
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid-solution surface alloying of Cu nanocubes with platinum-group metals: pathway switching and catalyst stabilization in CO2 reduction
Hirokazu Kobayashi *a,
Sachie Hikinoa,
Akihiko Anzai†
b,
Takahiro Matsuuc,
Mahiru Umenoc,
Tomohiro G. Noguchib,
Masaki Donoshitab,
Tomokazu Yamamotod,
Yasukazu Murakamide,
Kenichi Katof,
Takeharu Sugiyamag,
Hiroyuki Setoyamah,
Tetsuroh Shirasawai,
Yuya Shimohataj,
Takayoshi Ishimotoj and
Miho Yamauchi*abckl
*a,
Sachie Hikinoa,
Akihiko Anzai†
b,
Takahiro Matsuuc,
Mahiru Umenoc,
Tomohiro G. Noguchib,
Masaki Donoshitab,
Tomokazu Yamamotod,
Yasukazu Murakamide,
Kenichi Katof,
Takeharu Sugiyamag,
Hiroyuki Setoyamah,
Tetsuroh Shirasawai,
Yuya Shimohataj,
Takayoshi Ishimotoj and
Miho Yamauchi*abckl
aResearch Center for Negative Emissions Technologies, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: kobayashi@k-nets.kyushu-u.ac.jp
bInstitute for Materials Chemistry and Engineering (IMCE), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: yamauchi@ms.ifoc.kyushu-u.ac.jp
cDepartment of Chemistry, Faculty of Science, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
dThe Ultramicroscopy Research Center, Kyushu University, Motooka 744, Nishi-ku, Fukuoka 819-0395, Japan
eDepartment of Applied Quantum Physics and Nuclear Engineering, Kyushu University, Motooka 744, Nishi-ku, Fukuoka 819-0395, Japan
fRIKEN SPring-8 Center, 1-1-1 Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5148, Japan
gResearch Center for Synchrotron Light Applications, Kyushu University, 6-1 Kasuga Park, Kasuga-shi, Fukuoka 816-8580, Japan
hBeamline Group, SAGA Light Source, 6-1 Kasuga Park, Kasuga-shi, Fukuoka 816-8580, Japan
iNational Institute of Advanced Industrial Science and Technology (AIST), Research Institute for Measurement and Analysis Instrumentation, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
jSmart Innovation Program, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-4-1 Kagamiyama, Higashihiroshima, Hiroshima 739-8527, Japan
kInternational Institute for Carbon-Neutral Energy Research (WPI-I2CNER), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
lAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, 980-8577, Japan
First published on 26th May 2026
Abstract
Precise control over morphology and alloy configuration is essential for addressing complex reactions, such as the electrochemical CO2 reduction reaction (CO2RR), which proceeds through multiple intermediates and requires enhanced selectivity, activity and stability. However, achieving simultaneous regulation of these two structural features remains a formidable challenge. Here we report novel shape-controlled Cu-based solid-solution surface-alloy nanocrystals composed of Cu nanocube (NC) cores surrounded by atomically alloyed platinum-group metal shells (Cu/Cu1−xMx NCs, M = Pd, Pt, Ir, Ru) that alter CO2RR performance of Cu. In particular, surface alloying of Cu NCs with Ir switched product selectivity from C2H4 to HCOOH. Cu/Cu1−xIrx NCs exhibited superior HCOOH activity and stability compared with a Sn catalyst, which is a well-known element for producing HCOOH. Furthermore, Ir surface alloying preserved the cubic morphology of Cu NCs, whereas pure Cu degraded into nanograins. Our findings highlight a valuable approach to controlling reaction pathways through heteroatom interfaces and to designing highly active and stable electrocatalysts.
Introduction
Nanoscale alloy catalysts exhibit distinct catalytic selectivity, activity and durability depending on their size, shape and composition.1,2 Solid solution alloys, in which constituent elements are homogeneously mixed at the atomic level, are particularly effective for modulating electronic states and tuning material performance through the controlled composition and combination of elements.2,3 In contrast, core–shell structures—typically composed of a low-cost metal core and a precious-metal shell—offer synergistic functionalities by controlling the spatial arrangement at the core–shell interface while ensuring the efficient use of scarce elements.4,5 Shape-controlled core–shell structures further highlight the impact of specific atomic arrangements on well-defined crystal facets.6 Thus, integrating the advantages of solid solution and core–shell configurations with shape control provides a promising way to achieve another dimension of catalytic functions. However, simultaneous control of morphology and surface alloy configuration in core–shell structures remains highly challenging, as both features are highly sensitive to synthetic parameters. In particular, shape-controlled solid-solution alloys composed of elements that are immiscible in the bulk state are rarely achieved. Here, we report the synthesis of a series of shape-controlled Cu-based solid-solution surface alloy nanocrystals incorporating platinum-group metals (PGMs): Cu nanocube (NC) cores surrounded by atomically alloyed PGM shells (Cu/Cu1−xMx NCs, M = Pd, Pt, Ir, Ru) as catalysts for CO2 reduction. Notably, we demonstrate the formation of shape-controlled solid-solution surface alloys in Cu–Ir7 and Cu–Ru8 systems, despite their intrinsic immiscibility in the bulk state.The electrochemical CO2 reduction reaction (CO2RR) converts CO2 into carbon-based chemicals,9–11 and Cu is unique among metals in its ability to catalyse the CO2RR to form a wide range of products, including methane (CH4), ethylene (C2H4) and ethanol (C2H5OH).9 However, precise control over selectivity toward specific products remains a significant challenge. Strategies to improve product selectivity on Cu have focused on faceting control,12 defect13 and strain modulation,14 heteroatom doping,15 alloying,11,16,17 and surface functionalization.18 For example, (100)-faceted Cu NCs have exhibited higher selectivity toward C2H4 compared to (111)-faceted nano-octahedra.12 Single-atom Cu catalysts supported on materials such as oxides have been reported to promote the formation of CH4.19,20 Bimetallic Cu–Ag systems have been shown to enhance the formation of C2 products, such as C2H4 and C2H5OH, compared to pure Cu.21,22 To date, most approaches have relied on tuning the stability of CO* and CO–CO* intermediates during the CO2RR.23 In this study, through a comprehensive investigation of CO2 reduction on Cu/Cu1−xMx NCs (M = Pd, Pt, Ir, Ru), we demonstrate that solid-solution surface alloying with only 0.7 at% hydrogen evolution reaction (HER)-active Ir switches product selectivity from C2H4 to HCOOH. This alloying enables a distinct CO2 reduction pathway that bypasses CO* formation entirely. Remarkably, Cu/Cu1−xIrx NCs achieve higher activity toward HCOOH production than Sn nanoparticles (NPs), a benchmark HCOOH-selective metal catalyst, while retaining superior stability under reaction conditions. Moreover, Ir surface alloying effectively stabilizes the cubic morphology of Cu NCs, preventing the structural degradation into metallic nanograins that has been observed for pure Cu.24,25 Our multiple structural control strategy provides a powerful framework for tailoring nanocatalysts, addressing not only the CO2RR but also other electrocatalytic reactions.
Results and discussion
We synthesized Cu/Cu1−xMx (M = Pd, Pt, Ir, Ru), in which the shell portion of Cu NCs is atomically alloyed with M through a galvanic replacement reaction. First, uniform Cu NCs with an average edge length of 50.5 ± 5.3 nm were synthesized by a previously reported method (Fig. S1a and S2a).12 Subsequently, a controlled amount of an acetylacetonate complex in oleylamine, which serves as a metal precursor, was added dropwise to the solution and stirred at an appropriate synthesis temperature and for an appropriate time.Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images revealed that the morphologies of the obtained Cu/Cu1−xPdx, Cu/Cu1−xPtx and Cu/Cu1−xIrx NCs were cubic as was that of Cu NCs, indicating that the cubic shapes of Cu NCs were retained after the galvanic replacement reaction with Pd, Pt and Ir ions (Fig. S1b–d and S2b–d). The average edge length of Cu/Cu1−xPdx, Cu/Cu1−xPtx and Cu/Cu1−xIrx NCs was estimated to be 50.3 ± 6.5, 56.3 ± 7.2 and 51.0 ± 8.1 nm, respectively. The average edge length of Cu/Cu1−xIrx NCs is in good agreement with an edge length distribution of 49.8 ± 7.6 nm estimated from small-angle X-ray scattering (SAXS) (Fig. S3). From SEM–energy-dispersive X-ray (EDX) analyses, the atomic percentages of Pd, Pt and Ir included in Cu/Cu1−xPdx, Cu/Cu1−xPtx and Cu/Cu1−xIrx NCs were calculated to be 1.1, 1.0 and 0.7%, respectively.
To investigate the alloying states in the obtained NCs, high-resolution annular high-angle dark-field scanning TEM (HR-HAADF-STEM) and EDX elemental mapping of Cu and M were performed. HR-HAADF-STEM images of Cu/Cu1−xMx NCs (M = Pd, Pt, Ir) clearly display atomic planes with a d-spacing of approximately 0.36 nm in the cubic core region, consistent with the (100) planes of the face-centered cubic (fcc) Cu structure (Fig. S4, S5 and 1a, b). The brighter atomic columns observed in the shell region, due to the higher atomic number of M compared to Cu, suggest that M atoms are atomically dispersed in the surface region of the Cu matrix (Fig. S4, S5 and 1a, b). The thickness of the Cu1−xMx shell ranges from 4 to 8 atomic layers (1–2 nm) (Fig. S4, S5 and 1a, b). The STEM–EDX elemental mapping of Cu and the corresponding M elements (M = Pd, Pt, Ir) for Cu/Cu1−xMx NCs, shown in Fig. 1c–n, demonstrated that Pd, Pt and Ir atoms are homogeneously distributed near the surface of the Cu NCs, while Cu atoms are distributed across the entire NCs, forming a solid-solution surface alloy. The atomic percentages of Pd, Pt and Ir contained in Cu/Cu1−xPdx, Cu/Cu1−xPtx and Cu/Cu1−xIrx NCs were calculated to be 1.9, 1.3 and 1.0%, respectively, which are in agreement with the atomic percentages estimated from the SEM–EDX data. The STEM–EDX mapping data of the selected Cu1−xMx shell regions revealed that the alloy compositions were estimated to be Cu0.91Pd0.09, Cu0.92Pt0.08, and Cu0.94Ir0.06, respectively (Fig. S6–S8 and Tables S1–S3).
 | ||
| Fig. 1 (a and b) High-resolution HAADF-STEM images of Cu/Cu1−xIrx NCs. (c–f) HAADF-STEM images and EDX maps of Cu/Cu1−xPdx, (g–j) Cu/Cu1−xPtx and (k–n) Cu/Cu1−xIrx NCs. In the EDX maps, Cu, Pd, Pt and Ir are presented in blue, red, green and yellow, respectively. | ||
In the synchrotron X-ray diffraction (XRD) patterns of Cu/Cu1−xPdx, Cu/Cu1−xPtx and Cu/Cu1−xIrx NCs, in addition to the diffraction peaks derived from fcc Cu NCs as the major component, broad fcc diffraction peaks appeared at angles lower than those of Cu NCs as the minor component (Fig. 2a–e and S9–S12). From Rietveld refinements, the lattice constants of the minor components were estimated to be 3.640(2), 3.641(1), and 3.631(1) Å for Cu/Cu1−xPdx, Cu/Cu1−xPtx and Cu/Cu1−xIrx NCs, respectively (Fig. S9–S12). Assuming that the lattice constants follow Vegard's law, these minor components correspond to Cu0.91Pd0.09, Cu0.92Pt0.08, and Cu0.92Ir0.08 solid-solution alloys, respectively, which are highly consistent with the alloy compositions of Cu1−xMx shell regions estimated from the STEM–EDX data (Fig. S6–S8 and Tables S1–S3). These results indicate the formation of the Cu1−xMx solid-solution alloys on the surface of Cu NCs.
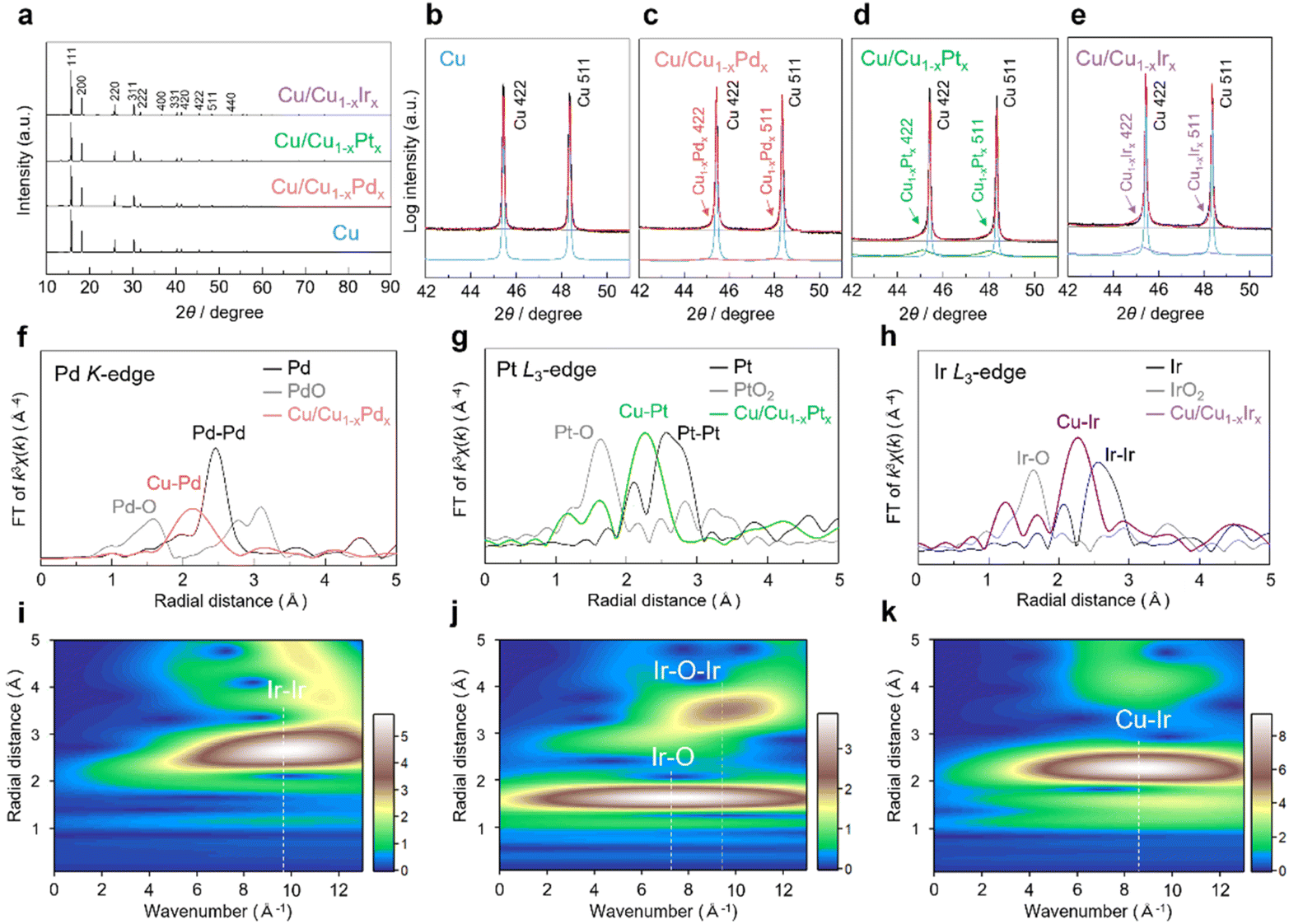 | ||
| Fig. 2 (a) XRD patterns and the expanded XRD patterns of (b) Cu, (c) Cu/Cu1−xPdx, (d) Cu/Cu1−xPtx and (e) Cu/Cu1−xIrx NCs. EXAFS spectra at (f) the Pd K-edge of Cu/Cu1−xPdx NCs, (g) the Pt L3-edge of Cu/Cu1−xPtx NCs, and (h) the Ir L3-edge of Cu/Cu1−xIrx NCs. Ir L3-edge wavelet transform EXAFS of (i) Ir foil, (j) IrO2 powder and (k) Cu/Cu1−xIrx NCs. The colour bar represents the intensity range. | ||
We performed X-ray absorption fine structure (XAFS) measurements to further characterize Cu/Cu1−xMx (M = Pd, Pt, Ir). Fig. 2f–h show the Fourier-transformed (FT) Pd K-edge, Pt L3-edge, and Ir L3-edge extended X-ray absorption fine structure (EXAFS) spectra for Cu/Cu1−xPdx, Cu/Cu1−xPtx and Cu/Cu1−xIrx NCs, respectively. In the R-space, prominent peaks were observed in the region of approximately 2.1–2.3 Å for Cu/Cu1−xPdx NCs and 2.2–2.3 Å for Cu/Cu1−xPtx and Cu/Cu1−xIrx NCs. These peaks are located between those of the corresponding standard metal and the oxide samples, indicating the formation of Cu–M bonds, while no typical peaks appear for the corresponding Pd–Pd, Pt–Pt, and Ir–Ir bonds. On the other hand, in the Cu K-edge FT-EXAFS spectra of Cu/Cu1−xMx, the prominent peak positions of the Cu–Cu contribution were similar to that of Cu NCs (2.2 Å) due to the strong influence of the Cu core and/or the Cu-rich alloy composition (approximately 90 at% Cu) of the shell (Fig. S13). The wavelet transform (WT) of Ir L3-edge EXAFS for Cu/Cu1−xIrx NCs was conducted to resolve the overlapping contributions from different scattering paths by simultaneously analyzing the k- and R-spaces. As shown in Fig. 2i and j, the WT contour plots of the Ir foil and IrO2 reference samples exhibited intensity maxima at 9.8 Å and 7.4 Å, corresponding to Ir–Ir and Ir–O scattering, respectively. In contrast, Cu/Cu1−xIrx NCs showed a single intensity maximum at 8.6 Å (Fig. 2k), which is attributed to Cu–Ir scattering. Similar WT features were observed for Cu/Cu1−xPdx and Cu/Cu1−xPtx NCs, confirming the solid-solution alloying in these systems as well (Fig. S14 and S15).
The electronic structure modification induced by the solid-solution alloying of Cu and M was investigated by X-ray photoelectron spectroscopy (XPS) measurements (Fig. S16–S18). The Pd 3d binding energy of Cu/Cu1−xPdx shifts positively relative to Pd powder, indicating that Pd is partially oxidized (Fig. S16). In contrast, the Pt 4f and Ir 4f binding energies of Cu/Cu1−xPtx (Fig. S17) and Cu/Cu1−xIrx NCs (Fig. S18) exhibit negative shifts relative to their bulk counterparts, indicating that Pt and Ir are partially reduced. Although the Cu 2p binding energy shifts are less distinguishable due to Cu surface oxidation upon exposure to air (Fig. S16–S18), the binding energy shifts of M suggest the occurrence of charge transfer from Pd to Cu in Cu/Cu1−xPdx NCs and from Cu to Pt or Ir in Cu/Cu1−xPtx and Cu/Cu1−xIrx NCs by solid-solution alloying, in agreement with density functional theory (DFT) calculations (Fig. S19 and Table S4).
The functions of solid-solution alloys can be continuously tuned by varying the compositions and combinations of their constituent elements.3,26 Most research on solid-solution alloy NPs has focused on combinations of elements that are miscible in the bulk phase under ambient conditions.27 Recently, non-equilibrium solid-solution alloy NPs, such as Cu–Ir,28 Cu–Ru,29 and Cu–Rh,30 have been reported due to advances in non-equilibrium solution-phase methods. However, achieving solid-solution alloying of immiscible combinations while retaining shape control remains a challenge in non-equilibrium synthesis methods that involve the rapid simultaneous reduction of metal precursors. In this study, we successfully synthesized shape-controlled solid-solution alloys of immiscible combinations using a galvanic replacement reaction. This approach includes not only the Cu–Ir system but also the Cu–Ru system (Fig. S20–S24), where the constituent elements are immiscible even above their melting points in the bulk phase, analogous to the immiscibility of oil and water.
The CO2RR performances of Cu/Cu1−xMx NCs, (M = Pd, Pt, Ru, Ir) were evaluated in a three-electrode flow cell. We deposited the catalyst onto a carbon gas-diffusion electrode (GDE) via spray coating of a material ink and tested the samples in 1 M KOH electrolyte. As shown in Fig. 3a, Cu NCs exhibited high faradaic efficiency of C2+ products (FEC2+) with applied potentials and achieved around 70% at −0.71 V vs. RHE which is consistent with previous reports.12 The CO2RR performance of Cu/Cu1−xMx NCs was strongly dependent on the M species (Fig. 3b, d and S25–S27), and replacing Cu with only 1 at% of Pd, Pt, Ru, and Ir atoms in sequence resulted in a gradual decrease in the selectivity for C2+ products and an increase in the selectivity for C1 components, particularly HCOOH at similar applied potentials (−0.65 to −0.70 V vs. RHE) (Fig. 3b). Remarkably, Cu/Cu1−xIrx NCs exhibited a high C1/CO2RR product ratio across the applied potential range of −0.6 to −0.9 V vs. RHE (Fig. 3c) with high HCOOH selectivity (Fig. 3d). To date, most alloying strategies to improve CO2RR performance involve combining a primary product-forming metal with a secondary one – for example, Cu-based alloys for C2+ products31 or p-block-based alloys for HCOOH.32 Here we demonstrate successful product switching from C2H4 to HCOOH by solid-solution surface alloying of Cu with HER-active Ir. This result motivated a comparative study with a representative Sn catalyst for CO2 to HCOOH conversion.9,32 Although the Sn NPs exhibited slightly higher selectivity for HCOOH than the Cu/Cu1−xIrx NCs (Fig. S28), the different three Cu/Cu1−xIrx NCs provided higher partial current densities over the entire potential range, together with a ∼0.3 V lower overpotential than the Sn NPs (Fig. 3e and S29). In addition, stability tests were performed at a conversion rate of −100 mA cm−2 for Sn NPs and Cu/Cu1−xIrx NCs, respectively (Fig. 3f). The Sn NPs exhibited a rapid decrease in both current density and HCOOH selectivity over time, accompanied by an increase in the HER. This degradation results from the leaching of Sn from the GDE, caused by its dissolution into the electrolyte through the reaction with strongly alkaline KOH. In contrast, Cu/Cu1−xIrx NCs retained both high HCOOH selectivity and current density. Notably, whereas p-block element-based catalysts are typically unstable under alkaline conditions, Cu/Cu1−xIrx NCs exhibit robust stability and activity.
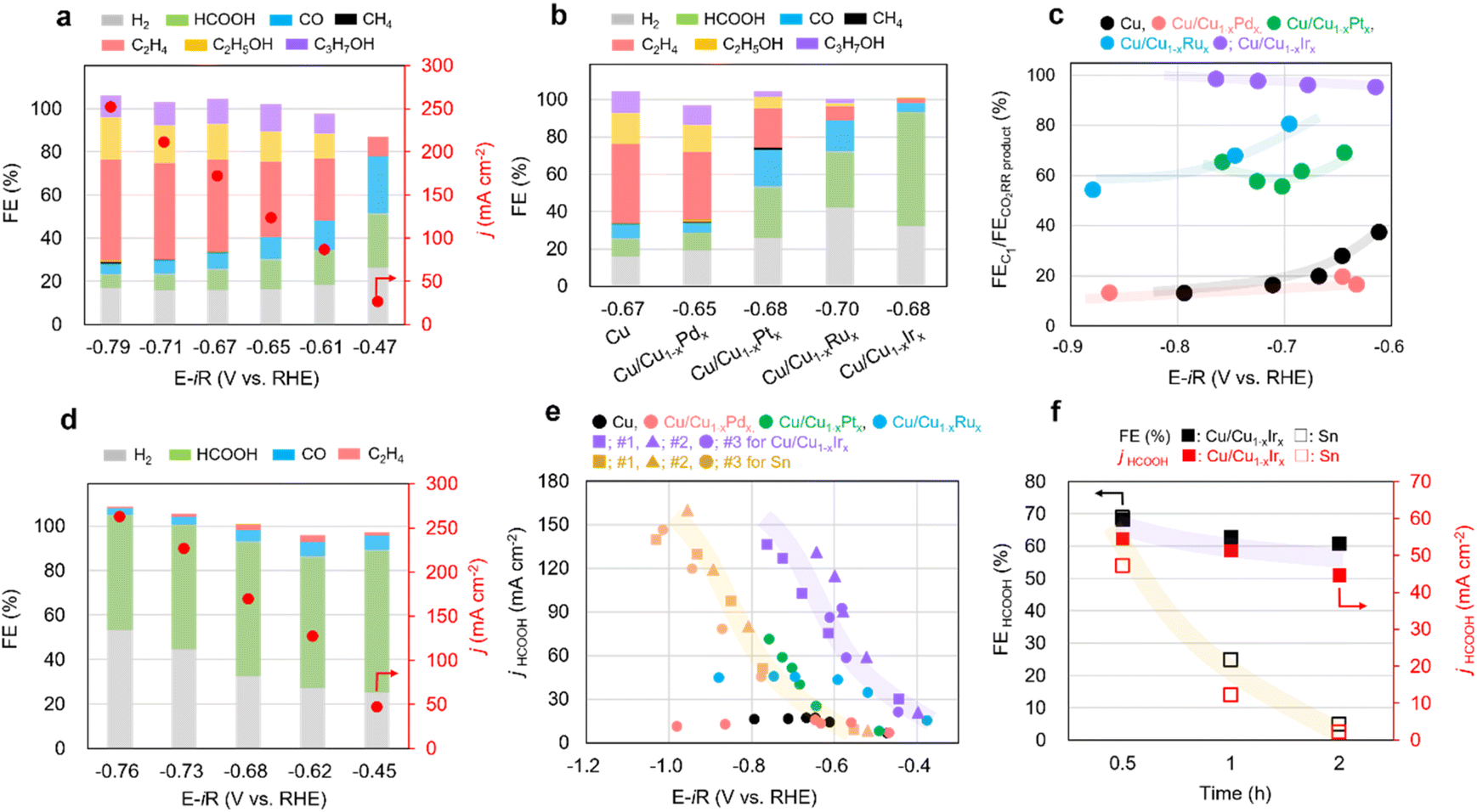 | ||
| Fig. 3 Faradaic efficiency of products on (a) Cu and (b) Cu/Cu1−xMx NCs at similar applied potentials (−0.65 to −0.70 V vs. RHE). (c) C1/CO2RR product selectivity ratio on Cu/Cu1−xMx NCs. (d) Faradaic efficiency of products on Cu/Cu1−xIrx NCs in a range of applied potentials. (e) Plot of partial current density of HCOOH versus applied potential on Cu NC, Sn NPs and Cu/Cu1−xMx. (f) Stability tests of Sn NPs and Cu/Cu1−xIrx NCs at 100 mA cm−2 current density for over 2 h. | ||
To gain insight into the mechanism underlying the shift in product selectivity from C2H4 to HCOOH, and the remarkable activity and stability in HCOOH production on Cu/Cu1−xIrx NCs, we first investigated their structure after CO2RR testing. SEM images before and after the CO2RR indicated that the cubic morphology of Cu NCs degraded substantially, accompanied by particle growth from 50 nm to 200 nm (Fig. 4a, b and S30). In contrast, the morphology and particle size of Cu/Cu1−xIrx NCs were relatively retained (Fig. 4c and d). STEM–EDX mapping indicated the preservation of the Cu–Ir solid-solution surface alloy (Fig. 4e–h) after the CO2RR. Furthermore, in situ FT-EXAFS analyses at the Ir L3-edge and Cu K-edge, along with its WT, revealed that the Ir–Cu (2.2–2.3 Å) and Cu–Cu (2.2 Å) bond lengths and a single intensity maximum at approximately k = 8 Å−1 were retained during the CO2RR (Fig. S31, S32 and 4i–k), demonstrating the persistence of the Cu–Ir solid-solution alloy structure on cubic Cu NCs under reaction conditions.
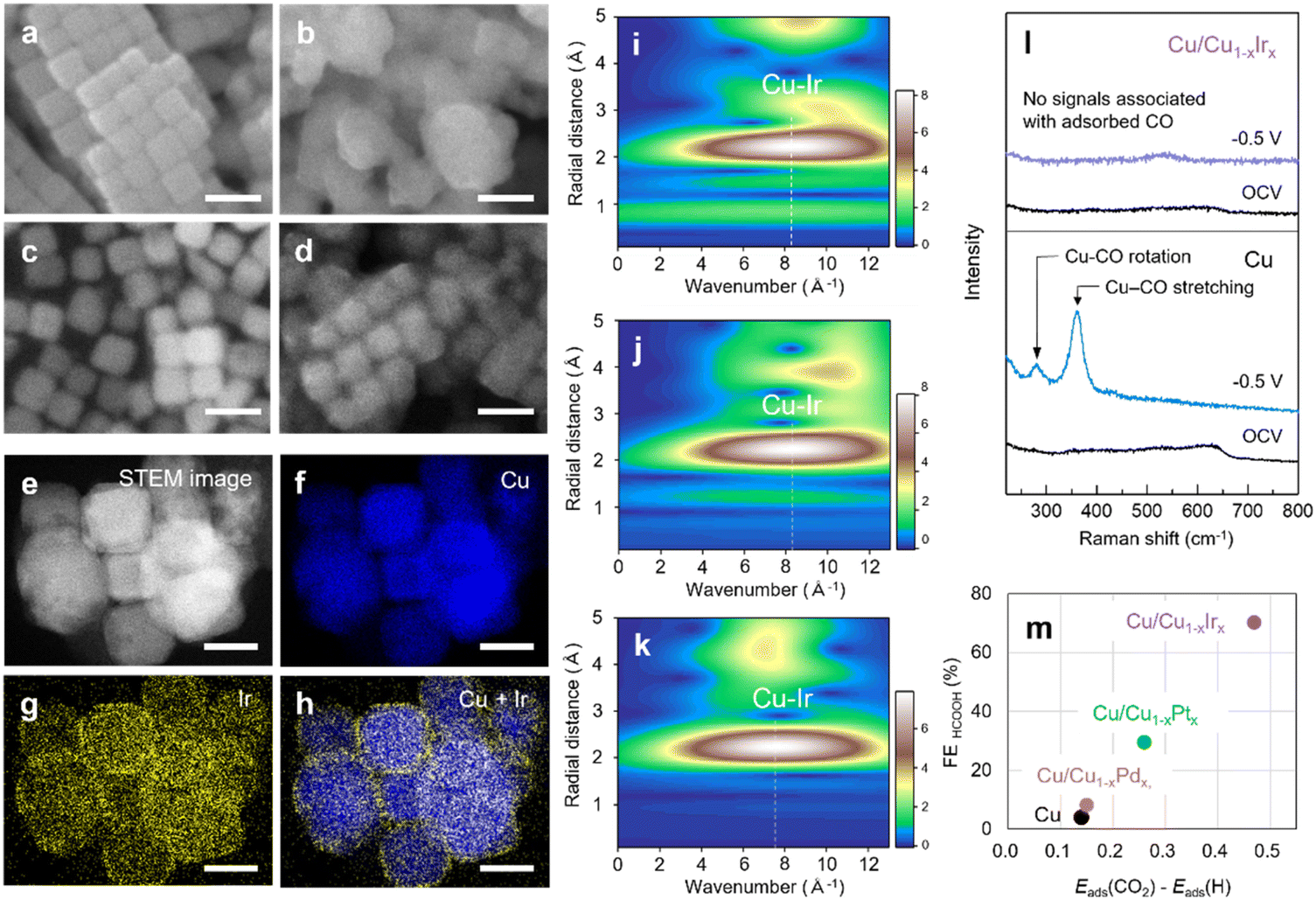 | ||
| Fig. 4 SEM images of (a and b) Cu and (c and d) Cu/Cu1−xIrx NCs (a and c) before and (b and d) after the CO2RR. Scale bars, 100 nm. (e–h) STEM images and EDX maps of Cu/Cu1−xIrx NCs after the CO2RR. Scale bars, 50 nm. Ir L3-edge wavelet transform EXAFS of Cu/Cu1−xIrx NCs at (i) OCV, open-circuit voltage, (j) −0.5 V and (k) OCV after −0.5 V. (l) In situ Raman spectra recorded at OCV and −0.5 V vs. RHE for Cu and Cu/Cu1−xIrx NCs, and (m) the differential adsorption energies (CO2 vs. H*) on Cu NC and the Cu/Cu1−xMx NCs (M = Pd, Pt, Ir). | ||
As CO2RR performance is highly sensitive to catalyst features such as facet orientation, particle size, and alloying state, strategies to preserve these structural parameters under reaction conditions are crucial. Although a few studies have explored approaches such as hetero-element incorporation through alloying33,34 or encapsulation with alumina shells,35 strategies to stabilize catalyst structures during the CO2RR remain scarce. In particular, retaining the size and morphology of Cu NCs at high current densities (>100 mA cm−2) is rarely addressed, since (100)-oriented Cu NCs typically undergo drastic reconstruction into crystalline domains even at low current densities (<10 mA cm−2).25 Here, we show the first demonstration that Ir incorporation effectively stabilizes both the size and morphology of Cu NCs, thereby contributing to the sustained stability in HCOOH production (Fig. 3f).
Given that Ir NPs produced only H2 via the HER (Fig. S33) and that a physical mixture of Ir NPs and Cu NCs resulted in low HCOOH selectivity (Fig. S34), the shift in product selectivity from C2H4 to HCOOH observed for Cu/Cu1−xIrx NCs arises from atomic-level surface alloying of Cu and Ir. To investigate CO2RR pathways, in situ Raman spectroscopy was conducted on the different catalysts. For Cu NCs, distinct peaks appeared at ∼280 and 360 cm−1 at an applied potential of −0.5 V vs. RHE (Fig. 4l), corresponding to the restricted rotation of adsorbed CO and Cu–CO stretching, respectively.36 *CO is the key intermediate that has been proposed for C–C coupling towards C2+ or further hydrogenation to C1 species such as CH4.23 Similar peaks were observed on Cu/Cu1−xPdx NCs (Fig. S35), which also exhibit C2+ product selectivity at the same potential (Fig. S25). In contrast, no CO*-related peaks were detected on Cu/Cu1−xIrx or Cu/Cu1−xPtx NCs at −0.5 V vs. RHE (Fig. 4l and S36). These results suggest that alloying Cu with Ir alters the reaction pathway from C2+ formation toward HCOOH production through a CO*-free pathway.23
From SEM measurements on Cu/Cu1−xPdx and Cu/Cu1−xPtx NCs after the CO2RR (Fig. S37), Cu/Cu1−xPdx exhibited morphological degradation, similar to that observed for Cu NCs after the CO2RR. In contrast, Cu/Cu1−xPtx NCs retained their cubic shape, similar to Cu/Cu1−xIrx NCs. We also confirmed this trend by STEM–EDX mapping analyses (Fig. S38 and S39). Furthermore, in situ XAFS measurements demonstrated that the Cu–Pt solid-solution alloy structure was preserved on the cubic Cu NCs under reaction conditions (Fig. S40). Recently, it has been reported that the degradation of cubic Cu nanocrystals into nanograins is driven by the adsorption of CO intermediates, followed by the formation of Cu carbonyl species at an applied potential.24 Therefore, the distinct HCOOH formation pathway via a CO*-free mechanism on Cu/Cu1−xPtx and Cu/Cu1−xIrx NCs is considered to play an important role in stabilizing both the size and morphology of the cubic Cu NCs during the CO2RR.
COOH* and HCOO* are critical intermediates in determining the product distribution in the CO2RR, where COOH*—formed by the reaction of chemisorbed CO2* with water-derived protons—leads to CO and C2+ products via an asymmetric coupling between CO* and another intermediate such as CH2*,37 while HCOO*—formed by the direct reaction of physisorbed CO2 with surface H*—drives HCOOH formation,38,39 and high H* coverage on the catalyst surface promotes HCOOH formation.40 To elucidate the origin of HCOOH formation on Cu/Cu1−xIrx NCs, we evaluated the adsorption energies of key intermediates (COOH*, HCOO* and H*) and CO2 on Cu NCs and Cu/Cu1−xMx NCs (M = Pd, Pt, Ir) using DFT calculations. The Cu/Cu1−xMx NCs were modelled using a Cu(100) slab, where three surface Cu layers were partially replaced with M atoms (Fig. S19). The results show that COOH* adsorption is more stable than HCOO* adsorption on Cu/Cu1−xIrx or Cu/Cu1−xPtx NCs, whereas HCOO* is more stable on Cu NCs and Cu/Cu1−xPdx NCs (Fig. S41–S45 and Table S6). This stabilization of the COOH* intermediate implies that CO or C2 formation is more favourable on Cu/Cu1−xIrx or Cu/Cu1−xPtx surfaces than on Cu NCs and Cu/Cu1−xPdx NCs, in contrast to the experimental results (Fig. 3b). These discrepancies highlight the importance of considering the adsorption of CO2 and H* prior to intermediate formation. The *H adsorption energy becomes stronger from Cu (−0.39 eV) and Cu/Cu1−xPdx (−0.45 eV) to Cu/Cu1−xPtx (−0.57 eV) and Cu/Cu1−xIrx (−0.85 eV), while the CO2 adsorption energies range from −0.25 to −0.37 eV over Cu1−xMx surfaces (Table S5). As shown in Fig. 4m, the differential adsorption energies (CO2 vs. H*) strongly correlate with FE for HCOOH, suggesting that strong H* adsorption on Cu/Cu1−xIrx NCs plays a key role in promoting HCOO* formation via a direct reaction with physisorbed CO2. While Sn in p-block metals requires a high overpotential for H* supply, isolated HER-active Ir atoms on Cu/Cu1−xIrx NCs efficiently provide H*, thereby decreasing the overpotential and enhancing catalytic activity compared with Sn NPs (Fig. 3e).
Conclusions
In this work, we have designed and developed Cu/Cu1−xMx NCs, M = Pd, Pt, Ru, Ir, for the first comprehensive investigation of CO2 reduction on Cu-based NCs with HER-active platinum group metals. The Cu/Cu1−xMx NCs were successfully synthesized through a galvanic replacement reaction, with Cu1−xMx solid-solution surface alloying confirmed by XRD, EXAFS and HAADF-STEM. Among them, Cu/Cu1−xIrx NCs achieved highly selective HCOOH production at an overpotential ∼0.3 V lower than that of conventional Sn catalysts. The active H* adsorbed on Cu/Cu1−xIrx NCs plays a key role in promoting HCOOH production with the low overpotential. Furthermore, Cu/Cu1−xIrx NCs demonstrate high operational stability, retaining high activity without degradation, in contrast to the rapid deactivation of Sn catalysts. The alloying with Ir effectively stabilized both the size and morphology of Cu NCs. These findings present a versatile strategy for tuning reaction pathways through isolated heteroatom interfaces, offering broad applicability to other elemental combinations and electrocatalytic reactions.Author contributions
H. K. and M. Y. designed the research. S. H. performed the synthesis, most of the structural characterization studies, and electrochemical tests. H. K., A. A., T. M., M. U., T. G. N. and M. D. performed the XAS measurement and analysed the EXAFS and XANES data under the supervision of T. S. and H. S. H. K. and S. H. collected and analysed the STEM data under the supervision of T. Y. and Y. M. H. K., M. D. and T. G. N. collected and analysed the synchrotron X-ray diffraction data under the supervision of K. K. T. S. collected and analysed the SAXS data. Y. S. and T. I. conducted DFT calculations. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
All relevant data are presented in the main text and supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6sc02600a.Acknowledgements
The authors acknowledge funding from the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (18H05517, 22K19088, 23H00313, 24H02202, 24H02205 and 25K01788), Moonshot Research and Development Program (JPNP18016) and Kinoshita Foundation for Science Research Grant FY 2024. XAFS experiments were performed at the Kyushu University Beamline (SAGA-LS/BL06, BL07) with the proposals of no. 2023IIK004, 2023IIIK011, 2024IIK014 and 2309057T. The synchrotron radiation experiments were performed at BL44B2 of SPring-8 with the approval of RIKEN.Notes and references
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- Y. Nakaya and S. Furukawa, Chem. Rev., 2023, 123, 5859–5947 CrossRef CAS PubMed.
- H. Kobayashi, K. Kusada and H. Kitagawa, Acc. Chem. Res., 2015, 48, 1551–1559 CrossRef CAS PubMed.
- S. Das, J. Pérez-Ramírez, J. Gong, N. Dewangan, K. Hidajat, B. C. Gates and S. Kawi, Chem. Soc. Rev., 2020, 49, 2937–3004 RSC.
- S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Nat. Mater., 2008, 7, 333–338 CrossRef CAS PubMed.
- J. Gu, Y.-W. Zhang and F. Tao, Chem. Soc. Rev., 2012, 41, 8050–8065 RSC.
- P. Franke and D. Neuschütz, in Binary Systems. Part 5: Binary Systems Supplement 1: Phase Diagrams, Phase Transition Data, Integral and Partial Quantities of Alloys, ed. P. Franke and D. Neuschütz, Springer, Berlin, Heidelberg, 2007, pp. 1–3 Search PubMed.
- H. Okamoto, J. Phase Equilib., 1992, 13, 440 Search PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS PubMed.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- R. P. Jansonius, L. M. Reid, C. N. Virca and C. P. Berlinguette, ACS Energy Lett., 2019, 4, 980–986 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- M. Yamauchi, Chem. Lett., 2021, 50, 1901–1908 CrossRef CAS.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold and M. Yamauchi, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- M. Sun, J. Cheng and M. Yamauchi, Nat. Commun., 2024, 15, 491 CrossRef CAS PubMed.
- A. Anzai, M. Fukushima, D. S. Rivera Rocabado, T. Ishimoto, T. Sugiyama, B. Ohtani, H. Kobayashi, M.-H. Liu, M. Donoshita, T. G. Noguchi, S. K. Maurya, K. Kato, C. Y. Sit, P. J. A. Kenis and M. Yamauchi, ACS Appl. Mater. Interfaces, 2025, 17, 22665–22676 CrossRef CAS PubMed.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- P. Iyengar, M. J. Kolb, J. R. Pankhurst, F. Calle-Vallejo and R. Buonsanti, ACS Catal., 2021, 11, 4456–4463 CrossRef CAS.
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S.-Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- Y. Yang, J. Feijóo, M. Figueras-Valls, C. Chen, C. Shi, M. V. Fonseca Guzman, Y. Murhabazi Maombi, S. Liu, P. Jain, V. Briega-Martos, Z. Peng, Y. Shan, G. Lee, M. Rebarchik, L. Xu, C. J. Pollock, J. Jin, N. E. Soland, C. Wang, M. B. Salmeron, Z. Chen, Y. Han, M. Mavrikakis and P. Yang, Nat. Catal., 2025, 8, 579–594 CrossRef CAS.
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef PubMed.
- Y. Yao, Q. Dong, A. Brozena, J. Luo, J. Miao, M. Chi, C. Wang, I. G. Kevrekidis, Z. J. Ren, J. Greeley, G. Wang, A. Anapolsky and L. Hu, Science, 2022, 376, eabn3103 CrossRef CAS PubMed.
- Binary Alloy Phase Diagrams, ed. T. B. Massalski, H. Okamoto, P. R. Subramanian and L. Kacprzak, ASM International, Materials Park, OH, 1990 Search PubMed.
- F. Wang, K. Kusada, D. Wu, T. Yamamoto, T. Toriyama, S. Matsumura, Y. Nanba, M. Koyama and H. Kitagawa, Angew. Chem., Int. Ed., 2018, 57, 4505–4509 CrossRef CAS PubMed.
- B. Huang, H. Kobayashi, T. Yamamoto, T. Toriyama, S. Matsumura, Y. Nishida, K. Sato, K. Nagaoka, M. Haneda, W. Xie, Y. Nanba, M. Koyama, F. Wang, S. Kawaguchi, Y. Kubota and H. Kitagawa, Angew. Chem., Int. Ed., 2019, 58, 2230–2235 CrossRef CAS PubMed.
- T. Komatsu, H. Kobayashi, K. Kusada, Y. Kubota, M. Takata, T. Yamamoto, S. Matsumura, K. Sato, K. Nagaoka and H. Kitagawa, Chem.–Eur. J., 2017, 23, 57–60 CrossRef CAS PubMed.
- J. Dai, J. Zhu, Y. Xu, X. Liu, D. Zhu, G. Xu, H. Liu and G. Li, ChemSusChem, 2025, 18, e202402184 CrossRef CAS PubMed.
- D. S. Tran, N.-N. Vu, H.-E. Nemamcha, C. Boisvert, U. Legrand, A. G. Fink, F. Navarro-Pardo, C.-T. Dinh and P. Nguyen-Tri, Coord. Chem. Rev., 2025, 524, 216322 CrossRef CAS.
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, J. Am. Chem. Soc., 2022, 145, 5370–5383 CrossRef PubMed.
- X. K. Lu, B. Lu, H. Li, K. Lim and L. C. Seitz, ACS Catal., 2022, 12, 6663–6671 CrossRef CAS.
- P. P. Albertini, M. A. Newton, M. Wang, O. S. Lecina, P. B. Green, D. C. Stoian, E. Oveisi, A. Loiudice and R. Buonsanti, Nat. Mater., 2024, 23, 680–687 CrossRef CAS PubMed.
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed.
- M. Sun, D. S. Rivera Rocabado, J. Cheng, T. G. Noguchi, M. Donoshita, T. Matsuu, M. Higashi, T. Fujigaya, T. Ishimoto and M. Yamauchi, Angew. Chem., Int. Ed., 2025, 64, e202502740 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard III, J. Am. Chem. Soc., 2016, 138, 13802–13805 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- Z. Liu, R. D. E. Krösschell, I. A. W. Filot and E. J. M. Hensen, Electrochim. Acta, 2024, 493, 144409 CrossRef CAS.
Footnote |
| † Current address: Institute for Catalysis, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan. |
| This journal is © The Royal Society of Chemistry 2026 |