
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light-induced bromine radical enhanced hydrogen atom transfer (HAT) reactions in organic synthesis
Barakha
Saxena
,
Roshan I.
Patel
and
Anuj
Sharma
 *
*
Department of Chemistry, Indian Institute of Technology Roorkee, Roorkee-247667, India. E-mail: anujsfcy@iitr.ac.in; anujsharma.mcl@gmail.com; Tel: +91-1332-284751
First published on 1st July 2024
Abstract
Hydrogen atom transfer (HAT) reactions have gained prominence in organic synthesis for providing a straightforward approach towards C–H bond activation for the formation of C-centered radical intermediates. However, halogen radical-assisted hydrogen atom transfer (HAT) reactions have become an interesting tool for C–H bond activation, facilitating the formation of C–C and C–X bonds. In particular, the bromine radical (Br˙) has garnered attention because of its remarkable capability as a hydrogen acceptor, which abstracts an H-atom from a C–H bond and generates a C-centered radical intermediate. Typically, transition metal- and organo-photocatalysts are commonly used to generate a bromine radical (Br˙) from a bromine anion (Br−). This newly generated bromine radical (Br˙) is useful in several organic transformations via C–H bond activation. In this review, we provide recent updates on bromine radical (Br˙) assisted hydrogen atom transfer (HAT) reactions with their scope, mechanism, and limitations.
 Barakha Saxena | Barakha Saxena obtained her BSc (2015) and MSc degrees (2017) in Organic Chemistry from Mahatma Jyotiba Phule Rohilkhand University, Bareilly, Uttar Pradesh, India. She received the Junior Research Fellowship Award (CSIR-JRF) just after completing her Master's. She joined as a PhD scholar in the research group of Prof. Anuj Sharma in the Department of Chemistry, Indian Institute of Technology (IIT), Roorkee, India. She received the Senior Research Fellowship Award (CSIR-SRF). Her current research is focused on novel strategies for C–C and C–X bond formation. |
 Roshan I. Patel | Roshan I. Patel obtained his BSc (2015) and MSc degrees (2017) in Organic Chemistry from Sardar Patel University, India. He was the gold medalist during his bachelor's degree study. He later joined as a research scholar in Prof. Anuj Sharma's group in the Department of Chemistry, Indian Institute of Technology (IIT), Roorkee, India. His current research is focused on efficient methods for C–H/X functionalization using contemporary methods. |
 Anuj Sharma | Prof. Anuj Sharma earned his PhD from the Institute of Himalayan Bioresource Technology (IHBT) in 2006. Afterward, he undertook two short postdoctoral assignments at UFSM, Santa Maria, in Brazil, in 2006, and KU Leuven, in Belgium, in 2007, before moving to the University of Arizona as a NIH postdoctoral fellow in Prof. Laurence Hurley's group. He is currently working as a professor in the Department of Chemistry, Indian Institute of Technology (IIT), Roorkee, India. His group focuses on developing green organic synthetic methodologies, including visible light driven C–H functionalization. |
Sustainability spotlightIn the past few decades, the chemistry of bromine radicals has been well studied, and especially transition-metal-based and heat-assisted radical-based strategies have been well developed. Furthermore, recent advancements in the field of radical chemistry have enabled chemists to explore direct functionalization via HAT. Unfortunately, these previous strategies require expensive metals and ligands, high temperatures, and toxic radical initiators, thus making them unsustainable. The development of simple and mild methodologies for bromine radical-enhanced hydrogen atom transfer reactions for the formation of C-centered radical intermediates under visible light is highly desirable. Visible light as an energy source for the reaction reduces the need for harmful UV light and aligns with green sustainable principles for the formation of C–C and C–X bond formation via bromine-assisted HAT. Moreover, the developed photocatalytic systems enhance the reaction efficiency and selectivity, making it more sustainable. Photocatalysts like transition metal complexes or organic dyes can activate bromine under visible light irradiation, facilitating the HAT process, which further facilitates C–C and C–X bond formation. Our review emphasizes the importance of the following UN sustainable development goals: affordable and clean energy (SDG 7), industry, innovation, and infrastructure (SDG 9), chemical and waste (SDG 12). |
1. Introduction
In synthetic organic chemistry, photocatalysis has undergone significant growth, providing a wide range of useful transformations for accessing valuable organic compounds.1 Typical photoredox catalysts such as metal photocatalysts (Ru- and Ir-complexes) and organic dyes (eosin Y, rose bengal acridinium-based, etc.) have served as popular photocatalysts by harnessing the oxidative/reductive capabilities of both organo-photocatalysts and transition metal (TM) photocatalysts in their activated state. These catalysts efficiently facilitate a broad range of reactions through single electron transfer (SET) or energy transfer (EnT) mechanisms due to their long-excited state lifetimes and useful photoredox potential.2 Moreover, using substrates or additives capable of forming electron-donor acceptor (EDA) complexes in the ground state have also emerged as an efficient method for C–C and C–X bond formation.3 In this regard, a new platform has recently emerged that combines photoredox catalysis with halide ion catalysis. They are coordinatively saturated and stable avoiding any ligand exchange with reaction partners.4 The proposed mechanism is based on a single electron transfer (SET) from the photo-excited catalyst to the halide ion (X−) to generate a halide radical (X˙) that promotes HAT from a substrate C–H bond. A hydrogen atom transfer (HAT) reaction involves the transfer of a hydrogen atom from a hydrogen donor (C–H) to a hydrogen acceptor (X˙) and has attracted considerable attention due to its capability for functionalizing complex molecules via C–H bond activation.5 HAT reactions are particularly relevant in the context of radical chemistry, where a radical species (X˙) abstracts a hydrogen atom from another molecule, resulting in the formation of a new radical and a different compound. The driving force behind HAT reactions is typically the difference in the stability of the radical intermediates formed before and after the transfer. This process is important in various chemical reactions, including those involved in organic, inorganic, and biological systems (Scheme 1).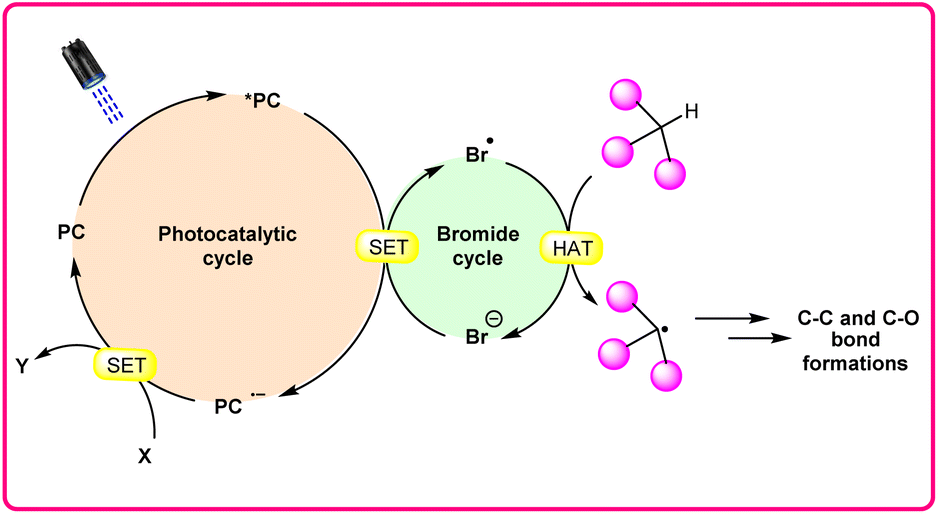 | ||
| Scheme 1 General approach toward the generation of a bromine radical and the hydrogen atom transfer process. | ||
Consequently, the utilization of a radical reagent capable of forming bonds that are more stable than those to be cleaved can enable direct functionalization via HAT. Various radical species (X˙), such as O-, N-, S-, B-, and halogen-centered radicals, have been identified as suitable HAT reagents.6 Among them, halogen radicals have garnered significant interest due to their ease of accessibility and high reactivity.
Halogen radicals, especially the chlorine radical (Cl˙) and bromine radical (Br˙), serve as potent hydrogen acceptors and play a crucial role as agents for facilitating hydrogen atom transfer (HAT).7 The bromine radical (Br˙) is a highly reactive species with an unpaired electron, which makes it capable of abstracting a hydrogen atom from a nearby molecule, leaving behind hydrogen bromide (HBr). The X–H (X= C, Si) bond dissociation energies (BDEs) of the hydrocarbons listed in Scheme 2 are generally in the vicinity of the bond formation energy of H–Br (87 kcal mol−1).8 Therefore, the bromine radical (Br˙) demonstrates a facile ability to cleave the X–H (X= C, Si) bonds. Conversely, iodine radicals (I˙) exhibit minimal to no capability for engaging in C–H bond activation.8b There have been significant efforts devoted to C–H bond activation via hydrogen atom transfer (HAT), which can be seen in a few excellent review articles.9 Some of the notable ones are by Ravelli and co-workers in 2020, who discussed direct, indirect, and remote photocatalytic HAT reactions for the formation of C-centered radical intermediate.9a However, photocatalytic hydrogen atom transfer (HAT), where halogen radicals are involved in the activation of the C–H bonds, was not discussed by them. Next, in 2022, Capaldo and co-workers published an article on photocatalytic halogen-radical assisted HAT reactions; however, HAT reactions via the bromine radical were merely covered.10 Later, in 2023, Itabashi, Asahara, and Ohkubo presented an excellent review on chloride radical (Cl˙)-mediated C–H oxygenation strategies via HAT for the formation of the C–O bond.11
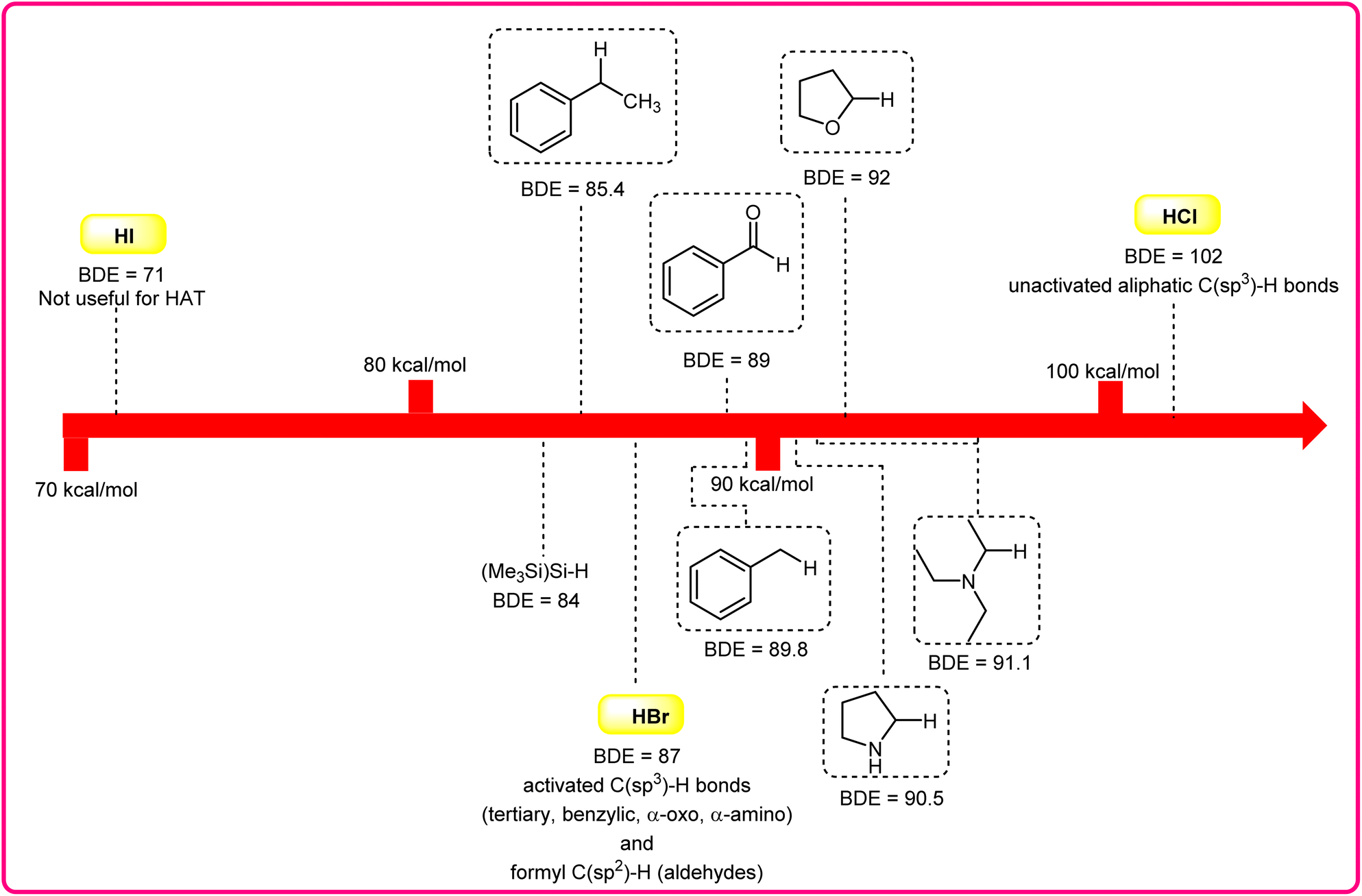 | ||
| Scheme 2 Bond dissociation energies (kcal mol−1) of C–H bonds and those of H–X bonds. | ||
As per our knowledge, there has been no review article exclusively focused on visible light-mediated hydrogen atom transfer (HAT) reactions using the bromine radical (Br˙). Within this framework, the present article is written, addressing strategies involving bromine radical (Br˙) mediated hydrogen atom transfer (HAT) under visible light irradiation for the generation of C–C and C–O bonds. The contents of the review are categorized into various types of hydrogen atom transfer reactions from the bromine radical (Br˙), such as (1) hydrogen atom transfer from aldehydes, (2) hydrogen atom transfer from benzylic carbons, (3) hydrogen atom transfer from α-hetero carbons, (4) hydrogen atom transfer from (Me3Si)3SiH, (5) hydrogen atom transfer from allylic carbons and (6) miscellaneous.
2. Hydrogen atom transfer from aldehydes
Hydrogen atom transfer (HAT) from aldehydes involves the transfer of a hydrogen atom from an aldehyde molecule to another molecule or radical intermediate. The hydrogen atom attached to the carbonyl carbon in aldehydes is relatively acidic due to the electron-withdrawing nature of the carbonyl group. Initially, the HAT reaction is initiated by the generation of a bromine radical (Br˙), often through the excited photocatalyst (*PC) via reductive quenching. This resulting bromine radical (Br˙) abstracts a hydrogen atom bound to the carbon atom adjacent to the carbonyl group. The abstraction occurs due to the high reactivity of the bromine radical species (Br˙), which seeks to stabilize itself by pairing its unpaired electron with an electron from the hydrogen atom, thus forming a new covalent bond. Moreover, the abstraction of the hydrogen atom leads to the formation of an acyl radical intermediate, which contains an unpaired electron, making it highly reactive and prone to further chemical reactions. The generation of an acyl radical via bromine radical-mediated HAT is shown in Scheme 3.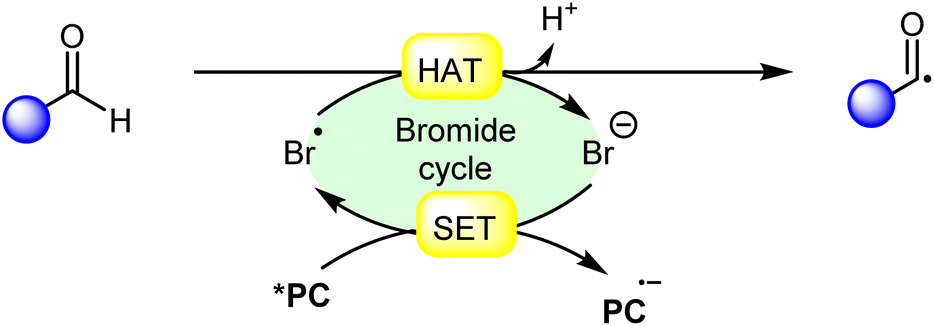 | ||
| Scheme 3 General approach for acyl radical intermediate formation via bromine radical mediated HAT. | ||
In 2020, Huang and co-workers reported photoredox Minisci-type alkylation of pyridines 1 with aliphatic and aromatic aldehydes 2 as an umpolung alkylating reagent (Scheme 4).12 Under optimized conditions, the authors were able to demonstrate the Minisci-type alkylation of some bioactive molecules such as quinoxyfen and fasudil in 40% and 50% yields, respectively. Various mechanistic studies such as H/D exchange, kinetic isotopic effect (KIE) experiments, and Stern–Volmer quenching studies were performed to understand the reaction mechanism. Mechanistically, the photoexcited catalyst *Ir(III) undergoes a single electron transfer (SET) process with a bromide ion (Br−) to generate a bromine radical (Br˙) and Ir(II). This bromine radical (Br˙) would abstract a hydrogen atom from aldehyde 2via deprotonated electron transfer (DPET) to access the acyl radical 4A. The resulting intermediate 4A adds to the N-heterocycle 1, accompanied by deprotonation, and subsequently spin-center shift (SCS) occurs to generate the hydroxyalkyl radical intermediate 4D. Next, the SET process from the reductive Ir(II) to the intermediate 4D furnishes the intermediate 4E. Furthermore, the reduction of 4E with aldehyde 2 affords the final product 3. With a change in reaction conditions, the authors further demonstrated the benzylation of nitrogen-containing heteroarenes 1 using benzaldehydes 2 by employing 4CzIPN as the photocatalyst, with CF3SO3H and (PhO)2PO2H as acids, and LiBr as the additive in chlorobenzene (PhCl) under visible light irradiation, affording the desired products 3 in 21–83% yield.
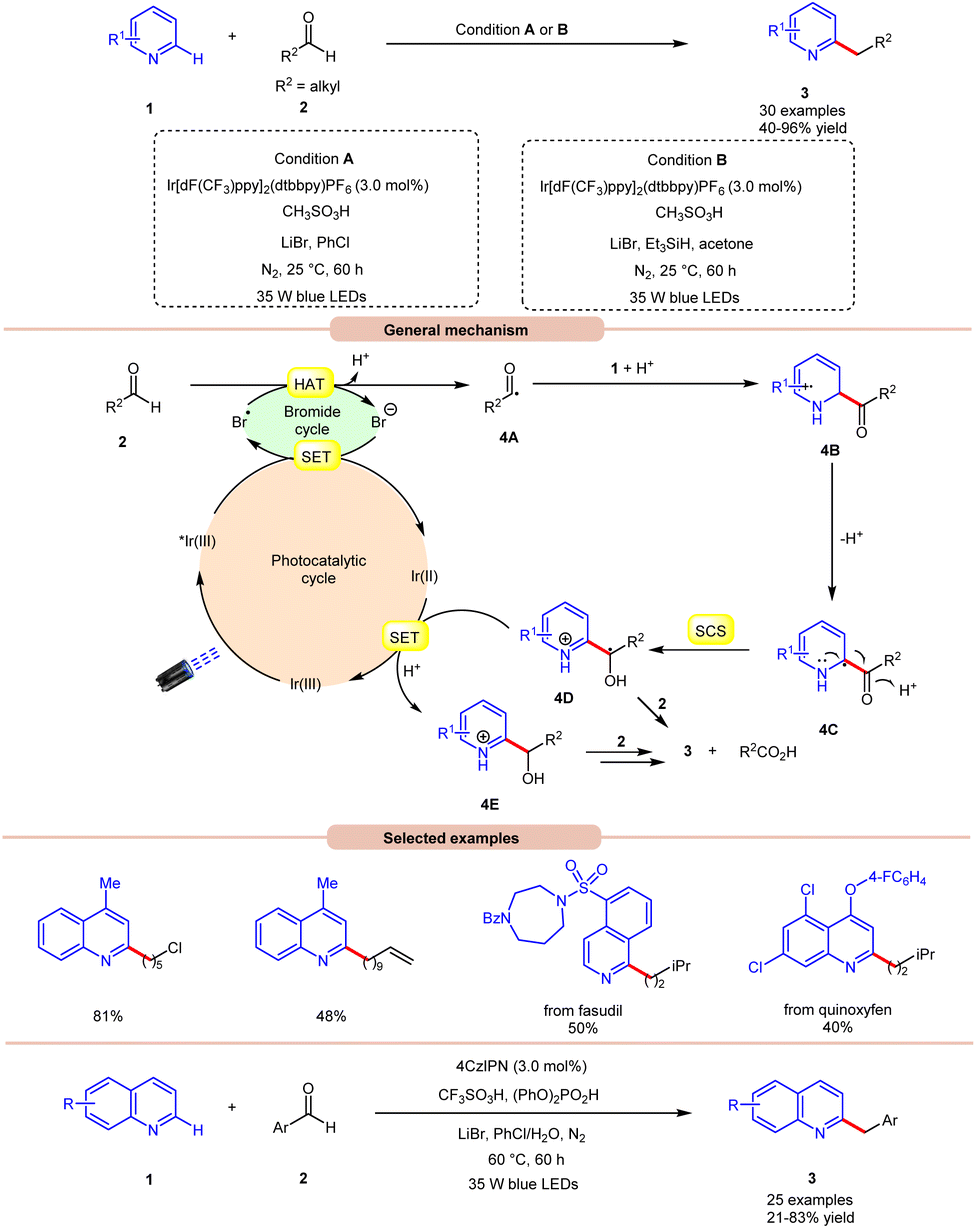 | ||
| Scheme 4 Ir-catalyzed photoredox Minisci-type alkylation of pyridines. | ||
Wang, Huang, and co-workers in 2020 illustrated dual photoredox/bromide catalyzed Minisci hydroxyalkylation of quinolines 4 using aldehydes 2 under visible light irradiation (Scheme 5).13 Inexpensive LiBr was chosen as an efficient moderator for this protocol with 4CzIPN as an organo-photocatalyst under visible light irradiation.
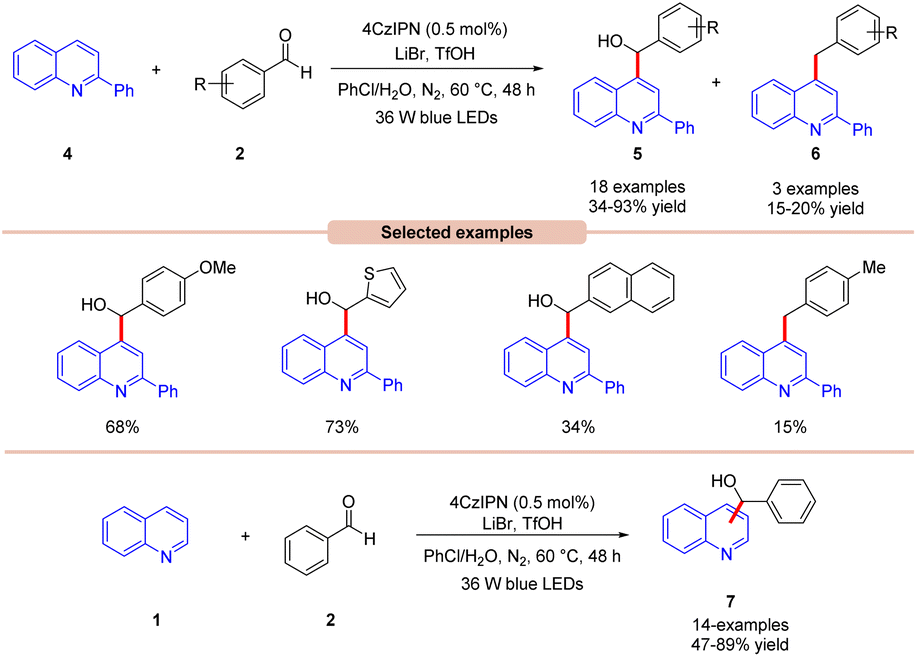 | ||
| Scheme 5 Dual 4CzIPN/LiBr mediated Minisci hydroxyl alkylation of quinolines with aryl aldehydes. | ||
A range of substituted aldehydes 2 reacted well under the optimized conditions, affording the desired products in moderate to excellent yields. Notably, aliphatic aldehydes 2 remained inactive under the reaction protocol. Deuterium studies indicated the formation of acyl radical via C–H scission of aldehyde 2, which was further proven by the TEMPO trapping experiment. The Stern–Volmer experiment reveals that 4CzIPN could efficiently oxidize the bromide anion, thus forming a bromine radical intermediate. Finally, the light-on-off experiment found that the visible light irradiation is crucial for the reaction protocol. Based on these findings, the authors proposed that the generated bromine radical intermediate undergoes HAT with the aldehyde 2 to form an acyl radical intermediate, which reacts with the quinoline 4, followed by a similar mechanistic pathway to that described in Scheme 4 to give the final product 5. Under the same reaction conditions, a broad range of quinolines 1 were also successfully converted to yield the desired hydroxyalkylated product 7. This protocol is limited to quinolines as it fails to undergo a Minisci-type reaction with benzothiazole, benzo-oxazole, and quinoxalinone.
Oxindoles are quintessential moieties in pharmaceuticals, organic materials, and natural products.14 Great attention has been given to their synthesis. Deng and co-workers in 2022 established visible light-induced radical alkylation of N-arylacrylamides 8 with aliphatic aldehydes 2 (Scheme 6).15 The authors employed 4CzIPN as a photocatalyst with NaBr as an additive and TBHP as an oxidant under visible light irradiation. Both electron-donating and electron-withdrawing groups at various positions of the aryl ring of N-arylacrylamides 8 were well tolerated. Moreover, the aliphatic aldehydes 2 containing cyclic and acyclic groups showed good reactivities under the reaction conditions. A key feature of this strategy was illustrated in the late-stage functionalization of naproxen, ibuprofen ciprofibrate, and probenecid drug molecules. Radical trapping experiments with TEMPO, BHT, or 1,1′-diphenylethylene completely suppressed the reaction, which supported the involvement of a radical mechanistic pathway. Based on these findings and several other controlled experiments, the authors proposed a plausible mechanism, as illustrated in Scheme 6. The photoexcited catalyst 4CzIPN* oxidizes the bromide anion to generate a bromine radical via a SET process, which participates in HAT with the aldehyde 2 to form acyl radical intermediate 6A, followed by carbon monoxide (CO) removal to form an alkyl radical intermediate. Next, the alkyl radical adds to the N-arylacrylamides 8, accompanied by cyclization to give access to radical intermediate 6C. Lastly, the SET/deprotonation of 6C yields the required alkylated product 9. Furthermore, under similar photocatalytic conditions, the authors demonstrated the acylation of N-arylacrylamides 8 using aromatic aldehydes 2 to afford the required acylated products 10. Notably, replacing aliphatic aldehydes with aromatic aldehydes under the same optimized conditions provides the acyl-substituted oxindoles 10 in 30–74% yields. Probably secondary and tertiary aliphatic aldehydes undergo decarbonylation due to the formation of stable alkyl radicals.
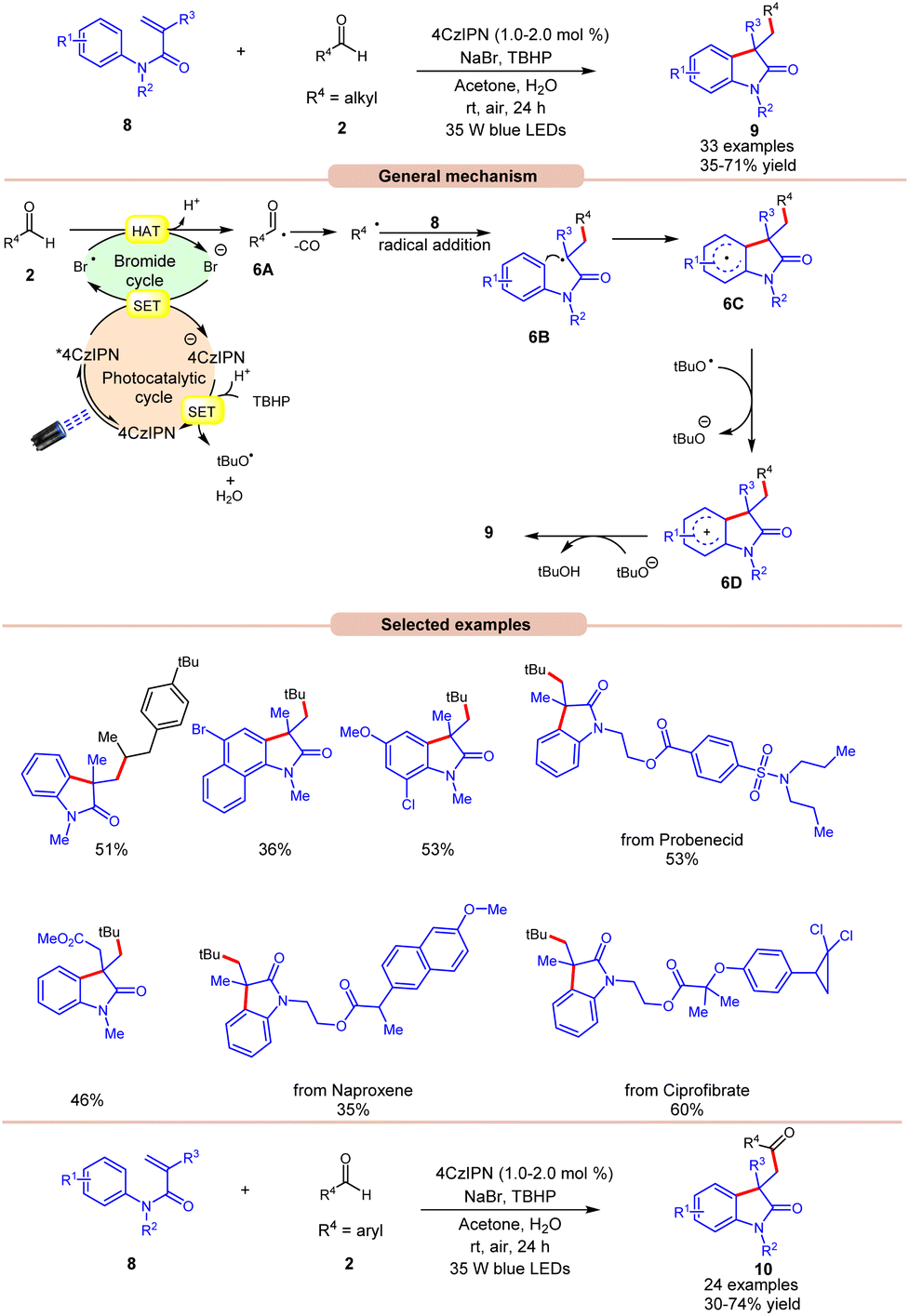 | ||
| Scheme 6 4CzIPN-catalyzed radical acylation of N-arylacrylamides with aliphatic or aromatic aldehydes. | ||
A highly effective approach towards the hydroacylation of vinyl arenes 11 was accomplished by Wu and co-workers using N-bromosuccinimide (NBS) as a catalyst under visible light irradiation (Scheme 7a).16a The striking feature of this protocol is that it does not require any external photocatalyst to generate the bromine radical. The ideal reaction conditions comprised NBS in fluorobenzene at 80 °C under blue light irradiation. Notably, the application of a non-aromatic solvent in the reaction mixture failed to give the targeted product. A broad range of styrene derivatives 11 reacted well with alkyl and aryl aldehydes 2, affording the hydroacylated product 12 in 32–75% yield. A TEMPO trapping experiment inhibited the product formation, indicating a radical mechanistic pathway. Light on/off studies indicated that visible light irradiation is crucial for the homolysis process. As shown in Scheme 7, in the presence of visible light irradiation, NBS undergoes N–Br bond homolysis to give a bromine radical in equilibrium with molecular bromine. Subsequently, the aldehyde group 2 undergoes HAT with the bromine radical to give HBr and acyl radical intermediate 7A, which adds to the terminal alkene position to give a benzyl radical intermediate 7B. Lastly, the desired product 12 is obtained via HAT with HBr. Product formation was further supported by DFT calculations. This strategy could also be applicable to the deuteration of aldehydes 2 with D2O 13, affording the deuterated product 14 in 72–97% yield. The compatibility of this protocol was further seen in the radical cascade reaction involving acrylates 15 and styrene 11, giving the desired product 16 in 41–79% yield.
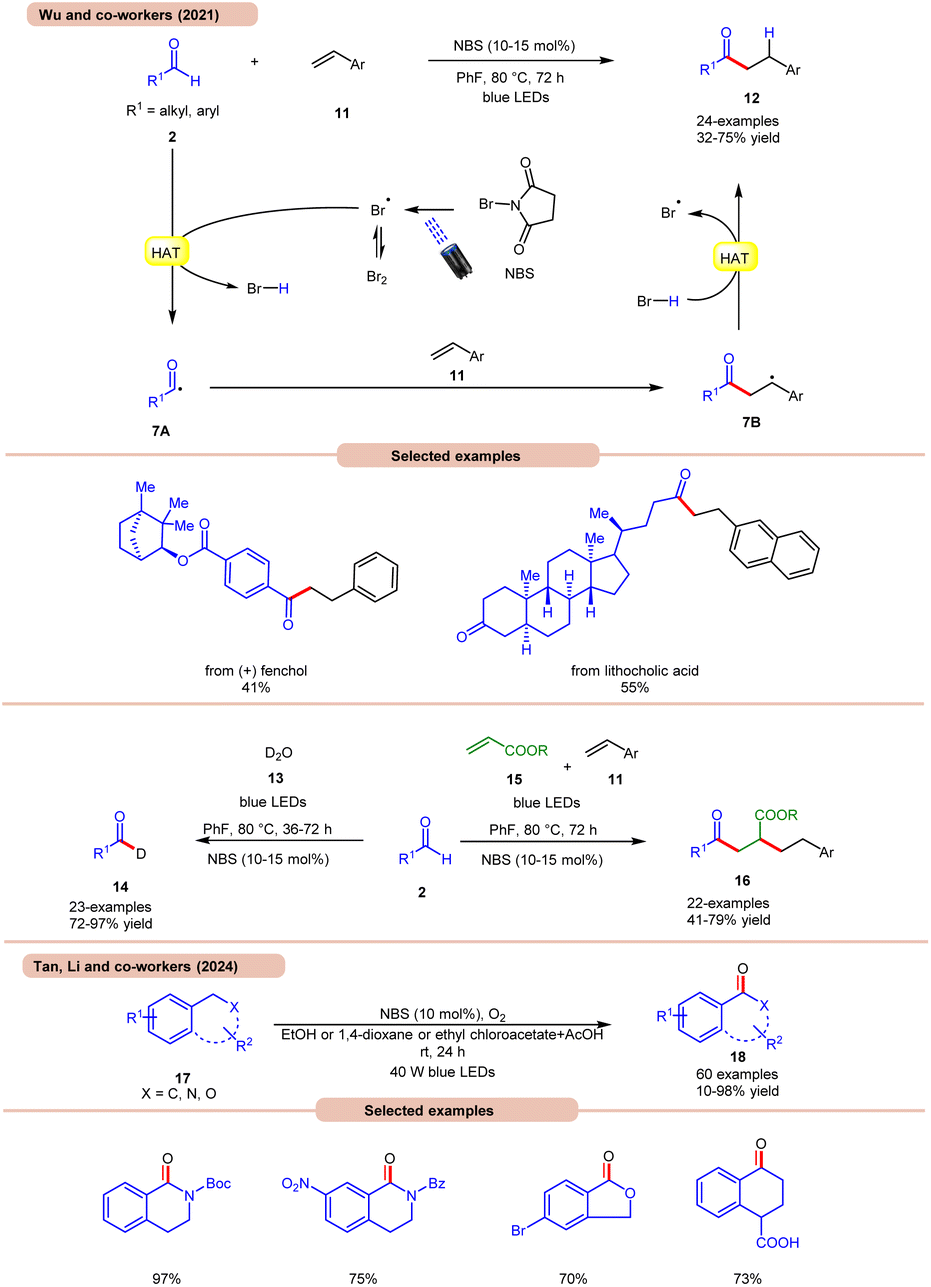 | ||
| Scheme 7 Bromine-radical catalyzed hydroacylation of vinyl arenes. | ||
Most recently, Tan, Li, and co-workers developed a metal-free photocatalytic system for aerobic oxygenation of the benzylic C(sp3)–H bonds of amines, ethers, alkylarenes, and hetero-aromatics 17 (Scheme 7b).16b In this method, the authors employed readily available NBS as a bromine radical source and O2 as an oxidant. The method has been successfully applied to the synthesis of bioactive and drug-valued targets, including corydaldine, ketoprofen, and isoflavone, providing good opportunities for applications in drug discovery and development.
Impressively, a new methodology for the synthesis of ketones 20 was developed by Kawasaki, Ishida, and Murakami, in 2020, by using a dual Ir-photoredox/nickel catalytic system (Scheme 8).17 In this strategy, NiBr2(dtbbpy) plays a dual role in the generation of a bromine radical and in the cross-coupling of benzylic and acyl radical intermediates. A variety of alkylarenes 19 were well tolerated with alkyl aldehydes 2 under the optimized conditions to yield the corresponding ketones 20 in 22–80% yields. A TEMPO trapping experiment supported the formation of benzylic and acyl radical intermediates. Mechanistically, an anion exchange interaction between Ir-photoredox and NiBr2(dtbbpy) catalytic systems gives [Ir(III)][Br]-complex 8A, which in the presence of visible light irradiation gives access to a bromine radical and Ir(II)-complex 8Bvia a SET event between photoexcited [Ir(III)]* and bromide anions.
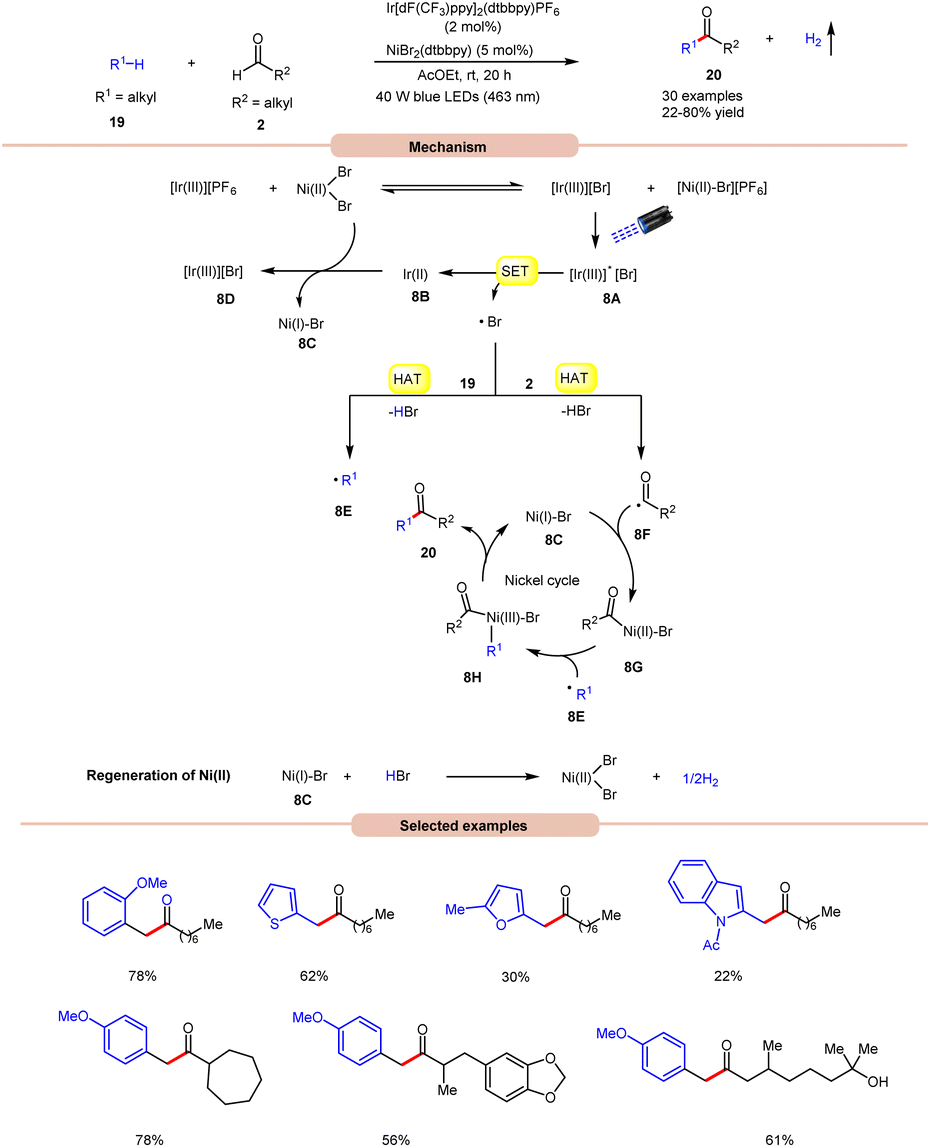 | ||
| Scheme 8 Bromine radical-mediated dehydrogenative coupling reaction for the synthesis of aryl ketones. | ||
Subsequently, the bromine radical participates in a HAT reaction with the aldehyde 2 and benzylic 19 C–H bond to give acyl radical intermediate 8F and benzylic radical intermediate 8E along with HBr. The resulting acyl radical intermediate 8F interacts with Ni(I)Br-species 8C to give access to Ni(II)–Br species 8G. Next, Ni(II)–Br species 8G interacts with benzylic radical intermediate 8E to give Ni(III)–Br complex 8H. Lastly, reductive elimination generates the final product 20 with the regeneration of 8C. Moreover, the interaction between 8C and HBr regenerates the corresponding Ni-catalyst along with H2. Notably, benzaldehydes did not yield successful outcomes in this protocol, primarily due to the challenge in abstracting the aldehydic hydrogen. This difficulty arises from the electron-withdrawing nature of the phenyl group attached to the aldehyde.
With a slight change in the optimized conditions, the same group later reported a visible light-mediated acylation of phenols 21 with aldehydes 2 for the synthesis of esters 22 in a dual Ir-photoredox/nickel bromide catalytic system (Scheme 9a).18a A broad range of aromatic phenols 21 and alkyl and aryl aldehydes 2 reacted well to afford the corresponding esters 22 in 46–99% yield. The robustness of this protocol made it compatible with naturally occurring phenols such as β-arbutin and α-tocopherol. In continuation of their work, the same group later reported the acylation of phenols 21 using alkyl alcohols 23 to afford the required acylated products 22 in 47–91% yields under similar reaction conditions (Scheme 9b).18b
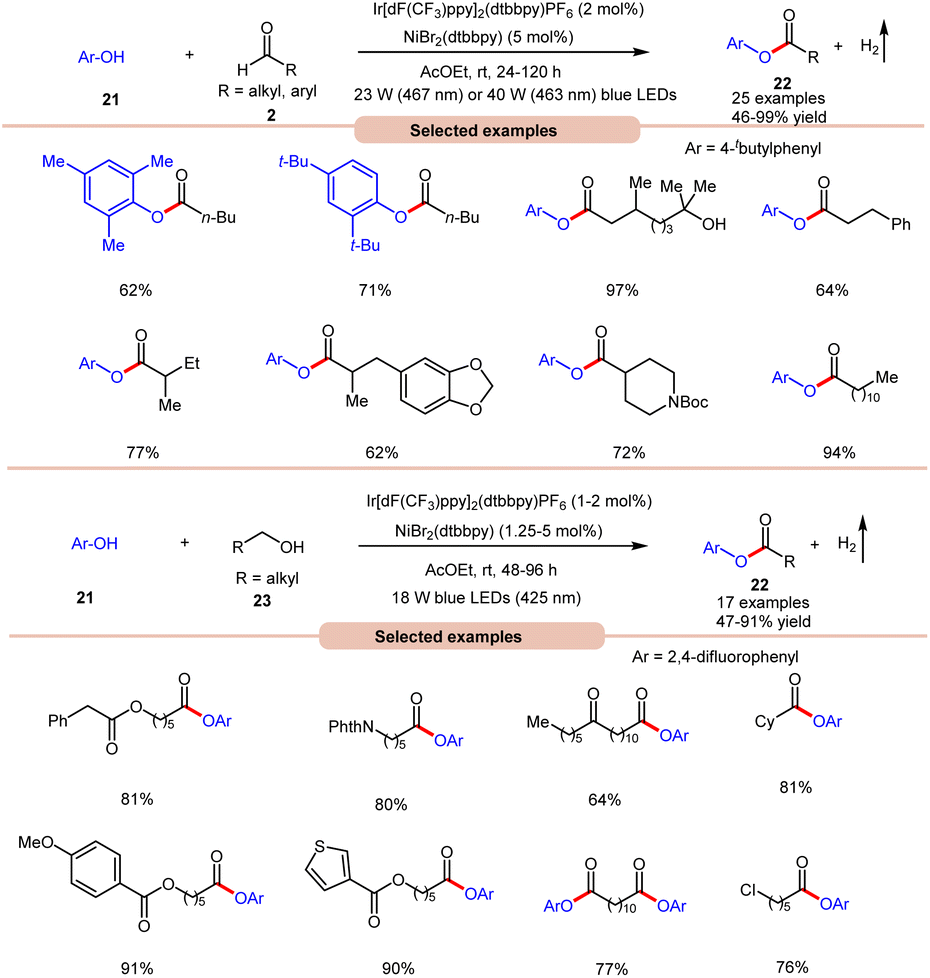 | ||
| Scheme 9 Bromine radical-mediated acylation of phenol with aldehydes and alcohols. | ||
3. Hydrogen atom transfer from benzylic carbons
Hydrogen atom transfer (HAT) from a benzylic carbon refers to the abstraction of a hydrogen atom bonded to a carbon atom adjacent to a benzene ring. When a bromine radical species interacts with a substrate containing a benzylic carbon, it can abstract a hydrogen atom from this position, leading to the formation of a benzylic radical intermediate. Notably, benzylic hydrogen atoms are more readily abstracted in comparison to other hydrogen atoms in a molecule due to the stability of the resulting benzylic radical, which is resonance-stabilized by the delocalization of the unpaired electron into the aromatic ring (Scheme 10). | ||
| Scheme 10 General approach for benzyl radical intermediate formation via bromine radical-mediated HAT. | ||
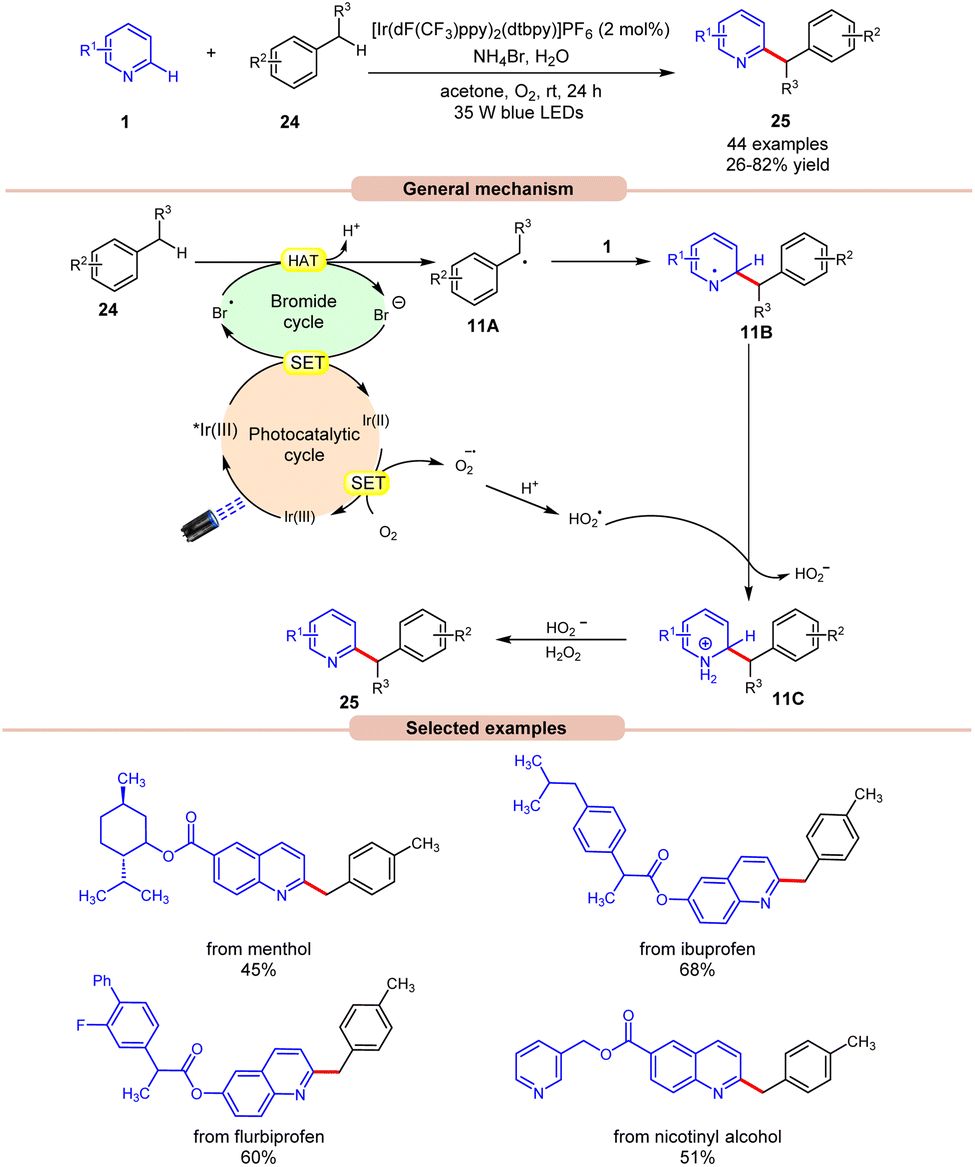
In this regard, an aerobic cross-dehydrogenative coupling (CDC) reaction was achieved by Huang, Deng, and co-workers in 2021 by a reaction between quinolines 1 derivatives and toluene 24 under visible light irradiation (Scheme 11).19 Initial investigations were carried out on p-xylene and 4-methylquinoline as model substrates for this Minisci-type CDC reaction. Optimized conditions revealed that light irradiation, a photocatalyst, and a bromide source are crucial for the transformation of this reaction. Out of the various bromide additives screened, NH4Br as a bromide source displayed the highest efficiency in reaction yield. A library of substituted quinoline derivatives 1 with electron-donating and withdrawing functionalities reacted well with toluene under the optimized conditions to afford the desired products 25 in 26–82% yield. Notably, toluene derivatives 24 bearing electron-donating groups gave a higher product yield in comparison to those with electron-donating ones. Besides, ethylbenzene and 2-methylnaphthalene also worked well to afford the desired product in 33–45% yield. A radical trapping experiment with TEMPO and 1,1-diphenylethylene supported the involvement of a radical mechanistic pathway. A quantum yield experiment (Φ = 0.52) eliminated the possibility of the radical chain pathway. As shown in Scheme 11, the photoexcited catalyst undergoes SET with the bromide anion to form a bromine radical, which undergoes HAT with the toluene 24 to form a benzyl radical intermediate 11A. This intermediate adds to the quinoline 1 to form an N-centered radical intermediate 11B. Subsequently, the oxidized catalyst Ir(II) is reduced by oxygen (O2) to form a superoxide radical 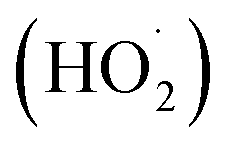 along with the completion of the catalytic cycle. This superoxide radical
along with the completion of the catalytic cycle. This superoxide radical 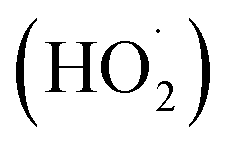 oxidizes the radical intermediate 11B to generate a cation intermediate 11C, which finally upon deprotonation yields the desired product 25. The compatibility of this strategy was further demonstrated in the late-stage functionalization of biologically active molecules.
oxidizes the radical intermediate 11B to generate a cation intermediate 11C, which finally upon deprotonation yields the desired product 25. The compatibility of this strategy was further demonstrated in the late-stage functionalization of biologically active molecules.
 | ||
| Scheme 11 Dual Ir-photoredox/NH4Br catalyzed cross-dehydrogenative coupling of N-heteroarenes with toluenes. | ||
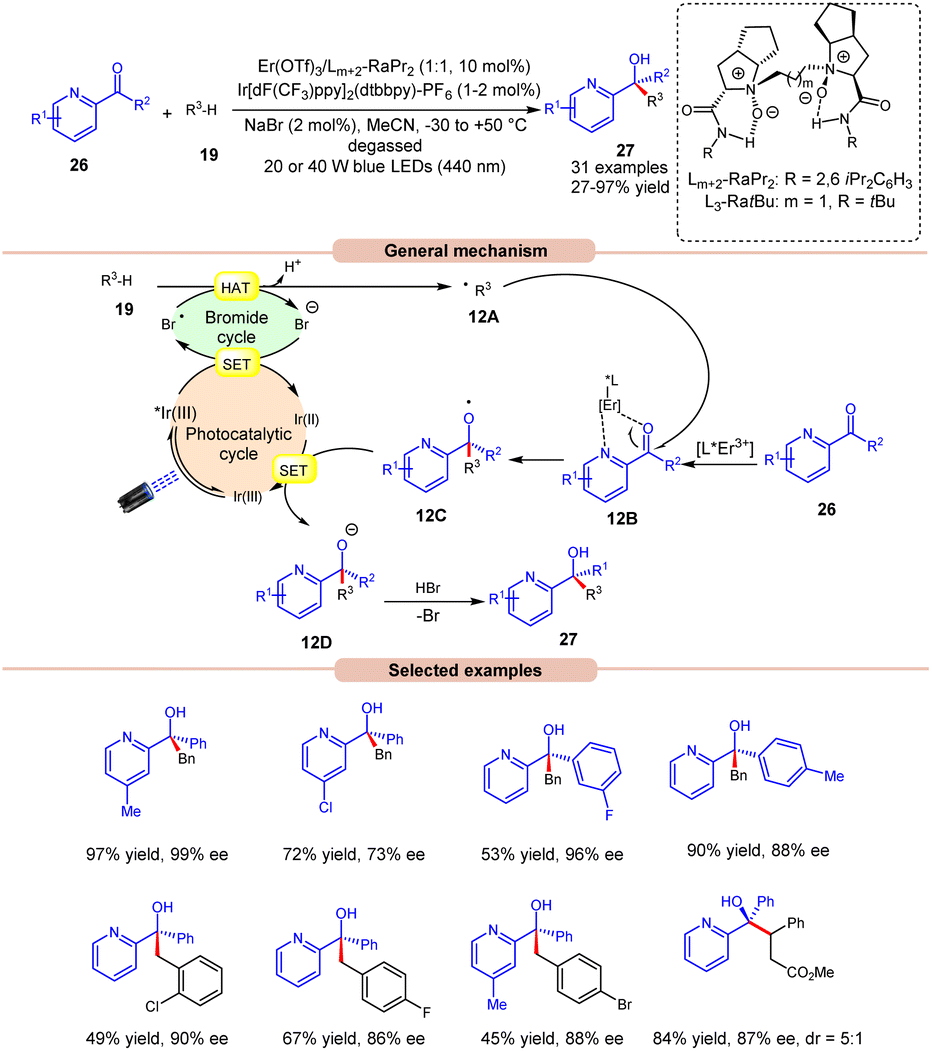
The following year, Liu, Feng and co-workers reported bromine radical-mediated enantioselective photoreduction of ketones 26 and hydrocarbons 19 for the synthesis of tertiary alcohols 27 (Scheme 12).20 Both electron-rich and electron-withdrawing groups on pyridine-based ketones 26 were converted into tertiary alcohols in good yields. However, ortho-substituted diarylketones gave a lower yield than the meta- and para-substituted ones, probably due to steric hindrance. Moreover, allylic and saturated cyclic hydrocarbons gave the required products 27 in good yields and enantioselectivity. Mechanistically, it is proposed that the photoexcited *Ir(III) undergoes SET with the bromide ion, generating the bromine radical along with Ir(II). This bromine radical participates in HAT with the benzylic C(sp3)–H partner 19, producing an alkyl radical intermediate 12A. Next, the pyridine-based ketones 26 coordinate with the chiral Er(III)-complex to generate the complex 12B. Therefore, the complex 12B undergoes spatial-selective radical addition with the alkyl radical 12A, generating the chiral radical intermediate 12C. Lastly, the resulting radical intermediate 12C undergoes SET/protonation to access the desired tertiary alcohols 27 with bromide ions, which again initiate the catalytic cycle.
 | ||
| Scheme 12 Bromine radical enhanced photoreduction of ketones and hydrocarbons for synthesizing tertiary alcohols. | ||
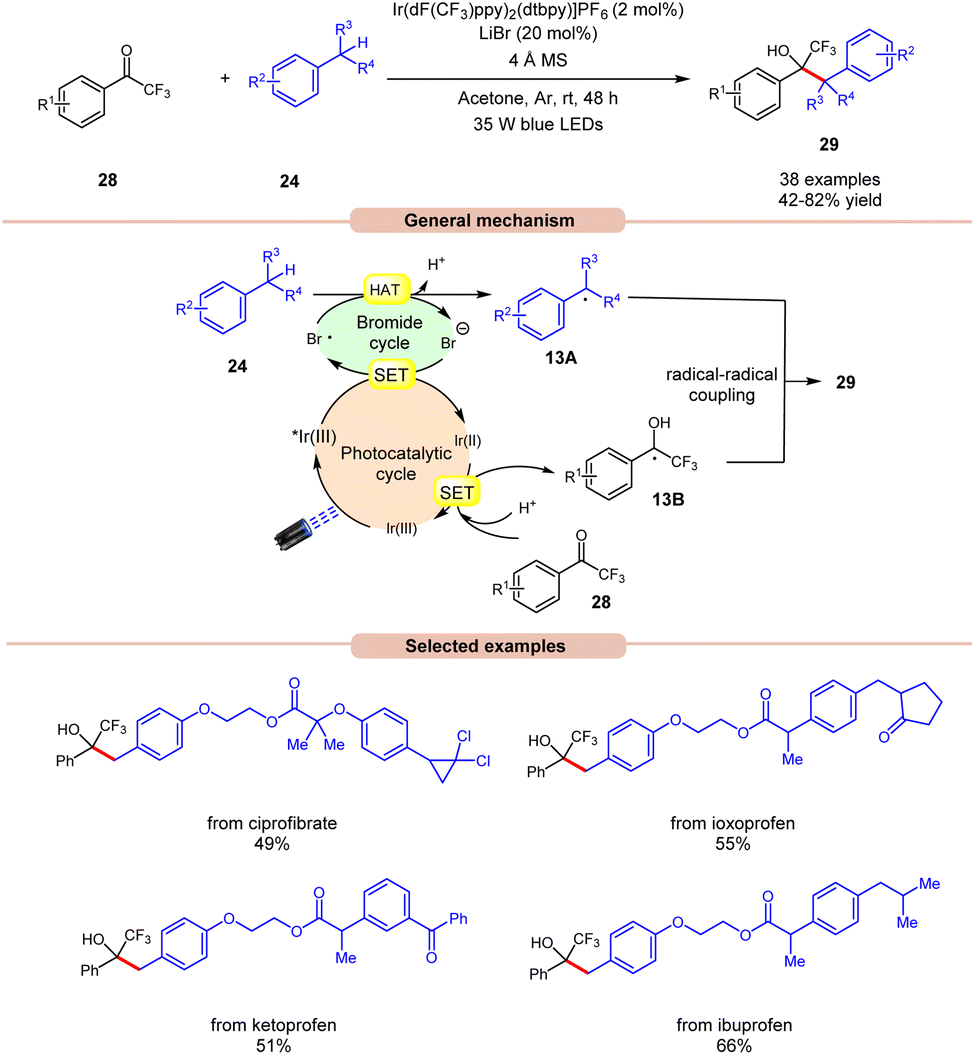
Recently, Deng and co-workers established visible light-induced benzylic C(sp3)–H functionalization of petroleum-derived alkylarenes 24 with trifluoromethyl ketones 28 to give trifluoromethyl alcohols 29 (Scheme 13).21 Substituted toluenes 24 with electron-donating and electron-withdrawing groups at various positions were well tolerated, accessing the desired products 29 in 42–82% yields. Furthermore, the meta- and ortho-substituted toluenes 24 gave the required products 29 in relatively low yields. In addition, methyl heteroarenes such as 2-thienyl, 2-furyl, and 2-methylnaphthalene reacted smoothly and gave the corresponding products in moderate yields. The robustness of this protocol is further demonstrated by the late-stage functionalization of complex bioactive molecules such as isoxepac, ciprofibrate, ibuprofen, flurbiprofen, ketoprofen, and naproxen. Radical trapping experiments using TEMPO, BHT and hydroquinone suggest the involvement of a radical pathway. Mechanistically, the photoexcited *Ir(III) complex oxidizes the bromine anion to generate the bromine radical, which abstracts a hydrogen atom in alkylarenes 24 to give benzyl radical 13A. Next, trifluoromethyl ketones 28 undergo a proton-coupled electron transfer (PCET) process with the reducing Ir(II)-complex to give the ketyl intermediate 13B. Finally, radical–radical coupling between the benzyl radical 13A and ketyl intermediate 13B affords the final desired product 29.
 | ||
| Scheme 13 Ir-catalyzed benzylic C(sp3)–H functionalization of alkylarenes with trifluoromethyl ketones. | ||
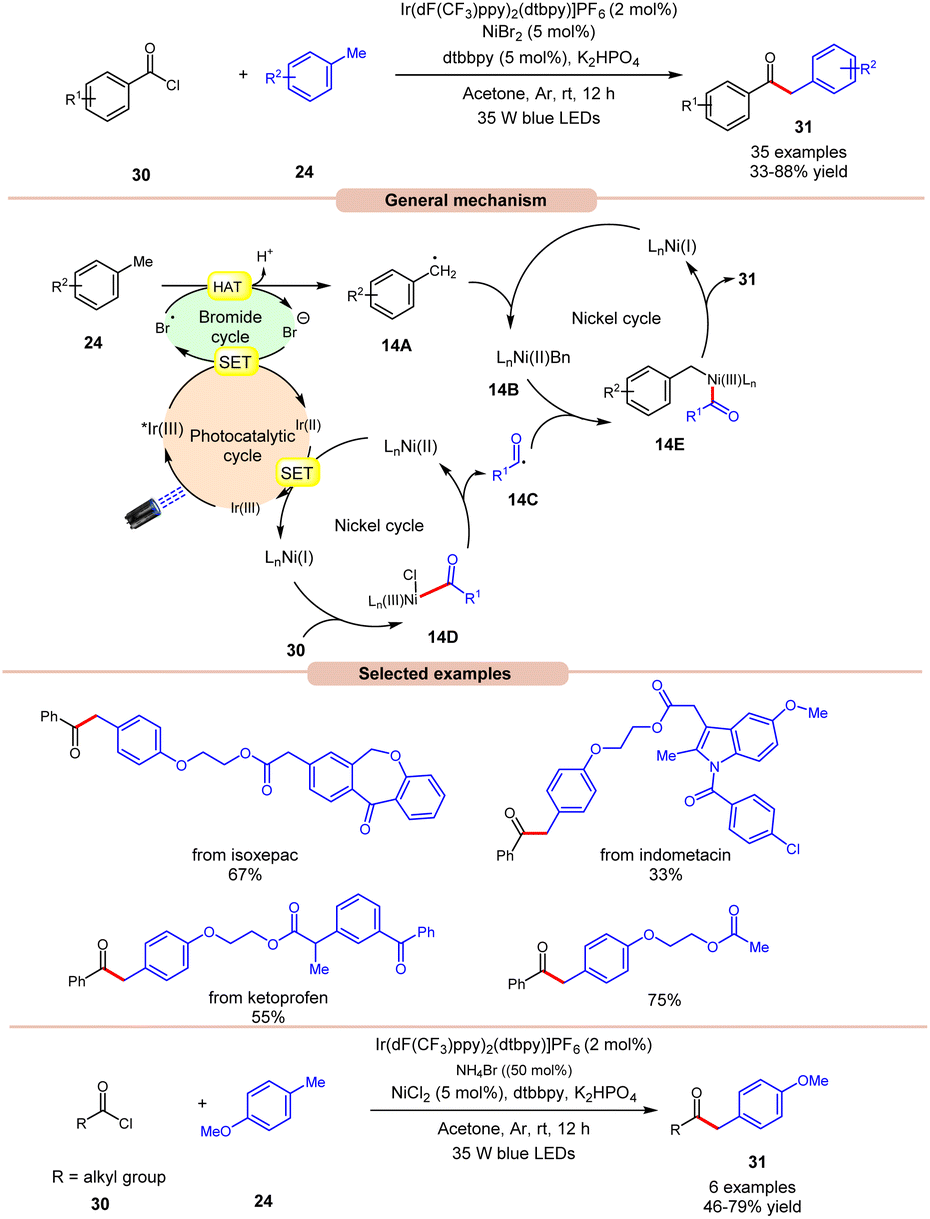
In 2022, Huang, Deng and co-workers revealed a dual photoredox/nickel-catalyzed coupling of methyl arenes 24 with acid chlorides 30via a bromine radical enhanced HAT pathway (Scheme 14).22 The authors utilized an Ir-complex photocatalyst with NiBr2 as a co-catalyst and bromine radical source in acetone. In this protocol, acid chloride 30 with electron-rich groups delivered better yields than the ones having electron-deficient groups.
 | ||
| Scheme 14 Dual Ir/nickel-catalyzed coupling of methyl arenes with acid chlorides. | ||
Unfortunately, 4-nitro benzoyl chloride did not work in this reaction. In the case of methylarenes, para-substituted derivatives displayed better reactivity compared to meta- and ortho-substituted derivatives due to steric hindrance. The compatibility of this method was demonstrated in the late-stage functionalities of complex drug molecules such as ioxoprofen, ketoprofen, and flurbiprofen, etc. Radical trapping experiments suggested the involvement of a radical pathway and their trapped adduct confirmed the formation of an acyl radical. The authors proposed that reductive quenching of the photoexcited *Ir(III) catalyst with bromine anions generates Ir(II) and bromine radicals. Simultaneously, Ni(I) species were generated by the interaction between Ir(II) and Ni(II) species, which react with acid chlorides 30 to give acyl radical intermediate 14Cvia intermediate 14D. In turn, the methyl arene 24 undergoes HAT with the generated bromine radical to give radical intermediate 14A, which upon oxidative addition with Ni(I) species generates Ni(II) intermediate 14B. Lastly, the trapping of acyl radical 14C and Ni(II) intermediate 14B, followed by reductive elimination, yields the final product 31. With a change in reaction conditions, the authors demonstrated the utility of this method in the acylation of methyl arenes 24 using alkyl acid chlorides 30 in good yields.
In 2022, Deng and co-workers reported dual Ir/Ni-catalyzed arylation of benzylic C–H bonds of toluene derivatives 24 with aryl bromides 32 (Scheme 15).23 Here, cross-coupling reactions were successfully conducted with a wide range of aryl- and heteroaryl bromides 32 containing electron-deficient, neutral, or electron-donating functional groups. It was further observed that substrates with electron-rich groups resulted in reduced yields of the corresponding coupling products 33. Mechanistically, the Ni(II) aryl bromide intermediate 15A was generated through the oxidative addition of the Ni(0) complex to aryl bromide 32. Next, the photoexcited *Ir(III) undergoes SET with 15A to give an aryl Ni(II)-species 15B and bromine radical (Br˙). This bromine radical abstracts the hydrogen atom from toluene derivative 24, to produce benzyl radical 15C. Following this, the Ni(II) aryl species 15B reacts with the benzylic radical 15C, yielding the Ni(III) aryl alkyl species 15D. Moreover, upon reductive elimination, the desired 1,1-diaryl alkane 33 is formed with the generation of the Ni(I) species, which transforms into the Ni(0) species via SET with the Ir(II) complex, thus concluding both catalytic cycles. The synthetic versatility of this method was further demonstrated in the late-stage arylation or benzylation of numerous drug-like and complex molecules.
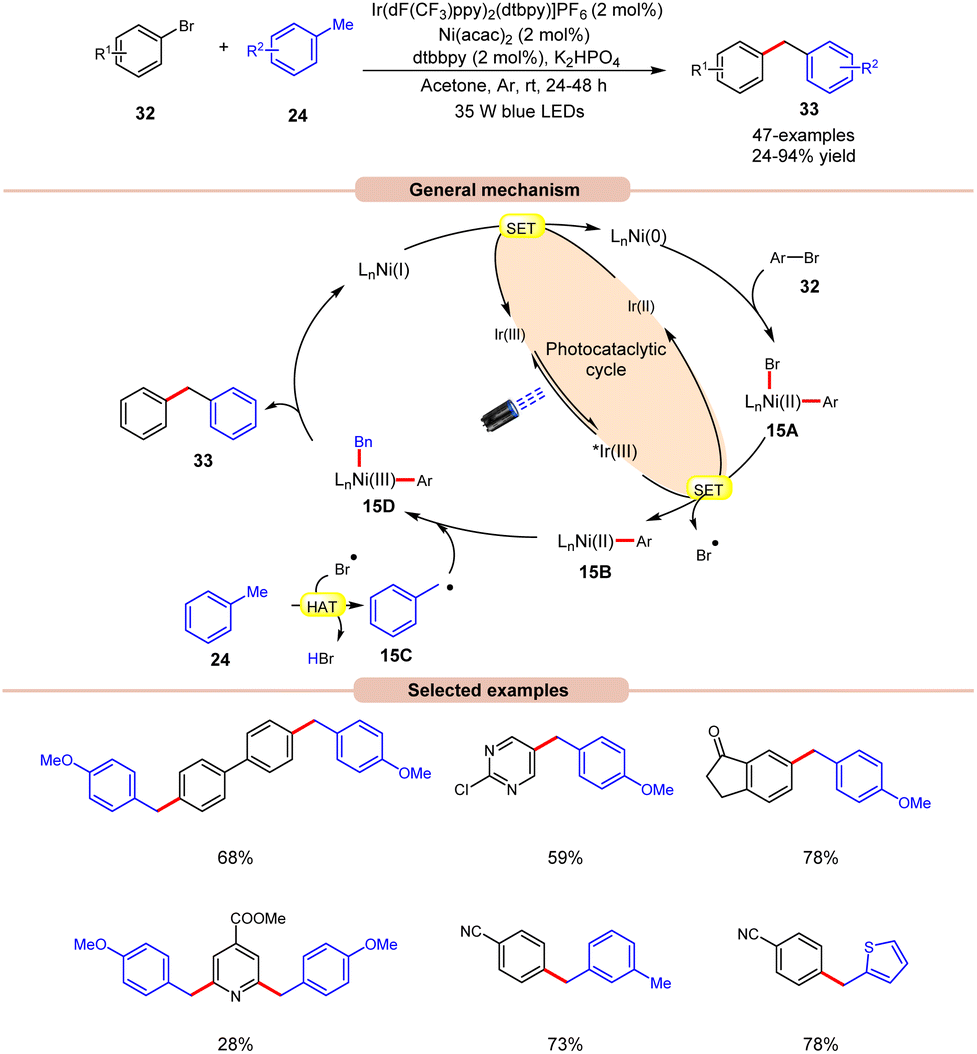 | ||
| Scheme 15 Dual Ir/Ni-catalyzed arylation of the benzylic C–H bonds of toluene derivatives with aryl bromides. | ||
Alkenylation reactions are versatile tools in organic synthesis, enabling the formation of carbon–carbon double bonds with high efficiency and selectivity. They find wide application in the preparation of pharmaceuticals, agrochemicals, and materials science.24 In this context, Lu and coworkers demonstrated stereo- and enantio-selective benzylic C(sp3)–H alkenylation, using a dual Ir/nickel catalytic system for the synthesis of chiral allylic compounds 35 (Scheme 16).25 Easily accessible alkylbenzenes 24 and alkenyl bromides 34, including complex molecules, participated in this cross-coupling reaction to afford the desired allylic compounds 35 with up to 93% ee and a >20/1 E/Z ratio under mild reaction conditions with good functional group tolerance. Alkenyl bromides 34 with electron-rich or electron-deficient aryl substituents were well tolerated. It is crucial to underscore the significance of stilbene as an additive since it acts as a triplet energy transfer inhibitor. This function effectively blocks the direct energy transfer event from the excited state photocatalyst to the product and eliminates the isomerization of the double bond. Furthermore, this methodology has been effectively utilized in bromides derived from natural products, affording the desired products 35 in moderate yields. The mechanism for product formation occurs in the manner as discussed in previous Scheme 15.
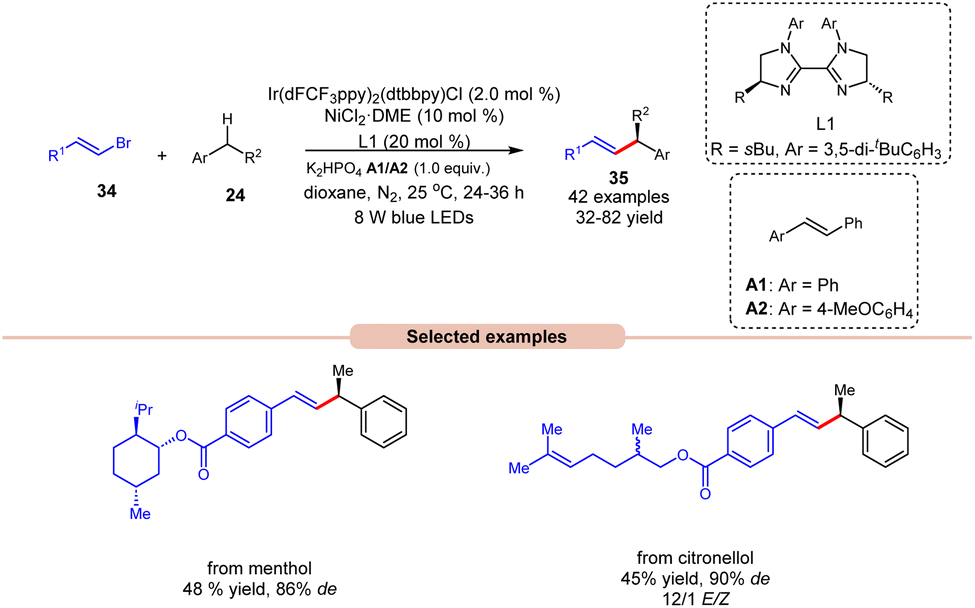 | ||
| Scheme 16 Dual Ir/nickel-catalyzed synthesis for stereo- and enantio-selective benzylic C(sp3)–H alkenylation. | ||
An unprecedented approach towards the selective alkylation of unactivated C(sp3)–H bonds 19 was achieved by Wu and co-workers in 2020, using a stop-flow micro-tubing reactor (SFMT) under visible light irradiation (Scheme 17).26 The authors observed that the utilization of CH2Br2 as a solvent, as well as a bromine radical source, was best suited for this alkylation reaction of the tertiary unactivated C(sp3)–H 19 bond by using a metal-free acridinium catalyst (Mer-AcrClO4). A series of tertiary alkanes 19 bearing electron-donating and electron-withdrawing alkyl and aryl groups reacted well, affording the corresponding products 37 in 41–95% yields. This reaction also achieved higher reactivity when conducted under flow microtubing reactors, compared to batch reactors. The screening of different photocatalysts revealed that Mer-AcrClO4 is highly reactive in catalyzing the reaction and a bromine radical could be effectively generated from CH2Br2 under photocatalytic conditions. Mechanistically, the photoexcited catalyst oxidizes CH2Br2, which eventually gives a bromine radical. Furthermore, the tertiary alkyl group 19 undergoes HAT with the bromine radical to give a radical intermediate 17A. This intermediate interacts with the Michael acceptor 36 to give radical intermediate 17B, accompanied by SET/protonation to yield the final product 37. Moreover, this technology was also applied in the amination of C(sp3)–H bonds 19 using dialkyl azodicarboxylates 38 as an aminating reagent, affording the corresponding products 39 in 49–78% yield. Notably, this method worked well with secondary and tertiary unactivated alkyl groups, whereas it failed with primary alkyl groups due to the lower stability of the alkyl radical.
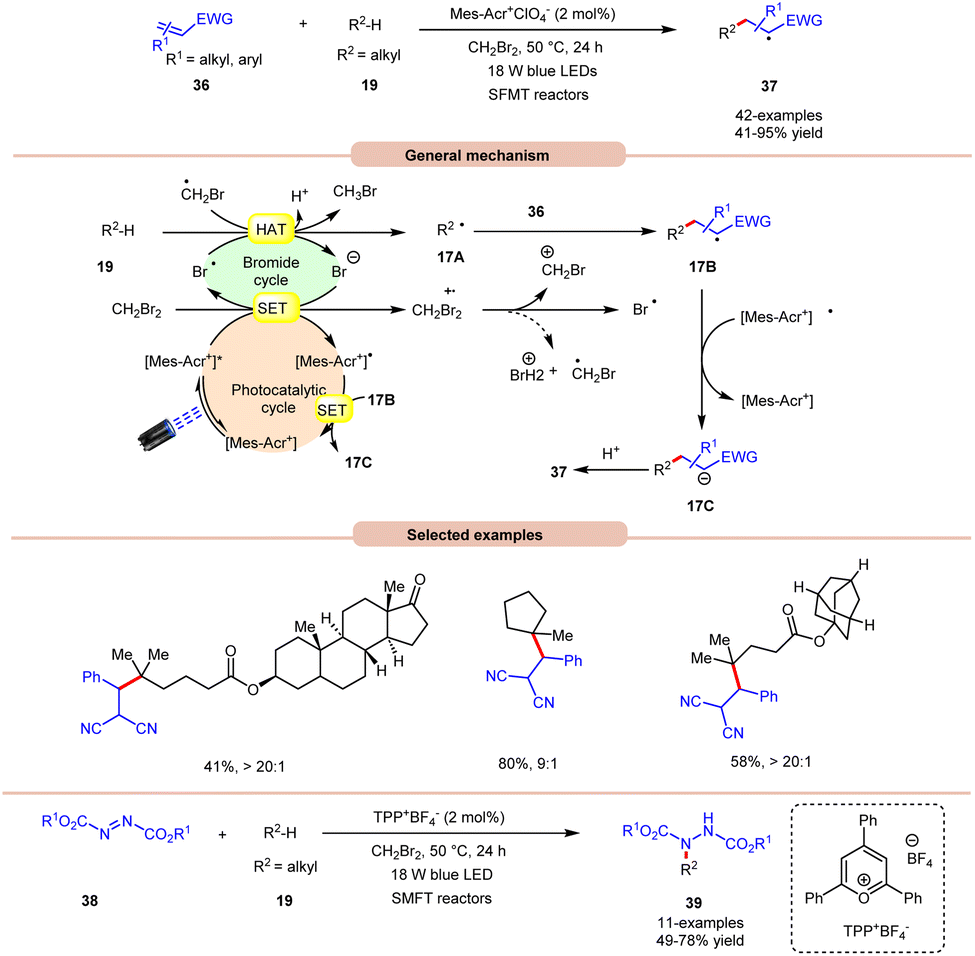 | ||
| Scheme 17 Bromine radical-assisted alkylation and amination of C(sp3)–H bonds. | ||
4. Hydrogen atom transfer from α-heterocarbons
Hydrogen atom transfer (HAT) involving an α-heteroatom refers to the abstraction of a hydrogen atom bonded to a carbon atom adjacent to a heteroatom (such as nitrogen, oxygen, sulfur, etc.). This type of reaction commonly occurs in organic chemistry and plays a significant role in various chemical transformations. In a molecule containing an α-heteroatom, the heteroatom can influence the reactivity of the adjacent carbon–hydrogen bonds. The electronegativity or the presence of lone pairs on the heteroatom can affect the bond strength and polarity of the adjacent C–H bond, making it more susceptible to abstraction by the bromine radical (Br˙) (Scheme 18).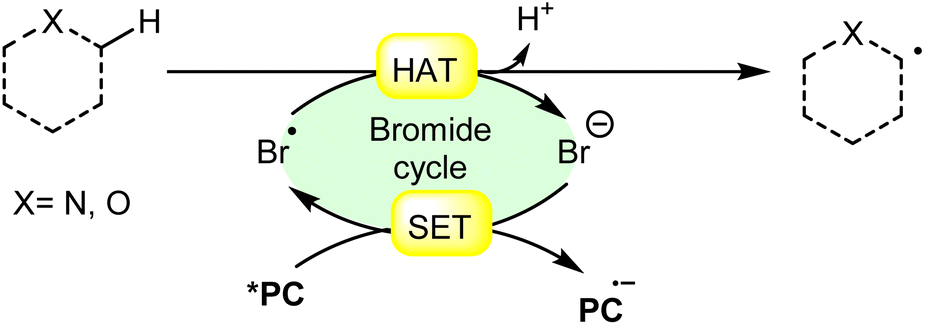 | ||
| Scheme 18 General approach for α-heteroatom radical intermediate formation via bromine radical-mediated HAT. | ||
In 2019, Huang and co-workers reported visible light Ir-photoredox catalyzed Minisci-type alkylation of quinolines 1 with ethers 40 serving as the alkylating agents (Scheme 19).27 The reaction demonstrates a wide range of functional group compatibility for both C2 and C4 coupling/alkylation of quinolines 1. Notably, the C-4 substituted and unsubstituted quinolines and benzoquinolines provided the desired C-2 alkylated products 41 in 30–91% yields.
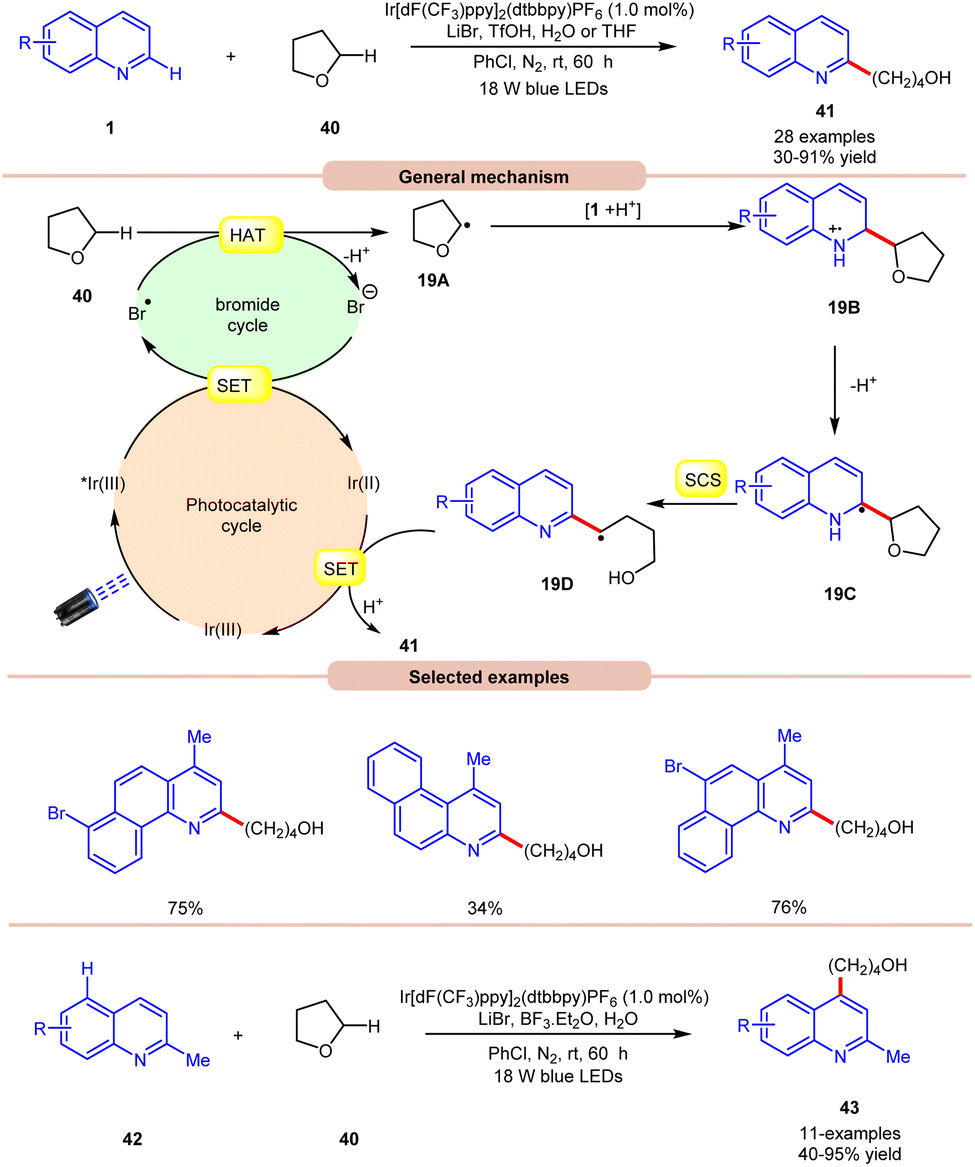 | ||
| Scheme 19 Ir-photoredox catalyzed Minisci-type alkylation of quinolines with ethers. | ||
A radical trapping experiment with TEMPO supports the involvement of a radical pathway. Mechanistically, the generated bromine radical (Br˙) abstracts a hydrogen atom from THF 40, resulting in the generation of an alkyl radical 19A, which adds to the charged compound 1, leading to the formation of the radical cation 19B. Subsequently, the radical cation 19B undergoes deprotonation, followed by a spin-centre shift (SCS) process to ultimately give rise to an alkyl radical intermediate 19Dvia C–O bond cleavage. Conclusively, the SET/protonation of 19D ultimately yields the desired alkylated quinoline product 41, while simultaneously regenerating the Ir(III) species.
In this work, an extension of C-2 blocked quinoline 42 gave the C-4 alkylated quinolines 43 in 40–95% yield by replacing TfOH with the Lewis acid BF3·Et2O. The substrate scope revealed that 2-methylquinolines 42 with different functional groups were smoothly reacted to give the C-4 alkylated product 43.
On the other hand, Deng and co-workers developed a visible-light Ir-photoredox catalyzed C(sp3)–H monofluoroalkenylation of ethers 40 for the synthesis of multi-substituted monofluoroalkenes 45via selective HAT and radical–radical cross-coupling (Scheme 20a).28a This reaction shows high regioselectivity for the α-carbon atoms of THF 40, thus allowing the synthesis of monofluoroalkenes 45 in yields of up to 92%. The substrate scope demonstrated a good functional group tolerance and could be carried out even on a gram scale.
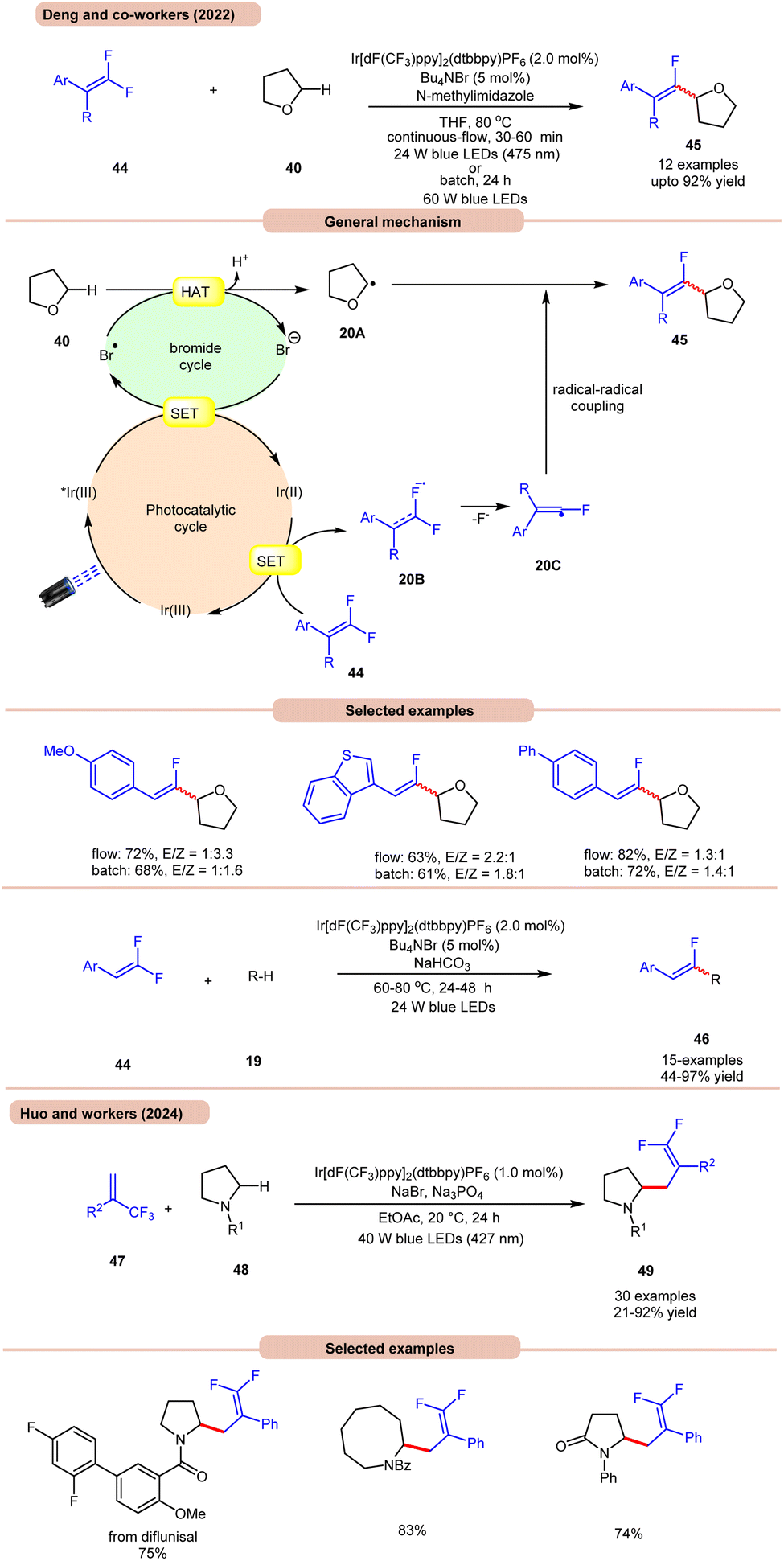 | ||
| Scheme 20 Ir-photoredox catalyzed C(sp3)–H monofluoroalkenylation of ethers. | ||
Radical trapping experiments suggested that the reaction proceeds via a radical pathway. The TEMPO-THF adduct was further confirmed by GCMS and the 1,1-diphenylethylen-THF adduct was also isolated. As shown in Scheme 20, the key alkyl radical intermediate 20A is formed via HAT between 40 and the bromine radical. At the same time, the fluoroalkenyl radical 20C was generated via SET reduction of 44 by the Ir(II)-complex and cleavage of the C–F bond. Finally, targeted product 45 is generated by the radical–radical cross-coupling of 20A and 20C.
The authors also illustrated the monofluoroalkenylation of cyclic and acyclic ethers, aliphatic aldehydes, and amides 19 as substrates, to afford the desired products 46 in 44–97% yields. It is worth noting that in Scheme 19, ring-opening of THF occurs, while in Scheme 20a, no ring-opening occurs in the reaction medium. This is due to the use of triflic acid (TfOH) in the reaction mixture, while in the absence of TfOH, no C–O cleavage occurs.
Huo and co-workers in 2024 reported photoredox-catalyzed, bromine-radical-mediated C(sp3)–H difluoroallylation of amides 48 (Scheme 20b).28b The authors used Ir[dF(CF3)ppy]2(dtbbpy)PF6 as a photocatalyst, NaBr as a HAT reagent, and Na3PO4 as a base in EtOAc under blue LEDs. This method incorporates both acyclic and cyclic-amino C(sp3)–H bonds 48 with a broad range of readily available trifluoromethyl alkenes 47, providing difluoroallylated amine derivatives 49 in good to excellent yields.
Polychloromethylated hydrocarbons are omnipresent in various bioactive compounds and their synthesis has gained some attention in the scientific community.29 In this regard, the synthesis of 1,1-dichloroalkane products 51 was achieved by Ji, Huang, and co-workers in 2022, using a dual Ir-photoredox/LiBr reaction system under visible light irradiation (Scheme 21).30 Initial studies were carried out on N-phenylmethacrylamide 8 and chloroform 50 at room temperature. Out of the various bases screened, the application of Et3N was proven to be highly efficient, affording the desired product 51 in 31–96% yield with H2O as a proton source and LiBr as an additive. N-phenylmethacrylamide 8 bearing various functional groups at the para-position of the benzene ring reacted well with chloroform 50, affording the targeted products 51 in 62–96% yield. On the other hand, α-naphthyl and β-naphthyl substituted methylacrylamides 8 resulted in the targeted product in moderate yields (47–54% yield). Besides, N-phenylmethacrylamide 8 with internal and terminal alkenes smoothly reacted under the ideal conditions to yield the desired products in 39–86% yield. The compatibility of this reaction was further demonstrated in the late-stage functionalization of naproxen and ibuprofen derivatives, resulting in the desired dichloromethylated products 51 in 77% and 91% yields, respectively. The generation of the ˙CHCl2 radical intermediate is confirmed by a radical trapping experiment with 1,1-diphenylethylene. Deuterated studies with D2O supported the incorporation of a proton source from H2O into the α-position of the carbonyl group of the dichloromethylated product. As shown in Scheme 21, the photoexcited catalyst *Ir(III) oxidizes the bromide anion to a bromine radical intermediate, which participates in HAT with Et3N to give radical intermediate 21A. This intermediate abstracts a chlorine atom from CHCl3 to give the key ˙CHCl2 radical intermediate 21C, which adds to the terminal alkene 8 to give radical intermediate 21D. Finally, the desired product 51 is obtained via SET/protonation.
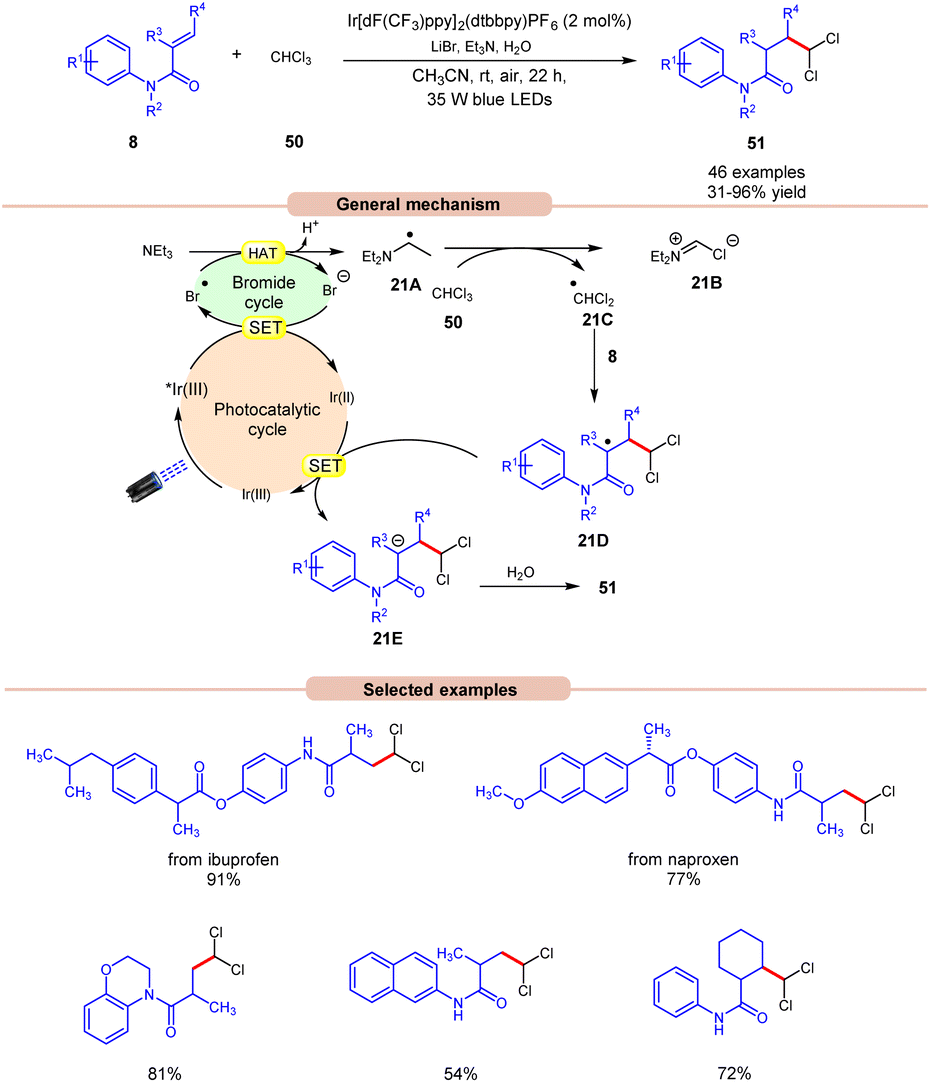 | ||
| Scheme 21 Dual Ir-photoredox/LiBr catalyzed 1,1-dichloromethylation of alkenes using chloroform. | ||
Giedyk and co-workers, in 2020, reported facile Ir-photoredox catalyzed Minisci-type alkylation of N-heteroarenes 1 with non-activated alkyl bromides 52 as radical precursors (Scheme 22).31 This photochemical system has proven to be robust and versatile, accommodating a wide range of primary and secondary alkyl bromides 52 with a variety of N-heterocycles 1. Expectedly, secondary alkyl bromides 52 produced higher yields than primary ones, due to the higher thermodynamic stability of the radical intermediate. Under the optimized conditions, the photocatalyst *Ir(III) oxidizes the bromide ion to generate a bromine radical and Ir(II). This Ir(II) complex subsequentially absorbs a second photon, leading to the generation of a highly reducing state of the Ir complex *Ir(II). Next, the *Ir(II) complex undergoes a SET event with alkyl bromide 52, followed by fragmentation forming an alkyl radical 22A and bromide anion, which participates in the catalytic cycle. The resulting alkyl radical 22A adds to the N-heteroarenes 1, followed by HAT with the bromine radical, which leads to the formation of the cation intermediate 22C. Finally, the desired product 53 is obtained via work-up. Moreover, the authors demonstrated the C–H alkylation of heteroarene 1 by using cetyl trimethyl ammonium bromide (CTAB) as a cationic surfactant with NaBr in the reaction medium to give the desired product 53 in 39–92% yield. It is worth noting that the reaction worked well with primary alkyls containing free hydroxy, chloride, amide, and –CF3 groups. Unfortunately, alkyl bromides bearing acetal groups remained inactive in this protocol.
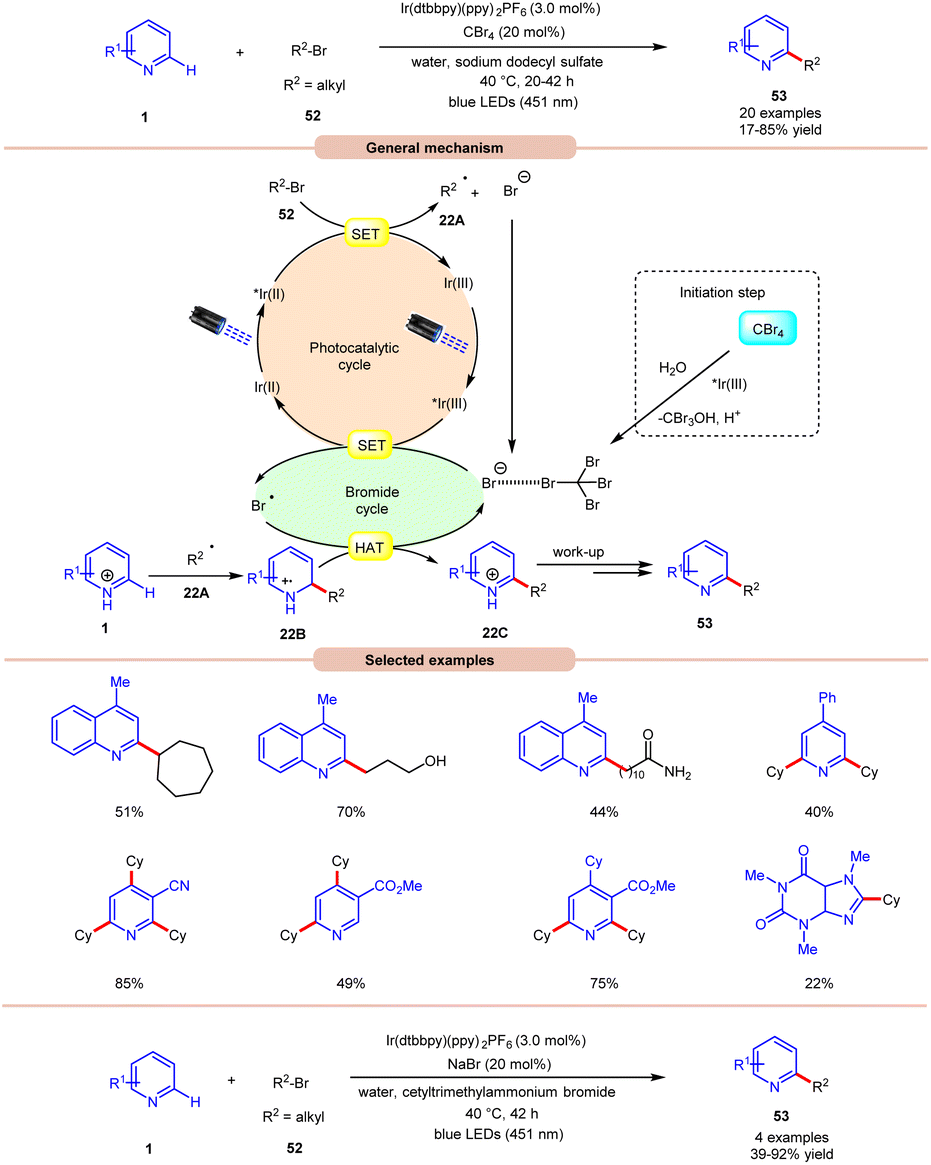 | ||
| Scheme 22 Ir-photoredox catalyzed Minisci-type alkylation of N-heteroarenes with non-activated alkyl bromides. | ||
A visible light-mediated dual Ni/organophotoredox catalyzed C(sp3)–H cross-coupling reaction with aryl bromide 52 was disclosed by König and co-workers in 2020, using NaBr as a bromide source (Scheme 23).32 A variety of linear and branched alkyl bromides 52 reacted well with the cyclic ether to afford the C(sp3)–H cross-coupling products 55 in moderate to good yields. Interestingly, benzyl chloride was also found to be an excellent coupling partner for this transformation. Moreover, the reaction could also be conducted on a gram scale. Based on previous reports, the authors postulated that the oxidative addition of Ni(0) into an aryl bromide 52 produces Ni(II) aryl bromide intermediate 23A. Next, the photoexcited catalyst 4CzIPN* oxidizes 23A to produce Ni(III) intermediate 23B. The photolysis of 23B results in the generation of a bromine radical and Ni(II) species 23C. The resulting bromine radical rapidly abstracts a hydrogen atom from α-carbon to oxygen, resulting in a C-centered radical intermediate, which adds to the Ni(II)-complex to produce Ni(III) species 23D. Subsequently, reductive elimination yields the desired C(sp3)–H cross-coupling product 55. Lastly, the SET reduction of the resulting Ni(I) intermediate by the highly reducing 4-CzIPN˙− species regenerates the Ni(0)-complex, thus completing the catalytic cycle.
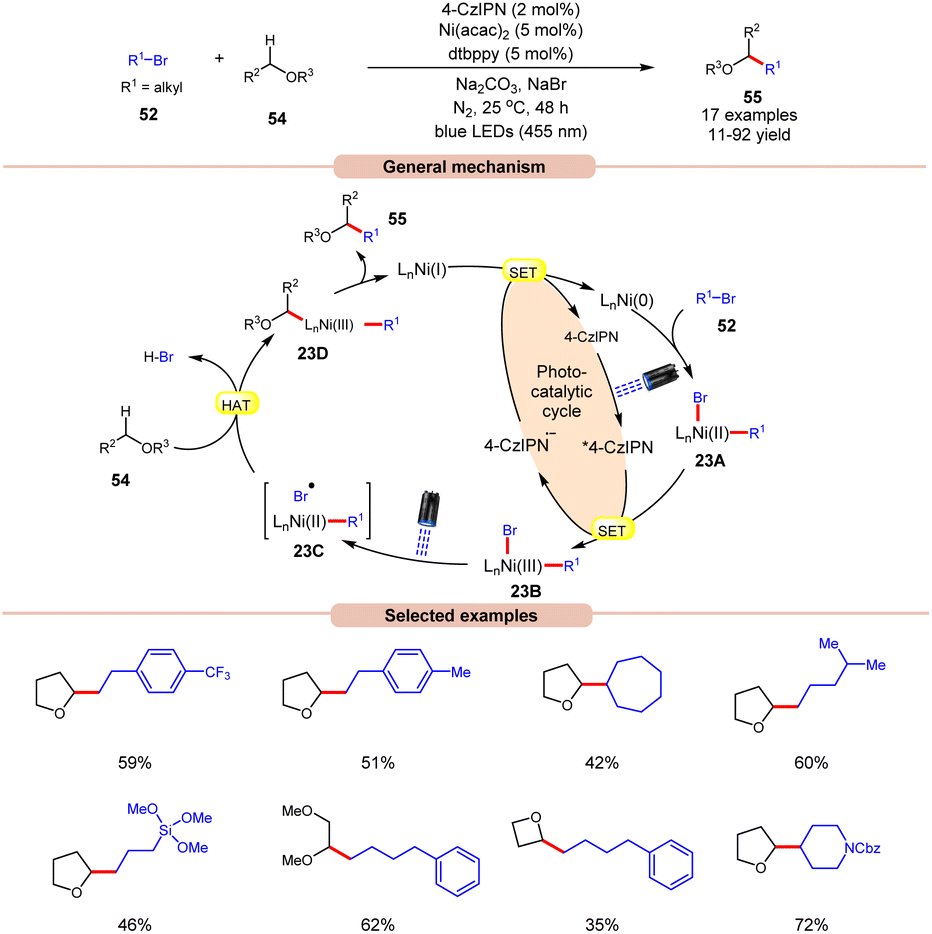 | ||
| Scheme 23 Dual Ni/organophotoredox catalyzed C(sp3)–H cross-coupling reaction with aryl bromide. | ||
In 2022, Song and co-workers reported visible light-mediated direct C–H difluoroalkylation of quinoxalinones 56 with bromodifluoroacylarenes 57via C(sp3)–Br bond homolytic cleavage (Scheme 24a).33a In this method, quinoxalin-2(1H)-ones 56 bearing electron-rich or electron-deficient functionalities readily afforded the desired difluoroalkylated products 58 in 30–87% yields. Several control experiments were performed to determine the reaction mechanism. Under visible light irradiation, the C(sp3)–Br bond undergoes homolysis to generate a difluoroalkyl radical intermediate 24A, which was further supposed by radical trapping experiments, and their adducts were confirmed by using HRMS analysis. Mechanistically, visible light-promoted C(sp3)–Br bond homolysis from ArCOCF2Br 57 generated the difluoroalkyl radical intermediate 24A and bromine radical. The bromine radical abstracts the hydrogen atom from quinoxalinone 56 to produce radical intermediate 24B with the release of HBr. The generated radical intermediate 24B undergoes radical coupling with difluoroalkyl radical 24A to furnish the desired products 58. The authors further expanded their work to the C(sp2)–H functionalization of aldehyde-derived hydrazones 59 with bromodifluoroacylarenes 57 for the direct preparation of functionalized α-iminodifluoroalkylated products 60. The substrate scope revealed that a variety of (hetero)aryl aldehyde-derived hydrazones 59 bearing either electron-rich or electron-deficient groups were efficiently transformed into the required products 60 with good to excellent yields. In this protocol, unsaturated aldehyde-derived hydrazones 59 such as alkynals, alkenals, and ester aldehydes were also compatible to obtain a series of complex difluoroalkylated products 60.
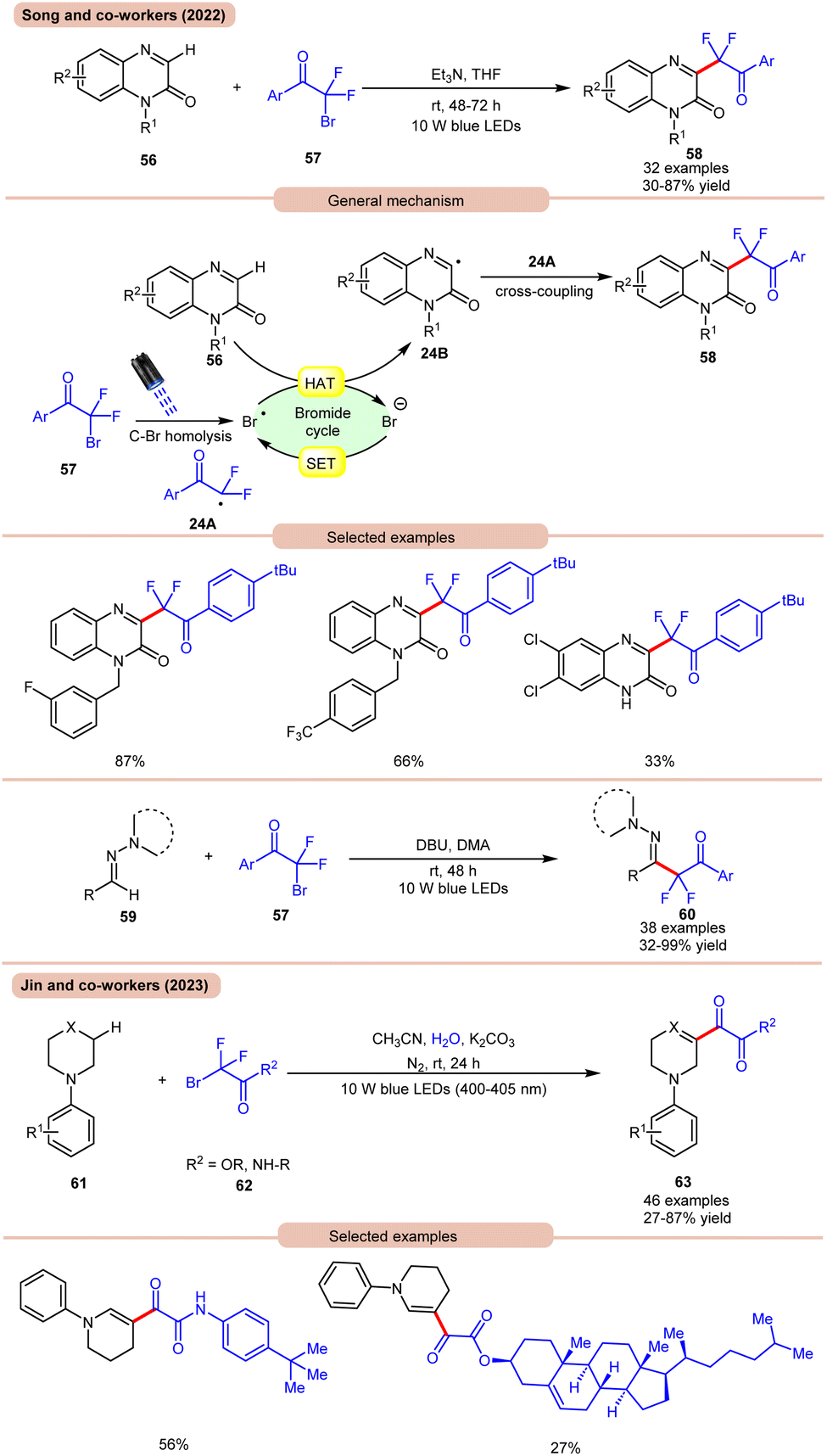 | ||
| Scheme 24 Visible light mediated direct C–H difluoroalkylation of quinoxalinones with bromodifluoroacylarenes. | ||
On the other hand, using difluoromethyl bromide 62 and H2O, Jin and co-workers in 2023 reported the β-alkoxyoxalylation of N-aryl cyclic amines 61 for the synthesis of β-ketoester/ketoamide substituted enamines 63 in 27–87% yields (Scheme 24b).33b It was proposed that difluoromethyl bromide undergoes homolysis to give a bromine radical, which participates in HAT and gives the key difluoroalkylated intermediate. This intermediate further reacts in the presence of H2O to give the desired product 63. In 2021, Huang and co-workers disclosed a visible light-mediated photoredox Minisci alkylation reaction of 4-hydroxyquinazolines 65 with ethyl acetate 64 (Scheme 25).34 In this protocol,4-hydroxyquinazolines 65 bearing various electron-donating and electron-withdrawing functionalities reacted well with ethyl acetate 64 to afford the desired products 66.
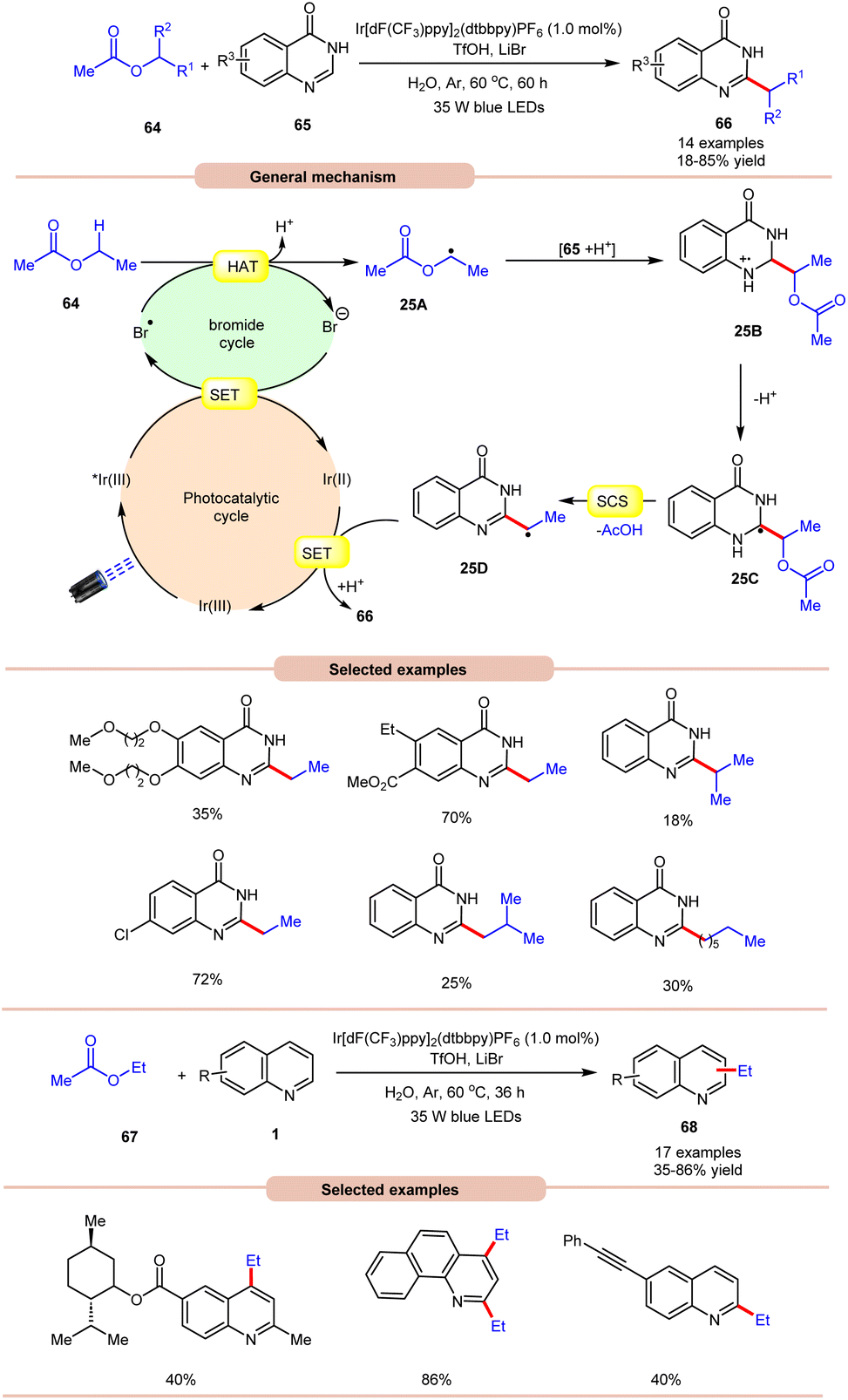 | ||
| Scheme 25 Ir-catalyzed Minisci alkylation reaction of 4-hydroxyquinazolines with ethyl acetate. | ||
Other alkylating reagents 64 such as isobutyl acetate, isopropyl acetate, n-pentyl acetate, n-butyl acetate, and n-heptyl acetate were also well tolerated, affording the required product 66 in low to moderate yields (18–55%). The reaction was completely suppressed in the presence of TEMPO, BHT, and DPE, which supported the involvement of a radical mechanistic pathway. Mechanistically, the photoexcited catalyst *Ir(III) is reduced by the bromide ion, leading to the generation of a bromine radical (Br˙). The bromine radical then initiates the abstraction of a hydrogen atom from ethyl acetate 64, resulting in the formation of an alkyl radical 25A. This alkyl radical 25A undergoes a radical addition reaction with 4-hydroxyquinazoline 65, accompanied by deprotonation, leading to the formation of radical intermediate 25C. Following this, a spin-center shift (SCS) process takes place, with the elimination of acetic acid (AcOH), resulting in the production of the radical intermediate 25D. Finally, upon SET/protonation, the required alkylated product 66 is obtained. Simultaneously, this step regenerates the Ir(III) species, completing the catalytic cycle. The applicability of this method was further demonstrated in the alkylation of quinoline and pyridine 1 derivatives under the optimized conditions, giving the required product 68 in 35–86% yield.
Doyle and co-workers in 2022, reported a dual Ir/Ni-photoredox catalyzed alkylation and (deutero)methylation of aryl halides 69 by using benzaldehyde di(alkyl) acetals 70 (Scheme 26).35 This protocol was found to be compatible with the late-stage functionalization of fenofibrate, which possesses an aryl chloride. The authors proposed that the photoexcited *Ir(III) oxidizes the bromide ion to a bromine radical, which undergoes HAT with the tertiary C–H bond of the acetal 70, followed by β-scission to afford an alkyl radical 26A and alkyl benzoate. Thereafter, the Ni(0)-complex undergoes oxidative addition with an aryl chloride and produces a Ni(II) aryl chloride intermediate 26B. The alkyl radical 26A is captured by 26B, generating Ni(III)–(Ar)(Me) species 26C, accompanied by reductive elimination to yield the desired product 71 and Ni(I)-complex. The reduced photocatalyst Ir(II) can then reduce Ni(I) to regenerate both Ir(III) and Ni(0)-catalysts. The authors further demonstrated the C(sp2)–C(sp3) cross-coupling reaction between acetals 70 as the source of aliphatic coupling partners with aryl bromides 32 to give the desired products 72 in 21–98% yield.
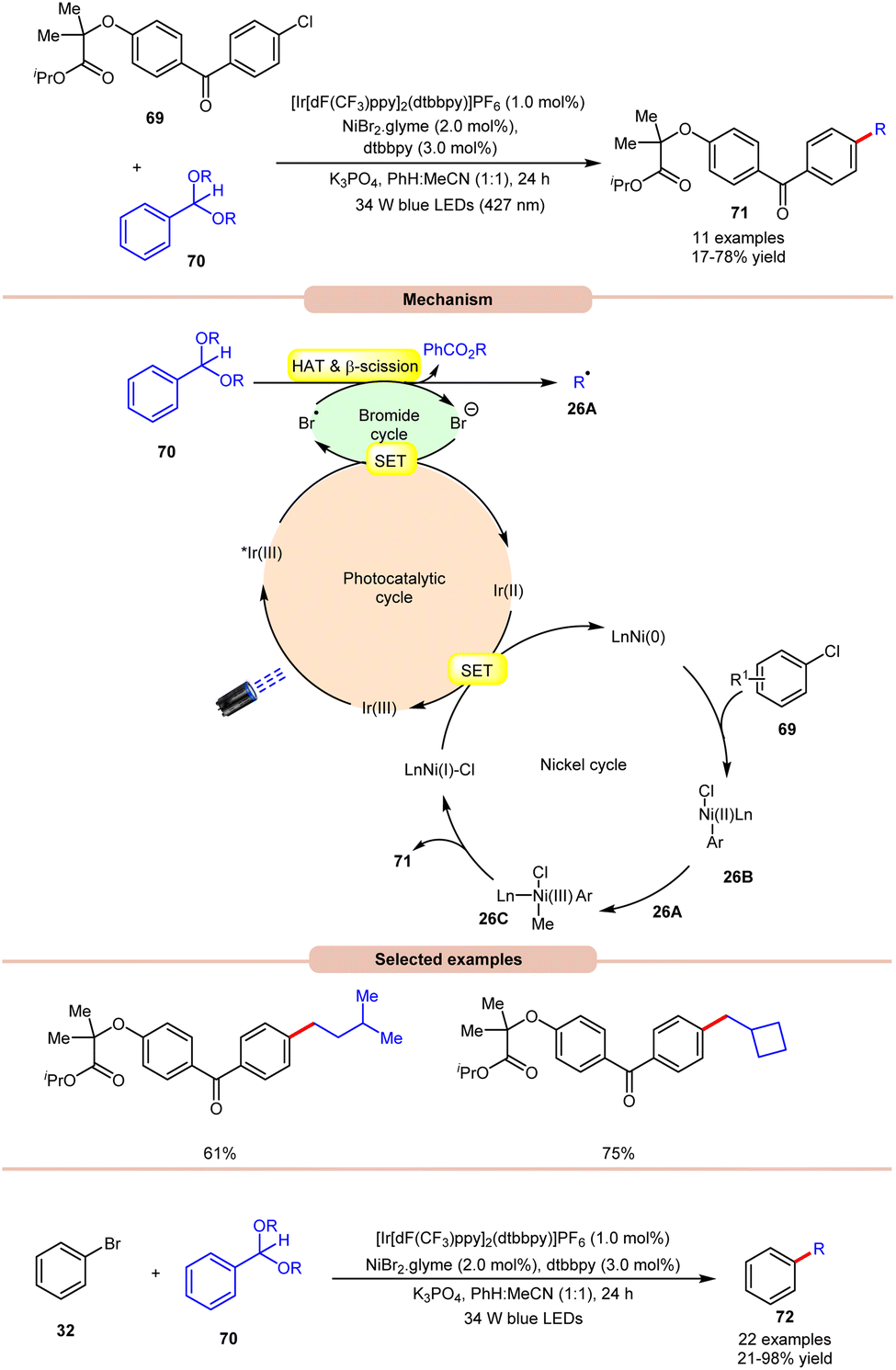 | ||
| Scheme 26 Dual Ir/Ni-photoredox-catalyzed alkylation and (deutero)methylation of aryl halides. | ||
5. Hydrogen atom transfer from (Me3Si)3SiH
Hydrogen atom transfer (HAT) from (Me3Si)3SiH (trimethylsilyl hydride) induced by a bromine radical is a well-known reaction in radical chemistry. Trimethylsilyl hydride is commonly used as a hydrogen atom donor in radical reactions due to its relatively weak Si–H bond and its ability to efficiently transfer a hydrogen atom to radical species. When a bromine radical encounters trimethylsilyl hydride, it can abstract a hydrogen atom from the Si–H bond, leading to the formation of a trimethylsilyl radical (Me3Si)3Si˙) and hydrogen bromide (HBr). This process can be represented by the following equation (Scheme 27):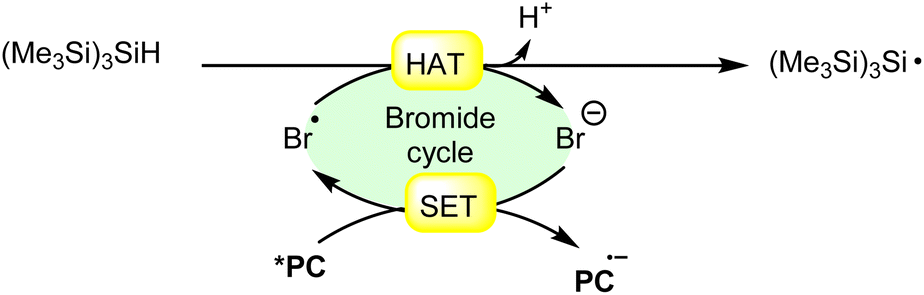 | ||
| Scheme 27 General approach for trimethylsilyl radical intermediate formation via bromine radical-mediated HAT. | ||
The resulting trimethylsilyl radical can further participate in various radical reactions, such as radical chain reactions or radical-mediated functionalization reactions. This type of hydrogen atom transfer is widely utilized in organic synthesis for the generation of radicals and the construction of complex molecules.
In 2018, ElMarrouni, Balsells and co-workers introduced a mild procedure for the direct visible light-initiated Giese addition of unactivated alkyl bromides 52 to electron-poor olefins (Michael acceptor) 73 or 74 (Scheme 28).36 In this process, visible light prompts the generation of an alkyl radical as a crucial intermediate from unactivated alkyl halides. This C(sp3)–C(sp3) bond formation proved to be highly efficient, accommodating a range of alkyl bromides successfully applied in the reaction of cyclic or acyclic α,β-unsaturated esters and amides. Mechanistic investigations suggested that the photoexcited *Ir(III) complex oxidizes the bromide ion to form a bromine radical, which abstracts a hydrogen atom from (Me3Si)3SiH to generate stabilized silyl radical intermediate 28A. This intermediate 28A rapidly undergoes halogen atom abstraction with the alkyl bromide 52, giving rise to the alkyl radical 28B and a bromosilane by-product. The alkyl radical 28B adds to the double bond of the Michael acceptor 73, generating another carbon radical intermediate 28C. A single-electron transfer (SET) between radical intermediate 28C and the reductant species Ir(II) provides an anion intermediate 28D, which upon protonation furnishes the desired product 75. Alternatively, hydrogen atom abstraction from (Me3Si)3SiH yields the corresponding product 75, thereby regenerating stabilized silyl radical species 28A. Notably, this methodology could be leveraged to access a key intermediate of vorinostat, an HDAC inhibitor employed in combating cancer and HIV.
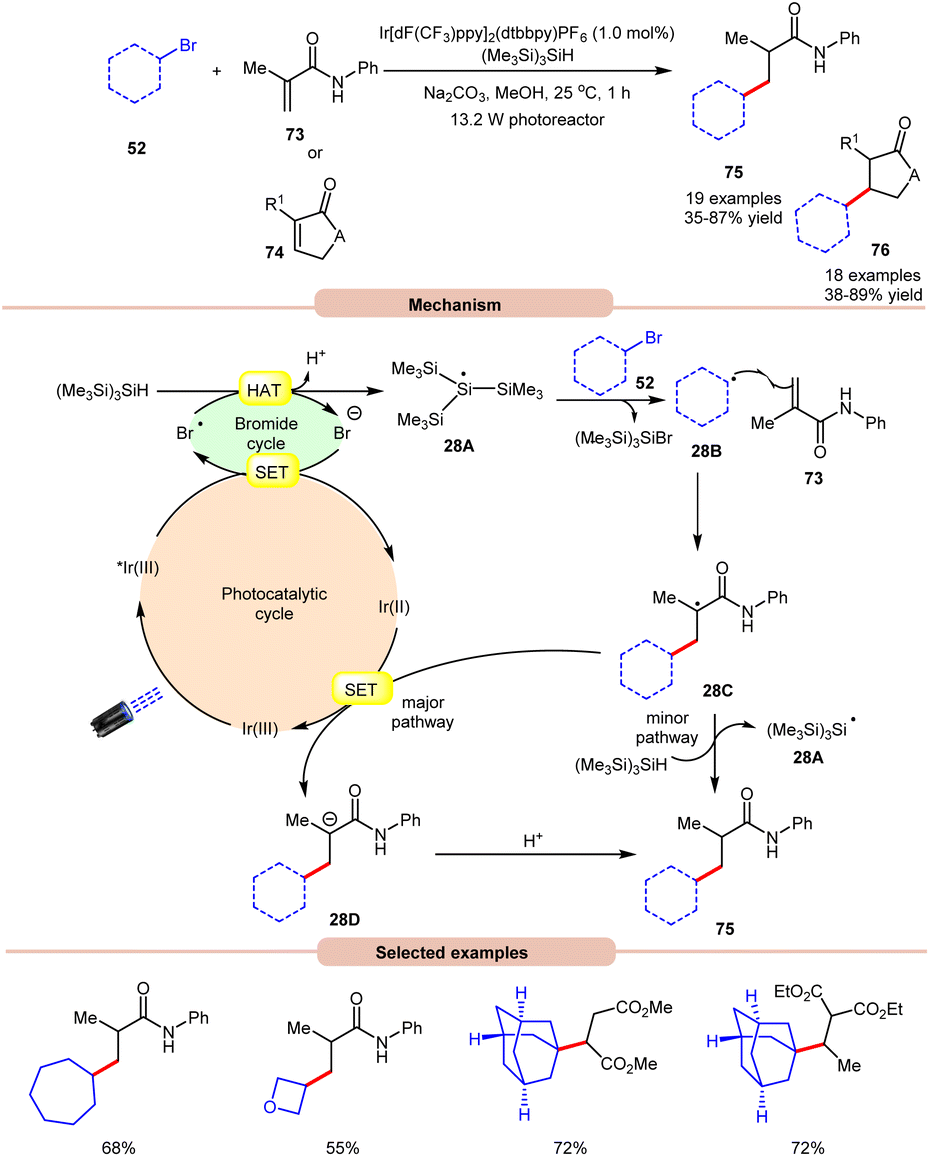 | ||
| Scheme 28 Visible light-mediated Giese addition of unactivated alkyl bromides to electron-poor olefins. | ||
In 2019, Wang and co-workers introduced an Ir-photoredox catalyzed Minisci C–H alkylation of heteroarenes 1 with unactivated primary, secondary, and tertiary alkyl bromides 52 by employing visible light irradiation (Scheme 29).37 This method demonstrated the efficient incorporation of a diverse array of cyclic and acyclic, unactivated primary, secondary, and tertiary alkyl groups 52 into N-heteroarenes 1 under optimized reaction conditions. Impressively, the protocol exhibited scalability, successfully performing on a gram scale. Additionally, its robustness was demonstrated in the late-stage functionalization of complex N-containing natural products and drugs.
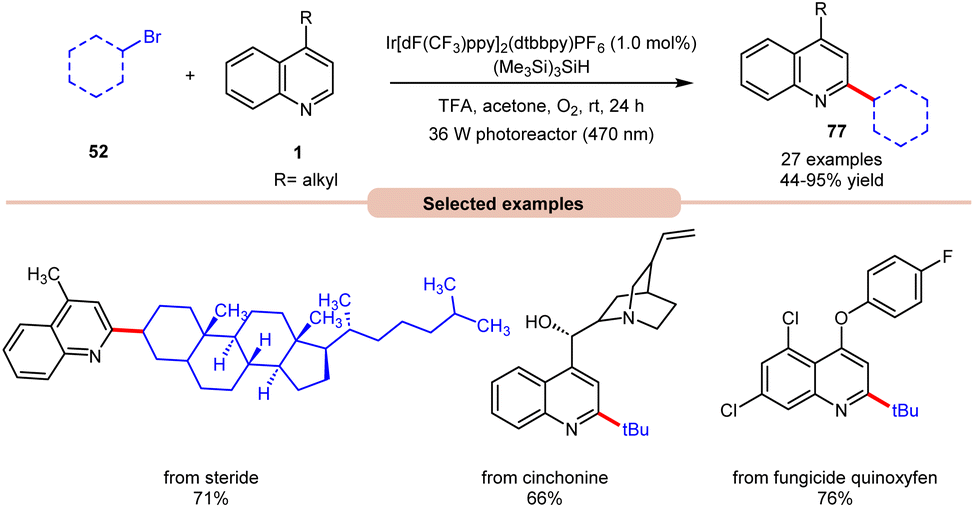 | ||
| Scheme 29 Ir-photoredox catalyzed Minisci C–H alkylation of heteroarenes with unactivated alkyl halides. | ||
Lately, Zhao, Shi, and co-workers revealed bromine radical-mediated hydrosilylation of alkene 36 using (TMS)3SiH and (n-Bu)4NBr as HAT reagents (Scheme 30).38 A wide range of electron-deficient alkenes and electron-rich alkenes 36 were reacted under this protocol to give the desired hydrosilylated products 78 in moderate to excellent yields. The compatibility of this method was further demonstrated in the late-stage functionalization of bioactive molecules in good yields.
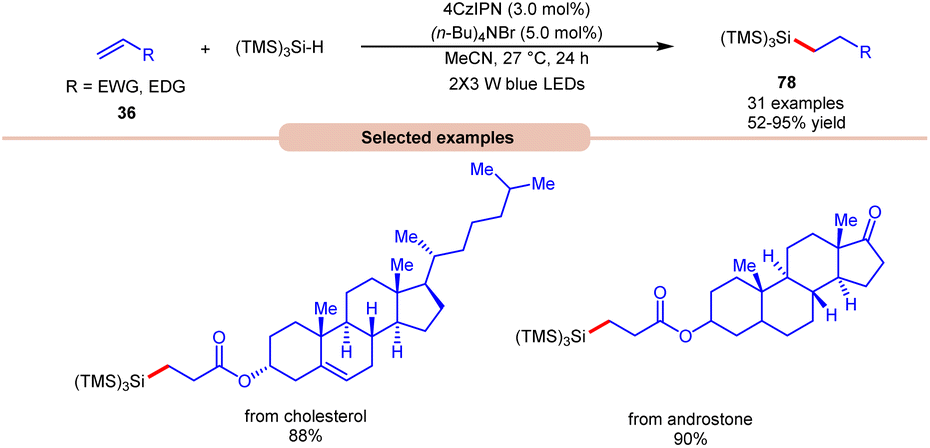 | ||
| Scheme 30 Bromine radical-mediated hydrosilylation of alkenes using (TMS)3SiH. | ||
MacMillan and co-workers, in 2016, developed a dual Ir/Ni-catalyzed approach towards the formation of a C(sp3)–C(sp2) bond via cross-electrophile coupling between alkyl 52 and aryl bromides 32 in good to excellent yields (Scheme 31).39 The study of substrate scope showed that aryl bromide 32 featuring electron-rich and electron-deficient substituents was compatible with the given reaction conditions. Interestingly, an unprotected 2-bromo aniline substrate can be employed directly and give the corresponding product in 65% yield. Additionally, N-containing heterocycles such as pyrazine, pyrimidine, pyridazinequinolone, isoquinoline, and pyrazole-substituted pyrazine were well tolerated under the reaction conditions. Furthermore, cyclic and acyclic alkyl bromide 52 worked well. Mechanistically, the photoexcited *Ir(III)-complex oxidizes the bromide anion to generate a bromine radical via the SET process, which upon HAT from tris(trimethylsilyl)silane (TTMS) forms a stabilized silyl radical intermediate 31A. Subsequently, silyl radical intermediate 31A rapidly abstracts a halogen atom from the alkyl bromide 52 to form alkyl radical 31B and the silyl bromide by-product. Moreover, a Ni(0)-complex undergoes oxidative addition with the aryl bromide 32 to form Ni(II) species 31C, which then interacts with an alkyl radical 31B to yield the Ni(III)-complex 31D. The reductive elimination of 31D delivers the desired C(sp3)–C(sp2) cross-coupling product 79 and a Ni(I) intermediate, which continues the catalytic cycle.
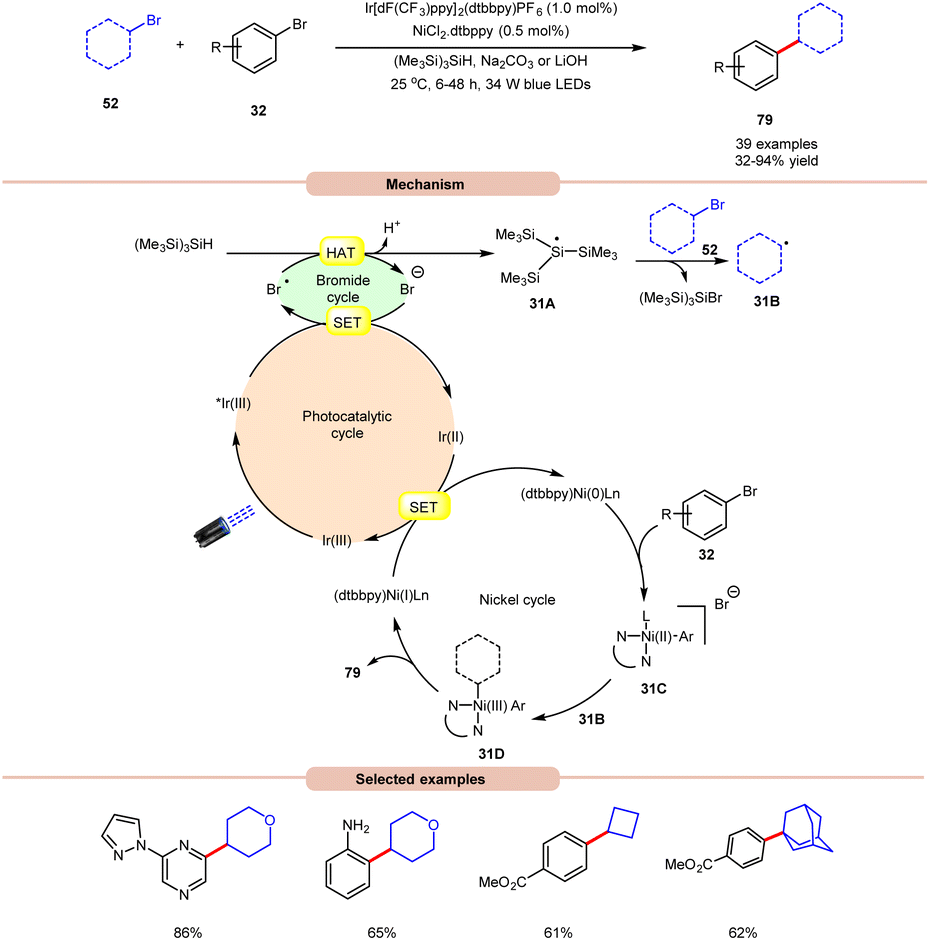 | ||
| Scheme 31 Dual Ir/Ni-catalyzed approach towards the formation of a C(sp3)–C(sp2) bond via cross-electrophile coupling between alkyl and aryl bromides. | ||
In the extension of their previous work, MacMillan and co-workers in 2021 reported dual photoredox Ir/Ni-catalyzed cross-electrophile coupling between an alkyl halide 80 and a diverse range of aryl bromides 32 to produce artificial analogues of tryptophan, phenylalanine, and histidine derivatives (Scheme 32).40 Various aryl bromides 32, featuring electron-donating and electron-withdrawing groups, were successfully utilized, leading to the desired products 81 in moderate to excellent yields.
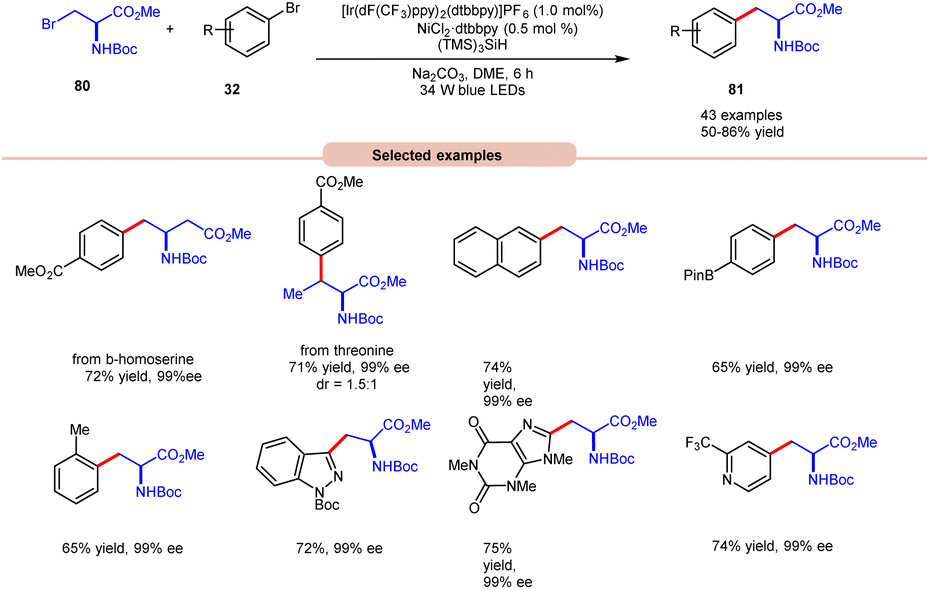 | ||
| Scheme 32 Dual Ir/Ni-catalyzed cross-electrophile coupling between alkyl halides and a diverse range of aryl halides. | ||
Additionally, substitution at the ortho-, meta-, and para-positions was well-accommodated, although ortho-substituted aryl halides 32 necessitated a higher loading of the nickel catalyst (5 mol%). Hong and co-workers achieved the synthesis of C4-alkylated pyridines 83 through an EDA-complex between pyridinium salts 82 and bromide ions under visible-light irradiation (Scheme 33).41
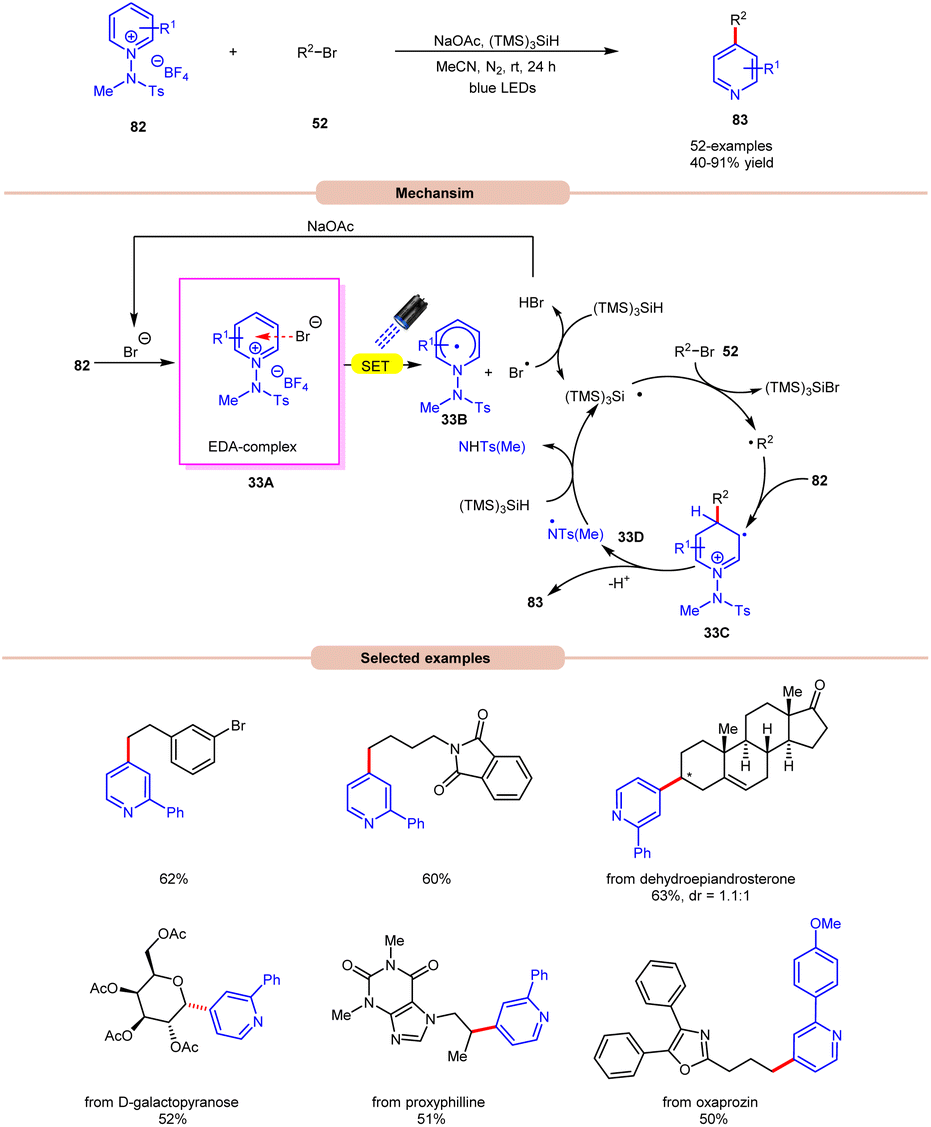 | ||
| Scheme 33 Bromide anion catalyzed alkylation of a pyridinium salt. | ||
A wide range of primary, secondary, and tertiary alkyl bromides 52 efficiently participated under the optimized conditions, yielding the desired products 83 in 40–91% yield. Initial experiments with a pyridinium salt and bromocyclohexane indicated that the in situ generated bromide anion effectively initiated a radical chain reaction under visible light irradiation, eliminating the need for an external bromide source. However, employing tetrabutylammonium bromide (TBAB) as a bromide anion source resulted in reduced yields of the targeted product. UV-visible absorption studies confirmed the formation of an EDA complex between the bromine anion and pyridinium salt 82. Stern–Volmer experiments revealed the formation of a bromine radical via reductive quenching of pyridinium salt 82 with the bromine anion. Moreover, the high quantum yield (Φ = 19.0) supported the involvement of a radical chain pathway. Mechanistically, an EDA-complex 33A is formed between the pyridinium salt (acceptor) 82 and bromide anion (donor). Visible light irradiation triggered a SET event, producing a bromine radical that initiated the chain reaction by abstracting an H-atom from (TMS)3SiH. The resulting silyl radical captures the bromine atom from alkyl bromides 52 to generate the corresponding alkyl radical (R˙). This alkyl radical regioselectively adds to the C4-position of the pyridinium salt 82, followed by deprotonation and homolytic N–N bond cleavage to yield the desired C4-alkylated pyridine product 83 and the regeneration of the silyl radical via N-centered radical intermediate 33D, which propagates the chain.
6. Hydrogen atom transfer from allylic carbons
The allylic position is particularly reactive due to the stability of the resulting allylic radical, which is resonance-stabilized. Allylic hydrogen atom transfer can occur through radical reactions, where a bromine radical abstracts a hydrogen atom from the allylic carbon as shown in Scheme 34.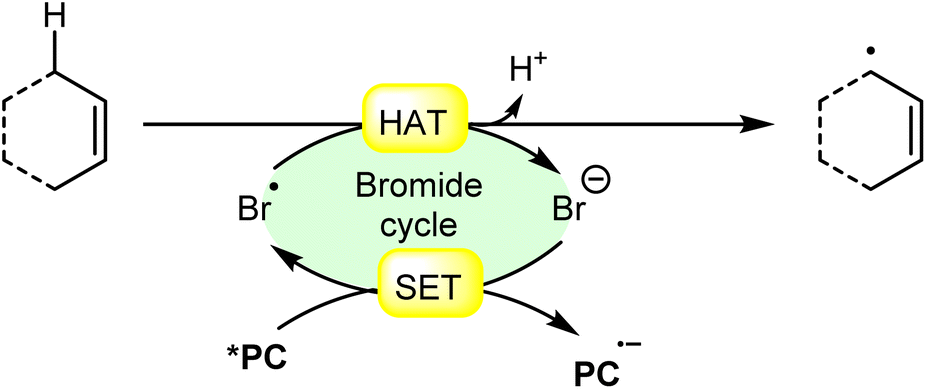 | ||
| Scheme 34 General approach for allylic radical intermediate formation via bromine radical-mediated HAT. | ||
An acridinium and Ni-catalyzed cross-coupling reaction between allylic compounds 84 and arylbromides 32 was reported by Rueping and coworkers in 2018 (Scheme 35).42 Under optimized reaction conditions, the oxidative addition of aryl bromide 32 to the Ni(0) complex gives a Ni(II) complex 35A. Next, triplet–triplet energy transfer (EnT) occurred from the photoexcited catalyst *Mes-Acr-Me+ and Ni(II) complex 35A to give intermediate 35B. After that, the excited form of Ni(II) complex 35B undergoes homolytic bond cleavage of the Br–Ni bond and generates a bromine radical and a Ni(I) complex 35C. The weak allylic C–H bond of alkene 84 undergoes H-atom transfer to the bromine atom and gives HBr and allylic radical 35D. Lastly, reductive elimination occurs to give the final product 85. This reaction is also compatible with vinyl bromides 34 to yield the desired products 86 in 51–64% yield.
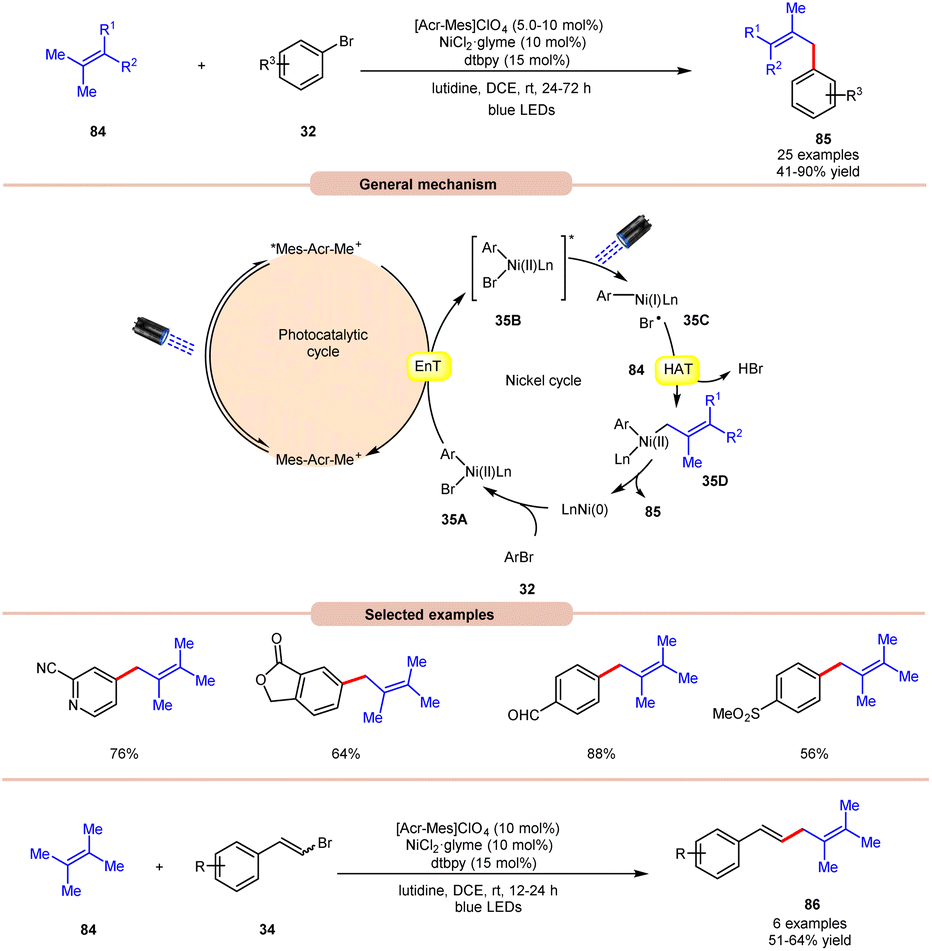 | ||
| Scheme 35 Dual acridinium/Ni-catalyzed cross-coupling reaction between allylic compounds and aryl bromides. | ||
Thereafter, a novel and efficient approach towards aerobic allylic C–H bond oxidation was achieved by Cai and co-workers in 2022 using nBu4NBr as a bromide source, with rose bengal as a photocatalyst under visible light irradiation (Scheme 36).43 This strategy was applicable to a wide range of tertiary cyclohexenol derivatives 87 as well as those containing 5, 7, and 8 membered cyclic ring and acyclic derivatives, affording the desired enone products 88 in 33–91% yield. The robustness of this photocatalytic aerobic reaction was also found compatible with a wide range of pharmaceutically active steroids and triterpene derivatives, resulting in the required products 88. Detailed mechanistic studies were performed to gain insights into the reaction mechanism. Radical quenching with TEMPO and DABCO (singlet oxygen quencher) confirmed the involvement of singlet oxygen in the reaction protocol. Stern–Volmer experiments supported the higher quenching efficiency of molecular oxygen with rose bengal compared to other reactants. When performed in the absence of nBu4NBr, the reaction resulted in the formation of a keto-acid product under similar conditions, indicating that singlet oxygen is formed in the reaction protocol and nBu4NBr is important for product formation. Based on these studies, the authors proposed that the photoexcited catalyst *RB undergoes an EnT process with O2 to form singlet oxygen (1O2), which itself participates in SET with a bromide anion to form a bromine radical and superoxide radical intermediate. Thereafter, substrate 87 undergoes HAT with the bromine radical to form C-centered radical intermediate 36A. This intermediate reacts with the superoxide radical to form an anion intermediate 36B. Finally, the protonation and elimination of H2O give access to the final enone product 88. This strategy offers a straightforward method for the allylic C–H oxidation of structurally challenging and complex molecules.
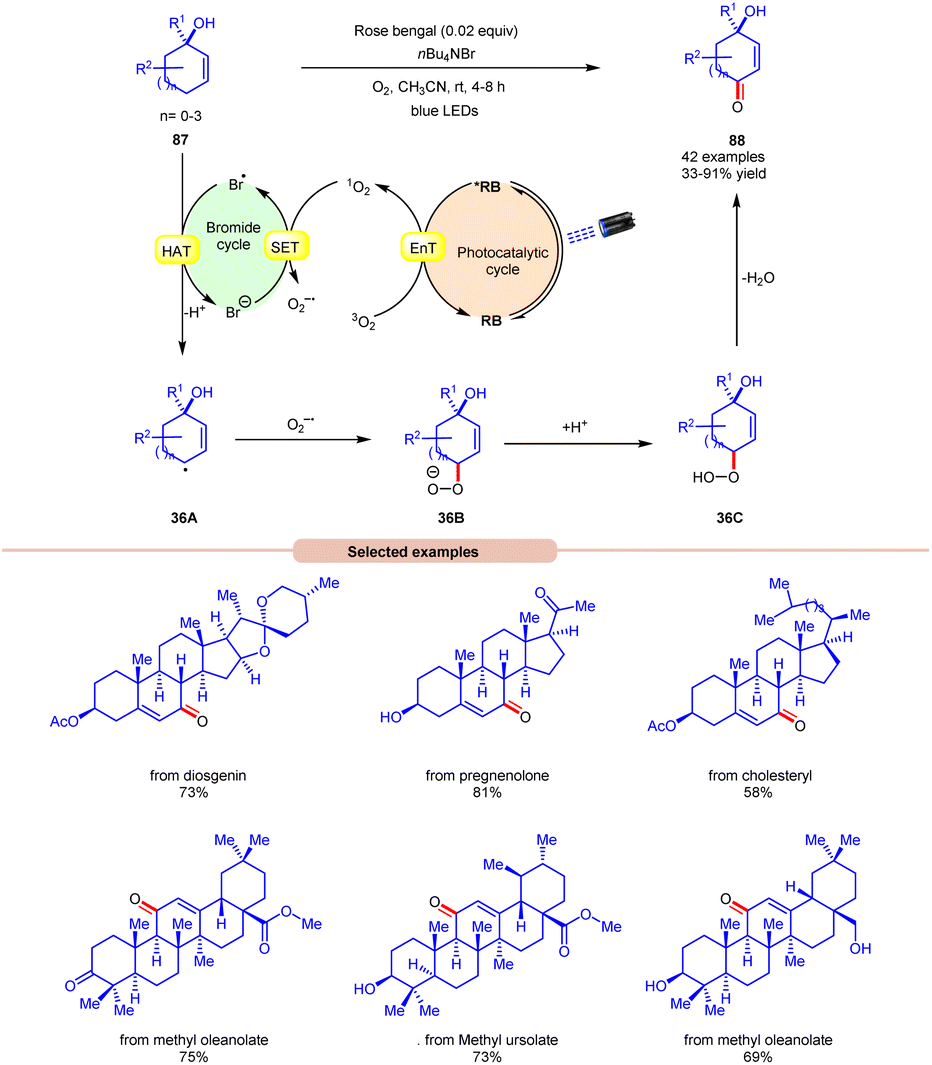 | ||
| Scheme 36 Rose bengal mediated allylic oxidation for the synthesis of enones. | ||
7. Miscellaneous
Li and co-workers, in 2021, demonstrated bromide-catalyzed C–H functionalization of amide carbonyl carbon for the synthesis of oxazolines, pyrrolidines, and dihydrooxazines (Scheme 37).44 For the synthesis of oxazolines, N-phenethylbenzamide derivative 89 was employed as a standard substrate with LiBr as a bromide catalyst with F-TEDA-BF4 (selectfluor) as an oxidant and Y(OTf)3 as an additive under visible light irradiation. An array of electron-donating and withdrawing functionalities on the N-phenethylbenzamide derivative 89 reacted smoothly to afford the desired product 90 in 19–70% yield. Notably, substrates bearing bulky arene groups such as 2,4,6-trimethyl or o-nitro or 3,5-CF3 groups afforded the targeted products in diminished yields. Mechanistic studies reveal that visible light, selectfluor, and air were indispensable for the reaction. The generation of an N-centered radical intermediate was ruled out from selectfluor (N–F bond homolysis) since no product bearing fluorine groups were observed. From these observations, the authors proposed the formation of intermediate 37C through the interaction between LiBr and selectfluor, which in the presence of light generates N-centered cation radial intermediate 37Dvia fragmentation. Next, the N-phenethylbenzamide derivative 89 undergoes intermolecular HAT with the bromine radical to form C-centered radical intermediate 37A. Next, bromination, accompanied by the subsequent cyclization and deprotonation, yields the desired oxazoline product 90 with the regeneration of the bromide catalyst. High loading of the catalyst and additive was the only limitation of this method. The authors further demonstrated the synthesis of dihydroxazines 91 and pyrroline 92 using this reaction protocol.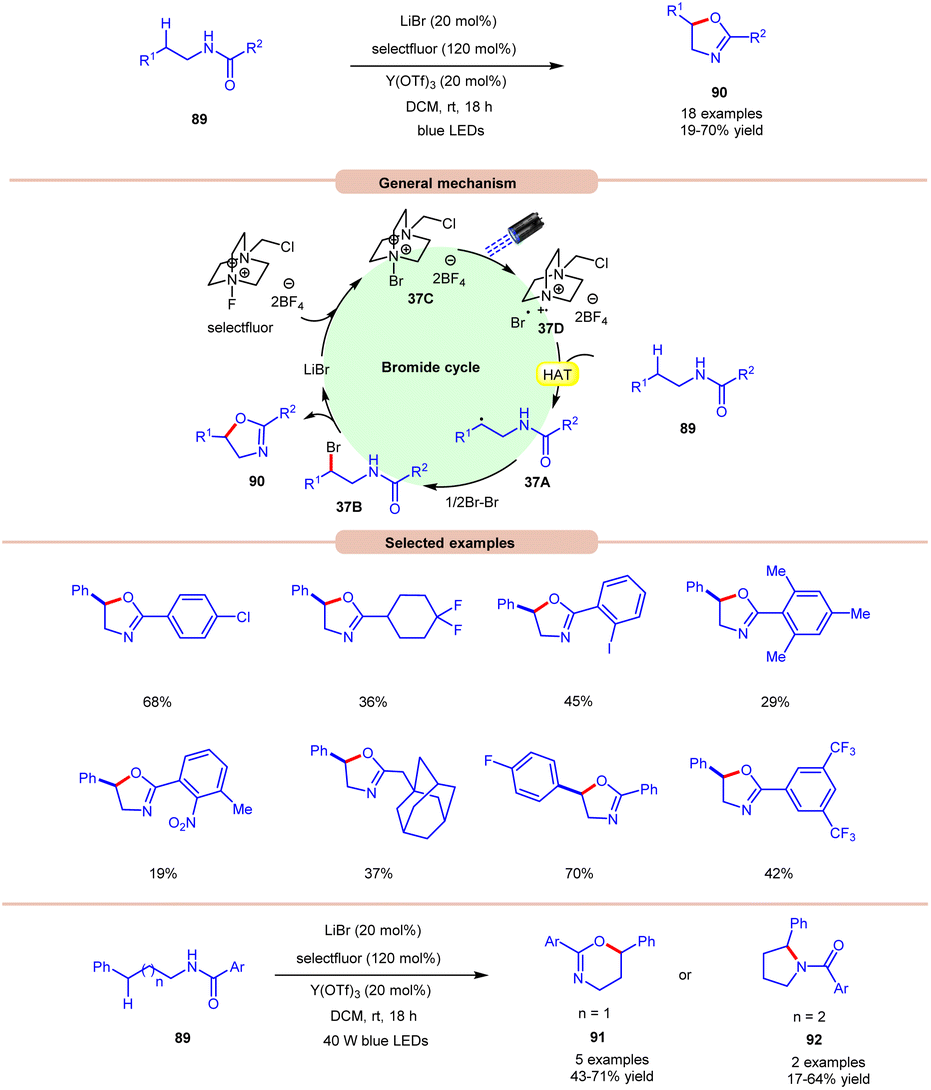 | ||
| Scheme 37 Visible light-mediated bromine radical catalyzed synthesis of oxazolines, pyrrolidines, and dihydrooxazines. | ||
8. Conclusions
This article provides an overview of C–H activation reactions initiated by Br˙ radicals generated through photocatalytic interactions with a catalyst under visible light irradiation. These Br˙ radicals serve as appealing hydrogen atom transfer (HAT) reagents and electron acceptors due to their robust oxidative properties and the formation of strong covalent bonds with hydrogen atoms. Moreover, the efficient generation of Br˙ radicals via light irradiation offers advantages in terms of reaction control. Compared to conventional synthetic methods, it can offer a more efficient and resource-conserving approach to constructing complex organic compounds. Despite the significant advancements made in this area, there are still many opportunities and challenges remaining, such as: (1) in this review article, the majority of the bromine radical mediated strategies are accelerated by expensive Ir-photoredox catalysis, sometimes in combination with nickel/ligand systems. This leaves room to explore other inexpensive metal and non-metal catalysts for their successful execution and ease the pathway to generate bromine radicals, which take part in the HAT reaction; (2) exploring electron-donor–acceptor (EDA) complex strategies and strategies involving visible light-mediated homolysis without the need for a photocatalyst for the generation of bromine radicals is highly desirable; (3) mainly, C–C, and C–O bond formation has been reported under these strategies; however, methods for the construction of C–N, C–S, C–P, C–X (X = halides) are still unexplored by bromine radical (Br˙) mediated HAT strategies. We hope that this review will stimulate interest in C–H activation reactions involving bromine radicals (Br˙) via HAT and deepen the understanding of their reactivity, thereby aiding in their application across various academic and industrial levels.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors have no conflicts of interest.Acknowledgements
Financial support from UCOST (UCS & T/R & D-35/20-21) Govt. of Uttarakhand, and SERB (CRG/2022/002691), India, is gratefully acknowledged. B. S. and R. P. thank CSIR and UGC for the SRF Fellowship, respectively.References
- (a) C. R. J. Stephenson, T. Yoon and D. W. C. Macmillan, in Visible Light Photocatalysis in Organic Chemistry, Wiley-VCH, German, 2018 CrossRef; (b) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS; (c) D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577–18580 CrossRef CAS; (d) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 ( Angew. Chem. , 2018 , 130 , 10188–10228 ) CrossRef CAS; (e) D. Ravelli, S. Protti and M. Fognoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (f) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS; (g) R. I. Patel, S. Sharma and A. Sharma, Org. Chem. Front., 2021, 8, 3166–3200 RSC; (h) N. Kvasovs and V. Gevorgyan, Chem. Soc. Rev., 2020, 50, 2244–2259 RSC; (i) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (j) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS; (k) T. Constantin, F. Julia and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef; (l) A. R. Allen, E. A. Noten and C. R. J. Stephenson, Chem. Rev., 2022, 122, 2695–2751 CrossRef CAS PubMed; (m) K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef; (n) S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed; (o) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS; (p) K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Chem. Rev., 2022, 122, 2353–2428 CrossRef CAS; (q) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS; (r) A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 60, 19526–19549 CrossRef CAS; (s) A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 133, 19678–19701 CrossRef; (t) R. I. Patel, J. Singh and A. Sharma, ChemCatChem, 2022, 14, e202200260 CrossRef CAS; (u) B. Saxena, R. I. Patel, J. Tripathi and A. Sharma, Org. Biomol. Chem., 2023, 21, 4723–4743 RSC.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC; (c) K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS; (d) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS; (e) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC; (f) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS; (g) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed; (h) S. Sharma and A. Sharma, Org. Biomol. Chem., 2019, 17, 4384–4405 RSC; (i) W. M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS; (j) R. I. Patel, A. Sharma, S. Sharma and A. Sharma, Org. Chem. Front., 2021, 8, 1694–1718 RSC.
- (a) C. G. S. Lima, T. D. M. Lima, M. Duarte, I. D. Jurbery and M. W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS; (b) Y. Q. Yuan, S. Majumder, M. H. Yang and S. R. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS; (c) P. Garra, J. P. Fouassier, S. Lakhdar, Y. Yagci and J. Lalevée, Prog. Polym. Sci., 2020, 107, 101277 CrossRef CAS; (d) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS; (e) B. Saxena, R. I. Patel and A. Sharma, Adv. Synth. Catal., 2023, 365, 1538–1564 CrossRef CAS.
- (a) L. Troian-Gautier, M. D. Turlington, S. A. M. Wehlin, A. B. Maurer, M. D. Brady, W. B. Swords and G. J. Meyer, Chem. Rev., 2019, 119(7), 4628–4683 CrossRef CAS; (b) S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed., 2018, 57, 15664–15669 CrossRef CAS; (c) M. Zidan, A. O. Morris, T. McCallum and L. Barriault, Eur. J. Org Chem., 2020, 2020, 1453–1458 CrossRef CAS; (d) H. P. Deng, Q. Zhou and J. Wu, Angew. Chem., 2018, 130, 12843–12847 CrossRef.
- (a) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed; (b) M. K. Nielsen, B. J. Shields, J. Liu, M. J. Williams, M. J. Zacuto and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 7191–7194 CrossRef CAS; (c) S. K. Kariofillis, B. J. Shields, M. A. Tekle-Smith, M. J. Zacuto and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 7683–7689 CrossRef CAS PubMed; (d) L. K. G. Ackerman, J. I. M. Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS PubMed; (e) Y. Jin, Q. Zhang, L. Wang, X. Wang, C. Meng and C. Duan, Green Chem., 2021, 23, 6984–6989 RSC.
- (a) J. M. Mayer, Acc. Chem. Res., 2011, 44(1), 36–46 CrossRef CAS; (b) L.-M. Zhao, Q.-Y. Meng, X.-B. Fan, C. Ye, X.-B. Li, B. Chen, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 3020–3024 CrossRef CAS; (c) T. Ide, J. P. Barham, M. Fujita, Y. Kawato, H. Egami and Y. Hamashima, Chem. Sci., 2018, 9, 8453–8460 RSC; (d) E. L. Saux, M. Zanini and P. Melchiorre, J. Am. Chem. Soc., 2022, 144, 1113–1118 CrossRef PubMed; (e) T. Uchikura, K. Tsubono, Y. Hara and T. Akiyama, J. Org. Chem., 2022, 87, 15499–15510 CrossRef CAS; (f) G. Kumar, S. Pradhan and I. Chatterjee, Chem.–Asian J., 2020, 15, 651–672 CrossRef CAS.
- (a) H. P. Deng, X. Z. Fan, Z. H. Chen, Q. H. Xu and J. Wu, J. Am. Chem. Soc., 2017, 139, 13579–13584 CrossRef CAS PubMed; (b) C. Y. Huang, J. Li and C. J. Li, Nat. Commun., 2021, 12, 4010 CrossRef CAS PubMed.
- (a) S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed; (b) Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS; (c) J. A. Kerr, Chem. Rev., 1966, 66(5), 465–500 CrossRef CAS; (d) X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117(13), 8622–8648 CrossRef CAS PubMed.
- (a) L. Capaldo, L. L. Quadri and D. Ravelli, Green Chem., 2020, 22, 3376–3396 RSC; (b) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122(2), 1875–1924 CrossRef CAS PubMed; (c) H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem Catal., 2021, 1, 523–598 CrossRef CAS; (d) H. Chena and S. Yu, Org. Biomol. Chem., 2020, 18, 4519–4532 RSC.
- S. Bonciolini, T. Noël and L. Capaldo, Eur. J. Org Chem., 2022, e202200417 CrossRef CAS.
- Y. Itabashi, H. Asahara and K. Ohkubo, Chem. Commun., 2023, 59, 7506–7517 RSC.
- Z. Wang, Q. Liu, X. Ji, G.-J. Deng and H. Huang, ACS Catal., 2020, 10, 154–159 CrossRef CAS.
- X. Ji, Q. Liu, Z. Wang, P. Wang, G.-J. Deng and H. Huang, Green Chem., 2020, 22, 8233–8237 RSC.
- (a) K. Ding, Y. Lu, Z. Nikolovska-Coleska, G. Wang, S. Qiu, S. Shangary, W. Gao, D. Qin, J. Stuckey, K. Krajewski, P. P. Roller and S. Wang, J. Med. Chem., 2006, 49, 3432–3435 CrossRef CAS; (b) S. Peddibhotla, Curr. Bioact. Compd., 2009, 5, 20 CrossRef CAS; (c) A. Millemaggi and R. J. K. Taylor, Eur. J. Med. Chem., 2010, 2010, 4527–4547 Search PubMed; (d) M. Kaur, M. Singh, N. Chadha and O. Silakari, Eur. J. Med. Chem., 2016, 123, 858–894 CrossRef CAS; (e) B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 763–798 CrossRef; (f) N. Ye, H. Chen, E. A. Wold, P. Y. Shi and J. Zhou, ACS Infect. Dis., 2016, 2, 382–392 CrossRef CAS.
- Z. Sun, H. Huang, Q. Wang, C. Huang, G. Mao and G.-J. Deng, Org. Chem. Front., 2022, 9, 3506–3514 CAS.
- (a) H. Wang, H. Liu, M. Wang, M. Huang, X. C. Shi, T. Wang, X. Cong, J. Yan and J. Wu, iScience, 2021, 24(6), 102693 CrossRef CAS; (b) T. Ye, Y. Li, Y. Ma, S. Tan and F. Li, J. Org. Chem., 2024, 89, 534–540 CrossRef CAS.
- T. Kawasaki, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2020, 142, 3366–3370 CrossRef CAS PubMed.
- (a) T. Kawasaki, N. Ishida and M. Murakami, Angew. Chem., Int. Ed., 2020, 59, 18267–18271 CrossRef CAS; (b) T. Kawasaki, T. Tosaki, N. Ishida and M. Murakami, Org. Lett., 2021, 23, 7683–7687 CrossRef CAS PubMed.
- Q.-L. Wang, H. Huang, Z. Sun, Y. Chen and G.-J. Deng, Green Chem., 2021, 23, 7790–7795 RSC.
- H. Yu, T. Zhan, Y. Zhou, L. Chen, X. Liu and X. Feng, ACS Catal., 2022, 12, 5136–5144 CrossRef CAS.
- Q.-L. Wang, H. Huang, M. Zhu, T. Xu, G. Mao and G.-J. Deng, Org. Lett., 2023, 25, 3800–3805 CrossRef CAS PubMed.
- Q.-L. Wang, H. Huang, G. Mao and G.-J. Deng, Green Chem., 2022, 24, 8324–8329 RSC.
- Q.-L. Wang, Z. Sun, H. Huang, G. Mao and G.-J. Deng, Green Chem., 2022, 24, 3293–3299 RSC.
- (a) M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Hoboken, NJ, 2013 Search PubMed; (b) R. Álvarez, B. Vaz, H. Gronemeyer and A. R. de Lera, Chem. Rev., 2014, 114, 1–125 CrossRef PubMed; (c) M. Hassam, A. Taher, G. E. Arnott, I. R. Green and W. A. L. Van Otterlo, Chem. Rev., 2015, 115, 5462–5569 CrossRef CAS PubMed; (d) B. E. Maryanoff and A. B. Reitz, Chem. Soc. Rev., 1989, 89, 863–927 CrossRef CAS; (e) P. A. Byrne and D. G. Gilheany, Chem. Soc. Rev., 2013, 42, 6670–6696 RSC; (f) H.-T. Chang, T. T. Jayanth, C.-C. Wang and C.-H. Cheng, J. Am. Chem. Soc., 2007, 129, 12032–12041 CrossRef CAS PubMed; (g) C. W. Cheung, F. E. Zhurkin and X. Hu, J. Am. Chem. Soc., 2015, 137, 4932–4935 CrossRef CAS PubMed; (h) B. Saxena, R. I. Patel, S. Sharma and A. Sharma, Green Chem., 2024, 26, 2721–2729 RSC; (i) Y. Ikeda, T. Nakamura, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2002, 124, 6514–6515 CrossRef CAS; (j) W. Affo, H. Ohmiya, T. Fujioka, Y. Ikeda, T. Nakamura, H. Yorimitsu, K. Oshima, Y. Imamura, T. Mizuta and K. Miyoshi, J. Am. Chem. Soc., 2006, 128, 8068–8077 CrossRef CAS PubMed; (k) K. Zhu, J. Dunne, M. P. Shaver and S. P. Thomas, ACS Catal., 2017, 7, 2353–2356 CrossRef CAS; (l) S. J. Meek, R. V. O'Brien, J. Liaveria and R. R. Schrock, Nature, 2011, 471, 461–466 CrossRef CAS; (m) T. Di Franco, A. Epenoy and X. Hu, Org. Lett., 2015, 17, 4910–4913 CrossRef CAS; (n) Q. Liu, X. Dong, J. Li, J. Xiao, Y. Dong and H. Liu, ACS Catal., 2015, 5, 6111–6137 CrossRef CAS.
- X. Cheng, T. Li, Y. Liu and Z. Lu, ACS Catal., 2021, 11, 11059–11065 CrossRef CAS.
- P. Jia, Q. Li, W. C. Poh, H. Jiang, H. Liu, H. Deng and J. Wu, Chem, 2020, 6, 1766–1776 CAS.
- Z. Wang, X. Ji, T. Han, G.-J. Deng and H. Huang, Adv. Synth. Catal., 2019, 361, 5643–5647 CrossRef CAS.
- (a) C.-L. Cao, G.-X. Zhang, F. Xue and H.-P. Deng, Org. Chem. Front., 2022, 9, 959–965 RSC; (b) Y. Lin, X. Shu and H. Huo, Synthesis, 2024, 56, 1702–1710 CrossRef CAS.
- (a) A. Butler and J. V. Walker, Chem. Rev., 1993, 93, 1937–1944 CrossRef CAS; (b) D. Kahne, C. Leimkuhler, W. Lu and C. Walsh, Chem. Rev., 2005, 105, 425–448 CrossRef CAS; (c) F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef CAS; (d) C. Paul and G. Pohnert, Nat. Prod. Rep., 2011, 28, 186–195 RSC; (e) J. Latham, E. Brandenburger, S. A. Shepherd, B. R. K. Menon and J. Micklefield, J. Chem. Rev., 2018, 118, 232–269 CrossRef CAS.
- Y. Chen, Z. Qu, S. Chen, X. Ji, G.-J. Deng and H. Huang, Adv. Synth. Catal., 2022, 364, 1573–1579 CrossRef.
- M. S. Santos, M. Cybularczyk-Cecotka, B. König and M. Giedyk, Chem.–Eur. J., 2020, 26, 15323–15329 CrossRef CAS PubMed.
- M. S. Santos, A. G. Corrêa, M. W. Paixão and B. König, Adv. Synth. Catal., 2020, 362, 2367–2372 CrossRef CAS.
- (a) C.-H. Qu, R. Huang, Y. Liu, T. Liu and G.-T. Song, Org. Chem. Front., 2022, 9, 4135–4145 RSC; (b) B. Sun, P.-X. Li, Y. Jiang, L.-L. Yang, P.-Y. Huang, R.-P. Shen, M.-J. Chen, J.-Y. Wang and C. Jin, Org. Lett., 2023, 25, 6773–6778 CrossRef CAS.
- C. Wang, H. Shi, G.-J. Deng and H. Huang, Org. Biomol. Chem., 2021, 19, 9177–9181 RSC.
- S. K. Kariofillis, S. Jiang, A. M. Żurański, S. S. Gandhi, J. I. M. Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 1045–1055 CrossRef CAS PubMed.
- A. ElMarrouni, C. B. Ritts and J. Balsells, Chem. Sci., 2018, 9, 6639–6646 RSC.
- J. Dong, X. Lyu, Z. Wang, X. Wang, H. Song, Y. Liua and Q. Wang, Chem. Sci., 2019, 10, 976–982 RSC.
- Y. Zhao, K. Zhang, Y. Bai, Y. Zhang and S. Shi, Chin. J. Inorg. Chem., 2023, 43, 2837–2847 CAS.
- P. Zhang, C. Chip Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138(26), 8084–8087 CrossRef CAS PubMed.
- T. M. Faraggi, C. Rouget-Virbel, J. A. Rincón, M. Barberis, C. Mateos, S. García-Cerrada, J. Agejas, O. de Frutos and D. W. C. MacMillan, Org. Process Res. Dev., 2021, 25, 1966–1973 CrossRef CAS.
- S. Jung, S. Shin, S. Park and S. Hong, J. Am. Chem. Soc., 2020, 142, 11370–11375 CrossRef CAS PubMed.
- L. Huang and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 10333–10337 CrossRef CAS.
- C. Liu, H. Liu, X. Zheng, S. Chen, Q. Lai, C. Zheng, M. Huang, K. Cai, Z. Cai and S. Cai, ACS Catal., 2022, 12, 1375–1381 CrossRef.
- N. Kaur, E. C. Ziegelmeyer, O. N. Farinde, J. T. Truong, M. M. Huynh and W. Li, Chem. Commun., 2021, 57, 10387–10390 RSC.
| This journal is © The Royal Society of Chemistry 2024 |