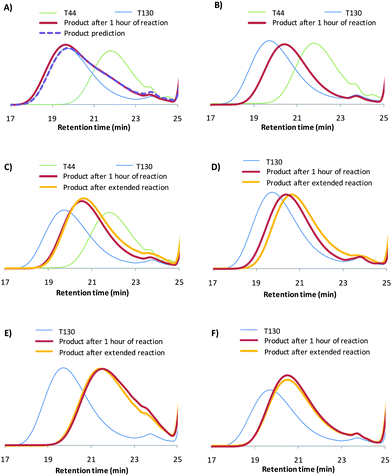 | ||
| Fig. 5 SEC results of the disulfide exchange reaction after mixing of the reactants for 1 hour at 60 °C (product line), and extended reaction (product final line): (a) equimolar amounts of T44 and T130, where prediction is the modeled product line if no exchange occurs, (b) equimolar amounts of T44 and T130 with 1 wt% of DMAP, (c) equimolar amounts of T44 and T130 with 1 wt% of DMAPA, (d) equimolar amounts of T130 and DBDS with 1 wt% of DMAPA, (e) equimolar amounts of T130 and PTM1 with 1 wt% of DMAPA and (f) equimolar amounts of T130 and PTM1 with 1 wt% of DTDA. | ||
T44 (low Mn) and T130 (high Mn), both polysulfides with thiol end-groups resembling the backbone structure of the self-healing thermoset, were mixed in order to investigate if and under which conditions exchange occurs. Fig. 5a shows that no significant exchange occurs when no additives are added. This can be seen from the prediction curve, which is modeled using the summation of the curves of the starting products multiplied with their mass fraction. When the same experiment was performed with the addition of 1 wt% of a base, DMAP (Fig. 5b) and DMAPA (Fig. 5c), a product peak is observed, which lies between the peak of T44 and T130. This indicates good exchange. It furthermore demonstrates that when the DMAPA experiment is allowed to react for another 5 days at 20 °C and 24 hours at 60 °C, the peak does not shift significantly, which implies nearly full exchange after 1 hour at 60 °C.
The exchange of T130 with DBDS was also tested to investigate whether and how fast this exchange would occur. Without the addition of DMAPA, no reaction occurred (see ESI S4†). When DMAPA is used, significant exchange occurs within 1 hour at 60 °C (Fig. 5d). However, it can be seen from the product final line (product after 7 days at 20 °C and 25 hours at 60 °C) that additional exchange occurred after this period, indicative of the influence of the disulfide structure in combination with the used base also observed in the aliphatic disulfide exchange experiments. Furthermore, the exchange reaction of T130 with PTM1 (tetrafunctional-thiol-crosslinker used in the thermoset system) was investigated (Fig. 5e). This system shows full exchange within 1 hour at 60 °C. Finally, dithiodianiline (DTDA), which has a disulfide connected to an aromatic ring, was used in combination with T130. This was done because aromatic structures are mostly used to obtain high Tg systems. The reaction between DTDA and T130 appears to be fully completed after 1 hour at 60 °C.
Besides disulfides, formaldehyde acetals (–CH2–O–CH2–O–CH2–) can also participate in an exchange reaction.56 To investigate if this mechanism plays a role in the self-healing mechanism, exchange experiments were done using T130, formaldehyde diethyl acetal (FDA) and possible DMAPA and PrOH as catalysts. However, none of these experiments resulted in a significant shift of the product peak compared to T130 (see ESI S4†).
Computational calculations of the relative bond strengths of used thiols and disulfides
From the model experiments described above, a discrepancy between the model and the self-healing thermoset seems to be present. In order to assess the effect of neighboring atoms on disulfide and thiol bond strength, a limited computational study was conducted. Computational calculations were performed in order to elucidate the sulfur–sulfur bond strength in DBDT. From these calculations it was found that the free energy (p = 1 atm and T = 298 K) of homolytic dissociation (Fig. 6) is 158.61 kJ mol−1.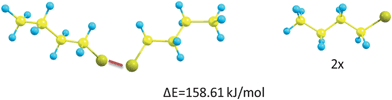 | ||
| Fig. 6 Conformation and energy difference of dibutyl disulfide and the product obtained when the disulfide bond (red) is broken homolytically. | ||
Similar calculations were done on a Thioplast® molecule containing three repeating units, where the bond was homolytically dissociated at the central disulfide bond. This resulted in a free energy of 106.02 kJ mol−1. These bond energies cannot be used quantitatively, since simulation in the gas phase does not take into account the effects of neighbouring molecules (present in a liquid system). However, the results can be used qualitatively, showing a significant difference in bond energy between the disulfide bond in DBDS and Thioplast®.
For the thiol groups, the free energy for heterolytic dissociation (resulting in a cation and anion) was calculated, in the gas phase at 1 bar and 298 K, for PTM1 and PT. For PTM1 this resulted in the conformations and free energies shown in Fig. 7, which shows a heterolytic dissociation energy of 1384.23 kJ mol−1. For PT, a heterolytic dissociation energy of 1472.78 kJ mol−1 was computed. Because ions have a very high energy in the gas phase, the heterolytic dissociation energies of the thiols are much higher than the homolytic dissociation energies calculated for the disulfide groups. The difference in dissociation energy between PT and PTM1 shows the higher acidity of the thiol-group of PTM1 relative to PT.
 | ||
| Fig. 7 Conformation and energy of pentaerythritol tetrakis(2-mercaptoacetate) and the product obtained when a sulfur–hydrogen bond is heterolytically dissociated. | ||
Effect of structure on disulfide exchange
In the model experiments, it is shown that the time scale of disulfide exchange through a radical mechanism is much larger than the time scale of a thiol–disulfide exchange. However, the rate of the thiol–disulfide exchange appears to be highly dependent on the basicity of the system, the type of disulfide and the thiol used. All these can be explained by dividing the reaction into three steps, deprotonation of the thiol, nucleophilic attack of the formed thiolate, and dissociation of the intermediate product in one of the two possible thiolates (Fig. 8).57 The reaction rate is determined by either the formation of the thiolate or the nucleophilic attack of the thiolate on the disulfide bond. Both these steps are determined by the stability of the thiolate. Large stability of the thiolate means that the deprotonation rate is high (a large amount of thiolates present in the system), but the rate of nucleophilic attack is low. Furthermore, the stability also determines which of the chains of the intermediate product is the most likely to be the leaving group. | ||
| Fig. 8 Exchange reaction divided into three steps: deprotonation of the thiol group, nucleophilic attack of the thiolate on one of the sulfur atoms of the disulfide, and dissociation of the intermediate product. | ||
The stability of the thiolate is mostly determined by the bond strength between the sulfur and the hydrogen/sulfur. Next to this, the pH of the system will also contribute to the stability of the thiolate (higher pH will result in higher stability). It appears that the rate for thiol–disulfide exchange is highest when the pH of the system is equal to the pKa of the thiol.58,59 In this case, the stability of the thiolate is such that the rates of deprotonation and nucleophilic attack are similar.
It has already been shown that the thiol–disulfide exchange rate is very high for the system containing DBU (pKa = 12). For the systems containing DMAPA (pKa = 10.38) and DMAP (pKa = 9.8) the reaction rate is much lower. However, we also showed that the thiol–disulfide reaction of Thioplast® structures proceeds rapidly using DMAP and DMAPA. This suggests that the pKa of the aliphatic thiols is much higher than the pKa of Thioplast® structures. This was also proven by the DFT calculations, which yielded a higher bonding strength for DBDS and PT compared to the Thioplast® disulfide and PTM1, meaning that these aliphatic thiols/thiols of dissociated disulfides have a higher pKa. Furthermore, the fact that the aliphatic disulfide exchange shows a ratio of 0.25 DPDS, 0.5 PBDS, and 0.25 DBDS (Fig. 3) means that there is a random statistical mixture and not a mixture favoring one of the compounds more than the other. In this case it can be explained by the fact that when the intermediate product is formed between PT and DBDS, the chance of PT and 1-butanethiol functioning as the leaving group is practically equal. This is caused by the similarity of the sulfur–hydrogen bond of these molecules. When using different thiols/disulfides (e.g. the ones in Fig. 8), a different equilibrium will be formed.
From a material point of view, it is important that there is no difference in the leaving group character of the thiolates, since the system should keep a steady exchange rate and should not form a more thermodynamically stable system. Therefore, in the ideal case, the attacking thiol should have the same pKa as the thiols of the disulfide that it attacks. In this case an attack is always 50% successful and therefore the exchange rate will be maximized.
Stress relaxation of the thermoset systems
The idea of self-healing systems is the ability to become temporarily mobile, which can lead to the closing of a defect or crack. The ability of a system to heal can therefore be related to the ability of a network to rearrange, which can be expressed by the ability of the material to flow or to relax stress. Self-healing systems deform under loading. Therefore, Dynamic Mechanical Thermal Analysis (DMTA) could not be used. Next to this, rheology measurements could not be used, since samples have to be cured in a compression molding press to obtain the right dimensions, which could not be operated under an inert atmosphere. This will lead to different properties because the samples are exposed to air (vide infra) and can therefore not be compared with the tensile samples. For that reason, stress relaxation was chosen as the method to investigate the ability of the different thermoset systems to rearrange their network. If a network is stretched fast, it can relax stress through changing its conformation, i.e. moving of crosslinks and chain segments without rearranging the network. If the network is able to rearrange, it will go to a lower state of energy, which will lead to more relaxation. This relaxation mechanism can lead to complete relaxation of the stress. In order to investigate this phenomenon, different thermoset systems were produced of which the compositions are shown in Table 1.| Thermoset | Molar ratios wt% | T cure (°C) | Time (h) | ||||
|---|---|---|---|---|---|---|---|
| T25 | MDA | PTM1 | PTM2 | DMAPA | |||
| T1 | 2 | 1 | 0 | 0 | 0 | 100 | 120 |
| T2 | 2 | 0 | 1 | 0 | 1 | 60 | 2 |
| T3 | 2 | 0 | 0 | 1 | 1 | 60 | 2 |
| T4 | 2 | 0 | 0 | 1.1 | 1 | 60 | 2 |
The curing reactions of thermosets T2–T4 were followed by FTIR spectroscopy and complete curing was observed after around 60 min for T2 and around 30 min for T4. More details on the FTIR measurements can be found in the ESI.† Thermoset T1 could not be measured with FTIR, since the peaks of the aromatic ring showed overlap with the vibrational band of the epoxy group. Therefore, T1 was checked for full curing by measuring the Young's modulus after 5 and 6 days of curing. Since the modulus did not change, full curing was assumed.
The (change in) ability of the system to rearrange was further investigated by using stress relaxation experiments. When a system has this ability, it can release stress through rearrangement of the network structure in a less stretched state. The objective of these experiments was to investigate which stress relaxation mechanisms are present. The stress relaxation can be modeled using the Maxwell model:

When plotted as a log(stress)–time plot, a linear curve should be obtained. However, if there are multiple stress relaxation mechanisms at work, multiple regions can be observed. In these experiments, T3 samples were stretched to 10% and subsequently the stress was measured for a period of five minutes. This was done for samples two hours, 1 day (26 hours), and 2 days (50 hours) after production. From these results, it can clearly be observed that there is almost complete stress relaxation within the time frame of the experiment (Fig. 9). Furthermore, the material appears to have two regions, especially at day 0. This indicates multiple stress release mechanisms. Most likely, the dominant stress relaxation mechanism is the thiol–disulfide exchange, since the stress relaxes completely back to zero.
 | ||
| Fig. 9 Stress–time curves for thermoset T3 stress relaxation experiments at different times and stress–time curves for thermoset T1 (reference thermoset). | ||
It can clearly be seen that the rate of stress relaxation decreases significantly from day 0 to day 2. This can be explained by the disappearance of thiol-groups by oxidation, leading to fewer possibilities for thiol–disulfide to occur, which will lead to less stress relaxation (see later discussion). As a reference, T1 was used in stress relaxation experiments, resulting in significantly less stress relaxation in the tested time (Fig. 9). Furthermore, the plot does not clearly show multiple regions within the time frame of the experiment. The relatively slow stress relaxation can be explained by the slow disulfide–disulfide exchange rate compared to the thiol–disulfide exchange rate of T3.
When thermoset T4 was tested for its tensile properties, large stress relaxation was observed (Fig. 10), which was only limited by the end settings of the equipment used. The material did not appear to be stretched, but appeared to flow at the testing speed (10 mm min−1).
 | ||
| Fig. 10 Stress–strain curves of thermoset T4, at day 0 and day 1, resulting in maximum strains limited by equipment parameters. | ||
Healing efficiency of thermoset systems
To investigate the self-healing properties of thermosets T1, T2, and T3, tensile properties of these cut (cut reference), cut and healed (healed), and uncut (reference) samples were compared. As can be observed in Fig. 11, the healed samples of T1 show some recovery of mechanical properties with an average healing efficiency of 0.16. The fact that the samples heal partially can be explained by the small ability the system has to rearrange by disulfide–disulfide exchange and the interdiffusion of dangling chains over the surface of the crack. | ||
| Fig. 11 Tensile properties of the reference thermoset (T1) in a healing experiment. | ||
Next, T2 was investigated for its healing properties (Fig. 12). T2 shows relatively good healing efficiencies ranging from 0.61 to 1.04 with an average of 0.81. The relatively large deviation in efficiencies can be attributed to the large experimental error in the healing setup, which does not have the possibility to apply exactly equal pressure on the samples. Furthermore, a minor displacement of the crack surfaces can cause a decrease in contact, which will lead to less healing.
 | ||
| Fig. 12 Tensile properties of T2 in a healing experiment. | ||
Besides these healing experiments for T3, samples that were tested after one day for stress relaxation were pulled apart and put back together manually. The samples were left for 1 day at room temperature and their tensile properties were measured. The samples that were used for stress relaxation after two days were used as reference. This set of samples shows very good healing (Fig. 13) with efficiencies of 0.51 and 0.99. The difference in healing efficiency can clearly be explained from the scar (i.e. the former crack) of the samples. As far as can be seen, the two surfaces of the sample with a healing efficiency of 0.99 were attached perfectly together, while the surfaces of the sample with a healing efficiency of 0.51 were slightly misaligned, thereby not matching both surface structures.
 | ||
| Fig. 13 Tensile properties of T3 in a healing experiment. Healing experiments done at samples first used for stress relaxation 24 hours after production. Pictures show the scar of the samples tested. | ||
Influence of oxygen
Next to the extremely large deformations, the samples measured at the same day of production and one day after production show quite a large difference in the maximum-stress. All samples were stored under atmospheric conditions in the laboratory. Because of the change between day 0 and day 1, further attention was paid to the history of the thermosets after production. Comparison of tensile test results of samples that after production were cooled down in air or nitrogen, and tested after 1 day, reveals a higher maximum stress and lower maximum strain for the sample cooled down in air (Fig. 14). This result indicates that the sample that is cooled down in air has more additional crosslinking after production. However, as shown by FTIR spectroscopy, no further curing occurs after one hour of reaction. Therefore, the additional crosslinking is most likely an effect of oxidative coupling of thiol groups, forming extra crosslinks. This also results in a system containing less thiol groups that can participate in the exchange reactions. | ||
| Fig. 14 Stress–strain curves of thermoset T4, where after curing the sample is cooled down in air or nitrogen. | ||
Analysis of self-healing thermoset systems
The thermosets synthesized in this study, which show a good ability to rearrange (T2–T4), were all based on a thiol curing agent. It is evident that the thiol curing agent has good exchange with the Thioplast® molecules (Fig. 5e). This means that exchange has already taken place upon curing, leading to a different network topology. Furthermore, thermoset systems practically never reach 100% conversion. This means that some free thiol groups are left after curing. These thiol groups are essential for the ability of the network to rearrange. This is visualized in Fig. 15, where after curing a network is formed. A thiol group (yellow) can exchange with a disulfide (red), which rearranges the network. If stress is applied, chains will stretch, which increases the free energy (decrease in entropy). The network will therefore rearrange to a system with a lower free energy, which will lead to stress relaxation. | ||
| Fig. 15 Representation of the curing reaction and subsequent ability of the network to rearrange. | ||
This exchange mechanism is also the mechanism responsible for the healing of the thermosets. It can be seen that thermoset T1 barely heals (Fig. 11), which is caused by the fact that there are no free thiol groups available. T2 shows much better healing properties (Fig. 12). The variation in efficiency between the samples is large, which is caused by the difficult reproducibility of the healing experiments. However, when the samples are not cut, but pulled apart, the thermoset shows very good healing when the two parts are put together aligned (Fig. 13). There are several possibilities for this observation. First of all, it is possible that surfaces are deformed due to the cutting, which will result in a mismatch. For the samples that are pulled apart, the rupture is formed in a fraction of second, so the surfaces will align very well. Furthermore, the ruptured samples are put together manually. In this process, a small force is applied which will make the surfaces match better. For the cut samples, this was not done. Secondly, oxidation of the thiol group can play a role. When surfaces do not match well, the crack surface is more susceptible to oxygen. As can be seen from the stress relaxation experiments (Fig. 9), the relaxation rate, and thus the ability of the system to rearrange, decreases over time. The ability to rearrange is caused by the thiol–disulfide exchange. There are only a few thiol groups in the material compared to the disulfide groups. Consequently, a decrease in thiol concentration will lead to a decrease in the ability of the system to rearrange. There are two possibilities for a decrease of the thiol concentration, i.e. additional post curing and thiol–thiol oxidative coupling. The FTIR spectra of the curing reaction show no additional curing after one hour of curing and therefore it can be assumed the reaction is fully completed, while the actual curing of the material was done for two hours. It is unlikely that this amount of post-curing occurs at room temperature over the next day. Furthermore, if the samples that are cooled down in air or nitrogen after curing are compared, it can be seen that the sample cooled in air (exposed to oxygen at a higher temperature) loses the ability to rearrange to a larger extent than the sample cooled under nitrogen. Next to this, the samples of T4 (which have an excess of thiol) also lost this ability fast, which cannot be caused by additional crosslinking, since thiols are in excess.
Thiols are sensitive towards oxidation. The process is pH dependent and appears to have the highest rate if the pH of the system is equal to the pKa of the thiol. These are also the conditions for which the thiol–disulfide exchange reaction proceeds best. It can therefore be expected that systems that promote thiol–disulfide exchange, also promote thiol–thiol oxidation.
Conclusions
In previous work we used a disulfide-based epoxy resin to produce a healable thermoset rubber that regains 100% of the original mechanical properties after healing for a short time (1 hour).20 The healing mechanism was ascribed to the ability of the disulfide to reversibly disconnect and connect. However, the time scale of disulfide exchange appears to be much longer.38,60 From the aliphatic disulfide exchange experiments, it can be concluded that the dominant mechanism is not the disulfide–disulfide exchange reaction, but the thiol–disulfide exchange reaction. However, the reaction is highly pH dependent, since thiol–disulfide exchange only proceeded to its full extent if DBU (pKa = 12) was used as a base and not when DMAPA (pKa = 10.38) or DMAP (pKa = 9.8) were used. Furthermore, it was shown that the addition of functional groups, which are present in the thermoset system, does not show a significant change in exchange rate.The disulfide exchange experiments using Thioplast® structures (which resemble the structure of the thermoset) confirmed that thiol–disulfide exchange is highly dependent on the pH of the system. The exchange of different Thioplast® structures did not proceed without a base, but when DMAP or DMAPA was added, the exchange did occur. It was also shown that aliphatic disulfides could exchange with Thioplast® structures, although at a slower rate.
The DFT calculations showed higher bond strengths for the disulfide bond in DBDS compared to the disulfide bond in a Thioplast® molecule. Furthermore, it was shown that the deprotonation energy for PT is higher than for PTM1. From this it can be concluded that the difference in bond strength, and thus pKa, is responsible for the difference in exchange rate for the different exchange experiments described above.
The exchange rate is probably the highest when the pKa of the thiols (and thiols of dissociated disulfides) is the same as the pH of the system, which is caused by the balance between the ability of the thiol to deprotonate and to nucleophilic attack the disulfide.
The thermoset systems produced were tested for stress relaxation and self-healing properties. T25 cured with MDA resulted in small stress relaxation and healing efficiencies (0.16), probably caused by the interdiffusion of dangling chains and small disulfide exchange through a radical mechanism.
The thermosets cured with thiols (PTM2) showed much faster stress relaxation. The observation of multiple stress relaxation mechanisms and the ability of the sample to fully relax demonstrate that the thermoset network can rearrange through thiol–disulfide exchange. T25/PTM2 thermosets showed fast stress relaxation. However, the stress relaxation became slower for older samples. This indicates the disappearance of thiol groups. The thiol groups probably undergo oxidative coupling, since it is likely that the conditions that promote thiol–disulfide exchange also promote thiol–thiol oxidation. This is emphasized by the higher ability of samples cooled down in nitrogen, instead of air, to rearrange after curing. Because of possible oxidation, it is very important to take into account the history of a material.
The stress relaxation and concomitant self-healing mechanism is based on disulfide–thiol exchange reactions and as such has a strong similarity with the vitrimer systems as proposed by Leibler, Tournilhac and co-workers (see ref. 39–43), although the behavior of our systems is oxygen sensitive.
Acknowledgements
The authors would like to thank Bob Fifield, Maria Eriksson, Weizhen Li, Chunxia Sun and Martin Ottink for their help with analysis and preparation. Dr Sjef Vekemans and Prof. Ton Peijs are acknowledged for the stimulating discussions. BK acknowledges the support from the South African Research Chairs Initiative (SARChI) of the Department of Science and Technology (DST) and the National Research Foundation (NRF).References
- C. Dry, Smart Mater. Struct., 1994, 3, 118 CrossRef CAS.
- C. Dry, Compos. Struct., 1996, 35, 263–269 CrossRef.
- C. Dry and W. McMillan, Smart Mater. Struct., 1996, 5, 297 CrossRef CAS.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. R. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS.
- E. Brown, S. White and N. Sottos, J. Mater. Sci., 2006, 41, 6266–6273 CrossRef CAS.
- E. N. Brown, N. R. Sottos and S. R. White, Exp. Mech., 2002, 42, 372–379 CrossRef CAS.
- C. J. Hansen, W. Wu, K. S. Toohey, N. R. Sottos, S. R. White and J. A. Lewis, Adv. Mater., 2009, 21, 4143–4147 CrossRef CAS.
- S. A. Hayes, W. Zhang, M. Branthwaite and F. R. Jones, J. R. Soc., Interface, 2007, 4, 381–387 CrossRef CAS.
- X. Luo, R. Ou, D. E. Eberly, A. Singhal, W. Viratyaporn and P. T. Mather, ACS Appl. Mater. Interfaces, 2009, 1, 612–620 CAS.
- J.-L. Wietor, A. Dimopoulos, L. E. Govaert, R. A. T. M. van Benthem, G. de With and R. P. Sijbesma, Macromolecules, 2009, 42, 6640–6646 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS.
- D. Montarnal, F. Tournilhac, M. Hidalgo, J.-L. Couturier and L. Leibler, J. Am. Chem. Soc., 2009, 131, 7966–7967 CrossRef CAS.
- M. E. L. Wouters, J. G. P. Goossens and F. L. Binsbergen, Macromolecules, 2002, 35, 208–216 CrossRef CAS.
- M. E. L. Wouters, V. M. Litvinov, F. L. Binsbergen, J. G. P. Goossens, M. van Duin and H. G. Dikland, Macromolecules, 2003, 36, 1147–1156 CrossRef CAS.
- M. A. J. van der Mee, R. M. A. l'Abee, G. Portale, J. G. P. Goossens and M. van Duin, Macromolecules, 2008, 41, 5493–5501 CrossRef CAS.
- V. A. Friese and D. G. Kurth, Coord. Chem. Rev., 2008, 252, 199–211 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698 CrossRef CAS.
- W. W. G. J. van Pelt and J. G. P. Goossens, Anal. Chim. Acta, 2007, 604, 69–75 CrossRef CAS.
- M. A. J. van der Mee, J. G. P. Goossens and M. van Duin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1810–1825 CrossRef CAS.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- Y. Zhang, A. A. Broekhuis and F. P. Picchioni, Macromolecules, 2009, 42, 1906–1912 CrossRef CAS.
- J. Canadell, H. Fischer, G. de With and R. A. T. M. van Benthem, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3456–3467 CrossRef CAS.
- J. Y. Chang, S. K. Do and M. J. Han, Polymer, 2001, 42, 7589–7594 CrossRef CAS.
- J. W. Kamplain and C. W. Bielawski, Chem. Commun., 2006, 1727–1729 RSC.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125 CrossRef CAS.
- R. P. Wool, Soft Matter, 2008, 4, 400–418 RSC.
- V. R. Sastri and G. C. Tesoro, J. Appl. Polym. Sci., 1990, 39, 1439–1457 CrossRef CAS.
- J. Kamada, K. Koynov, C. Corten, A. Juhari, J. A. Yoon, M. W. Urban, A. C. Balazs and K. Matyjaszewski, Macromolecules, 2010, 43, 4133–4139 CrossRef CAS.
- B. M. Kiran and N. Jayaraman, Macromolecules, 2009, 42, 7353–7359 CrossRef CAS.
- V. V. Rajan, W. K. Dierkes, R. Joseph and J. W. M. Noordermeer, Prog. Polym. Sci., 2006, 31, 811–834 CrossRef CAS.
- D. Vietti, Polysulfides, in The Encyclopedia of Polymer Science and Technology, John Wiley & Sons, Inc., New York, 2001, vol. 3, pp. 732–744 Search PubMed.
- J.-M. Poudrel and E. R. Cole, Phosphorus, Sulfur Silicon Relat. Elem., 2001, 175, 79–86 CrossRef CAS.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS.
- R. Nicolaÿ, J. Kamada, A. Van Wassen and K. Matyjaszewski, Macromolecules, 2010, 43, 4355–4361 CrossRef.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS.
- J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J. P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS.
- U. Lafont, H. van Zeijl and S. van der Zwaag, ACS Appl. Mater. Interfaces, 2012, 4, 6280–6288 CAS.
- G. D. T. Owen, W. J. MacKnight and A. V. Tobolsky, J. Phys. Chem., 1964, 68, 784–786 CrossRef CAS.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- Y.-X. Lu, F. Tournilhac, L. Leibler and Z. Guan, J. Am. Chem. Soc., 2012, 134, 8424–8427 CrossRef CAS.
- R. Dobinson, W. Hofmann and B. P. Stark, The Determination of Epoxy Groups, Pergamon Press Ltd., London, 1st edn, 1969 Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford C. T., 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. Houk and G. M. Whitesides, J. Am. Chem. Soc., 1987, 109, 6825–6836 CrossRef CAS.
- R. Singh and G. M. Whitesides, J. Am. Chem. Soc., 1990, 112, 1190–1197 CrossRef CAS.
- H. G. Visser, W. Purcell and S. S. Basson, Transition Met. Chem., 2003, 28, 235–240 CrossRef CAS.
- S. K. Perera, W. A. Dunn and L. R. Fedor, J. Org. Chem., 1980, 45, 2816–2821 CrossRef CAS.
- D. H. Ripin and D. A. Evans, pKa Table of various organic and inorganic compound, 2005 Search PubMed.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Am. Chem. Soc., 2005, 127, 13666–13671 CrossRef CAS.
- I. M. Kolthoff, W. Stricks and R. C. Kapoor, J. Am. Chem. Soc., 1955, 77, 4733–4739 CrossRef CAS.
- D. A. Keire, E. Strauss, W. Guo, B. Noszal and D. L. Rabenstein, J. Am. Chem. Soc., 1992, 57, 123–127 CAS.
- G. M. Whitesides, J. E. Lilburn and R. P. Szajewski, J. Am. Chem. Soc., 1977, 42, 332–338 CAS.
- J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J. P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Self-healing setup, aliphatic disulfide exchange data, disulfide exchange of structures resembling thermoset structure, FTIR spectroscopy data. See DOI: 10.1039/c3py00087g |
| This journal is © The Royal Society of Chemistry 2013 |