
Regulating the miscibility of donors/acceptors to manipulate the morphology and reduce non-radiative recombination energy loss enables efficient organic solar cells†
Ziqi
Han‡
a,
Ke
Wang‡
b,
Yongqiang
Chai
c,
Rui
Zhang
 *d,
Jianqi
Zhang
e,
Dan
He
*a,
Chunru
Wang
c and
Fuwen
Zhao
*b
*d,
Jianqi
Zhang
e,
Dan
He
*a,
Chunru
Wang
c and
Fuwen
Zhao
*b
aCollege of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, P. R. China. E-mail: hedan@csu.edu.cn
bState Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, P. R. China. E-mail: zhaofuwen@csu.edu.cn
cBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
dDepartment of Physics, Chemistry and Biology (IFM), Linköping University, Linköping SE-58183, Sweden. E-mail: rui.zhang@liu.se
eCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, National Center for Nanoscience and Technology, Beijing 100190, P. R. China
First published on 14th February 2024
Abstract
Due to the high exciton binding energy and relatively low charge carrier mobilities of organic photovoltaic materials, it is crucial to optimize the active layer morphology of organic solar cells (OSCs) to well juggle exciton dissociation and charge carrier transport, and inhibit charge carrier recombination for high power conversion efficiencies (PCEs). Herein, we efficiently improve the crystallinity and miscibility of fused ring electron acceptors (FREAs) via lengthening the side chains and developing four FREAs, BTP-nC8, BTP-C8, BTP-C12 and BTP-C20. The dual functions of lengthening the side chains of FREAs make PM6:FREA blend films present the tendency of first improving then deteriorating in crystallinity, phase separation, domain purity and thus charge carrier dynamics, which leads the JSC and FF of PM6:FREA-based OSCs to show the same trend along with the side-chain length of FREAs. More importantly, enhancing the miscibility between PM6 and FREA facilitates the spatial registry to reduce the formation and recombination rate of triplet excitons in the PM6:FREA blend films, thus inhibiting the non-radiative recombination for decreased ΔEnr, and then increasing VOC in OSCs. Among them, PM6:BTP-C8 based OSCs well balance the multiple impacts of lengthening the side chains to achieve the highest PCE of 17.77%. This work demonstrates that it is important to finely control the crystallinity and miscibility of organic photovoltaic materials to achieve high PCEs in OSCs.
 Fuwen Zhao | Fuwen Zhao received her PhD degree in Physical Chemistry from Institute of Chemistry Chinese Academy of Sciences (ICCAS) in 2017, followed by a National Postdoctoral Program for Innovative Talents postdoc fellowship at ICCAS and a Marie Curie postdoc fellowship at Linköping University. Now she is a full professor in State Key Laboratory of Powder Metallurgy, Central South University. Her current research interests include high-performance organic/perovskite solar cells and organic photocatalysis. Her favorite sayings are “God rewards the diligent” and “no pain, no gain”. |
Introduction
Organic solar cells (OSCs) have attracted much attention over the last three decades, due to the distinctive advantages of low cost, light weight and mechanical flexibility.1–5 Currently, the mainstream OSC is a sandwich structure with the active layer, viz. donor/acceptor blend, in the middle part. Due to the high exciton binding energy and relatively low charge carrier mobilities of organic photovoltaic materials,6–8 the donor/acceptor blend in the active layer has to achieve a nano-scale interpenetrating network structure within the thickness of a hundred nanometers to concurrently ensure exciton diffusion to donor/acceptor interfaces for dissociation and then that the dissociated charge carriers transport to the electrodes efficiently, achieving high short-circuit current density (JSC) and fill factor (FF).9,10 Thus, it is important to optimize the phase separation of the active layer in OSCs. In addition, the aggregation state, including crystallinity,11,12 donor/acceptor interfaces,13 density of states,14,15etc. of the active layer materials also has important impacts on the exciton dynamics and charge carrier recombination in the blend, which would be reflected in the energy loss (Eloss) and thus open-circuit voltage (VOC) of OSCs, except for the difference between the highest occupied molecular orbital (HOMO) energy level of the donors and the lowest unoccupied molecular orbital (LUMO) energy level of the acceptors.16 So far, the relatively large Eloss, especially the non-radiative recombination energy loss (ΔEnr) (a part of Eloss), is considered as one of the main issues limiting the further improvement of the power conversion efficiencies (PCEs) for OSCs.17,18 Either the phase separation or the aggregation state of donor/acceptor materials is strongly related to the miscibility between the donor and acceptor in the active layer.19,20 Therefore, it is crucial to regulate the miscibility of the active layer to achieve highly efficient OSCs.In recent years, the emergence and rapid development of fused ring electron acceptors (FREAs), especially ITIC,21 Y622 and their derivatives, has significantly boosted the advance of OSCs.1 One of the main reasons for the breakthrough originates from ingeniously designing and introducing proper side chains, since the side chains of FREAs not only determine the solubility but also have great impacts on the molecular aggregation.23 Therefore, considerable efforts have been made to finely select and/or adjust the side chains of FREAs to control intramolecular/intermolecular interaction and thus the morphology of the active layer in OSCs.24 Taking Y6 derivatives as examples, side chains including various length alkyl,25 alkyl phenyl,26 alkyl thienyl,27etc. were attached to the terminal thiophene unit to regulate the solubility, molecular packing, crystallinity and aggregation of FREAs. However, the effect of the length of side chains, attached to the pyrrole unit, on the photovoltaic performance of FREAs, especially on VOC, was seldom systematically investigated.28 In addition, the larger dipole moment of the C–Cl bond than C–F bond can enhance the intramolecular charge transfer effect to reduce the bandgap, and the empty 3d orbitals of chlorine atoms can also bring about the noncovalent interactions of Cl⋯π and Cl⋯S to facilitate molecular stacking.29 Thus, chlorinated end groups are excellent alternatives for FREAs.30
Motivated by these points, we developed four FREAs, BTP-nC8, BTP-C8, BTP-C12 and BTP-C20 (Fig. 1a), who possess the same conjugated backbone, but different side chains attached onto the pyrrole units. Lengthening the side chains of the FREAs endows them with increased crystallinity and enhanced miscibility with the classical polymer donor, PM6. The OSCs based on PM6:FREA were further fabricated to investigate its impacts on the photovoltaic performance. The evolutions of charge carrier dynamics, active layer morphology, and triplet excitons (T1) in the PM6:FREA based devices were also studied to elucidate the inner relationship among the properties of FREAs, active layer morphology and device performance. Among them, PM6:BTP-C8 based OSCs well balanced the multiple impacts of lengthening the side chains of FREAs and achieved the highest PCE of 17.77%.
