
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Small molecule WDR5 inhibitors down-regulate lncRNA expression†
Jen-Yao
Chang‡
 ,
Cora
Neugebauer‡
,
Anne
Mues genannt Koers
and
Peter
't Hart
*
,
Cora
Neugebauer‡
,
Anne
Mues genannt Koers
and
Peter
't Hart
*
Chemical Genomics Centre of the Max Planck Society, Max Planck Institute of Molecular Physiology, Otto-Hahn-Strasse 11, 44227 Dortmund, Germany. E-mail: peter.t-hart@mpi-dortmund.mpg.de
First published on 10th January 2024
Abstract
WD repeat domain 5 (WDR5) plays an important role as a scaffold protein in both protein–protein and RNA–protein complexes involved in epigenetic gene regulation. In particular, some of these lncRNAs were reported to regulate the expression of genes in cis as well as themselves through binding WDR5. In this report, we investigate the two known binding sites of WDR5 in relation to lncRNA binding and expression. The WBM binding site mediates both protein–protein and lncRNA–protein interactions while the WIN site, which is on the opposite side of the protein, is only known to mediate protein–protein interactions. To dissect the function of different binding sites on WDR5, we characterized them with selective peptide ligands using fluorescence polarization and used these to demonstrate the selectivity of small molecule inhibitors of these two major binding sites. RNA immunoprecipitation experiments were performed to show that lncRNA–WDR5 complex formation could be interrupted using a WBM site inhibitor. Finally, we demonstrated that WDR5 regulated lncRNAs are down regulated with different sensitivity toward the corresponding inhibitors, demonstrating the potential of targeting lncRNA–protein interactions to reduce oncogenic lncRNA expression.
Introduction
WD repeat domain 5 (WDR5) is a protein consisting of seven repeating units of the WD domain, forming an iconic donut shaped protein structure (Fig. 1).1 It usually acts as a scaffold protein for larger protein complex formation, including various epigenetic modulating complexes such as the MLL-COMPASS complex, NSL complex, NuRD complex, and the MYC-MAX complex.2–6 In addition to being a scaffold in protein–protein complexes, it can also form long noncoding RNA (lncRNA)–protein complexes to modulate histone modifications, leading to changes in gene expression.7,8 For example, HOTTIP is a lncRNA that requires WDR5 to activate the late HOXA gene cluster as well as its own expression to maintain cell self-renewal and pluripotency.7,8 To do so, HOTTIP acts as a guide that recruits WDR5 to chromatin, followed by recruitment of the MLL complex components to initiate histone 3 lysine 4 methylation at the target genes.8,9 Dysregulation of HOTTIP is correlated to cancer progression and could become an interesting therapeutic target.8–11 Although small-interfering RNA (siRNA) can selectively silence their target lncRNAs, they are limited in their ability to distribute and permeate cells.12 An alternative strategy to influence oncogenic lncRNA levels such as HOTTIP is not through direct targeting, but rather by targeting the proteins that are part of their mode of action. This was highlighted in the report by Wang et al., who demonstrated that knock down of WDR5 lead to reduced expression levels of HOTTIP.7 The same group later reported how HOTTIP recognizes the WBM site on WDR5.8 Their results suggested that WDR5 is required for HOTTIP expression and that targeting the HOTTIP–WDR5 interaction with competitive inhibitors could be an interesting therapeutic strategy. Besides HOTTIP, a report by Subhash et al. demonstrated WDR5 binds many more lncRNAs including ones that have reported oncogenic activity further highlighting the potential of inhibiting such interactions.13 Unlike protein–protein interactions (PPI), RNA–protein interactions (RPI) often lack structural information to assist in the design process of inhibitors. In addition, WDR5 has two major binding pockets, the WIN site, and the WBM site, making the situation more complicated for designing effective and selective inhibitors. Several PPI inhibitors have been developed and studied for their ability to target protein–WDR5 interactions, e.g. Karatas et al. reported an optimized sequence that has sub-nanomolar binding affinity to the WIN site, while Grebien et al. demonstrated that OICR-9429, a small molecule inhibitor for the WIN pocket, can antagonize the WDR5-MLL interaction in cellulo.14,15 On the other hand, the WBM site is much less explored but is promising for its role in the MYC dependent pathway. Macdonald et al. demonstrated that their optimized WBM inhibitor 7k could reduce cMYC enrichment in WDR5 pull-down experiments,16 while Ding et al., reported that their WDR5-cMYC interaction inhibitor leads to growth inhibition in several cancer cells.17,18 Recently, we reported that targeting the WBM site with a macrocyclic peptide could reduce lncRNA enrichment by WDR5 pull-down in vitro.19 Here, we aim to bring these details together to provide a clear picture of the possibility of targeting lncRNA–WDR5 interactions as a therapeutic strategy by using well described inhibitors of the two binding sites on WDR5.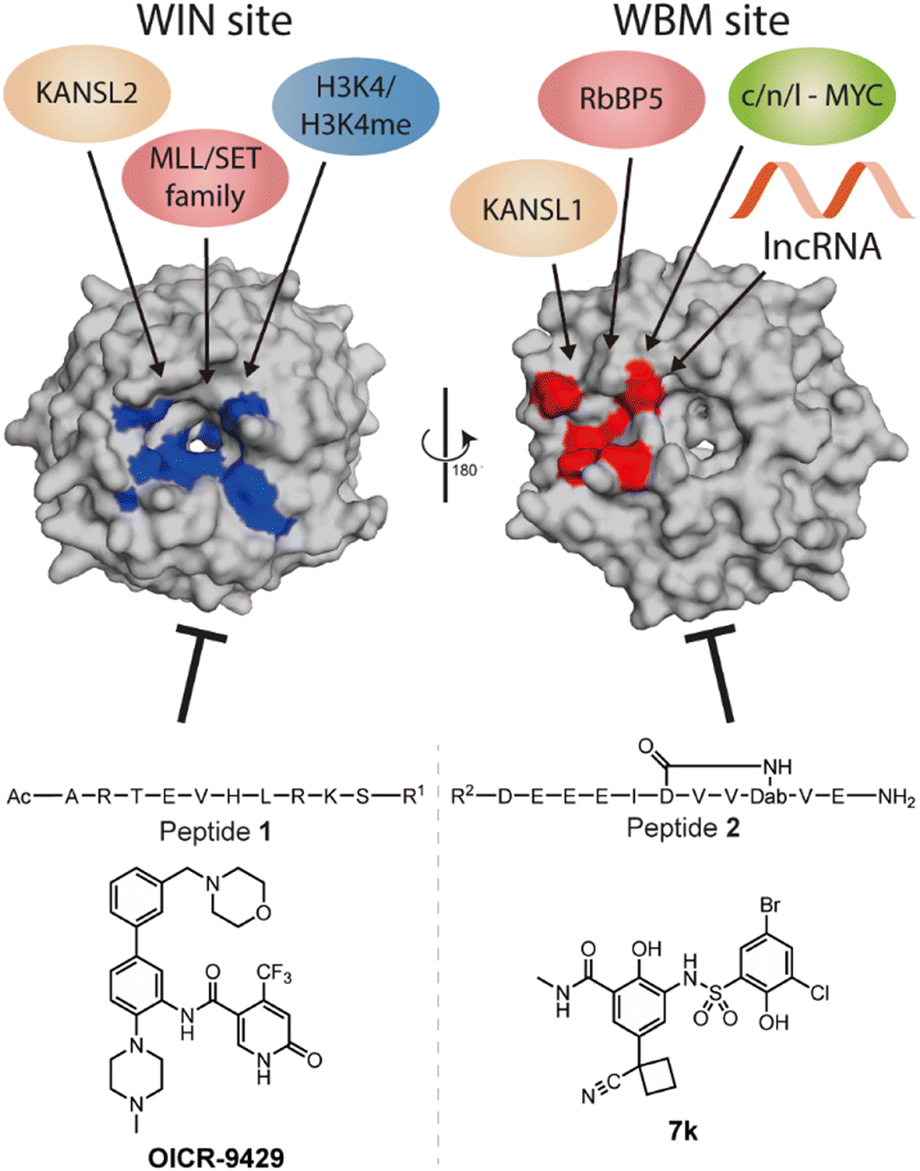 | ||
| Fig. 1 Crystal structure (PDB: 8Q1N) of WDR5 and the reported binding partners for the corresponding binding sites. A65, S91, D107, F133, Y191, Y260, F263 are coloured in blue to indicate the location of WIN binding pocket; N225, Y228, L240, F266, V268, Q289 are coloured in red to indicate the location of WBM binding pocket. | ||
Results
To characterize the binding affinity of the two major binding sites on wildtype FLAG-WDR5 (WDR5WT), a direct fluorescence polarization (FP) experiment was performed with two selective peptide binding probes, peptide 1F for the WIN site, and peptide 2F for the WBM site (Fig. 2A).14 The observed affinity for both binding sites was similar to the previously reported values (Fig. 2B–D). The commonly used mutant WDR5F266A was evaluated using the same protocol to reveal its effect on the two binding sites. This mutant was reported to have a lower lncRNA binding affinity, while retaining the ability for MLL-complex formation and the corresponding histone methylation potential (Fig. 2B–D).8 Since lncRNA binding takes place via the WBM site the reduced affinity of peptide 2 was expected. However, WDR5F266A also shows a 93-fold lower affinity for the WIN-site binding peptide when compared to WDR5WT, meaning that the mutation can also retard protein complex formation although the affinity is still in the nanomolar range (Fig. 2B and D). To rule out effects of the fluorescent labels we evaluated them using the unlabelled peptides 1NH2 and 2Ac as competitors (Fig. 2A). The results confirmed that the tracers recognized their target sites in a specific manner (Fig. 2E–G) as the obtained KI values of the unlabelled peptides were similar to the KD values of the labelled peptides. Furthermore, the acetylated WIN-site peptide 1NH2 does not inhibit the WBM-site and vice versa meaning that these two tracers were orthogonal.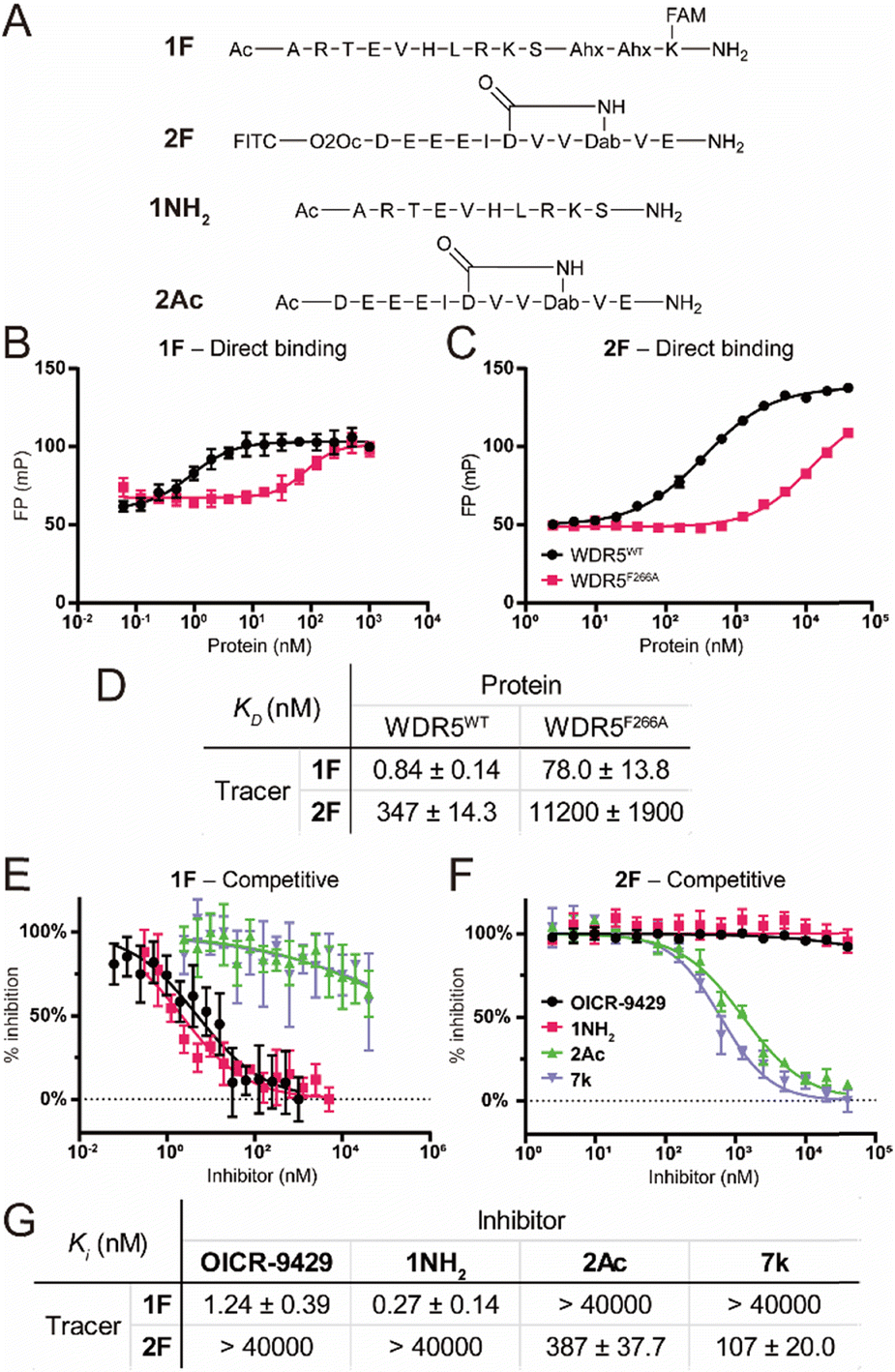 | ||
| Fig. 2 Fluorescence polarization assay using peptides 1F and 2F as tracers. A: Chemical structure of the used peptides. B: Titration of WDR5WT/F266A against 1F. C: Titration of WDR5WT/F266A against 2F. D: Binding affinities of peptides 1F and 2F. E: Competitive fluorescence polarization experiments at the WIN site. Inhibitors were titrated against a fixed concentration of 1F–WDR5WT complex. F: Competitive experiment at the WBM site. Inhibitors were titrated against a fixed concentration of 2F–WDR5WT complex. G: KI values measured for all compounds determined in the competitive fluorescence polarization experiments. All FP experiments were performed as two biological replicates of two technical replicates each. KI values are calculated as average of all four replicates and errors are reported as the standard deviation. | ||
Besides the site selective peptides, we also evaluated two small molecules, OICR-9429 and 7k, which were designed to be selective for the WIN and WBM site respectively, but their selectivity toward the other binding site was not evaluated (Fig. 2E–G).15,16 Indeed, we did not observe any crossover inhibition for the small molecules at the opposite binding sites (Fig. 2G).
After characterizing the binding site selectivity of OICR-9429 and 7k, we evaluated their ability to compete with lncRNA–WDR5 complex formation. In a report from our group, we demonstrated that peptides were able to inhibit lncRNA–protein interactions in vitro, and only inhibitors for targeting the WBM site can reduce RNA–WDR5 complex formation.19 Here the same strategy was applied using the small molecules, utilizing competitive in vitro RNA immunoprecipitation (iv-RIP) to verify the influence of small molecules on lncRNA–protein complex formation. To this end, cellular RNA extracts were incubated with either DMSO or compound, in the presence of FLAG-WDR5WT or FLAG-WDR5F266A, and immunoprecipitated using an anti-FLAG antibody. The coprecipitated RNA was then isolated and analysed using RT-qPCR. In this experiment, two lncRNAs, HOTTIP and HOXC13-AS, were monitored for their enrichment from RNA extracts from U-2 OS cells because they were both reported to be sensitive to WDR5 knockdown.7,13 The results demonstrated that molecule 7k, the direct inhibitor for the WBM site, could inhibit HOTTIP–WDR5WT and HOXC13-AS–WDR5WT complex formation (Fig. 3) but the effect was moderate. Although we did not observe inhibition of the WBM site from OICR-9429 in the FP experiments, it was able to reduce RNA binding albeit not significantly. A no-RNA control as well as a FLAG-GFP control were included to demonstrate that RNA enrichment was dependent on the presence of a WDR5 variant. WDR5F266A also showed a reduced capability of enrichment for RNA, even though WDR5F266A itself was more efficiently enriched than WDR5WT (Fig. S6†).
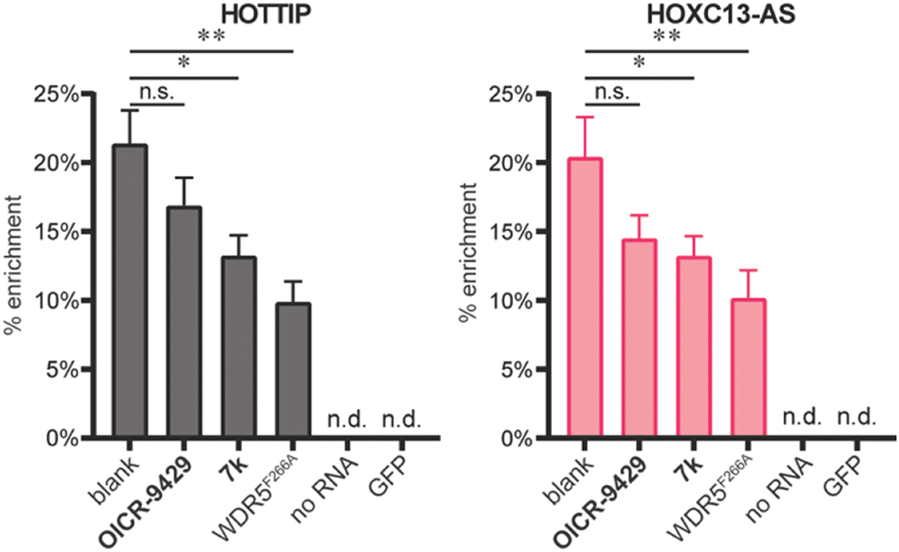 | ||
| Fig. 3 iv-RIP results using RNA isolates from U-2 OS cells and WDR5WT, WDRF266A, or GFP. Compounds OICR-9429 and 7k were tested at 10 μM in combination with WDR5WT. The no RNA control was tested with WDR5WT. n.s.: p > 0.05, *: 0.05 ≥ p > 0.01, **: 0.01 ≥ p > 0.001. | ||
Wang et al. reported that knocking down WDR5 could lead to the downregulation of HOTTIP and several genes around the late HOXA region.7 We hypothesized that we could use the small molecule inhibitors to verify if a direct lncRNA–WDR5 interaction is required for cells to maintain lncRNA expression levels. OICR-9429 and molecule 7k were used to antagonize the lncRNA–WDR5 complex in MDA-MB-231 cells, a triple-negative breast cancer cell line, and the expression level of HOTTIP was studied. Treatment with molecule 7k lead to down-regulation of HOTTIP after three days of treatment (Fig. 4A), showing that interrupting the HOTTIP–WDR5 interaction in cellulo indeed could lead to down-regulation of HOTTIP itself. After a one-day treatment no effect could be observed, indicating that the downregulation takes time to develop. Although OICR-9429 did not inhibit lncRNA–WDR5 complex formation as strongly as molecule 7k, it is still able to reduce HOTTIP expression after three days of treatment.21 In addition, one-day treatment of OICR-9429 has a small but significant effect. Taken together the data suggests that targeting WDR5 irrespective of the binding site leads to down-regulation of HOTTIP.
 | ||
| Fig. 4 lncRNA expression levels under various treatment conditions. RT-qPCR results were analysed using the 2−ΔΔCt method.20 A: Expression levels of HOTTIP after one/three-days incubation, Blank was treated with 0.1% DMSO while compounds were used at 10 μM. B: Expression levels of FOXD3-AS1 after one/three-days incubation time, Blank was treated with 0.1% DMSO while compounds were used at 10 μM. C: Expression level of HOTTIP after treatment with 1/5/10 μM compound of interest for three days. D: Expression level of FOXD3-AS1 after treatment with 1/5/10 μM compound of interest for three days. n.s.: p > 0.05, *: 0.05 ≥ p > 0.01, **: 0.01 ≥ p > 0.001, ***: 0.001 ≥ p > 0.0001, ****: 0.0001 ≥ p. | ||
Besides HOTTIP, a second lncRNA, FOXD3-AS1, was analysed under the same conditions as it was previously described to be bound by WDR5,13 while HOXC13-AS was not detectable in MDA-MB-231 (Table S3†). It is worth noting that FOXD3-AS1 does not show a time dependency after prolonging the treatment from one to three days (Fig. 4B). Treatment with OICR-9429 lead to a 20% reduction of expression of FOXD3-AS1, while treatment with molecule 7k did not provide significant changes, indicating that FOXD3-AS1 was not sensitive to the disruption of FOXD3-AS1–WDR5 complex, but still depend on a fully functional WDR5 with a free WIN binding pocket.
In addition to time dependency, concentration dependency was also tested. The treatment time was set to three days to ensure a clearly observable effect. The effect of treatment with OICR-9429 reached a maximum at 5 μM for both HOTTIP and FOXD3-AS1, while 1 μM of OICR-9429 was not enough to reduce the expression of HOTTIP significantly (Fig. 4C and D). On the other hand, a concentration dependent effect for molecule 7k could only be observed for HOTTIP, and 10 μM was required to provide significant changes. Meanwhile, FOXD3-AS1 is not sensitive to the treatment of molecule 7k in the concentration range used, similar to what we found in the time-dependent experiment.
Conclusions
In this report, we investigated the roles of the two known binding sites (WIN and WBM) of WDR5 by competitive FP assays. The WIN and WBM sites did not show any allosteric influence on each other when the peptide-based tracers were used that were derived from native WDR5 protein binding partners. Furthermore, we tested the mutant WDR5F266A to quantify the binding affinity and revealed that this mutation leads to a reduced affinity for its peptide binding partners similar as was reported for lncRNA. Although this was expected for the WBM site binding peptide, it is unclear why this mutation also affects binding of the WIN site targeting peptide 1F.As determined by iv-RIP, molecule 7k could inhibit lncRNA–WDR5 formation to a level almost similar to the WDR5F266A mutant. Based on previously described data, we hypothesized that interruption of the HOTTIP–WDR5 interaction could downregulate HOTTIP.8 Indeed, 7k was able to reduce HOTTIP expression but longer incubation times of three days were required to observe the effect. Interestingly, the same treatment did not influence FOXD3-AS1 expression although a previous study published by Subhash et al. demonstrated that silencing WDR5 could.13 These results suggest that FOXD3-AS1 may require the presence of WDR5 to maintain its expression, but not FOXD3-AS1–WDR5 complex formation.
In contrast to 7k, the WIN site inhibitor OICR-9429 was not able to reduce lncRNA binding significantly in the iv-RIP experiments. Albert et al. proposed a potential allosteric control between the WIN and WBM sites through the interaction on one of the WD40 blades,22 which indicates a possible explanation of how WIN sites could interfere with the binding at WBM sites. However, more extensive structural and biochemical experiments will be required to support this hypothesis. Nevertheless, the ability of OICR-9429 to antagonize the WDR5 supported histone methylation is likely the main reason for observing an overall downregulation of HOTTIP as well as FOXD3-AS1. A difference in time dependence was observed for the two lncRNAs (Fig. 4B) where the effect for FOXD3-AS1 showed no difference between a one-day or three-day treatment, while the effect for HOTTIP increased after prolonged treatment.
By combining the fact that OICR-9429 could downregulate FOXD3-AS1 while 7k could not, but that 7k was able to downregulate HOTTIP it seems possible to selectively downregulate specific lncRNAs based on the targeted binding site. Considering the role HOTTIP plays in cancer development and the oncogenes that it controls, it is very interesting to see that it could be downregulated without a direct RNA silencing strategy, providing new possibilities in targeting selected oncogenic lncRNAs. These strategies can involve small molecules targeting epigenetic scaffolding proteins rather than RNA silencing approaches which typically suffer from difficulties with delivery.
Author contributions
Jen-Yao Chang: conceptualization, methodology, validation, formal analysis, investigation, writing – original draft, visualization, supervision, funding acquisition Cora Neugebauer: methodology, validation, investigation Anne Mues: methodology, investigation Peter 't Hart: conceptualization, resources, writing – review & editing, visualization, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge Christiane Heitbrink (TU Dortmund) for measuring HRMS data. We also thank the Ministry of Education (Taiwan) for funding Jen-Yao Chang with a Government Scholarship for Overseas Study. Open Access funding provided by the Max Planck Society.Notes and references
- A. Schuetz, A. Allali-Hassani, F. Martín, P. Loppnau, M. Vedadi, A. Bochkarev, A. N. Plotnikov, C. H. Arrowsmith and J. Min, EMBO J., 2006, 25, 4245–4252 CrossRef CAS PubMed.
- A. duPuy Guarnaccia and W. P. Tansey, J. Clin. Med., 2018, 7, 21 CrossRef PubMed.
- X. Chen, J. Xu, X. Wang, G. Long, Q. You and X. Guo, J. Med. Chem., 2021, 64, 10537–10556 CrossRef CAS PubMed.
- J. Dias, N. Van Nguyen, P. Georgiev, A. Gaub, J. Brettschneider, S. Cusack, J. Kadlec and A. Akhtar, Genes Dev., 2014, 28, 929–942 CrossRef CAS PubMed.
- L. R. Thomas, Q. Wang, B. C. Grieb, J. Phan, A. M. Foshage, Q. Sun, E. T. Olejniczak, T. Clark, S. Dey, S. Lorey, B. Alicie, G. C. Howard, B. Cawthon, K. C. Ess, C. M. Eischen, Z. Zhao, S. W. Fesik and W. P. Tansey, Mol. Cell, 2015, 58, 440–452 CrossRef CAS PubMed.
- Y. Takahashi, G. H. Westfield, A. N. Oleskie, R. C. Trievel, A. Shilatifard and G. Skiniotis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20526–20531 CrossRef CAS PubMed.
- K. C. Wang, Y. W. Yang, B. Liu, A. Sanyal, R. Corces-Zimmerman, Y. Chen, B. R. Lajoie, A. Protacio, R. A. Flynn, R. A. Gupta, J. Wysocka, M. Lei, J. Dekker, J. A. Helms and H. Y. Chang, Nature, 2011, 472, 120–124 CrossRef CAS PubMed.
- Y. W. Yang, R. A. Flynn, Y. Chen, K. Qu, B. Wan, K. C. Wang, M. Lei and H. Y. Chang, eLife, 2014, 3, e02046 CrossRef PubMed.
- Z. Odho, S. M. Southall and J. R. Wilson, J. Biol. Chem., 2010, 285, 32967–32976 CrossRef CAS PubMed.
- S. Ghafouri-Fard, S. Dashti and M. Taheri, Biomed. Pharmacother., 2020, 127, 110158 CrossRef CAS PubMed.
- X. Chen, Y. Liu, Q. Zhang, B. Liu, Y. Cheng, Y. Zhang, Y. Sun, J. Liu and H. Gen, Front. Cell Dev. Biol., 2021, 8, 582723 CrossRef PubMed.
- B. Hu, L. Zhong, Y. Weng, L. Peng, Y. Huang, Y. Zhao and X.-J. Liang, Signal Transduction Targeted Ther., 2020, 5, 1–25 CrossRef PubMed.
- S. Subhash, K. Mishra, V. S. Akhade, M. Kanduri, T. Mondal and C. Kanduri, Nucleic Acids Res., 2018, 46, 9384–9400 CrossRef CAS PubMed.
- H. Karatas, E. C. Townsend, D. Bernard, Y. Dou and S. Wang, J. Med. Chem., 2010, 53, 5179–5185 CrossRef CAS PubMed.
- F. Grebien, M. Vedadi, M. Getlik, R. Giambruno, A. Grover, R. Avellino, A. Skucha, S. Vittori, E. Kuznetsova, D. Smil, D. Barsyte-Lovejoy, F. Li, G. Poda, M. Schapira, H. Wu, A. Dong, G. Senisterra, A. Stukalov, K. V. M. Huber, A. Schönegger, R. Marcellus, M. Bilban, C. Bock, P. J. Brown, J. Zuber, K. L. Bennett, R. Al-awar, R. Delwel, C. Nerlov, C. H. Arrowsmith and G. Superti-Furga, Nat. Chem. Biol., 2015, 11, 571–578 CrossRef CAS PubMed.
- J. D. Macdonald, S. Chacón Simon, C. Han, F. Wang, J. G. Shaw, J. E. Howes, J. Sai, J. P. Yuh, D. Camper, B. M. Alicie, J. Alvarado, S. Nikhar, W. Payne, E. R. Aho, J. A. Bauer, B. Zhao, J. Phan, L. R. Thomas, O. W. Rossanese, W. P. Tansey, A. G. Waterson, S. R. Stauffer and S. W. Fesik, J. Med. Chem., 2019, 62, 11232–11259 CrossRef CAS PubMed.
- J. Ding, L. Liu, Y.-L. Chiang, M. Zhao, H. Liu, F. Yang, L. Shen, Y. Lin, H. Deng, J. Gao, D. R. Sage, L. West, L. A. Llamas, X. Hao, S. Kawatkar, E. Li, R. K. Jain, J. A. Tallarico, S. M. Canham and H. Wang, J. Med. Chem., 2023, 66, 8310–8323 CrossRef CAS PubMed.
- J. Ding, G. Li, H. Liu, L. Liu, Y. Lin, J. Gao, G. Zhou, L. Shen, M. Zhao, Y. Yu, W. Guo, U. Hommel, J. Ottl, J. Blank, N. Aubin, Y. Wei, H. He, D. R. Sage, P. W. Atadja, E. Li, R. K. Jain, J. A. Tallarico, S. M. Canham, Y.-L. Chiang and H. Wang, ACS Chem. Biol., 2023, 18, 34–40 CrossRef CAS PubMed.
- J.-Y. Chang, C. Neugebauer, S. Schmeing, G. Amrahova and P. 't Hart, Chem. Commun., 2023, 59, 10656–10659 RSC.
- A. Fuentes-Iglesias, V. Garcia-Outeiral, J. A. Pardavila, J. Wang, M. Fidalgo and D. Guallar, STAR Protoc., 2020, 1, 100093 CrossRef PubMed.
- J. Zhang, Q. Zhou, K. Xie, L. Cheng, S. Peng, R. Xie, L. Liu, Y. Zhang, W. Dong, J. Han, M. Huang, Y. Chen, T. Lin, J. Huang and X. Chen, J. Exp. Clin. Cancer Res., 2021, 40, 203 CrossRef CAS PubMed.
- L. Albert, J. Nagpal, W. Steinchen, L. Zhang, L. Werel, N. Djokovic, D. Ruzic, M. Hoffarth, J. Xu, J. Kaspareit, F. Abendroth, A. Royant, G. Bange, K. Nikolic, S. Ryu, Y. Dou, L.-O. Essen and O. Vázquez, ACS Cent. Sci., 2022, 8, 57–66 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00605k |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |