
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuneable C3 product selectivity of glycerol electrooxidation on cubic and dendritic Pt nanocatalysts†
Irina
Terekhina
 and
Mats
Johnsson
*
and
Mats
Johnsson
*
Department of Materials and Environmental Chemistry, Arrhenius Laboratory, Stockholm University, Stockholm SE-106 91, Sweden. E-mail: mats.johnsson@mmk.su.se
First published on 10th June 2024
Abstract
Glycerol, being an abundant and cheap by-product of biodiesel production, has emerged as a raw material that can be recycled into value-added compounds. In the present study, Pt nanoparticles of cubic (PtCUBE) and dendritic (PtDEND) morphologies were investigated as catalysts for the glycerol electrooxidation reaction (GEOR) at 20 °C. To optimise the electrocatalytic performance and GEOR selectivity towards three-carbon (C3) products, namely lactate, glycerate, and tartronate, the effects of morphology, electrolyte composition, and applied potential were studied. At low glycerol concentrations, C–C bond cleavage was more favoured, especially on PtDEND. Both PtCUBE and PtDEND showed high C3 product selectivity up to 91% at 0.67 V vs. RHE, with lactate reaching a maximum selectivity of 68% on PtCUBE, which also exhibited the best mass and specific activities compared to PtDEND.
1. Introduction
In recent years, there has been a notable shift in focus within industrial and scientific circles toward the exploration of clean, renewable, and sustainable energy sources and fuels. One example is biodiesel—one of the common biofuels available that is produced from animal fats, vegetable oils, and their residues via the transesterification of methanol with triglycerides to fatty acid alkyl esters (biodiesel).1,2 However, the expansion of the biodiesel industry over the decades has resulted in the generation of an enormous excess of glycerol (GLY), a by-product, which is estimated to be produced in hundreds of millions of kilograms—nearly 10 wt% of the biodiesel produced.3,4 This eventually resulted in a significant drop in the price of glycerol due to it exceeding market demand.5,6 Thus, being a cheap and abundant raw material with a three-carbon chain makes glycerol a great candidate for valorisation into more valuable products. One promising approach is the glycerol electrochemical oxidation reaction (GEOR), which allows for the generation of valuable compounds on the anode and the simultaneous co-production of hydrogen on the cathode with a lower operating cell potential compared to water splitting.4,7Generally, the GEOR can yield compounds such as glycerol carbonates, acetals, ketals and esters,2 which have a wide range of uses from the food industry to cosmetics and pharmaceuticals. Obtaining three-carbon (C3) products like glyceraldehyde, dihydroxyacetone, glyceric acid/glycerate, lactic acid/lactate, and tartronic acid/tartronate is of the highest interest. However, taking into account the number of compounds produced, a predictable question arises: how can one tune the reaction's selectivity?
In the past decades, numerous studies8–11 have been devoted to investigating the glycerol electrooxidation mechanisms on noble and non-noble metal-based catalysts and their alloys in acidic and alkaline media. Platinum, palladium, and gold-based materials have been extensively investigated as catalysts for the GEOR. Due to their high activity and stability and remarkable selectivity towards C3 products, the desire to improve their performance is still driving the scientific community.11,12 Additionally, it has been found that not only does the catalyst material affect the GEOR performance but also its morphology and the facets exposed.13–16 In particular, extensive studies of the performance towards the glycerol (electro)oxidation reaction and/or oxidation product selectivity have been performed on pure Pt and Pt-based binary and ternary alloys with various morphologies like nanocubes,17,18 nanocubes with excavated and dendritic structures,19 porous and dendritic structures20–23 as well as irregular and spherical nanoparticles loaded on different supports like TiO2,24 ZrO2,25 carbon nanotubes,26 carbon black,27,28 and graphene nanosheets.29 Although the search for the best GEOR catalyst has been ongoing for many years, there is still a surprisingly low number of reports showing thorough quantitative analysis of the yield and selectivity of GEOR products as well as glycerol conversion using different media, electrolyte composition and reaction temperature in addition to studies on defined morphologies.
Herein, we report the electrocatalytic performance of cubic (PtCUBE) and dendritic (PtDEND) platinum nanoparticles (NPs) for the GEOR in an alkaline medium at three applied potentials and room temperature. Both PtCUBE and PtDEND were highly selective for C3 product formation, specifically, lactate at a low applied potential and the highest pH. Additionally, several C3–C1 selectivity trends were observed in electrolytes of different compositions. Overall, the cubic morphology of PtCUBE NPs provided the best performance over PtDEND not only in terms of electrochemical performance but also glycerol conversion and valuable GEOR product selectivity.
2. Experimental section
2.1. Chemicals and materials
Platinum(II) acetylacetonate (Pt(acac)2, Acros Organics, 98%), potassium tetrachloroplatinate(II) (K2PtCl4, Combi-Blocks Inc., 98%), potassium chloride (KCl, VWR chemicals), tungsten hexacarbonyl (W(CO)6, Merck, ≥98%), cetyltrimethylammonium bromide (CTAB, Sigma-Aldrich, >99.0%), hydrochloric acid (HCl, VWR chemicals, 37%), L-ascorbic acid (Sigma-Aldrich, >99.0%), potassium hydroxide (KOH, Honeywell, puriss. p.a., ≥86%), glycerol (bidistilled, VWR chemicals, ≥99.5%), oleylamine (OAm, Sigma-Aldrich, technical grade, 70%), oleic acid (OA, Tokyo Chemical Industry, >99.0%), hexane (Honeywell, >99%), ethanol (absolute, VWR chemicals, ≥99.7%), isopropanol (Carlo Erba Reagents, ≥99.5%), Nafion perfluorinated resin solution (5 wt% in lower aliphatic alcohols and water, contains 15–20% water) were used without further purification. Carbon fibre paper (CFP, H23, thickness 210 μm) was purchased from Freudenberg and subjected to hydrophilic treatment and catalyst modification.Ultrapure water with a resistivity of 18.2 MΩ cm from Millipore (Simplicity Water Purification System) was used throughout all the experiments and aqueous solution preparations.
2.2. Synthesis of Pt dendritic nanoparticles
Similar to a previously reported procedure,30 to synthesise Pt dendritic (PtDEND) NPs, 20 mL of 250 mM CTAB aqueous solution was placed into a 25 mL round-bottomed flask and heated to 95 °C under magnetic stirring (800 rpm). Then, 2 mL of 100 mM K2PtCl4 solution was added, followed by stirring for 5 minutes. After that, 320 μL of 1 M ascorbic acid solution was quickly injected, and the reaction mixture was stirred at 95 °C for 10 min. Finally, the resulting solution was washed by centrifugation three times with water and once with absolute ethanol at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 minutes, followed by redispersion in ethanol for storage.
000 rpm for 10 minutes, followed by redispersion in ethanol for storage.
2.3. Synthesis of Pt cubic nanoparticles
A modified previously reported procedure was utilised to synthesise Pt cubic (PtCUBE) NPs.31 A mixture of 80 mg of Pt(acac)2, 32 mL of OAm and 8 mL of OA in a 50 mL round-bottomed flask equipped with a rubber septum was flushed with argon gas for 15 minutes to remove excess oxygen while being heated to 130 °C under magnetic stirring (800 rpm). Then, 200 mg of W(CO)6 was added. After that, the reaction mixture temperature was increased to 220 °C, and it was allowed to react for 1 hour under the same stirring rate and argon stream. Finally, the resulting solution was washed by centrifugation four times with hexane at 12000 rpm for 15 minutes, followed by redispersion in hexane for storage.
2.4. Structural characterisation
Transmission electron microscopy (TEM) imaging was carried out on a JEOL JEM-2100F microscope operating at an acceleration voltage of 200 kV. TEM samples were prepared by depositing ∼20 μL of nanoparticle ethanol or hexane dispersion onto a 200-mesh carbon-coated copper grid and allowing the solvent to evaporate. Powder X-ray diffraction (PXRD) analysis was performed on a Bruker D8 Discover diffractometer using a Cu Kα X-ray source (λCuKα1 = 1.540598 Å and λCuKα2 = 1.544426 Å), equipped with a LYNXEYE XE-T detector. Diffractograms were collected within the 2θ range of 10–90° with a step size of 0.01° and a scan rate of 0.375° min−1.2.5. Electrochemical measurements
Details on the carbon fibre paper (CFP) and Nafion membrane treatments, catalyst ink preparation, and working electrode fabrication can be found in our previous study.15All electrochemical measurements were carried out using an SP-50 potentiostat (Biologic, France) at room temperature (20 °C). The setup comprised a typical three-electrode cell with the catalyst-modified CFP electrodes being working electrodes, a saturated calomel electrode (SCE) being a reference electrode and platinum mesh being a counter electrode. Catalyst performance was investigated in two anolytes: 1 M KOH + 0.1 M glycerol and 2 M KOH + 1 M glycerol, with 1 M KOH and 2 M KOH solutions being catholytes, respectively. All solutions were freshly prepared before use and were purged with argon for 30 min before measurements to remove oxygen, and purging was maintained throughout the experiments.
First, all electrodes were immersed in a catholyte solution in a single-compartment electrochemical cell with an electrolyte volume of 25 mL. To stabilise the catalysts and calculate the absolute electrochemical surface area (aECSA), 10 cycles were recorded using cyclic voltammetry (CV) in the potential range of −1.0–0.2 V vs. SCE. The hydrogen underpotential deposition region (HUPD) was used for the aECSA calculation using eqn (1):
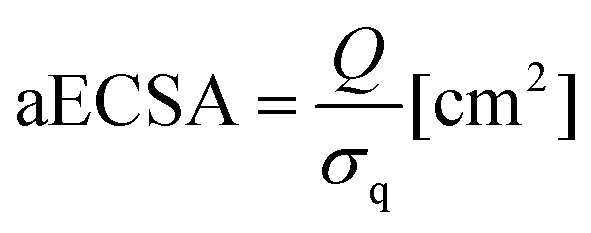 | (1) |
The ECSA was determined using eqn (2):
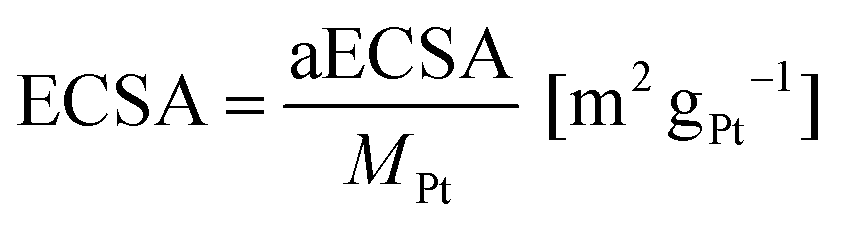 | (2) |
Then, for CV and chronoamperometric (CA) measurements in glycerol-containing solutions, the working and reference electrodes were inserted into the anolyte chamber and the counter electrode into the catholyte chamber of the divided electrochemical cell with a total cell volume of 30 mL (Scheme S1†). The chambers were separated by a Nafion (N-117) membrane. Glycerol electrolysis was carried out in potentiostatic mode at 0.67, 0.77 and 0.87 V vs. the reversible hydrogen electrode (RHE) for 2 hours with constant stirring at 500 rpm.
After that, to assess the performance of the catalysts after the CV and CA, all electrodes were thoroughly rinsed and immersed again in the single-compartment electrochemical cell with a volume of 25 mL and studied with CV first in the corresponding catholyte and finally in the corresponding anolyte by registering 5 cycles for each electrolyte.
To further analyse the GEOR products, ∼150 μL of the anolyte was sampled before and after the electrolysis. For 1 M KOH + 0.1 M glycerol anolyte samples, 100 μL aliquots were further neutralised by 100 μL of 0.5 M H2SO4 solution, and for the 2 M KOH + 1 M glycerol anolyte samples, 100 μL aliquots were neutralised by 100 μL of 1 M H2SO4 and additionally diluted with 800 μL of ultrapure water. Also, to monitor the crossover of glycerol and GEOR products through the membrane, ∼150 μL of the catholyte was sampled at the end of the electrolysis and neutralised using the same procedure. The migrated glycerol concentration was less than 1% of the initial concentration, and the concentrations of the GEOR products were at trace levels.
All the electrochemical data are presented with IR correction (at 85% compensation) due to some uncompensated resistance that was in the range of 3.4–5.6 Ω for 1 M KOH, 2.9–5.1 Ω for 2 M KOH, 1.9–5.4 Ω for 1 M KOH + 0.1 M glycerol and 1.7–4.6 Ω for 2 M KOH + 1 M glycerol electrolytes. The CV curves were recorded at a scan rate of 50 mV s−1, and the current was converted to mass activity (Jmass) and specific activity (JECSA) according to eqn (3) and (4):
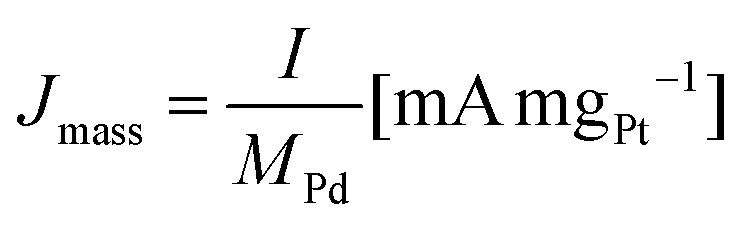 | (3) |
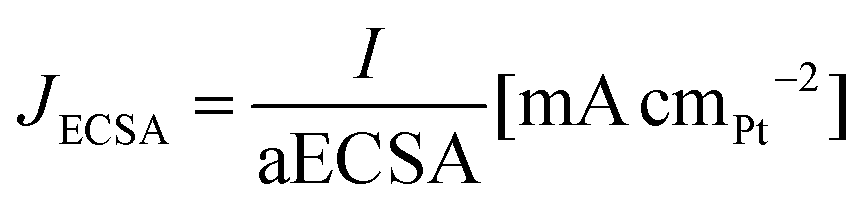 | (4) |
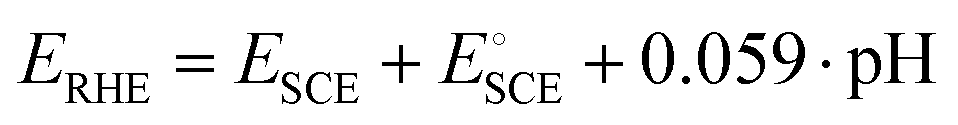 | (5) |
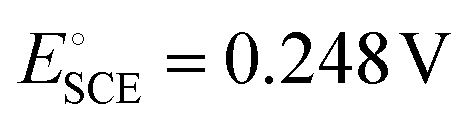 vs. the standard hydrogen electrode (SHE) at 20 °C.
vs. the standard hydrogen electrode (SHE) at 20 °C.
Details regarding the total faradaic efficiency (FE) and carbon balance (CB) calculations are presented in the ESI (Tables S1, 2 and eqn (S1)–(S4)).†
2.6. HPLC measurements
The GEOR product HPLC analysis was performed with an Agilent 1260 Infinity II isocratic pump, multisampler, and multicolumn thermostat equipped with a 1290 Infinity II refractive index detector. The analytical columns connected in series included a Bio-Rad guard column with a standard cartridge holder and a Micro-Guard cation H+ cartridge (4.6 × 30 mm), a Bio-Rad Aminex HPX-87H column (7.8 × 300 mm), and a Shodex Sugar SH1011 column (8 × 300 mm). The columns were maintained at 30 °C, and the eluents were 1 and 8 mM solutions of H2SO4. The injection volume was 20 μL, and the sample analysis time was 85 minutes at a flow rate of 0.25 mL min−1.The quantitative analysis of the possible GEOR products was based on the retention times and peak areas found from the calibration solution analysis. Calibration curves were plotted with peak area as a function of standard solution concentration in the range of 0.1–10 mM for the possible GEOR products and 0.1–100 mM for glycerol. In order to achieve the best peak separation, glycerol, glycerate, lactate and glycolate standard solutions were calibrated in 1 mM H2SO4 and the remaining ones were calibrated in 8 mM H2SO4.
3. Results and discussion
3.1. Structural characterisation of Pt catalysts
PtCUBE and PtDEND NPs were prepared via wet chemistry synthesis (Scheme 1). The PtCUBE and PtDEND NPs have well-defined shapes and a mean size of 10.5 ± 1.6 and 34.9 ± 3.0 nm, respectively, as can be seen from low-magnification TEM images (Fig. 1a and b). The PtCUBE particles have a cubic morphology, and the PtDEND particles have a dendritic structure with some porosity. The corresponding fast Fourier transform (FFT) patterns (Fig. 1c and d) indicate that the synthesised PtCUBE and PtDEND NPs are single crystalline. The d-spacings measured from the lattice fringes were 0.193 and 0.226 nm for PtCUBE and PtDEND, respectively, which is close to d200 ≈ 0.196 and d111 ≈ 0.226 nm of FCC Pt (PDF #00-072-0455), supported also by PXRD analysis (Fig. S1†). Both catalysts had diffraction peaks featuring the (111), (200), (220), (311) and (222) planes; nevertheless, neither PtCUBE nor PtDEND diffractograms showed intense (200) and (111) peaks with (111) being the most pronounced. This feature might be due to the preferred orientation.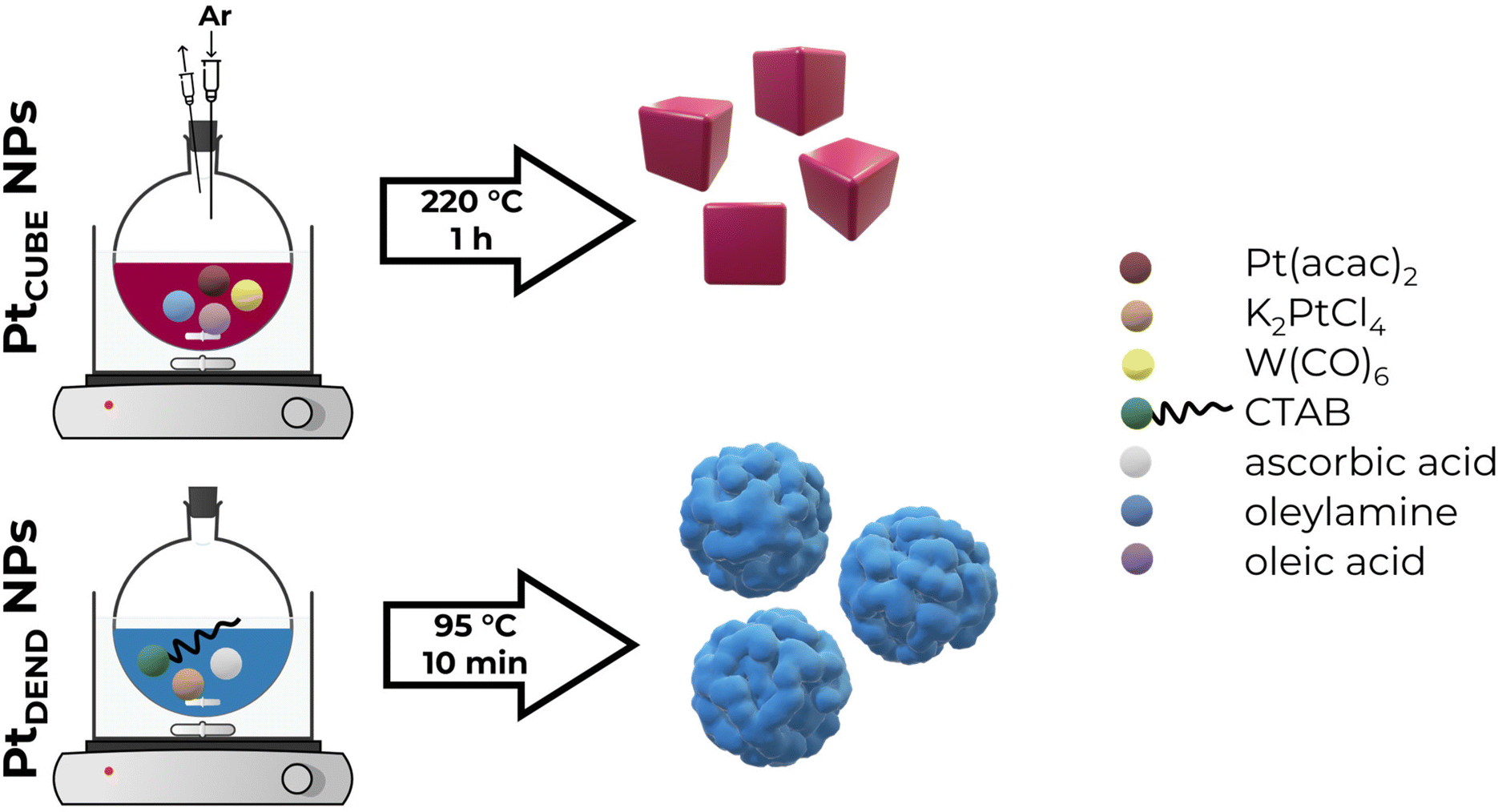 | ||
| Scheme 1 Schematic representation of the Pt nanoparticle synthesis. | ||
 | ||
| Fig. 1 TEM images of (a and c) PtCUBE and (b and d) PtDEND. Insets show particle size distributions and fast Fourier transform (FFT) patterns of the corresponding NPs. | ||
3.2. Electrocatalytic glycerol oxidation performance
Electrocatalytic performance and selectivity of the as-prepared PtCUBE and PtDEND NPs in the GEOR were examined by cyclic voltammetry (CV) and chronoamperometry (CA). Working electrodes were produced by drop-casting certain amounts of catalyst inks on carbon fibre paper. Typical cyclic voltammograms (CVs) of PtCUBE and PtDEND NPs in supporting electrolytes of 1 M KOH and 2 M KOH are shown in Fig. 2a. Overall, the CV curves have typical profiles characteristic of Pt with well-defined peaks in the hydrogen adsorption/desorption (∼0.15–0.45 V), platinum oxide film layer formation (∼0.8–1.4 V) and reduction (∼1.1–0.45 V) regions. The CV curves show that the catalysts retain almost the same mass activity in diluted and concentrated KOH solutions, with PtDEND having the highest values. Moreover, it remained almost unchanged even after the potentiostatic measurements (Fig. S2a†). However, in 2 M KOH electrolyte, a significant increase of the peak at ∼0.68–0.7 V was observed, which corresponds to hydroxide anion chemisorption on Pt. Clearly, after prolonged exposure to excess KOH, a certain amount of hydroxide anions was still present on the catalyst surface.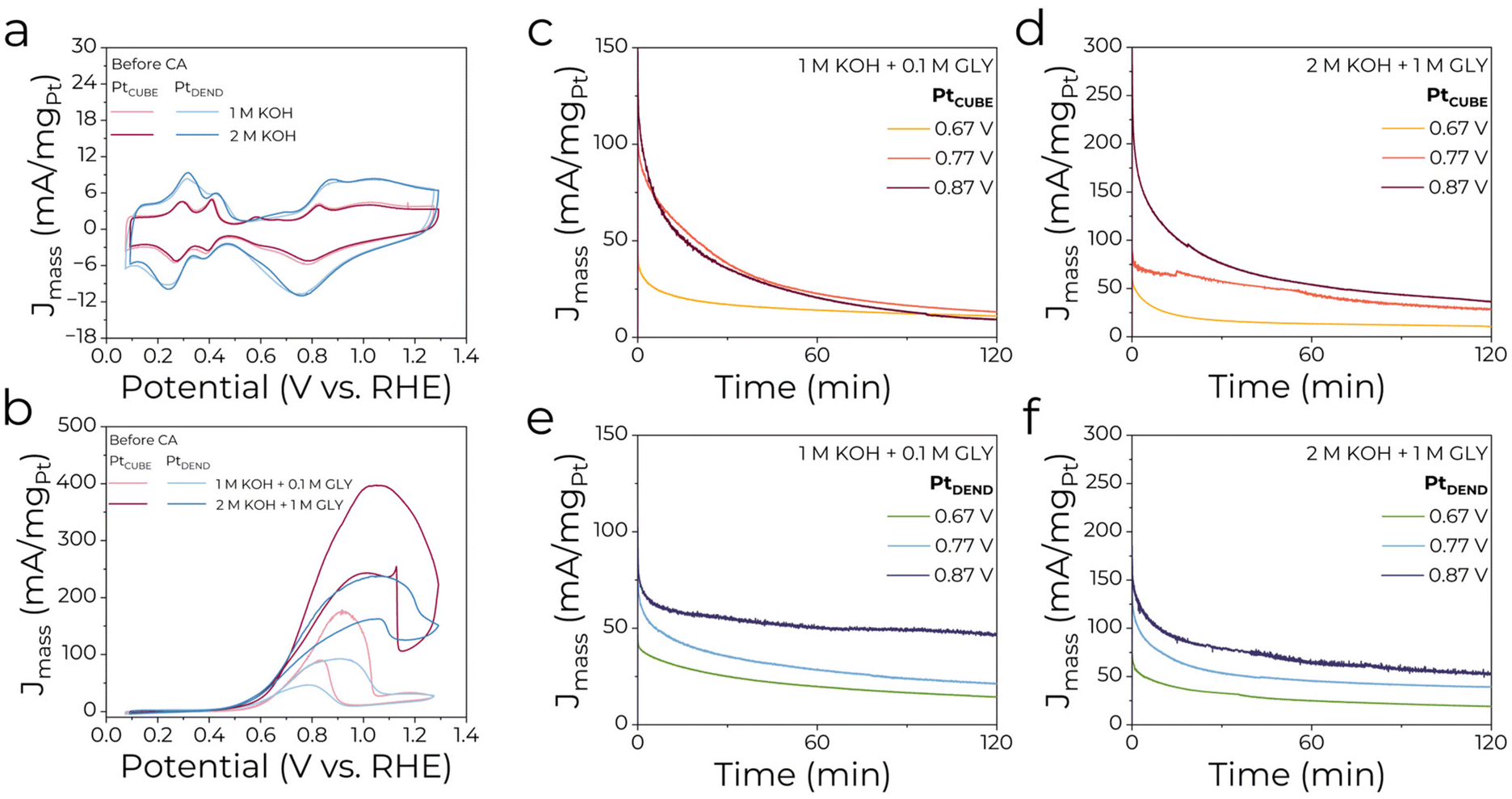 | ||
| Fig. 2 CV profiles of Pt nanocatalysts in (a) 1 M KOH and 2 M KOH and (b) 1 M KOH + 0.1 M GLY and 2 M KOH + 1 M GLY electrolytes. Potentiostatic electrolysis at 0.67, 0.77 and 0.87 V for (c and d) PtCUBE and (e and f) PtDEND in 1 M KOH + 0.1 M GLY and 2 M KOH + 1 M GLY electrolytes. The currents are normalised by the absolute Pt mass loading. | ||
Fig. 2b shows CV profiles for PtNPs/CFP electrodes in 1 M KOH + 0.1 M GLY and 2 M KOH + 1 M GLY electrolytes. Generally, in the forward scan, after reaching a maximum, the first GEOR peak indicates a sudden drop in mass activity due to deactivation from Pt oxidation. In the reverse scan, the second anodic peak is caused by Pt surface reactivation, resulting in glycerol oxidation. Mass activities for the concentrated glycerol solution doubled compared to the diluted one for both catalysts, showing that the higher glycerol concentration results in higher overall activity for the GEOR. The highest mass activities of 178 and 397 mA mgPt−1 were observed for PtCUBE in 1 M KOH + 0.1 M GLY and 2 M KOH + 1 M GLY electrolytes, while PtDEND achieved 93 and 237 mA mgPt−1, which can easily compete with the results from previously reported studies on Pt-based catalysts for the GEOR (Table S3†). Similar to pure KOH solutions, the catalytic response after the potentiometric measurements was assessed in the presence of glycerol (Fig. S2b†). Except for PtDEND in 2 M KOH + 1 M GLY electrolyte, the mass activity values dropped only slightly even after 2 hours of electrolysis in both electrolytes, showing the possibility of catalyst recycling for more than one electrolysis run.
In addition, the specific activity of the catalysts was assessed. The electrochemical surface area (ECSA) was determined by integrating the hydrogen underpotential deposition region and found to be 6.46 ± 0.27 and 12.4 ± 4.0 m2 gPt−1 for PtCUBE and PtDEND, respectively. After normalising the currents by aECSA (Fig. S3a†), it was revealed that PtCUBE was still superior to PtDEND in both electrolytes. The estimated specific activity at the forward GEOR peak maximum for PtCUBE was around 2.9 and 6.2 mA cmPt−2, and for PtDEND 0.75 and 1.9 mA cmPt−2 in 1 M KOH + 0.1 M GLY and 2 M KOH + 1 M GLY electrolytes, respectively. Furthermore, it almost did not change even after the electrolysis (Fig. S3b†).
In order to study the catalytic performance and selectivity of the GEOR, potentiostatic measurements were performed for 2 hours at 0.67, 0.77 and 0.87 V. Anolyte aliquots were taken before and after the electrolyses and neutralised to stabilise the GEOR products for further HPLC analysis. The chronoamperometric curves shown in Fig. 2c–f and Fig. S3c–f† show that the current first decreased and then gradually reached a stable value of ∼10–50 mA mgPt−1 (Fig. 2c–f). The decline can be caused by the decrease in glycerol concentration and/or the adsorption of inactive intermediates on the active sites of the catalysts.
3.3. HPLC results evaluation
The overall GEOR selectivity and fraction of valuable products were further evaluated. The GEOR reaction mechanism is thought to go through glycerol oxidation to three-carbon (C3) intermediates, dihydroxyacetone (DHA) and glyceraldehyde (GALD), which are further oxidised to other C3, C2 and C1 products via C–C and C–O bond cleavage.4 Increasing the applied potential and introducing high fractions of non-noble transition metals generally result in a higher degree of carbon chain cleavage and reduced selectivity for C3–C2 products. In order to identify possible GEOR products, calibration curves in the concentration range of 0.1–10 mM were made for standards of DHA, GALD, glycerate (GLE), lactate (LACT), hydroxypyruvate (HYDP), tartronate (TART), mesoxalate (MESOXA), glycolate (GLO), oxalate (OXA), glyoxylate (GLYOXY), acetate (ACET) and formate (FORM) and in the range of 0.1–100 mM for glycerol (Fig. S4†). Additionally, samples of pure 1 M and 2 M KOH were tested to identify peaks that were not associated with any of the species in the electrolysis samples (Fig. S5a†). Fig. S5b† shows the chromatograms of standard solutions of possible GEOR products with 1 mM H2SO4 and 8 mM H2SO4 as a mobile phase. The two mobile phases were used to achieve the best peak separation for glycerol, glycerate, lactate and glycolate. HPLC chromatograms of electrolysis samples (Fig. S6†) showed the presence of glycerate, lactate, tartronate, glycolate, oxalate, and formate with glyoxylate and dihydroxyacetone having the lowest concentrations and no traces of glyceraldehyde, hydroxypyruvate, mesoxalate and acetate.3.3.1.1. Effect of the KOH/glycerol concentration. The total yield of GEOR products after the electrolysis at PtCUBE and PtDEND is plotted in Fig. 3a for 1 M KOH + 0.1 M GLY and Fig. 3b for 2 M KOH + 1 M GLY electrolytes. The trends clearly show greater product generation up to ∼45 mM in 2 M KOH + 1 M GLY electrolyte compared to ∼14 mM in 1 M KOH + 0.1 M GLY with glycerate and lactate being generally dominant for both catalysts. By comparing Fig. 3c and d, one can see that the electrolyte composition also affects the ratio of the products generated. In the diluted electrolyte, 1 M KOH + 0.1 M GLY, the overall fraction of C2–C1 products is more prevalent than in the 2 M KOH + 1 M GLY electrolyte, in which C3 products dominate. This might be explained by a lower saturation of the active sites in diluted solutions. Since the number of glycerol molecules is relatively small and the active sites are mostly occupied by hydroxide anions, they are more available for glycerol adsorption followed by massive secondary adsorption, hence, further oxidation of C3–C2 species. Moreover, PtDEND facilitates the formation of C2–C1 products more strongly than PtCUBE. This phenomenon might be explained by the porous structure of PtDEND, which makes capturing freshly generated GEOR products and their subsequent oxidation much easier compared to that achieved with the non-porous morphology of PtCUBE. The C3–C2 selectivity as a function of the applied potential plateaus in 2 M KOH + 1 M GLY electrolyte (Fig. 4b) indicates the beginning of the saturation point for generation of GEOR products and glycerol conversion. Another consequence of the saturation of active sites is the reduced glycerol conversion. Even though PtCUBE and PtDEND exhibited comparable glycerol conversion up to ∼30–40 mM (Fig. 3e), it was much lower in concentrated solutions relative to the initial glycerol concentration. That is, for PtCUBE, it was found to be 14.8, 26.2, and 31.2% in 1 M KOH + 0.1 M GLY and 3.1, 3.3 and 3.4% in 2 M KOH + 1 M GLY electrolytes, whereas PtDEND reached 15.7, 21.4 and 31.5% and 1.2, 2.5 and 3.9%, respectively. Additionally, when the glycerol conversion was normalised by the aECSA to find the intrinsic performance (Fig. S7†), PtCUBE showed a nearly doubled conversion. At the same time, the faradaic efficiency grew with the increase of the glycerol concentration (Table S2†).
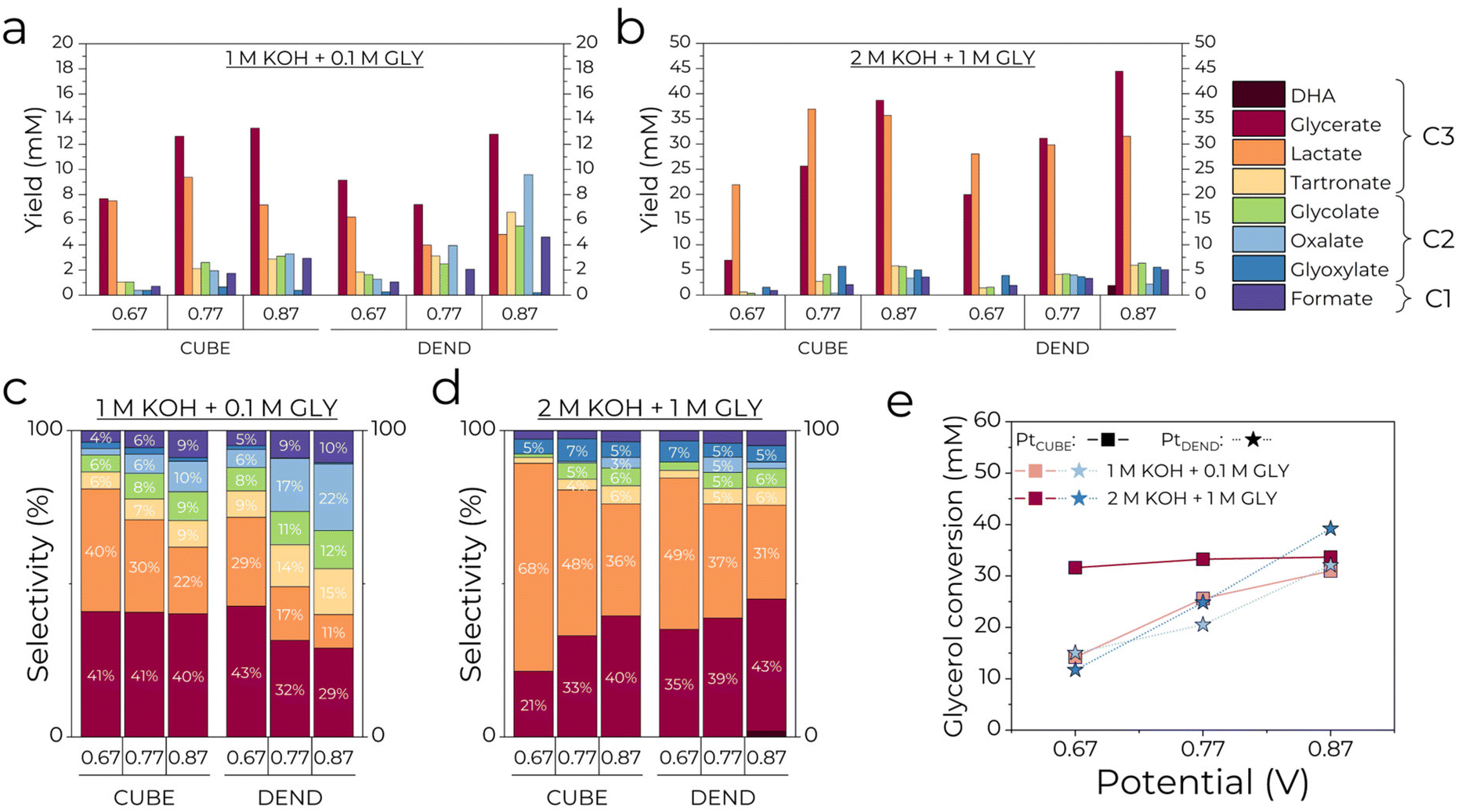 | ||
| Fig. 3 The yields of GEOR products after 2 hours of electrolysis at 0.67, 0.77 and 0.87 V and selectivity of GEOR products performed in (a and c) 1 M KOH + 0.1 M GLY and (b and d) 2 M KOH + 1 M GLY electrolytes. (e) Glycerol conversion in 1 M KOH + 0.1 M GLY and 2 M KOH + 1 M GLY electrolytes. Note that in (c and d), the selectivity of products less than or equal to 3% is not marked. | ||
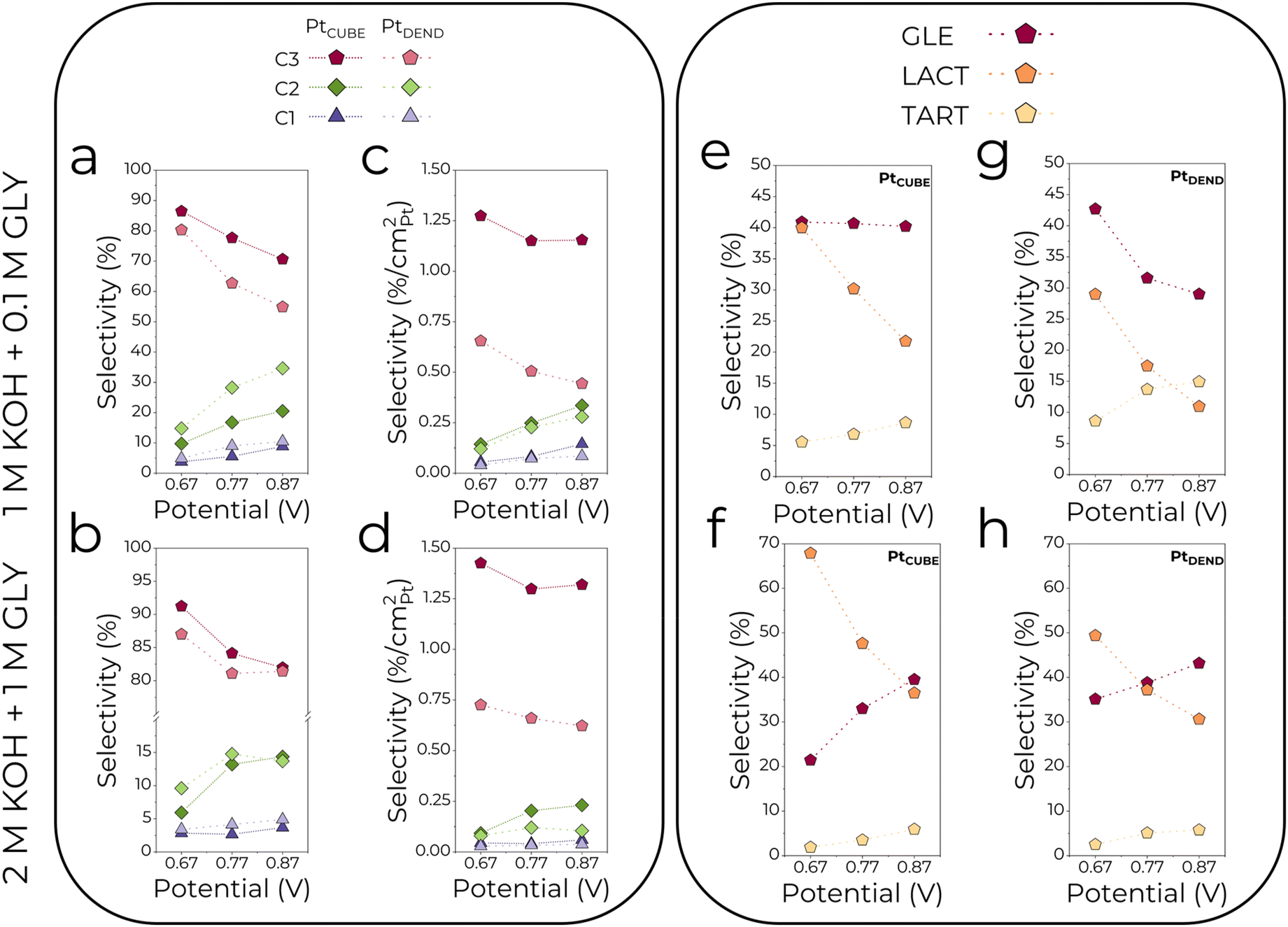 | ||
| Fig. 4 Selectivity of C3–C1 and individual C3 products as a function of applied potential in (a, c, e and g) 1 M KOH + 0.1 M GLY and (b, d, f and h) 2 M KOH + 1 M GLY electrolytes. Note that (c and d) have different y-axis scales, where the C3–C1 product selectivity is normalised by the aECSA. | ||
Behind the suggestions on the electrolyte composition effect on the performance and selectivity of the GEOR are studies performed by Dai et al. and White et al.28,33 Dai et al. investigated and optimised the performance of AuPt nanoparticles of different compositions for the GEOR by modifying the reaction conditions, such as applied potential, and the KOH and glycerol concentrations.28 The highest lactate selectivity was found at 0.45 V vs. RHE, and the AuPt (15% Ptsurf) catalyst exhibited the best performance, so the experiments on variable reaction parameters were carried out with the catalyst and applied potential fixed. A series of KOH concentrations (0.1, 0.5, 1, and 2 M) with 0.5 M GLY were compared; the change from 0.1 to 0.5 M caused a significantly improved lactate selectivity from 39% to 68%, respectively, which supports the base-catalysed nature of lactate formation. However, a further increase in pH did not show a significant lactate selectivity change, meaning that in the high pH range of more than 13, the influence of hydroxide anions on the GEOR is limited. Hence, one would think that the glycerol concentration might mainly affect the reaction. Nevertheless, in the same study, it was demonstrated that in a series of glycerol concentrations (0.05, 0.1, 0.5, and 1 M) with 1 M KOH, there was no significant change in the selectivity for lactate and other products, with the glycerol conversion declining with the glycerol concentration increase, which supports our hypothesis on the blockage of catalyst active sites. In a study by White et al., it was demonstrated that the ratio of NaOH to glycerol in an electrolyte greatly affects the performance of the GEOR and mass transport on a PdNi/NiRDE (Pd0.9Ni0.1) catalyst at 80 °C.33 A series of NaOH:glycerol molar ratios were investigated: 20, 10, 4, 2 and 1, where the studied NaOH concentrations were 1 and 2 M and glycerol concentrations were 0.1, 0.5, 1, and 2 M. In brief, the NaOH:glycerol ratio of 2 in the 1 M NaOH + 0.5 M GLY and 2 M NaOH + 1 M GLY electrolytes resulted in the most favourable conditions for the GEOR, with 2 M NaOH + 1 M GLY having the highest peak current density. At the same time, further doubling of the glycerol concentration to 1 M and 2 M in 1 M and 2 M NaOH solutions did not increase the current density, indicating that the concentration of hydroxide anions limits the GEOR. Even though the GEOR selectivity was not assessed, the study demonstrates that the stoichiometry of electrolyte composition strongly affects the mass transport, which is directly connected to the mechanism and selectivity of the GEOR.
3.3.1.2. Effects of the applied potential. Besides the electrolyte composition, the effect of applied potential was studied. As can be seen from Fig. 3c and d and 4a and b, low potentials of 0.67 V resulted in the highest selectivity for C3 products, which gradually drops when the potential is increased to 0.77 and 0.87 V. Simultaneously, the C2 selectivity rises substantially together with the slight growth of C1 (formate) selectivity. Interestingly, PtDEND exhibits the lowest selectivity towards C3 products in the 1 M KOH + 0.1 M GLY electrolyte at 0.87 V, making oxalate the second dominant product after glycerate. The highest intrinsic activity towards formation of C3 products was observed for PtCUBE (Fig. 4c and d) in both 1 M KOH + 0.1 M GLY and 2 M KOH + 1 M GLY electrolytes.
3.4. Insights into the GEOR mechanism
Numerous studies have been devoted to understanding the general GEOR pathway on noble metals in an alkaline medium.4,9,11,12 In brief, the glycerol oxidation mechanism involves two competing pathways: oxidative dehydrogenation (ODH) and dehydration (Scheme 2). First, a glycerol molecule is oxidised to glyceraldehyde or dihydroxyacetone, which exists in a dynamic chemical interconversion catalysed by OH− groups. The equilibrium is crucial to the selective GEOR since it heavily depends on the reaction conditions such as the type of catalyst, its morphology, temperature, pH, glycerol concentration and applied potentials. Then, glyceraldehyde or dihydroxyacetone may undergo either dehydration to lactate via an intramolecular Cannizzaro rearrangement of pyruvaldehyde under the influence of OH− groups or further ODH to glycerate. Other C3–C1 products like tartronate, mesoxalate, glycolate, oxalate, glyoxylate, acetate, formate, formaldehyde, and CO2 might be generated.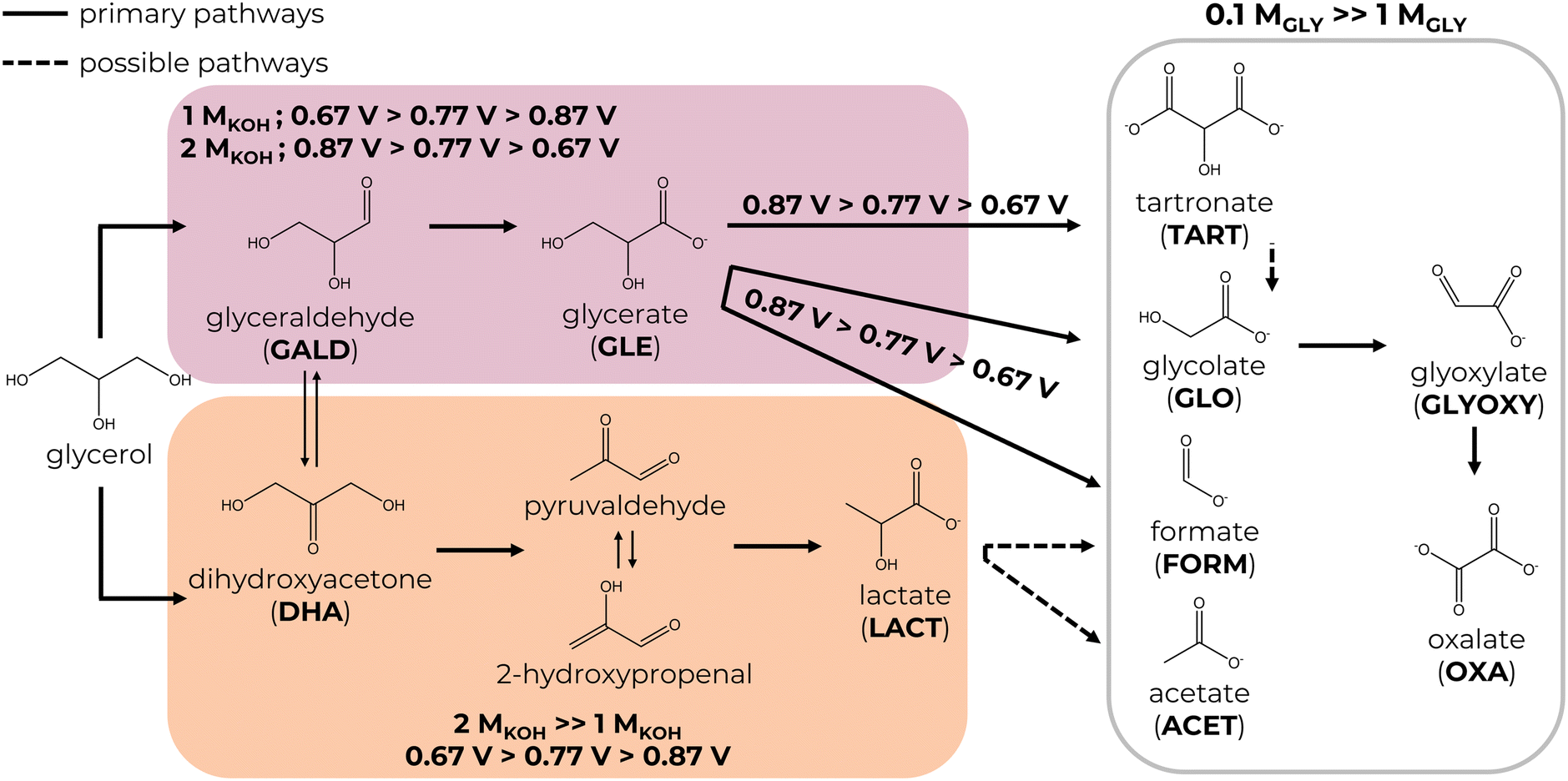 | ||
| Scheme 2 Proposed GEOR mechanism on the PtNPs/CFP electrodes in an alkaline medium. | ||
Examples of some studies on the GEOR mechanism and the effect of the catalyst and reaction conditions chosen can be seen in Table S3.† Zhou et al.18 have investigated the performance of Pd and Pt@Pd nanocubes towards the GEOR and the influence of the applied potential in an alkaline medium at room temperature. The study has concluded that the synergistic effect of Pd and Pt favours the C–C cleavage: not only did Pt@Pt nanocubes exhibit the highest specific activities compared to the Pd nanocubes and commercial Pt/C and Pd/C but they also yielded more glycolate up to 40% at 0.2 V vs. SCE after 2 hours of electrolysis. When the potential was lowered to −0.1 and −0.4 V vs. SCE, the glycolate selectivity dropped to 30%, resulting in glyceraldehyde becoming the dominant product (40%), meaning that the ODH pathway dominates for that catalyst, and it can be easily tuned to form larger fractions of C3 products.
Another study on Pt catalysts has been carried out by Anil et al.20 Here, Pt catalysts with three types of pore mesostructures were developed and studied, namely catalysts with hierarchical pores (HPC), cubic pores (CPC), and linear pores (LPC). Due to the best combination of large and small pore sizes, HPC was found to have the best intrinsic activity. For all three types of catalyst, glycerate was found to be the dominant product with selectivities up to 60% after 1 hour of electrolysis at 0.69 V vs. RHE and 60 °C, which shows that the ODH is promoted, and the results align with the findings of our study to a certain degree. For PtDEND, exhibiting nanoporous morphology, the electrolysis in 1 M KOH + 0.1 M GLY electrolyte at 0.67 V vs. RHE gives the highest glycerate selectivity compared to that for PtCUBE. However, the electrolyte composition and temperature effects on the diffusion cannot be completely neglected.
Another study was reported by Oh et al.,27 where the authors investigated the effect of the pH on the GEOR on Pt/C and PtCu/C nanoparticles. In general, PtCu/C exhibited the highest reaction rate and turnover frequency (TOF) values with pH increase. Nevertheless, the findings for pure Pt/C nanoparticles were quite close to those in the present study: when the potential of 1.0 V vs. RHE is applied at 60 °C, the increase of the pH from 13 to 14 dramatically increases lactate selectivity from 17 to 37% at pH 14, proving that the dehydration pathway is favoured at high alkalinity. However, on PtCu/C, the primary alcohol oxidation via the ODH is more pronounced than on Pt/C, causing increased fractions of tartronate and oxalate.
A slightly more detailed study on the GEOR dependence on the composition and electrolysis potential has been done by Dai et al.,28 as was mentioned in section 3.3.1.1. Their study revealed that the lower applied potential and higher OH− concentration improve the lactate selectivity on PtAu/C nanoparticles, and the maximum is reached for Au-enriched catalysts (Pt15%surfAu/C) with pure Pt/C nanoparticles showing the worst performance. The GEOR performed in 1 M KOH + 0.5 M GLY at 0.45 V vs. RHE for 2 hours at room temperature resulted in lactate selectivity of 72% with a glycerol conversion rate of approximately 26 mMGLY h−1, which is close to 68% for PtCUBE at 0.67 V in 2 M KOH + 1 M GLY with a glycerol conversion rate of 16 mMGLY h−1.
Finally, an interesting study was published by Zhou et al.29 A series of binary and ternary alloys of Pt-based nanoparticles with Ni, Ru and Rh supported on graphene nanosheets (GNS) were investigated. The best activity towards the GEOR at room temperature was exhibited by Rh-containing catalysts PtRh/GNS and PtRhNi/GNS. Interestingly, when the GEOR was carried out at −0.4, −0.1 and 0.2 V vs. SCE, the maximum of glycolate of ∼65% was found for Pt/GNS at 0.2 V. Another GEOR product, glycerate, achieved the maximum selectivity of ∼48% on PtNi/GNS at −0.1 V. Additionally, it was found that alloying with Ru facilitated formation of C3 products. In contrast, Rh-containing catalysts facilitated the formation of C2 products. In general, the studied catalysts favoured the ODH pathway of the GEOR. Thus, it has been extensively shown how essential are the choice of catalyst and its properties, as well as the reaction conditions, for optimal and efficient glycerol electrooxidation.
Herein, based on the thorough interpretation of HPLC data, we suggest the following descriptions for the features of the GEOR mechanism and variations of selectivity towards C3–C1 products on PtCUBE and PtDEND in 1 M KOH + 0.1 M GLY and 2 M KOH + 1 M GLY electrolytes at applied potentials of 0.67, 0.77 and 0.87 V at 20 °C:
i. Glycerate ODH to tartronate and further C–C cleavage to glycolate, glyoxylate, oxalate and formate are accelerated in a diluted electrolyte and generally are the most prominent at higher potentials due to increased adsorption of C3–C2 products on glycerol-unsaturated active sites of the catalysts. This means the GEOR selectivity towards C3 products is superior in the more concentrated electrolyte with a maximum value of 91%. In addition, the formation of C2–C1 products is favoured on PtDEND more than on PtCUBE, possibly due to the porous structure of PtDEND, which eases the capturing of freshly generated GEOR products and their subsequent oxidation.
ii. Glycerate formation through glyceraldehyde ODH is slightly more favourable in a concentrated electrolyte. In a diluted electrolyte, glycerate selectivity drops with potential increase, while in the more concentrated electrolyte, it follows an opposite trend, reaching maximum selectivity of 43%.
iii. Lactate selectivity is generally the greatest in the more concentrated electrolyte, with the highest selectivity of 68% achieved at the lowest potential.
iv. Comparing overall intrinsic performance, PtCUBE allow the highest selectivity towards C3 products to be reached in both electrolytes. As for the individual C3 products, their selectivity was found to be up to 3 times greater than that of PtDEND.
4. Conclusion
In this study, Pt NPs of cubic and dendritic morphology were synthesised and investigated as promising catalysts for glycerol electrooxidation in highly alkaline media. The novelty of the present study lies in the successful application of nanostructured cubic and dendritic electrocatalysts for the GEOR coupled with a thorough quantitative identification of the GEOR products by HPLC, which was used as a basis for understanding the reaction mechanism in electrolytes of different compositions and applied potentials. The comparison of the electrocatalytic performance has shown that both PtCUBE and PtDEND were highly selective for the formation of C3 products, reaching up to 86% in 1 M KOH + 0.1 M GLY and 91% in 2 M KOH + 1 M GLY electrolytes at 0.67 V vs. RHE. Meanwhile, the maximum selectivity of one of the dominant products, lactate, reached 68%. However, in 1 M KOH + 0.1 M GLY electrolyte, C–C bond cleavage was more favoured, especially for PtDEND, due to its porous structure that entraps C3 products and facilitates their further oxidation to C2–C1 species.Furthermore, PtCUBE exhibited mass activity and specific activity superior to that of PtDEND and the highest intrinsic glycerol conversion and selectivity towards C3 products. In addition, the selectivity of individual C3 products was found to be up to 3 times greater than that of PtDEND, making PtCUBE the overall best-performing catalyst.
These results support that these electrocatalysts, especially of a cubic morphology, have the potential for practical applications in glycerol electrocatalysis and the successful conversion of glycerol into more valuable substances.
Data availability
Data will be made available upon reasonable request.Author contributions
Irina Terekhina: conceptualisation, methodology, validation, formal analysis, investigation, visualisation, and writing—original draft. Mats Johnsson: supervision and writing—review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Foundation for Strategic Research (SSF, Grant No. EM16-0010). The authors thank Prof. Ann Cornell's research group at the Department of Chemical Engineering, KTH Royal Institute of Technology, Stockholm, Sweden, for providing access to the HPLC equipment, particularly Dr Jai White, for assistance with HPLC measurements.References
- G. Antolín, F. V. Tinaut, Y. Briceño, V. Castaño, C. Pérez and A. I. Ramírez, Bioresour. Technol., 2002, 83, 111–114, DOI:10.1016/s0960-8524(01)00200-0
.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30, 10.1039/b710561d
- M. A. Dasari, P. P. Kiatsimkul, W. R. Sutterlin and G. J. Suppes, Appl. Catal., A, 2005, 281, 225–231, DOI:10.1016/j.apcata.2004.11.033
- C. Coutanceau, S. Baranton and R. S. B. Kouamé, Front. Chem., 2019, 7, 100, DOI:10.3389/fchem.2019.00100
- R. Ciriminna, C. D. Pina, M. Rossi and M. Pagliaro, Eur. J. Lipid Sci. Technol., 2014, 116, 1432–1439, DOI:10.1002/ejlt.201400229
- C. A. G. Quispe, C. J. R. Coronado and J. A. Carvalho Jr., Renewable Sustainable Energy Rev., 2013, 27, 475–493, DOI:10.1016/j.rser.2013.06.017
- M. Simões, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124, DOI:10.1002/cssc.201200335
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333, DOI:10.1021/acscatal.8b01317
- Z. Fan, W. Zhang, L. Li, Y. Wang, Y. Zou, S. Wang and Z. Chen, Green Chem., 2022, 24, 7818–7868, 10.1039/d2gc02956a
- T. Ali, H. Wang, W. Iqbal, T. Bashir, R. Shah and Y. Hu, Adv. Sci., 2023, 10, 2205077, DOI:10.1002/advs.202205077
- J. Wu, X. Yang and M. Gong, Chin. J. Catal., 2022, 43, 2966–2986, DOI:10.1016/s1872-2067(22)64121-4
- M. S. E. Houache, K. Hughes and E. A. Baranova, Sustainable Energy Fuels, 2019, 3, 1892–1915, 10.1039/c9se00108e
- A. C. Garcia, M. J. Kolb, C. van Nierop, Y. Sanchez, J. Vos, Y. Y. Birdja, Y. Kwon, G. Tremiliosi-Filho and M. T. M. Koper, ACS Catal., 2016, 6, 4491–4500, DOI:10.1021/acscatal.6b00709
- J. Zhou, J. Hu, X. Zhang, J. Li, K. Jiang, Y. Liu, G. Zhao, X. Wang and H. Chu, J. Catal., 2020, 381, 434–442, DOI:10.1016/j.jcat.2019.11.019
- I. Terekhina, J. White, A. Cornell and M. Johnsson, ACS Appl. Nano Mater., 2023, 6, 11211–11220, DOI:10.1021/acsanm.3c01236
- J. Ouyang, X. Liu, B. H. Wang, J. B. Pan, S. Shen, L. Chen, C. T. Au and S. F. Yin, ACS Appl. Mater. Interfaces, 2022, 14, 23536–23545, DOI:10.1021/acsami.2c04608
- T. Song, F. Gao, Y. Zhang, C. Chen, C. Wang, S. Li, H. Shang and Y. Du, Nanoscale, 2020, 12, 9842–9848, 10.1039/d0nr00163e
- Y. Zhou and Y. Shen, Electrochem. Commun., 2018, 90, 106–110, DOI:10.1016/j.elecom.2018.04.012
- H. Du, K. Wang, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2019, 258, 117951, DOI:10.1016/j.apcatb.2019.117951
- A. Anil, J. White, E. C. dos Santos, I. Terekhina, M. Johnsson, L. G. M. Pettersson, A. Cornell and G. Salazar-Alvarez, J. Mater. Chem. A, 2023, 11, 16570–16577, 10.1039/d3ta01738a
- V. K. Landge, S. H. Sonawane, R. V. Chaudhari and G. U. B. Babu, Ind. Eng. Chem. Res., 2021, 60, 185–195, DOI:10.1021/acs.iecr.0c04626
- F. Ren, Z. Zhang, Z. Liang, Q. Shen, Y. Luan, R. Xing, Z. Fei and Y. Du, J. Colloid Interface Sci., 2022, 608, 800–808, DOI:10.1016/j.jcis.2021.10.054
- D. Wang, Y. Zhang, Z. Li, Z. Wu, S. Hata, F. Gao, Y. Shiraishi and Y. Du, J. Colloid Interface Sci., 2023, 636, 602–609, DOI:10.1016/j.jcis.2023.01.026
- M. Douthwaite, N. Powell, A. Taylor, G. Ford, J. M. López, B. Solsona, N. Yang, O. Sanahuja-Parejo, Q. He, D. J. Morgan, T. Garcia and S. H. Taylor, ChemCatChem, 2020, 12, 3097–3107, DOI:10.1002/cctc.202000026
- J. Ftouni, N. Villandier, F. Auneau, M. Besson, L. Djakovitch and C. Pinel, Catal. Today, 2015, 257, 267–273, DOI:10.1016/j.cattod.2014.09.034
- J. Qi, N. Benipal, C. Liang and W. Li, Appl. Catal., B, 2016, 199, 494–503, DOI:10.1016/j.apcatb.2016.06.055
- L. S. Oh, J. Han, E. Lim, W. B. Kim and H. J. Kim, Catalysts, 2023, 13, 892, DOI:10.3390/catal13050892
- C. Dai, L. Sun, H. Liao, B. Khezri, R. D. Webster, A. C. Fisher and Z. J. Xu, J. Catal., 2017, 356, 14–21, DOI:10.1016/j.jcat.2017.10.010
- Y. Zhou, Y. Shen and J. Piao, ChemElectroChem, 2018, 5, 1636–1643, DOI:10.1002/celc.201800309
- H. Lee, S. E. Habas, S. Kweskin, D. Butcher, G. A. Somorjai and P. Yang, Angew. Chem., Int. Ed., 2006, 45, 7824–7828, DOI:10.1002/anie.200603068
- J. Zhang and J. Fang, J. Am. Chem. Soc., 2009, 131, 18543–18547, DOI:10.1021/ja908245r
- T. Biegler, D. A. J. Rand and R. Woods, J. Electroanal. Chem. Interfacial Electrochem., 1971, 29, 269–277, DOI:10.1016/s0022-0728(71)80089-x
- J. White, A. Anil, D. Martín-Yerga, G. Salazar-Alvarez, G. Henriksson and A. Cornell, Electrochim. Acta, 2022, 403, 139714, DOI:10.1016/j.electacta.2021.139714
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01127a |
| This journal is © The Royal Society of Chemistry 2024 |