 | ||
| Fig. 1 (a) Molecular structures of PM6 and the FREAs. (b) Normalized absorption spectra of PM6 and FREA neat films. | ||
Results and discussion
The synthetic routes for FREAs, BTP-nC8, BTP-C8, BTP-C12, and BTP-C20 are presented in the ESI† (Fig. S1). The chemical structures of the intermediates and FREAs were confirmed by NMR spectra (Fig. S2–S13, ESI†) and mass spectra (Fig. S14–S17, ESI†). The electrochemical characteristic curves of BTP-nC8, BTP-C8, BTP-C12 and BTP-C20 were determined by cyclic voltammetry (Fig. S18, ESI†). According to the empirical equation, the HOMO/LUMO energy levels of BTP-nC8, BTP-C8, BTP-C12 and BTP-C20 were calculated to be −5.73/−3.82 eV, −5.74/−3.84 eV, −5.75/−3.83 eV and −5.76/−3.83 eV, respectively (Table S1, ESI†), which well agree with the reported values.25 The four FREAs almost have the same HOMO/LUMO energy levels, due to their identical conjugated backbones. The solution and thin-film absorption spectra of FREAs were recorded on a UV-Vis spectrophotometer. In solution, four FREAs also exhibit almost the same absorption with the maximum absorption peak at 745 nm (Fig. S19, ESI†). However, in thin-film states, the maximum absorption peaks of BTP-nC8, BTP-C8, BTP-C12 and BTP-C20 are 794 nm, 819 nm, 828 nm and 809 nm, respectively, which show the tendency of first increasing then decreasing along with the increment of the side chain length (Fig. 1b). The red-shifted absorption of BTP-C8 and BTP-C12, compared with BTP-nC8, probably results from their more ordered molecular stacking.28 Likewise, the absorption onsets of BTP-nC8, BTP-C8, BTP-C12 and BTP-C20 have a similar tendency as their maximum peaks, corresponding to the optical bandgaps of 1.40 eV, 1.39 eV, 1.39 eV and 1.42 eV, respectively (Table S1, ESI†). This implies that there must be some difference in the molecular packing of the four FREAs. To further investigate the molecular orientation and molecular stacking in the film, two-dimensional grazing-incidence wide-angle X-ray scattering (GIWAXS) was conducted. BTP-C8, BTP-C12 and BTP-C20 all prefer the face-on orientation relative to the substrate in neat films, while the BTP-nC8 thin film is dominated by the edge-on orientation (Fig. S20, ESI†), probably due to the linear alkyl chains attached to the pyrrole unit of BTP-nC8.28,31 The π–π stacking diffraction peaks of BTP-nC8, BTP-C8 and BTP-C12 neat films are located at 1.71–1.72 Å−1, corresponding to the d-spacings of 3.65–3.67 Å, while the BTP-C20 neat film exhibits two π–π stacking diffraction peaks at 1.72 Å−1 and 1.42 Å−1, corresponding to the d-spacings of 3.65 Å and 4.42 Å, respectively, indicating the hierarchical structure in the film. Besides, the π–π stacking diffraction intensities gradually increase from BTP-nC8, to BTP-C8, BTP-C12 and BTP-C20, which implies the enhanced crystallinity along with side-chain length. The GIWAXS results of the FREA neat films echo their thin-film absorption. In addition, we further chose the classical high-performance wide-bandgap polymer, PM6, as a donor,32 which possesses complementary absorption and matched energy levels with the FREAs (Fig. 1b and Fig. S18, ESI†), and evaluated the miscibility between PM6 and FREAs via investigating the Flory-Huggins interaction parameters (χ) between them based on the surface free energy (γ).33,34 The contact angles of water and glycerol on PM6, BTP-nC8, BTP-C8, BTP-C12 and BTP-C20 neat films are 98.8°/89.6°, 97.8°/82.2°, 96.6°/82.8°, 97.4°/83.3° and 99.9°/86.2°, respectively (Fig. 2). According to Wu's model,35 the γ values of PM6, BTP-nC8, BTP-C8, BTP-C12 and BTP-C20 were calculated to be 21.33 mN m−1, 26.59 mN m−1, 25.98 mN m−1, 25.37 mN m−1 and 23.89 mN m−1, respectively. Since the Flory-Huggins interaction parameter (χ) is directly proportional to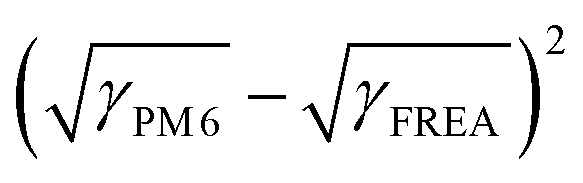 , the reduced surface-free-energy root difference between PM6 and FREAs means a smaller χ, as shown in Table S2 (ESI†). The smaller the χ value, the better the miscibility between PM6 and FREAs. Thus, it can be easily deduced that the miscibility between PM6 and the FREAs increases along with the side-chain length. In short, increasing the side-chain length of FREAs has negligible effects on their energy levels, but can change the molecular packing, improve the crystallinity and enhance the miscibility with the polymer donor, PM6.
, the reduced surface-free-energy root difference between PM6 and FREAs means a smaller χ, as shown in Table S2 (ESI†). The smaller the χ value, the better the miscibility between PM6 and FREAs. Thus, it can be easily deduced that the miscibility between PM6 and the FREAs increases along with the side-chain length. In short, increasing the side-chain length of FREAs has negligible effects on their energy levels, but can change the molecular packing, improve the crystallinity and enhance the miscibility with the polymer donor, PM6.
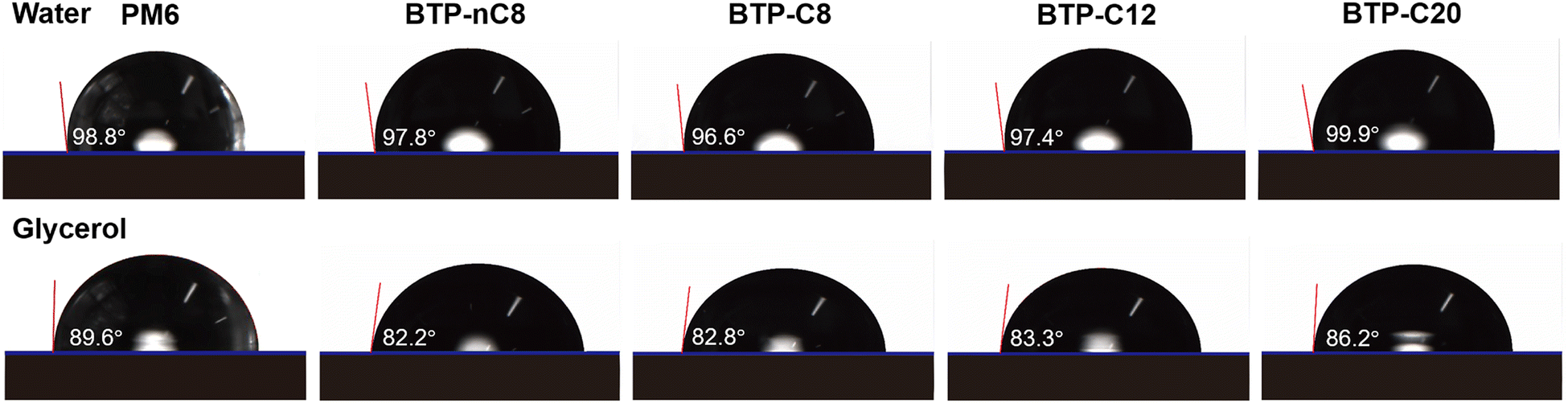 | ||
| Fig. 2 Contact angles of water and glycerol on PM6 and FREA neat films. | ||
To explore the effects of variational properties of FREAs on the photovoltaic performance, conventional OSCs with a structure of ITO/PEDOT:PSS/PM6:FREA/PDINN/Ag were fabricated. The device optimized processes and data are collected in Tables S3–S14 (ESI†). The current density–voltage (J–V) curves and photovoltaic parameters of their optimized OSCs are presented in Fig. 3a and Table 1, respectively. It can be found that the JSC and FF of the OSCs show the tendency of first increasing then decreasing, while the VOC of the OSCs increases from 0.854 ± 0.003 V (PM6:BTP-nC8), 0.877 ± 0.003 V (PM6:BTP-C8), and 0.889 ± 0.002 V (PM6:BTP-C12) to 0.905 ± 0.002 V (PM6:BTP-C20), successively. Among them, the PM6:BTP-C8 based OSC well balances the photovoltaic parameters and achieves the highest PCE of 17.77% with the best JSC of 26.23 mA cm−2 and FF of 77.0%. To further verify the JSC of the devices, external quantum efficiencies (EQEs) of the optimized OSCs were determined. The EQE spectra of the PM6:FREA based OSCs are as shown in Fig. 3b, and the calculated integration currents, obtained from the EQE curves, of the PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs are 25.58 mA cm−2, 26.19 mA cm−2, 25.68 mA cm−2, and 25.38 mA cm−2, respectively, which all agree well with the values from their J–V curves (<5% mismatch).
 | ||
| Fig. 3 (a) J–V curves and (b) EQE spectra of the optimized PM6:FREA based OSCs. (c) Photocurrent density versus effective voltage curves of the PM6:FREA based OSCs. (d) Dependence of VOC on the incident light intensity of the PM6:FREA based devices. (e) EQEEL curves of the PM6:FREA based devices. (f) Comparison of ΔEnr and Eloss of the PM6:FREA based OSCs. | ||
| PM6:FREA | V OC [V] | J SC [mA cm−2] | FF [%] | PCE [%] | J EQE [mA cm−2] |
|---|---|---|---|---|---|
| PM6:BTP-nC8 | 0.857 (0.854 ± 0.003) | 26.01 (26.23 ± 0.23) | 71.4 (70.7 ± 0.7) | 15.93 (15.77 ± 0.19) | 25.58 |
| PM6:BTP-C8 | 0.879 (0.877 ± 0.003) | 26.23 (26.00 ± 0.46) | 77.0 (76.5 ± 0.4) | 17.77 (17.37 ± 0.47) | 26.19 |
| PM6:BTP-C12 | 0.888 (0.889 ± 0.002) | 26.03 (26.08 ± 0.15) | 73.1 (71.8 ± 1.1) | 16.91 (16.40 ± 0.41) | 25.68 |
| PM6:BTP-C20 | 0.907 (0.905 ± 0.002) | 25.73 (25.75 ± 0.12) | 70.3 (69.8 ± 0.5) | 16.42 (16.14 ± 0.30) | 25.38 |
To investigate the reasons for the evolution of JSC and FF in the OSCs, the charge carrier dynamics of their devices were studied. The photocurrent density (Jph) versus the effective voltage (Veff) was determined to probe into the exciton dissociation properties in the PM6:FREA based OSCs (Fig. 3c).36,37Jph is denoted as Jph = JL − JD, where JL is the current density under illumination and JD is that in the dark. Veff is obtained by Veff = Vappl − V0, where V0 is the voltage when Jph is zero and Vappl is the applied bias voltage. Jsat represents the Jph under a high Veff (Veff = 2 V in this work), where all photo-generated excitons are assumed to dissociate into free charge carriers and be collected by the electrodes. The Jsats values of PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs are 26.89 mA cm−2, 26.71 mA cm−2, 26.56 mA cm−2 and 26.55 mA cm−2, respectively, due to the similar absorption of PM6:FREA blends. Then, under JSC conditions, the exciton dissociation probabilities (P(E,T) = Jph/Jsat) were estimated to be 96.9% (96.6 ± 0.4%), 98.2% (97.9 ± 0.3%), 98.0% (97.5 ± 0.4%) and 96.9% (96.5 ± 0.4%) for PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based devices, respectively, indicating the superior exciton dissociation in PM6:BTP-C8 and PM6:BTP-C12 based OSCs, which would contribute to higher JSC. The charge carrier recombination in the PM6:FREA based OSCs was further investigated via studying the dependence of JSC and VOC on incident light intensity, Plight. The JSCs of the PM6:FREA based OSCs under different Plight were fitted by using the power-law equation: JSC ∝ [Plight]α (Fig. S21, ESI†).38 The α values of PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs are 0.974 (0.967 ± 0.005), 0.995 (0.995 ± 0.003), 0.984 (0.980 ± 0.003) and 0.963 (0.959 ± 0.003), respectively. The closest to 1 for α indicates the weakest bimolecular recombination in the PM6:BTP-C8 based OSC. Their VOCs were also plotted against ln[Plight] to evaluate the monomolecular recombination in the devices.39 As shown in Fig. 3d, the slopes for PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based devices were calculated to be 1.17kT/q ((1.21 ± 0.03)kT/q), 1.13kT/q ((1.14 ± 0.01)kT/q), 1.09kT/q ((1.10 ± 0.02)kT/q) and 1.04kT/q ((1.06 ± 0.01)kT/q), respectively, where k is the Boltzmann constant, T is the Kelvin temperature and q is the elementary charge. The smaller slope implies the lower monomolecular recombination. Thus, the monomolecular recombination shows a decreasing tendency from PM6:BTP-nC8, to PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs. To further assess the charge carrier transport properties in the OSC, PM6:FREA based hole-only and electron-only devices were fabricated to determine the hole and electron mobilities by using the space-charge limited current method. As shown in Fig. S22 and Table S15 (ESI†), the hole mobilities (μh) of the PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based devices are 1.13 × 10−4 cm2 V−1 s−1, 7.97 × 10−4 cm2 V−1 s−1, 4.91 × 10−4 cm2 V−1 s−1 and 1.48 × 10−4 cm2 V−1 s−1, respectively, while the electron mobilities (μe) of those electron-only devices are 1.57 × 10−4 cm2 V−1 s−1, 6.86 × 10−4 cm2 V−1 s−1, 3.06 × 10−4 cm2 V−1 s−1 and 0.35 × 10−4 cm2 V−1 s−1, respectively. The PM6:BTP-C8 based devices exhibit the highest and most balanced charge carrier mobilities, which helps to achieve superior JSC and FF. In short, even though the monomolecular recombination in PM6:BTP-C12 and PM6:BTP-C20 based OSCs is slightly weaker than that in the PM6:BTP-C8 based one, other properties of PM6:FREA based devices, including P(E,T), bimolecular recombination and charge carrier mobilities, all show the evolution of first improvement then deterioration, which is highly consistent with the tendency of JSC and FF in OSCs. Among them, the PM6:BTP-C8 based devices have the most efficient exciton dissociation, weakest bimolecular recombination, and highest and most balanced charge carrier mobilities, thus contributing to the best JSC and FF in the OSC.40
To clarify the relationship between charge carrier dynamics in PM6:FREA based devices and material properties, the morphology of PM6:FREA blend films was further investigated by GIWAXS, resonant soft X-ray scattering (R-SoXS) and atomic force microscopy (AFM), respectively. As shown in Fig. 4, all blend films prefer the face-on orientation with the intense π–π stacking (010) diffraction peaks in the out-of-plane (OOP) direction located at ∼1.71 Å−1, corresponding to the d-spacing of ∼3.67 Å. The crystalline coherence lengths (CCLs) of the π–π stacking of PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 blend films are 18.6 Å, 19.8 Å, 17.3 Å and 15.9 Å, respectively, which shows the tendency of first increasing then decreasing. Such evolution of the morphology of the PM6:FREA blend films should originate from the double-edged sword effects of lengthening the side chains of the FREAs: on the one hand, lengthening the side chains improves the crystallinity to promote molecular packing in the PM6:FREA blend film; on the other hand, it also enhances the miscibility of FREA with PM6 to facilitate interpenetration and thus interfere with molecular stacking. The PM6:BTP-C8 blend film well balances the dual functions and thus achieves the most ordered molecular packing, facilitating charge carrier transport for the highest JSC and FF. R-SoXS analysis was further conducted to investigate phase separation in the PM6:FREA blend films, utilizing a photon energy of 284.30 eV to achieve optimal material contrast.41 The scattering profile revealed a mode of distribution (referred to as smode) that corresponds to the characteristic mode length scale, ξ. It is noted that the mode domain size is half that of ξ. According to the R-SoXS profiles (Fig. S23, ESI†), the domain sizes of PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 blend films were 41 nm, 26 nm, 14 nm and 18 nm, respectively. The scattering profiles were further processed to extract the domain purity through the total scattering intensity (TSI), according to Porod's invariant.42 The relative domain purities of the PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 blend films were 0.95, 1.0, 0.90 and 0.92, respectively, which also shows the tendency of first increasing then decreasing. AFM was also employed to investigate the phase separation in the PM6:FREA blend films. As shown in Fig. S24a–d (ESI†), the root-mean-square roughness values of the PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 blend films were 1.18 nm, 2.03 nm, 1.27 nm and 1.60 nm, respectively, showing the same tendency as the reported results.28 The PM6:BTP-C8 blend film exhibits the largest root-mean-square roughness, probably due to the superior crystallinity. In the AFM phase images (Fig. S24e–h, ESI†), all PM6:FREA blend films present nanofiber structures, but the PM6:BTP-C8 blend film has the longest and clearest nanofibers, due to its highest crystallinity and domain purity, which is in line with the GIWAXS and R-SoXS results.
 | ||
| Fig. 4 (a)–(d) 2D GIWAXS patterns of the PM6:FREA blend films. (e) Corresponding GIWAXS intensity profiles along the IP and OOP directions. | ||
To rationalize the increment in VOCs from PM6:BTP-nC8, PM6:BTP-C8, and PM6:BTP-C12 to PM6:BTP-C20-based OSCs, we firstly extracted the bandgaps of the blend films from the EQE spectra. The bandgaps of the PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 blend films are 1.440 eV, 1.430 eV, 1.435 eV and 1.441 eV, respectively.43 Thus, the Elosss of the PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs were calculated to be 0.583 eV, 0.551 eV, 0.547 eV and 0.534 eV, respectively. We then determined the ΔEnr of the PM6:FREA based OSCs via measuring the electroluminescence quantum efficiency (EQEEL).44 As shown in Fig. 3e, the EQEELs of the PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based devices are 2.26 × 10−5, 4.95 × 10−5, 5.60 × 10−5 and 1.12 × 10−4, respectively. According to the equation: ΔEnr = −kT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln[EQEEL],44 the calculated ΔEnr of the PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs decrease from 0.276 eV to 0.256 eV, 0.253 eV and 0.235 eV, successively. Thus, the reduction of Eloss from the PM6:BTP-nC8 to PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs is mainly ascribed to the decrement in ΔEnr (Fig. 3f). To clarify the decrement of ΔEnr in the PM6:FREA based OSCs along with the prolonged side chains, we further studied exciton dynamics via femtosecond transient absorption (femto-TA) to assess the formation of non-emissive T1 in the film, since ΔEnr is strongly related to it.44,45 We selectively excited FREAs in both neat and blend films by using a pump pulse at the maximum absorption wavelength. The 2D color plot of the femto-TA spectra of FREA neat films and PM6:FREA blend films are collected in Fig. S25a–d and S26a–d (ESI†), respectively. The selected delay times of the neat and blend films are presented in Fig. S25e–h and S26e–h (ESI†), respectively, for a better view. The initial femto-TA spectra of the FREAs after photoexcitation are characterized by a sharp ground-state bleaching (GSB) at 850 nm, a dominant excited-state absorption (ESA) at 900 nm, and a broad ESA band centered around 1500 nm (Fig. S25, ESI†).46 Afterwards, the initial GSB at 850 nm and EAS at 900 nm recover rapidly, while the maximum of the broad ESA band shifts from 1500 nm to 1450 nm, which is very similar to the formation of T1 in the literature.47 Essentially, similar spectra features evolve for the PM6:FREA blend films on the femto-TA timescale (Fig. S26, ESI†), implying the non-radiative recombination pathways through the T1 states in all systems.48 Given that all PM6:FREA blend films were excited by using the identical pump fluence, the populations of the initial singlet excitons should be the same. Indeed, the initial ESA peaks of all PM6:FREA blend films almost overlap with each other at 900 nm (Fig. 5a). However, as shown in Fig. 5b, the intensity of T1 at 1450 nm decreases from PM6:BTP-nC8 to PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 blend films, successively. We further investigated the decay kinetics of T1via extracting the signals at 1450 nm of the FREA neat films (Fig. 5c) and PM6:FREA blend films (Fig. 5d). It can be found that there is no obvious difference for the decay trace of T1 in the FREA neat films. However, by fitting the decay trace of T1, the lifetimes of T1 gradually increase from PM6:BTP-nC8 (406 ps) to PM6:BTP-C8 (460 ps), PM6:BTP-C12 (644 ps) and PM6:BTP-C20 (714 ps) blend films. In short, we can conclude that the increment of the side-chain length of the FREAs can efficiently inhibit the formation of T1 and reduce the recombination rate of T1 in the PM6:FREA blend films, thus suppressing the non-radiative recombination to decrease ΔEnr from PM6:BTP-nC8 to PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs, probably since prolonging the side chains of FREAs enhances the miscibility and then benefits the spatial registry between FREA with PM6 to allow for the closer intermolecular contacts and strengthened wave function interactions.48
ln[EQEEL],44 the calculated ΔEnr of the PM6:BTP-nC8, PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs decrease from 0.276 eV to 0.256 eV, 0.253 eV and 0.235 eV, successively. Thus, the reduction of Eloss from the PM6:BTP-nC8 to PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs is mainly ascribed to the decrement in ΔEnr (Fig. 3f). To clarify the decrement of ΔEnr in the PM6:FREA based OSCs along with the prolonged side chains, we further studied exciton dynamics via femtosecond transient absorption (femto-TA) to assess the formation of non-emissive T1 in the film, since ΔEnr is strongly related to it.44,45 We selectively excited FREAs in both neat and blend films by using a pump pulse at the maximum absorption wavelength. The 2D color plot of the femto-TA spectra of FREA neat films and PM6:FREA blend films are collected in Fig. S25a–d and S26a–d (ESI†), respectively. The selected delay times of the neat and blend films are presented in Fig. S25e–h and S26e–h (ESI†), respectively, for a better view. The initial femto-TA spectra of the FREAs after photoexcitation are characterized by a sharp ground-state bleaching (GSB) at 850 nm, a dominant excited-state absorption (ESA) at 900 nm, and a broad ESA band centered around 1500 nm (Fig. S25, ESI†).46 Afterwards, the initial GSB at 850 nm and EAS at 900 nm recover rapidly, while the maximum of the broad ESA band shifts from 1500 nm to 1450 nm, which is very similar to the formation of T1 in the literature.47 Essentially, similar spectra features evolve for the PM6:FREA blend films on the femto-TA timescale (Fig. S26, ESI†), implying the non-radiative recombination pathways through the T1 states in all systems.48 Given that all PM6:FREA blend films were excited by using the identical pump fluence, the populations of the initial singlet excitons should be the same. Indeed, the initial ESA peaks of all PM6:FREA blend films almost overlap with each other at 900 nm (Fig. 5a). However, as shown in Fig. 5b, the intensity of T1 at 1450 nm decreases from PM6:BTP-nC8 to PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 blend films, successively. We further investigated the decay kinetics of T1via extracting the signals at 1450 nm of the FREA neat films (Fig. 5c) and PM6:FREA blend films (Fig. 5d). It can be found that there is no obvious difference for the decay trace of T1 in the FREA neat films. However, by fitting the decay trace of T1, the lifetimes of T1 gradually increase from PM6:BTP-nC8 (406 ps) to PM6:BTP-C8 (460 ps), PM6:BTP-C12 (644 ps) and PM6:BTP-C20 (714 ps) blend films. In short, we can conclude that the increment of the side-chain length of the FREAs can efficiently inhibit the formation of T1 and reduce the recombination rate of T1 in the PM6:FREA blend films, thus suppressing the non-radiative recombination to decrease ΔEnr from PM6:BTP-nC8 to PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs, probably since prolonging the side chains of FREAs enhances the miscibility and then benefits the spatial registry between FREA with PM6 to allow for the closer intermolecular contacts and strengthened wave function interactions.48
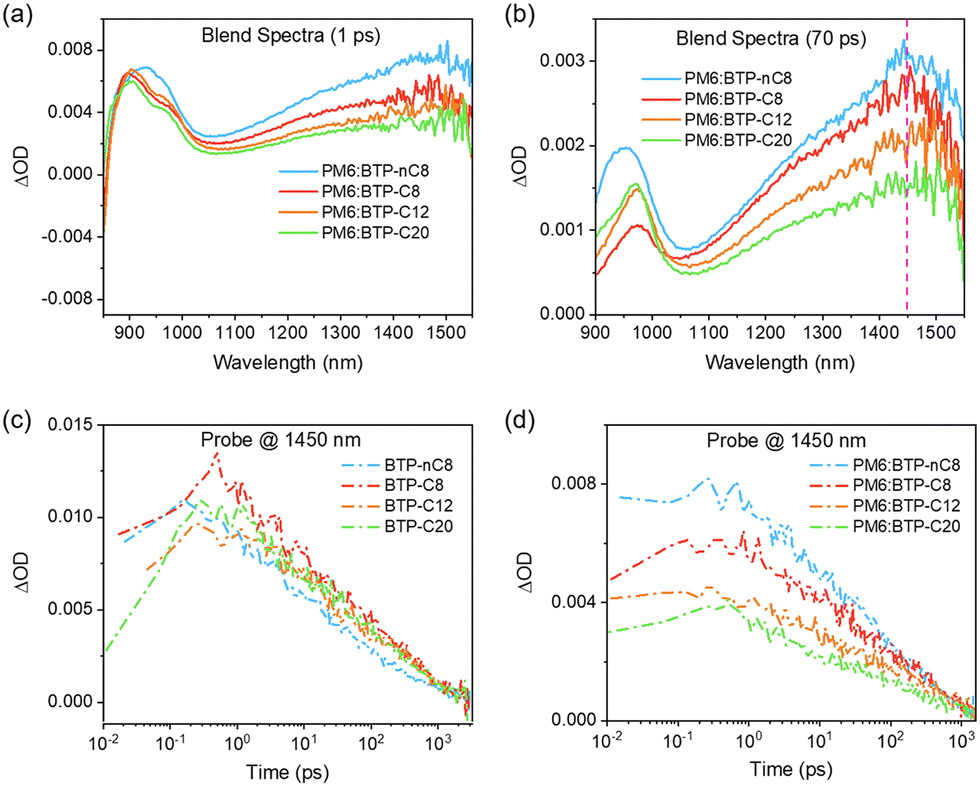 | ||
| Fig. 5 Femto-TA spectra at the delay of (a) 1 ps and (b) 70 ps for the PM6:FREA blend films. fs-Resolved kinetic curves of (c) FREA neat films and (d) PM6:FREA blend films probed at 1450 nm. The fs-resolved TA data are acquired with a pump at the maximum absorption wavelength of the FREAs. The signals are normalized at the maximal amplitude of the ESA signal at 900 nm. | ||
Conclusions
In this work, we developed four FREAs, BTP-nC8, BTP-C8, BTP-C12 and BTP-C20, which exhibit enhanced crystallinity and miscibility simultaneously along with side-chain length. The dual functions of lengthening the side chains of the FREAs make the morphology of PM6:FREA blend films show the tendency of first improving then deteriorating in crystallinity, domain purity, phase separation and thus charge carrier dynamics, which leads the JSC and FF of PM6:FREA based OSCs to afford the same trend with the side-chain length of the FREAs. In addition, enhancing the miscibility via prolonging the side chains of the FREAs facilitates the spatial registry to promote intermolecular contacts and strengthen wave function interactions between PM6 and the FREAs. This efficiently reduces the formation and recombination rate of T1 in the PM6:FREA blend films to suppress the non-radiative recombination to decrease ΔEnr and thus increase VOC from PM6:BTP-nC8 to PM6:BTP-C8, PM6:BTP-C12 and PM6:BTP-C20 based OSCs, successively. Among them, the PM6:BTP-C8 based OSCs well balance the multiple impacts to achieve the highest PCE of 17.77% with the best JSC and FF. Our work demonstrates that it is important to regulate the side chains of FREAs and finely control the crystallinity and miscibility to optimize the active layer morphology and reduce Eloss for high PCEs in OSCs.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant No. 52372056, 52272056, 52232003), the Science and Technology Innovation Program of Hunan Province (Grant No. 2023RC3044), and the Natural Science Foundation of Hunan Province (Grant No. 2022JJ40570) for financial support. X-ray data were acquired at beamlines 7.3.3 and 11.0.1.2 at the Advanced Light Source, supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.Notes and references
- J. Wang, P. Xue, Y. Jiang, Y. Huo and X. Zhan, Nat. Rev. Chem., 2022, 6, 614–634 CrossRef.
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- F. Zhao, J. Zhou, D. He, C. Wang and Y. Lin, J. Mater. Chem. C, 2021, 9, 15395–15406 RSC.
- K. Fukuda, K. Yu and T. Someya, Adv. Energy Mater., 2020, 10, 2000765 CrossRef CAS.
- W. Liu, S. Sun, L. Zhou, Y. Cui, W. Zhang, J. Hou, F. Liu, S. Xu and X. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202116111 CrossRef CAS PubMed.
- D. He, M. Zeng, Z. Zhang, Y. Bai, G. Xing, H.-M. Cheng and Y. Lin, SmartMat, 2023, 4, e1176 CrossRef CAS.
- F. Zhao, D. He, C. Zou, Y. Li, K. Wang, J. Zhang, S. Yang, Y. Tu, C. Wang and Y. Lin, Adv. Mater., 2023, 35, 2210463 CrossRef CAS PubMed.
- J. Wu, H. Cha, T. Du, Y. Dong, W. Xu, C.-T. Lin and J. R. Durrant, Adv. Mater., 2022, 34, 2101833 CrossRef CAS PubMed.
- G. Yu, J. Gao, J. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- F. Zhao, H. Zhang, R. Zhang, J. Yuan, D. He, Y. Zou and F. Gao, Adv. Energy Mater., 2020, 10, 2002746 CrossRef CAS.
- J. Zhou, D. He, Y. Li, F. Huang, J. Zhang, C. Zhang, Y. Yuan, Y. Lin, C. Wang and F. Zhao, Adv. Mater., 2023, 35, 2207336 CrossRef CAS PubMed.
- Q. He, W. Sheng, M. Zhang, G. Xu, P. Zhu, H. Zhang, Z. Yao, F. Gao, F. Liu, X. Liao and Y. Chen, Adv. Energy Mater., 2021, 11, 2003390 CrossRef CAS.
- K. Vandewal, J. Widmer, T. Heumueller, C. J. Brabec, M. D. McGehee, K. Leo, M. Riede and A. Salleo, Adv. Mater., 2014, 26, 3839–3843 CrossRef CAS PubMed.
- G. Garcia-Belmonte, P. P. Boix, J. Bisquert, M. Lenes, H. J. Bolink, A. La Rosa, S. Filippone and N. Martín, J. Phys. Chem. Lett., 2010, 1, 2566–2571 CrossRef CAS.
- D. He, Y. Li, F. Zhao and Y. Lin, Chem. Commun., 2024, 60, 364–373 RSC.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef.
- X.-K. Chen, D. Qian, Y. Wang, T. Kirchartz, W. Tress, H. Yao, J. Yuan, M. Hülsbeck, M. Zhang, Y. Zou, Y. Sun, Y. Li, J. Hou, O. Inganäs, V. Coropceanu, J.-L. Bredas and F. Gao, Nat. Energy, 2021, 6, 799–806 CrossRef CAS.
- D. He, F. Zhao, C. Wang and Y. Lin, Adv. Funct. Mater., 2022, 32, 2111855 CrossRef CAS.
- L. Ye, H. Hu, M. Ghasemi, T. Wang, B. A. Collins, J.-H. Kim, K. Jiang, J. H. Carpenter, H. Li, Z. Li, T. McAfee, J. Zhao, X. Chen, J. L. Y. Lai, T. Ma, J.-L. Brédas, H. Yan and H. Ade, Nat. Mater., 2018, 17, 253–260 CrossRef CAS PubMed.
- Y. Cai, Y. Li, R. Wang, H. Wu, Z. Chen, J. Zhang, Z. Ma, X. Hao, Y. Zhao, C. Zhang, F. Huang and Y. Sun, Adv. Mater., 2021, 33, 2101733 CrossRef CAS PubMed.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- D. He, F. Zhao, L. Jiang and C. Wang, J. Mater. Chem. A, 2018, 6, 8839–8854 RSC.
- Z. Luo, T. Xu, C. Zhang and C. Yang, Energy Environ. Sci., 2023, 16, 2732–2758 RSC.
- C. Li, J. Zhou, J. Song, J. Xu, H. Zhang, X. Zhang, J. Guo, L. Zhu, D. Wei, G. Han, J. Min, Y. Zhang, Z. Xie, Y. Yi, H. Yan, F. Gao, F. Liu and Y. Sun, Nat. Energy, 2021, 6, 605–613 CrossRef CAS.
- G. Chai, Y. Chang, J. Zhang, X. Xu, L. Yu, X. Zou, X. Li, Y. Chen, S. Luo, B. Liu, F. Bai, Z. Luo, H. Yu, J. Liang, T. Liu, K. S. Wong, H. Zhou, Q. Peng and H. Yan, Energy Environ. Sci., 2021, 14, 3469–3479 RSC.
- Y. Chang, J. Zhang, Y. Chen, G. Chai, X. Xu, L. Yu, R. Ma, H. Yu, T. Liu, P. Liu, Q. Peng and H. Yan, Adv. Energy Mater., 2021, 11, 2100079 CrossRef CAS.
- D. Mo, H. Chen, J. Zhou, N. Tang, L. Han, Y. Zhu, P. Chao, H. Lai, Z. Xie and F. He, J. Mater. Chem. A, 2020, 8, 8903–8912 RSC.
- P. Chao, L. Liu, J. Zhou, J. Qu, D. Mo, H. Meng, Z. Xie, F. He and Y. Ma, ACS Appl. Energy Mater., 2018, 1, 6549–6559 CrossRef CAS.
- M. Pu, C. Cao, H. Chen, Y. Zhu, P. Tan, X. Lai and F. He, Chem. Eng. J., 2022, 437, 135198 CrossRef CAS.
- I. Osaka, M. Saito, T. Koganezawa and K. Takimiya, Adv. Mater., 2014, 26, 331–338 CrossRef CAS PubMed.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655–4660 CrossRef CAS PubMed.
- K. H. Kim, H. Kang, H. J. Kim, P. S. Kim, S. C. Yoon and B. J. Kim, Chem. Mater., 2012, 24, 2373–2381 CrossRef CAS.
- F. Zhao, D. He, J. Xin, S. Dai, H. Xue, L. Jiang, Z. Wei, W. Ma, X. Zhan, Y. Li and C. Wang, Sci. China: Chem., 2019, 62, 790–796 CrossRef CAS.
- J. Comyn, Int. J. Adhes. Adhes., 1992, 12, 145–149 CrossRef CAS.
- V. Mihailetchi, L. Koster, J. Hummelen and P. Blom, Phys. Rev. Lett., 2004, 93, 216601 CrossRef CAS PubMed.
- D. He, J. Zhou, Y. Zhu, Y. Li, K. Wang, J. Li, J. Zhang, B. Li, Y. Lin, Y. He, C. Wang and F. Zhao, Adv. Mater., 2024, 36, 2308909 CrossRef CAS PubMed.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B, 2010, 82, 245207 CrossRef.
- W. L. Leong, S. R. Cowan and A. J. Heeger, Adv. Energy Mater., 2011, 1, 517–522 CrossRef CAS.
- Y. Zeng, D. Li, H. Wu, Z. Chen, S. Leng, T. Hao, S. Xiong, Q. Xue, Z. Ma, H. Zhu and Q. Bao, Adv. Funct. Mater., 2022, 32, 2110743 CrossRef CAS.
- E. Gann, A. T. Young, B. A. Collins, H. Yan, J. Nasiatka, H. A. Padmore, H. Ade, A. Hexemer and C. Wang, Rev. Sci. Instrum., 2012, 83, 045110 CrossRef CAS PubMed.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2013, 3, 65–74 CrossRef CAS.
- Y. Wang, D. Qian, Y. Cui, H. Zhang, J. Hou, K. Vandewal, T. Kirchartz and F. Gao, Adv. Energy Mater., 2018, 8, 1801352 CrossRef.
- U. Rau, Phys. Rev. B, 2007, 76, 085303 CrossRef.
- B. Geffroy, P. le Roy and C. Prat, Polym. Int., 2006, 55, 572–582 CrossRef CAS.
- Z. Chen, X. Chen, Z. Jia, G. Zhou, J. Xu, Y. Wu, X. Xia, X. Li, X. Zhang, C. Deng, Y. Zhang, X. Lu, W. Liu, C. Zhang, Y. Yang and H. Zhu, Joule, 2021, 5, 1832–1844 CrossRef CAS.
- J. M. Marin-Beloqui, D. T. W. Toolan, N. A. Panjwani, S. Limbu, J. S. Kim and T. M. Clarke, Adv. Energy Mater., 2021, 11, 2100539 CrossRef CAS.
- A. J. Gillett, A. Privitera, R. Dilmurat, A. Karki, D. Qian, A. Pershin, G. Londi, W. K. Myers, J. Lee, J. Yuan, S.-J. Ko, M. K. Riede, F. Gao, G. C. Bazan, A. Rao, T.-Q. Nguyen, D. Beljonne and R. H. Friend, Nature, 2021, 597, 666–671 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc04810a |
| ‡ Z. Han and K. Wang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |