
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ generation of organocalcium compounds for a calcium-based Grignard-type chemistry
Simon
Sengupta
 ,
Sven
Krieck
and
Matthias
Westerhausen
*
,
Sven
Krieck
and
Matthias
Westerhausen
*
Institute of Inorganic and Analytical Chemistry, Friedrich Schiller University Jena, Humboldtstraße 8, D-07743 Jena, Germany. E-mail: m.we@uni-jena.de
First published on 5th August 2024
Abstract
Organocalcium compounds are highly reactive reagents whereas the alkaline-earth metal itself is a weak reductant. This discrepancy hampered a straightforward development of an organocalcium chemistry. The in situ generation of the highly reactive organocalcium reagent and immediate metalation of a H-acidic compound (iGMM) or addition onto a polar π-system (iGAM) offers not only a loophole to organocalcium reagents but opens the entry to a rich organic chemistry of this non-toxic and globally abundant alkaline-earth metal, being competitive to the organolithium chemistry.
State-of-the-art of organic calcium-based chemistry
Widely used organic alkaline-earth metal chemistry was mainly restricted to the Grignard reaction, i.e. the reduction of halogeno hydrocarbons with magnesium, which is commonly taught today already on undergraduate level and represents a prominent tool in daily organic chemistry. The toxicity of beryllium strictly limits the use of this element in organic and coordination chemistry. The application of the homologous calcium metal as reductant in organic reactions was firstly probed by Beckmann in 1905,1 however, severe drawbacks hampered a straightforward development of a calcium-based Grignard-type chemistry.2 Contradictory reports on the outcome of the reaction of calcium with halogeno hydrocarbons, on the stability and on the reactivity significantly decelerated the establishment of a Grignard-type chemistry with this alkaline-earth metal and the interest in this calcium-based Grignard-type chemistry ceased. The main reasons were the inertness of calcium metal itself due to an atomization enthalpy of 178.2 kJ mol−1 being approx. 20% and 10% larger than of the homologous neighbors Mg and Sr (requiring metal activation prior to use and/or drastic reaction conditions),3 preparative challenges (such as cocondensation procedures)2 and the enormous reactivity of the produced organocalcium reagents (leading to side reactions like Wurtz-type coupling, ether degradation, or β-hydride elimination reactions).4 Nevertheless, the valuable use of organocalcium compounds in synthetic methods has been valued recently.5In the early 1990s, several research groups developed preparative procedures for highly soluble calcium bis[bis(trimethylsilyl)amide], Ca[N(SiMe3)2]2 or Ca(hmds)2, as entry into a homogeneous organic calcium-based chemistry. All these protocols had their disadvantages like the use of heavy metal substrates such as Sn(hmds)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 and Hg(hmds)27 in transmetalation protocols, ammonia-saturated organic solvents8 leading to sluggish reaction mixtures, salt metathesis methods with the initial synthesis of organopotassium compounds,9 metalation reactions with the previous synthesis of dibenzylcalcium,10 or direct reduction in the presence of BiPh3.11 Especially the formation of Ca(hmds)2 from the metathesis reaction of CaI2 with K(hmds) also yields the unrecognized calciate KCa(hmds)3 which exhibits a different reactivity than pure Ca(hmds)2.10 Nevertheless, the report on the first structurally authenticated organocalcium derivative [(dx)2Ca(CH(SiMe3)2)2] (dx = 1,4-dioxane) in 1991 via a cocondensation protocol revived the organocalcium chemistry.12 In addition, the synthesis of sparingly soluble dimethylcalcium verified that not intrinsic instability of the organocalcium reagent is responsible for the preparative challenges.13 Two decades ago, the Grignard-type reduction of halogeno arenes with activated calcium in THF, yielding arylcalcium halides of the type [(thf)nCa(Ar)X] with X = Br and I and n depending on the bulkiness of Ar, was established after initial attempts leading to oxide-centered cages due to provoked ether degradation processes.14 Schlenk-type equilibria allowed the isolation of diarylcalcium compounds,15 preferably via addition of KO(tBu) to the Ar–Ca–X reaction mixtures producing soluble Ca(Ar)2 and insoluble calcium tert-butanolates and potassium halide.16 Alkylcalcium reagents of the type H2n+1Cn–Ca–X without stabilizing aryl or trialkylsilyl substituents are not accessible by a straightforward reduction of the hydrocarbyl halide with calcium and therefore, bulky ligands have been employed to shield the reactive Ca–alkyl moiety in heteroleptic complexes,17 a concept that has also been employed successfully to prepare alkylstrontium compounds.18
6 and Hg(hmds)27 in transmetalation protocols, ammonia-saturated organic solvents8 leading to sluggish reaction mixtures, salt metathesis methods with the initial synthesis of organopotassium compounds,9 metalation reactions with the previous synthesis of dibenzylcalcium,10 or direct reduction in the presence of BiPh3.11 Especially the formation of Ca(hmds)2 from the metathesis reaction of CaI2 with K(hmds) also yields the unrecognized calciate KCa(hmds)3 which exhibits a different reactivity than pure Ca(hmds)2.10 Nevertheless, the report on the first structurally authenticated organocalcium derivative [(dx)2Ca(CH(SiMe3)2)2] (dx = 1,4-dioxane) in 1991 via a cocondensation protocol revived the organocalcium chemistry.12 In addition, the synthesis of sparingly soluble dimethylcalcium verified that not intrinsic instability of the organocalcium reagent is responsible for the preparative challenges.13 Two decades ago, the Grignard-type reduction of halogeno arenes with activated calcium in THF, yielding arylcalcium halides of the type [(thf)nCa(Ar)X] with X = Br and I and n depending on the bulkiness of Ar, was established after initial attempts leading to oxide-centered cages due to provoked ether degradation processes.14 Schlenk-type equilibria allowed the isolation of diarylcalcium compounds,15 preferably via addition of KO(tBu) to the Ar–Ca–X reaction mixtures producing soluble Ca(Ar)2 and insoluble calcium tert-butanolates and potassium halide.16 Alkylcalcium reagents of the type H2n+1Cn–Ca–X without stabilizing aryl or trialkylsilyl substituents are not accessible by a straightforward reduction of the hydrocarbyl halide with calcium and therefore, bulky ligands have been employed to shield the reactive Ca–alkyl moiety in heteroleptic complexes,17 a concept that has also been employed successfully to prepare alkylstrontium compounds.18
Preconditions for the use of organocalcium reagents
Calcium and lithium have similar Pauling electronegativity values of 1.0 and hence, comparable ionicity and reactivity might be expected whereas magnesium has a larger electronegativity of 1.2,3 responsible for a slightly more covalent character. Calcium is non-toxic and globally abundant whereas the resources of lithium are limited and desperately needed for batteries to ensure electromobility and global change to sustainable and non-fossil fuels. In addition, isoelectronic calcium and group III ions suggest that their d0 systems have chemically accessible empty d-orbitals (Lewis acidic orbitals) leading to unique structures such as pyramidal coordination spheres of tricoordinate metal centers as in molecular YR319 and [CaR3]− ions.20 The ions Ca2+ and Y3+ have comparable radii whereas isoelectronic Sc3+ is significantly smaller and organometallic compounds of Sc3+ form anionic scandate complexes like [R3ScX]− with X being a halide or methyl group due to the very strong Lewis acidic character of this group III ion.21 For a competitive use of calcium in daily preparative organic chemistry, especially in comparison to the established classical Grignard reagents and organolithium chemistry, several conditions are advantageous:
– A nitrogen atmosphere is sufficient during handling of the metals Mg and Ca whereas reduction with Li requires an argon atmosphere.
– Preparative simple protocols are highly beneficial without the need of metal activation, synthesis of metal–organic precursor compounds or sophisticated breadboarding.
– A one-step procedure without intermediate work-up and purification steps should allow the synthesis of pure compounds on a multi-gram scale within a few hours.
– The use of common organic solvents at anhydrous ambient reaction conditions is advantageous.
– Side reactions such as attack of solvents should play a neglectable role to allow the isolation of pure compounds.
In most cases, it is unnecessary to isolate the Grignard reagents prior to their use in subsequent conversion reactions such as metalation, addition, reduction, and metathetical applications. Similar reaction conditions are beneficial for the acceptance of a calcium-based Grignard-type chemistry to limit the retention time of organocalcium reagents in order to reduce variety and amount of side products.
In situ generation of calcium-based Grignard-type reagents
For an efficient procedure to utilize in situ generated organocalcium reagents, side reactions must be negligible and/or their by-products must be easily removable. On the one hand, ether degradation processes commonly start with an α-deprotonation (β-deprotonation plays a minor role) followed by cycloreversion and formation of volatile ethene and ethenolate from THF or ethanolate from diethyl ether, respectively.22 Alkoxides of calcium are sparingly soluble and precipitate from ethereal reaction mixtures. On the other hand, Wurtz-type coupling reactions form R–R and CaX2 from R–X and Ca via the reaction or intermediate R–Ca–X with R–X.4 Selection of an adequate substrate R–X yields volatile coupling products R–R, whereas CaX2 represents an innocent by-product because it forms in any case via a Schlenk-type equilibrium besides CaR2 from heteroleptic R–Ca–X.23 The driving force of the Wurtz-type coupling is the thermodynamically favored salt elimination, whereas metalation reactions including ether degradation are kinetically controlled.In typical protocols excess or stoichiometric amounts of calcium granules are suspended in THF in the presence of a suitable substrate (Scheme 1).24 Activation or purification of calcium prior to use is unnecessary and commercially available calcium turnings are suitable. The inertness of calcium allows a large variety of substrates that are neither attacked nor reduced under these conditions. Then, bromoethane is added dropwise at room temperature to the stirred reaction mixture and within very few hours the calcium metal disappears, and the substrate is consumed by intermediately formed Et–Ca–Br. During this reaction, the solution turns cloudy due to precipitation of sparingly soluble calcium bromide [(thf)4CaBr2]. After removal of excess calcium and calcium bromide by filtration, the envisioned product can be isolated from the filtrate by common work-up procedures. Excess of volatile EtBr as well as the Wurtz-type coupling product butane are quantitatively removed under reduced pressure. Iodomethane is disadvantageous because produced dimethylcalcium is insoluble and covers the calcium turnings; therefore, the reaction decelerates or even ceases leading to an increasing amount of side products.
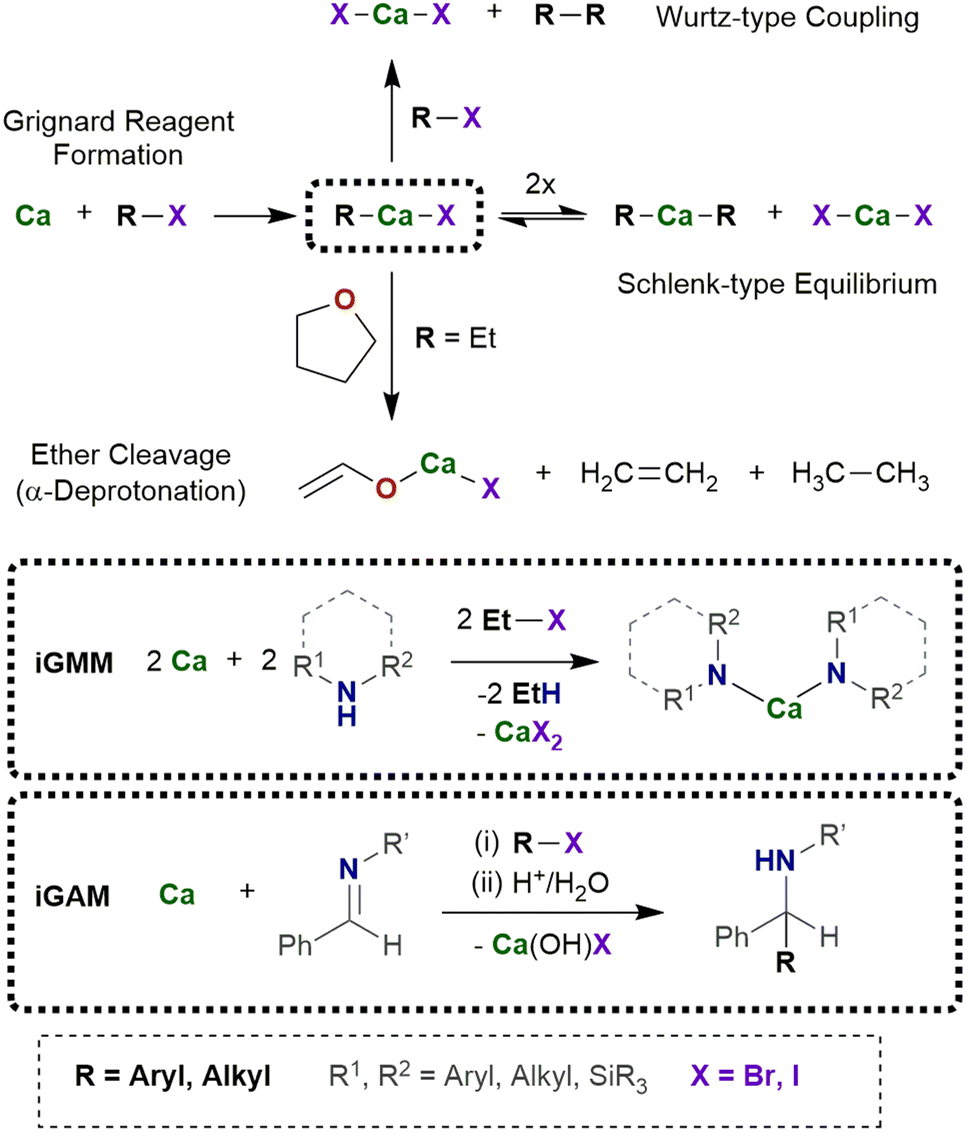 | ||
| Scheme 1 In situ formation of the calcium-based Grignard-reagent R–Ca–X and subsequent Schlenk-type equilibrium leading to homoleptic CaR2 and CaX2 as well as undesired Wurtz-type C–C coupling and ether degradation reactions. In the bottom, application of R–Ca–X in metalation and addition reactions is depicted. | ||
Scope and limitations of this in situ calcium Grignard preparation
The in situ calcium-based Grignard reaction is a heterogeneous reaction on the surface of the metal particles and mechanistically much more complex than homogeneous reactions. The reaction pathway includes adsorption of the substrate R–X on the metal surface, electron transfer and formation of the radical anion [R–X]˙−, dissociation into the halide ion and the radical R˙, formation of [Ca–R]+ and after capture of the halide ion finally R–Ca–X. Solvation and desorption yields the organocalcium compound [(thf)nCa(R)X], which may undergo subsequent reactions (Schlenk-type equilibrium, metalation, addition onto unsaturated compounds, or undesired Wurtz-type coupling). To enable electron transfer from calcium onto the substrate R–X, steric hindrance of the substrate can be low to moderate such as R = n-alkyl, Ph. As mentioned above, the small methyl halide leads to formation of insoluble precipitate of CaMe2 on the metal surface and the reduction reaction ceases. To avoid blocking of the metal surface, solvation of the calcium compounds with Lewis bases of sufficient basicity is mandatory. THF represents an appropriate solvent whereas weaker bases like diethyl ether and hydrocarbons are inapplicable. The reactions of R–Ca–X with another substrate must be faster than the formation of this calcium-based Grignard reagent to avoid increasing concentration of R–Ca–X and CaR2 in the reaction mixture.The in situ Grignard metalation method (iGMM) of e.g. amines in THF at room temperature is an established procedure yielding the corresponding amides of calcium, [(thf)nCa(NR2)2] as depicted in Scheme 2.23 In a typical protocol, calcium (approx. 1.2 equiv.) and amine HNR2 (1.0 equiv.) are suspended in THF (0.5–1 M solution) and then EtBr (1.0 equiv.) is added dropwise at ambient temperature. After an induction period, moderate gas evolution and turbidity of the solution are observed. In some cases, the yield can be increased by addition of a second batch of EtBr (0.5–1.0 equiv.). The calcium amides can easily be isolated on multi-gram scale by removal of the solvent and recrystallization from THF or a mixture of pentane and THF.
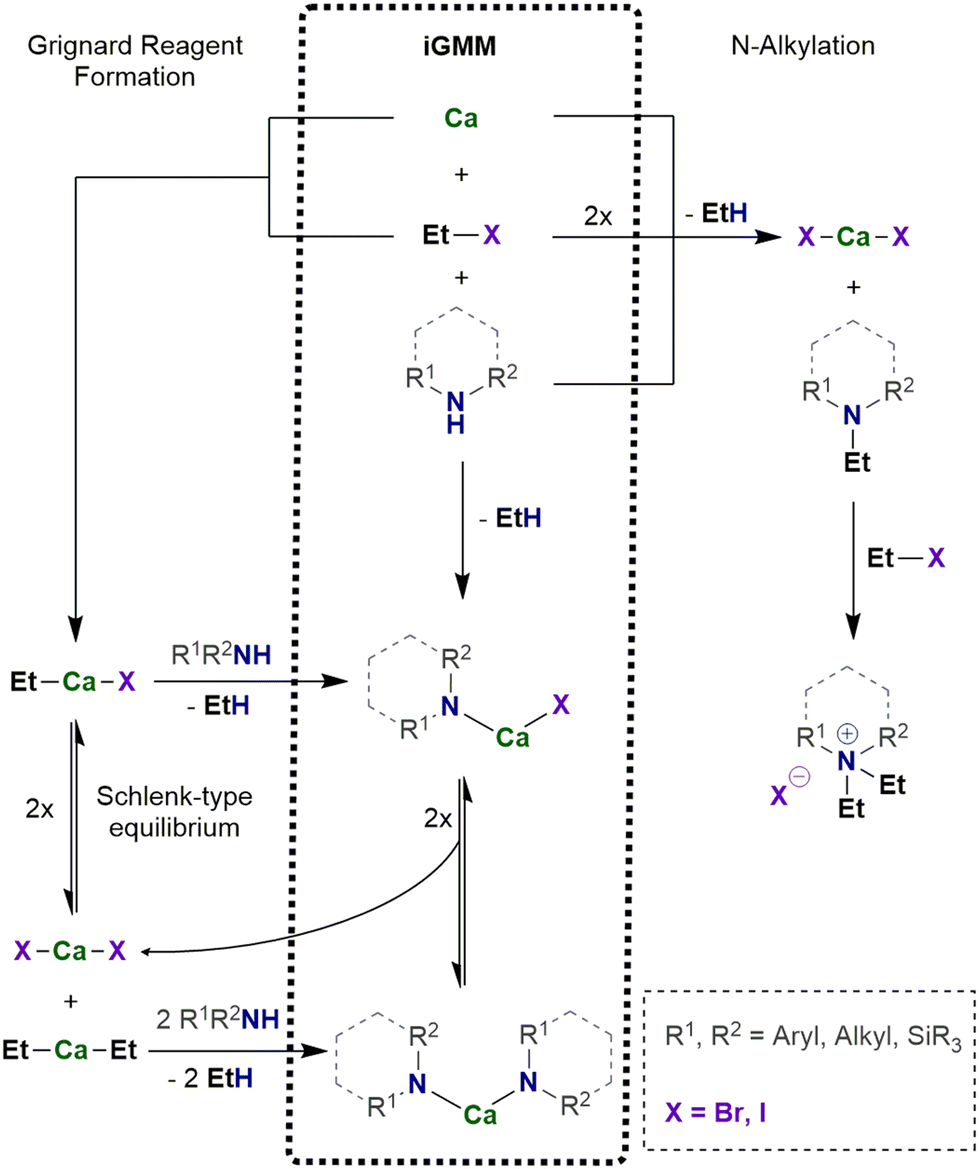 | ||
| Scheme 2 In situ Grignard Metalation Method (iGMM) of amines with intermediately prepared R–Ca–X. | ||
For the iGMM, the difference of the kinetic pKa values, i.e. the Brønsted acidity difference, is a dependable measure. A pKa value of 42 for ethane allows the metalation of numerous substrates with smaller pKa values (and hence larger Brønsted acidity) like alcohols, amines, phosphines, cyclopentadienes, and others. Another important factor is lack of steric hindrance. Whereas CpH (pKa = 16) and monosilylated cyclopentadienes are easily converted to their calcocene derivatives, the yields are significantly lower for 1,3-bis(trialkylsilyl)cyclopentadienes for steric reasons.23 Calciation of phenylated methane succeeds with low yield for diphenylmethane (pKa = 33) whereas the alkyl C–H moieties of toluene (pKa = 37, low acidity) and triphenylmethane (pKa = 30, steric shielding) do not react under these conditions. Contrary to this finding, the magnesium-based iGMM yields heteroleptic Grignard reagents R–Mg–Br and offers straightforward access to e.g. amidomagnesium bromides,25 known as Hauser bases with strong metalation power, and cyclopentadienylmagnesium halides26 with increasing interest as possible electrolytes in magnesium batteries.27
In the in situ Grignard addition method (iGAM), freshly generated R–Ca–X is used for addition reactions across imines allowing the synthesis of bulky calcium amides (Scheme 3).28 In a typical procedure calcium (1.5 eq.) and imine (1.0 eq.) are suspended in an ethereal solvent (0.25–0.5 M solution).29 The iodoalkane or -arene (1.5 eq.) is added at room temperature. The reaction mixture turns reddish brown within ten minutes and after stirring overnight, hydrolytic work-up yields bulky amines. To avoid significant amounts of the aza-pinacol coupling product, the iodoalkane or -arene must be added to the suspension shortly after its preparation. In this procedure, the iodoarene can additionally be substituted by e.g. alkyl, chloro, or methoxy substituents. The reaction of a suspension of an alkaline-earth metal and iodobenzene in THF at room temperature with (E)-N-phenyl-benzylideneamine PhN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(H)Ph and subsequent hydrolysis after 17 h yields a mixture of the reduction product Ph(H)N–CH2Ph, the aza-pinacol coupling Ph(H)N–C(H)Ph–C(H)Ph–N(H)Ph, and the projected addition compound Ph(H)N–C(H)Ph2. The performance of Mg is generally poor whereas Ca gives projected Ph(H)N–C(H)Ph2 with a yield of 82% and only traces of the reduction (2%) and aza-pinacol coupling (6%) side products. The heavier alkaline-earth metals shift the product distribution in favor of the aza-pinacol coupling compound which is the major product (78%) if barium metal has been applied.
C(H)Ph and subsequent hydrolysis after 17 h yields a mixture of the reduction product Ph(H)N–CH2Ph, the aza-pinacol coupling Ph(H)N–C(H)Ph–C(H)Ph–N(H)Ph, and the projected addition compound Ph(H)N–C(H)Ph2. The performance of Mg is generally poor whereas Ca gives projected Ph(H)N–C(H)Ph2 with a yield of 82% and only traces of the reduction (2%) and aza-pinacol coupling (6%) side products. The heavier alkaline-earth metals shift the product distribution in favor of the aza-pinacol coupling compound which is the major product (78%) if barium metal has been applied.
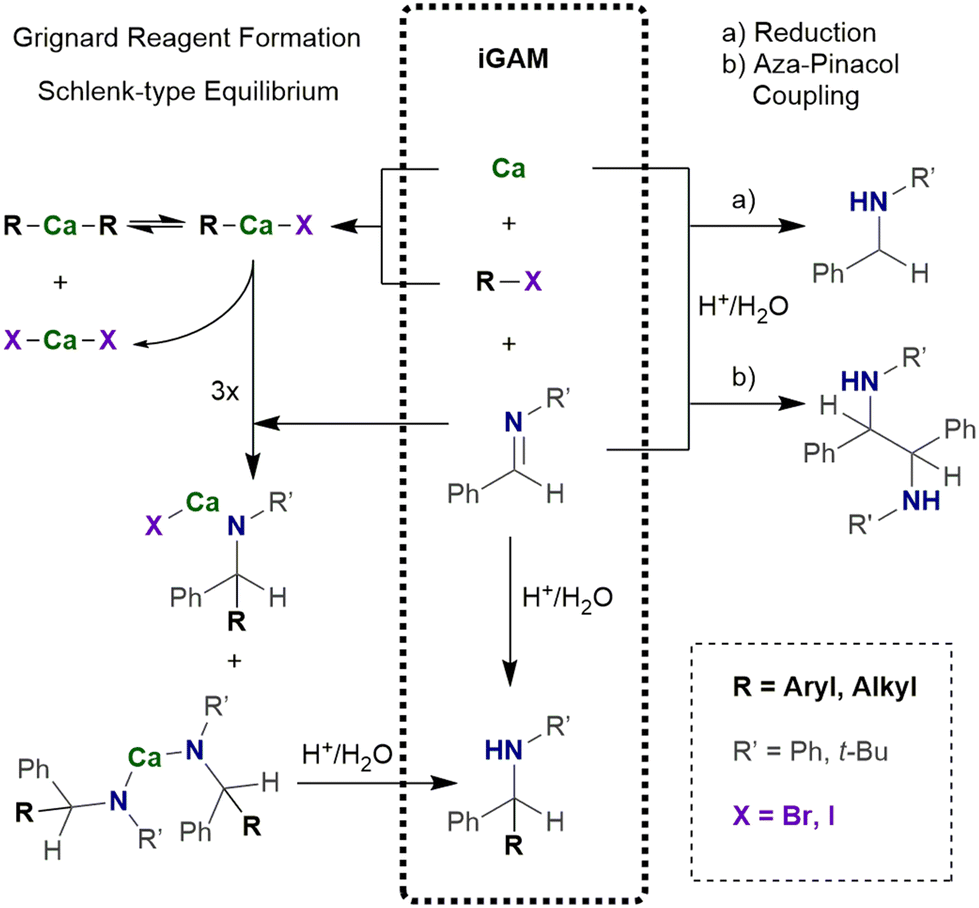 | ||
| Scheme 3 In situ Grignard Addition Method (iGAM) of imines with intermediately prepared R–Ca–X. | ||
In a mechanochemical alternative28,30 the iGAM can be performed in a ball mill. Calcium (approx. 1.5 mmol, 1.5 eq.) and imine (1.0 mmol, 1.0 eq.) are placed in a stainless-steel jar with 5 stainless steel balls (2 mm diameter) and flushed with argon.29 Ethereal solvent (0.2 mL, approx. 5 M) is added under argon. Iodoalkane or -arene (1.5 eq.) is added to this slurry and the reaction mixture shaken for 1 h at room temperature. The yields are slightly lower than in the common iGAM in THF. In this mechanochemical procedure, less ethereal solvent is used but an argon atmosphere is highly beneficial for protection of the reaction mixture against moisture and air to prevent hydrolysis and oxidation processes. Nevertheless, a protecting argon atmosphere is not mandatory for mechanochemically generated organocalcium compounds.31
Outlook
The straightforward accessibility of alkyl/aryl calcium halides will establish a broad organic chemistry of this alkaline-earth metal, competitive to or even outperforming organolithium and Grignard reagents. The simple synthetic protocol, allowing to produce large amounts in only very few hours, overcompensates the lack of storability of stock solutions of calcium-based Grignard-type reagents R–Ca–X. The in situ formation of these organocalcium reagents is a highly advantageous method because calcium metal and many substrates such as H-acidic or unsaturated compounds coexist in THF solution without or with slow interaction. Addition of bromoethane or other organic bromides and iodides at room temperature starts a mild and controllable reaction yielding intermediate organocalcium reagents that are immediately consumed by the substrates conveniently leading to pure compounds. Organic chlorides and fluorides are inapplicable. Even in the case that a conversion, i.e. a fast reaction of intermediately formed R–Ca–X with the substrate, fails the Wurtz-type coupling reaction predominates and R–R as well as CaX2 form in a hazard-free manner under mild conditions. Therefore, this regime is ideal to probe the suitability of organocalcium compounds in research and development to give kudos to this largely disregarded organocalcium chemistry based on the non-toxicity and global abundance of this alkaline-earth metal.For the iGMM steric requirements and pKa values (Brønsted acidity) represent the key factors for suitable substrates. Bulky substituents in the vicinity of the H-acidic functionality hinder the metalation. Therefore, it may be expected that calcium derivatives with small anions are the preferred compounds produced by this method. These compounds have already been used as anionic polymerization initiators, intermediately prepared by the reaction of [(thf)2Ca{N(SiMe3)2}2] with e.g. methanol because pure Ca(OMe)2 is an insoluble alkoxide once precipitated.
The iGAM requires a fast addition of intermediately prepared R–Ca–X across multiple bond systems. To ensure a fast addition reaction, polar π-systems are necessary that are not shielded by bulky substituents. Advantageously, a large variety of organic groups R are suitable for this reaction. Even 1,4-diidobenzene might be a suitable organic halide because calcium reacts only with one iodo functionality (contrary to lithium). One can envision a plethora of new organocalcium compounds via addition to diverse substrates such as nitriles, alkynes, alkenes, carbon monoxide, carbon dioxide and many other unsaturated compounds.
Another aspect concerns the heavier alkaline-earth metals strontium and barium. Due to enhanced reactivity of freshly grated Sr and Ba, metalation reactions commonly succeed via direct metalation of H-acidic compounds. The iGAM, however, could allow to intermediately prepare also alkyl and aryl derivatives of the heavier alkaline-earth metals for addition onto unsaturated substrates.
Conclusions
The organocalcium reagents exhibit a more polar metal–carbon bond than the classical Grignard reagents and hence, a higher reactivity related to an enhanced nucleophilicity of the carbanion is observed which requires shorter dwell time in ethereal solvents to prevent ether degradation reactions. This precondition can easily be achieved by an immediate consumption of in situ prepared organocalcium reagents by H-acidic substrates or unsaturated polar compounds via metalation and addition reactions. The use of a Lewis basic solvent is coercive to enable stripping of the calcium complexes from the surface of the calcium particles to maintain a continuing formation of R–Ca–X.Data availability
In this Frontier article with the title “In situ Generation of Organocalcium Compounds for a Calcium-Based Grignard-Type Chemistry” only previous work of us and others is cited. No unpublished data are presented in this manuscript.Conflicts of interest
There are no conflicts to declare.References
- E. Beckmann, Ber. Dtsch. Chem. Ges., 1905, 38, 904–906 CrossRef CAS.
- M. Westerhausen, M. Gärtner, R. Fischer, J. Langer, L. Yu and M. Reiher, Chem. – Eur. J., 2007, 13, 6292–6306 CrossRef CAS PubMed; M. Westerhausen, J. Langer, S. Krieck, R. Fischer, H. Görls and M. Köhler, Top. Organomet. Chem., 2013, 45, 29–72 CrossRef PubMed; M. Westerhausen, A. Koch, H. Görls and S. Krieck, Chem. – Eur. J., 2017, 23, 1456–1483 CrossRef PubMed.
- A. F. Holleman, E. Wiberg and N. Wiberg, Inorganic Chemistry, Academic Press, San Diego, 2001 Search PubMed.
- A. Koch, Q. Dufrois, M. Wirgenings, H. Görls, S. Krieck, M. Etienne, G. Pohnert and M. Westerhausen, Chem. – Eur. J., 2018, 24, 16840–16850 CrossRef CAS PubMed.
- D. O. Khristolyubov, D. M. Lyubov and A. A. Trifonov, Russ. Chem. Rev., 2021, 90, 529–565 CrossRef; S. Harder and J. Langer, Nat. Rev. Chem., 2023, 7, 843–853 CrossRef PubMed.
- M. Westerhausen, Inorg. Chem., 1991, 30, 96–101 CrossRef CAS.
- D. C. Bradley, M. B. Hursthouse, A. A. Ibrahim, K. M. Abdul Malik, M. Motevalli, R. Möseler, H. Powell, J. D. Runnacles and A. C. Sullivan, Polyhedron, 1990, 9, 2959–2964 CrossRef CAS.
- S. R. Drake, D. J. Otway and S. P. Perlepes, Main Group Met. Chem., 1991, 14, 243–256 CAS.
- P. S. Tanner, D. J. Burkey and T. P. Hanusa, Polyhedron, 1995, 14, 331–333 CrossRef CAS; E. D. Brady, T. P. Hanusa, M. Pink and V. G. Young, Inorg. Chem., 2000, 39, 6028–6037 CrossRef PubMed; X. He, B. C. Noll, A. Beatty, R. E. Mulvey and K. W. Henderson, J. Am. Chem. Soc., 2004, 126, 7444–7445 CrossRef PubMed.
- A. M. Johns, S. C. Chmely and T. P. Hanusa, Inorg. Chem., 2009, 48, 1380–1384 CrossRef CAS PubMed.
- M. M. Gillett-Kunnath, J. G. MacLellan, C. M. Forsyth, P. C. Andrews, G. B. Deacon and K. Ruhlandt-Senge, Chem. Commun., 2008, 4490–4492 RSC.
- F. Geoffrey, N. Cloke, P. B. Hitchcock, M. F. Lappert, G. A. Lawless and B. Royo, J. Chem. Soc., Chem. Commun., 1991, 724–726 Search PubMed.
- B. M. Wolf, C. Stuhl, C. Maichle-Mössmer and R. Anwander, J. Am. Chem. Soc., 2018, 140, 2373–2383 CrossRef CAS PubMed.
- C. Ruspic and S. Harder, Organometallics, 2005, 24, 5506–5508 CrossRef CAS; R. Fischer, H. Görls and M. Westerhausen, Inorg. Chem. Commun., 2005, 8, 1159–1161 CrossRef.
- R. Fischer, M. Gärtner, H. Görls, L. Yu, M. Reiher and M. Westerhausen, Angew. Chem., Int. Ed., 2007, 46, 1618–1623 CrossRef CAS PubMed.
- J. Langer, S. Krieck, H. Görls and M. Westerhausen, Angew. Chem., Int. Ed., 2009, 48, 5741–5744 CrossRef CAS PubMed.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168–1171 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, H. Elsen, C. A. Fischer, J. Langer, M. Wiesinger and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 5396–5401 CrossRef PubMed.
- M. Westerhausen, M. Hartmann and W. Schwarz, Inorg. Chim. Acta, 1998, 269, 91–100 CrossRef CAS.
- P. B. Hitchcock, A. V. Khvostov and M. F. Lappert, J. Organomet. Chem., 2002, 663, 263–268 CrossRef CAS.
- A. Mortis, D. Barisic, K. Eichele, C. Maichle-Mössmer and R. Anwander, Dalton Trans., 2020, 49, 7829–7841 RSC.
- A. Maercker, Angew. Chem., Int. Ed. Engl., 1987, 26, 972–989 CrossRef.
- P. Schüler, S. Sengupta, A. Koch, H. Görls, S. Krieck and M. Westerhausen, Chem. – Eur. J., 2022, 28, e202201897 CrossRef PubMed; P. Schüler, S. Sengupta, S. Krieck and M. Westerhausen, Chem. – Eur. J., 2023, 29, e202300833 CrossRef PubMed.
- S. Krieck, P. Schüler, J. M. Peschel and M. Westerhausen, Synthesis, 2019, 1115–1122 CAS.
- S. Sengupta, P. Schüler, H. Görls, P. Liebing, S. Krieck and M. Westerhausen, Chem. – Eur. J., 2022, 28, e202201359 CrossRef CAS PubMed.
- P. Schüler, H. Görls, S. Krieck and M. Westerhausen, Chem. – Eur. J., 2021, 27, 15508–15515 CrossRef PubMed.
- S. Zaubitzer, S. Dongmo, P. Schüler, S. Krieck, F. Fiesinger, D. Gaissmaier, M. van den Borg, T. Jacob, M. Westerhausen, M. Wohlfahrt-Mehrens and M. Marinaro, Energy Technol., 2022, 10, 2200440 CrossRef CAS; P. Schüler, S. Sengupta, S. Zaubitzer, F. Fiesinger, S. Dongmo, H. Görls, M. Wohlfahrt-Mehrens, M. van den Borg, D. Gaissmaier, S. Krieck, M. Marinaro, T. Jacob and M. Westerhausen, Eur. J. Inorg. Chem., 2022, e202200149 CrossRef; M. Cheng, Y. Wang, D. Zhang, S. Zhang, Y. Yang, X. Lv, J. Wang and Y. NuLi, J. Energy Chem., 2023, 76, 1–10 CrossRef For a review see e.g. J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720 CrossRef PubMed.
- S. Sengupta, P. Schüler, P. Liebing and M. Westerhausen, Chem. – Eur. J., 2023, 29, e202300035 CrossRef CAS PubMed.
- In the cited ref. 28 the stoichiometric ratio of calcium and imine are erroneously reversed in the general description, but the preparative procedures of specific compounds are presented correctly here.
- D. Tan and F. Garcia, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- P. Gao, J. Jiang, S. Maeda, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2022, 61, e202207118 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |