
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of dynamic bond concentration on the mechanical properties of vitrimers†
Harsh
Pandya
 a and
Fardin
Khabaz
*ab
a and
Fardin
Khabaz
*ab
aSchool of Polymer Science and Polymer Engineering, The University of Akron, Akron, OH 44325, USA
bDepartment of Chemical, Biomolecular, and Corrosion Engineering, The University of Akron, Akron, OH 44325, USA. E-mail: fkhabaz@uakron.edu
First published on 22nd August 2024
Abstract
The presence of dynamic covalent bonds allows vitrimers to undergo topology alterations and display self-healing properties. Herein, we study the influence of varying the concentration of dynamic bonds on the macroscopic properties of hybrid vitrimer networks by subjecting them to triaxial stretching tests using molecular simulations. Results show that the presence of dynamic bonds allows for continuous stress relaxation in the hybrid networks leading to delayed craze development and higher stretching as compared to permanently crosslinked networks. The work highlights the ability of glassy vitrimer networks to relax tensile stress during deformation successfully.
Vitrimers are crosslinked polymeric networks that undergo melt-like flow facilitated by associative bond exchange reactions.1–3 They undergo topology-altering reactions in response to stimuli, such as temperature, stress, pH, or light, allowing them to deform while simultaneously preserving network integrity, and showcasing versatile properties such as solvent resistance, welding, self-healing, and recyclability.4–14 These properties, although interesting, underscore intricate correlations between bond exchange rates on kinetics and their effect on the network's macroscopic properties.
The ability of vitrimers to relax applied stress by undergoing bond exchange adds another dimension to their mechanical behavior. However, a drawback is that these networks exhibit significant creep at low temperatures and have low melt viscosity at high temperatures, making them difficult to process using conventional thermoplastic processing techniques.15–18 A possible solution is to create hybrid vitrimer networks consisting of both dynamic and permanent covalent linkages. In this regard, a modified Flory–Stockmayer theory suggests that a hybrid vitrimer network typically requires at least 67 mol% of dynamic covalent bonds to suppress creep, yet recent experimental findings indicate this requirement can be reduced to as low as 20 mol%.17,19 Similar works have examined the effect of mechanical deformation on transient networks and self-healing gels containing exchangeable bonds highlighted the effect of associative bonds on the materials' response.20–23 The rate of topology rearrangements in vitrimers can be influenced by adjusting the kinetics of bond exchange reactions, which can be achieved by either varying the concentration of reactive sites or by modifying the network architecture.24 Network architecture dictates the spatial distribution of reactive sites within the network leading to the formation of reactive chain-ends, and network defects such as loops, dangling chain ends, and or free chains.25,26 It has been well established that a gradient or block arrangement of reactive sites, or incorporating loop defects, accelerates the stress relaxation in associative networks.24,25,27 The concentration of dynamic bonds, however, affects the rate at which the reactive sites find each other, a prerequisite for successful bond exchange, and thus their variation has a direct impact on the network's response to applied deformation.
In this study, we seek to answer the following questions: (1) how does varying the concentrations of these linkages affect the melt behavior of hybrid networks, an essential property for reprocessing? and (2) how does the concentration of dynamic linkages affect the stress response of the networks under tensile deformation? Our results demonstrate that increasing the concentration of reactive sites leads to a remarkable change in the properties of the resultant glassy hybrid networks, such as delayed development of craze fibrils, and also enhances the ability of the network to withstand higher strains before fracture. Exploring this correlation between the mechanical response of glassy vitrimers and the concentration of reactive sites holds significant promise for tailoring vitrimer networks for targeted applications.
A hybrid molecular dynamics (MD) and Monte Carlo (MC) methodology developed in our previous works is used for simulating the bond exchanges in hybrid vitrimer networks,28–31 and details of the model are given in the ESI.† To create these hybrid networks, different concentrations of bonds between the crosslinkers and polymer chains were randomly assigned as reactive as shown in Fig. 1. The ratio of the total reactive bonds to the total number of bonds between the polymer and crosslinker is termed as ξ. ξ values were varied between 0 and 1, where a ξ value of 0 represents a permanently crosslinked network, i.e., an ideal thermoset, whereas a ξ value of 1 represents an ideal vitrimer, where all the crosslink sites can freely participate in the bond exchange reactions.
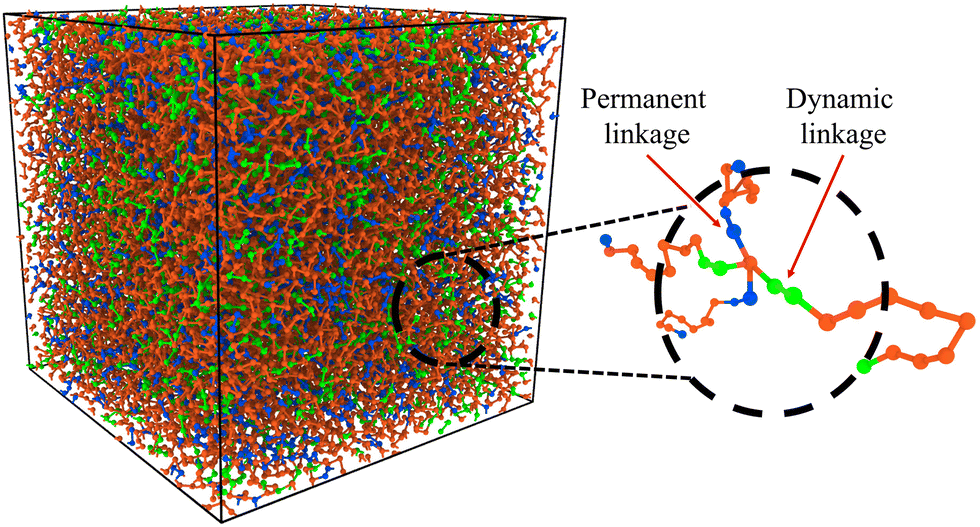 | ||
| Fig. 1 Simulation box snapshot of hybrid vitrimer network. The blue beads represent the segements with permanent crosslinks, and the green beads show the dynamic linkages. The orange beads represent the linear polymer chains rendered using OVITO.32 | ||
We begin the analysis by determining the primary and secondary transition temperatures for the hybrid networks. Here, we report the scaled volume V as a function of temperature as the hybrid networks are quenched from T = 2.0 down to T = 0.1 is shown in Fig. 2A. V increases as a function of temperature, and the hybrid networks at higher ξ values show larger increases in V in the melt state. An increase in ξ signifies enhanced vitrimer-like characteristics of the network that result in the observed increase of V. To elucidate this effect, we also report the excess volume VE, normalized with the volume of the thermoset network (VThermoset), as a function of ξ, calculated as VE = (VVitrimer − VThermoset)/VThermoset, at T = 2.0 (see the inset of Fig. 2A). Initially, the VE steadily increases with ξ and then plateaus around ξ ≅ 0.8. Determining the critical ξ beyond which the V for the hybrid networks' stabilizes is a crucial insight into the network strands' mobility and relaxation characteristics. The glass transition temperature, Tg, is determined as the point of intersection of the fitted lines in the glassy and rubbery regimes of V (Fig. 2A). Tg values of all the networks are about T ≅ 0.45 which is attributed to the absence of bond exchange reactions at lower temperatures. The restricted mobility of the network chains in the glassy regime hinders the interaction between the reactive sites and results in negligible topological rearrangements.
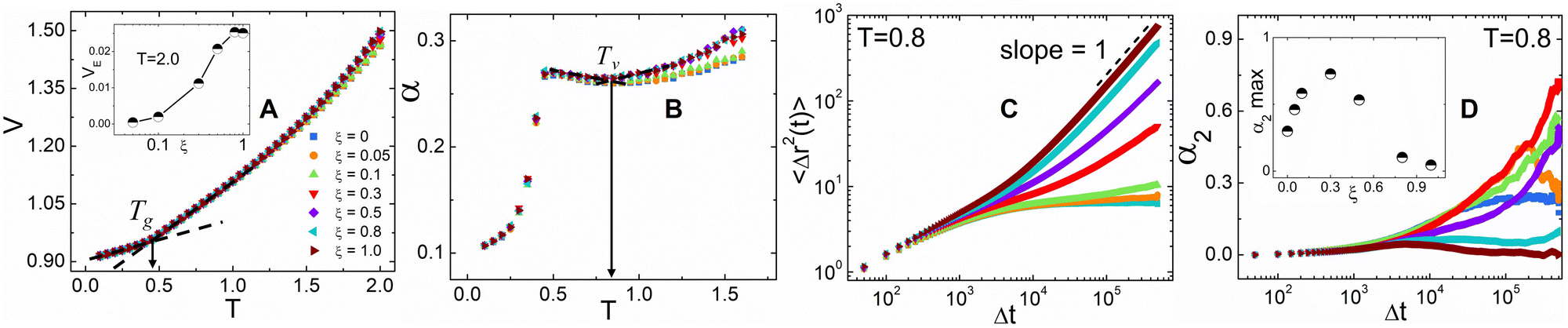 | ||
| Fig. 2 (A) Scaled volume, V, and (B) coefficient of thermal expansion, α, as a function of temperature, T, for hybrid vitrimer networks with different fractions of dynamic bonds, ξ. The inset in (A) shows the excess volume VE as a function of ξ at T = 2.0. Tv is determined as the local plateau in the α plot occurring beyond the Tg. (C) MSD of hybrid networks at different ξ, in the melting regime, T = 0.8. Here, diffusive motion is indicated by the dashed line representing a slope of unity on the log–log scale. (D) Time dependence of the non-Gaussian parameter, α2. The inset shows the maximum value for α2 as a function of ξ. | ||
Vitrimers also exhibit a secondary transition temperature, known as the topology freezing temperature, Tv, which demarcates the point at which the bond exchange reactions become ubiquitous throughout the network.1,33,34 In previous works, we have successfully established a method to determine Tv using the coefficient for thermal expansion α, given by 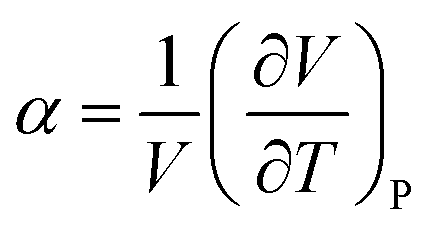 , where Tv is determined as the local plateau of the α as a function of T, as seen in Fig. 2B.30,35 Here, Tv is in the range of 0.85 ≤ Tv ≤ 0.9. The hybrid networks with lower ξ (i.e., 0.05 ≤ ξ ≤ 0.3) show a transition about Tv ≅ 0.9, whereas hybrid networks with a higher ξ have a Tv ≅ 0.85. The range of Tv, although narrow, corroborates that the secondary transition and, thus, the topology rearrangements are controlled by the rate at which the reactive sites encounter each other.27 At higher ξ, the reactive beads find each other at a faster rate, and thus, the hybrid networks show, although marginally, a reduced Tv.
, where Tv is determined as the local plateau of the α as a function of T, as seen in Fig. 2B.30,35 Here, Tv is in the range of 0.85 ≤ Tv ≤ 0.9. The hybrid networks with lower ξ (i.e., 0.05 ≤ ξ ≤ 0.3) show a transition about Tv ≅ 0.9, whereas hybrid networks with a higher ξ have a Tv ≅ 0.85. The range of Tv, although narrow, corroborates that the secondary transition and, thus, the topology rearrangements are controlled by the rate at which the reactive sites encounter each other.27 At higher ξ, the reactive beads find each other at a faster rate, and thus, the hybrid networks show, although marginally, a reduced Tv.
Using simulation trajectories of the central crosslinker beads in the melt state, i.e., at a temperature of T = 0.8, we compute the mean squared displacement (MSD) of the hybrid networks.31 The MSD is determined as 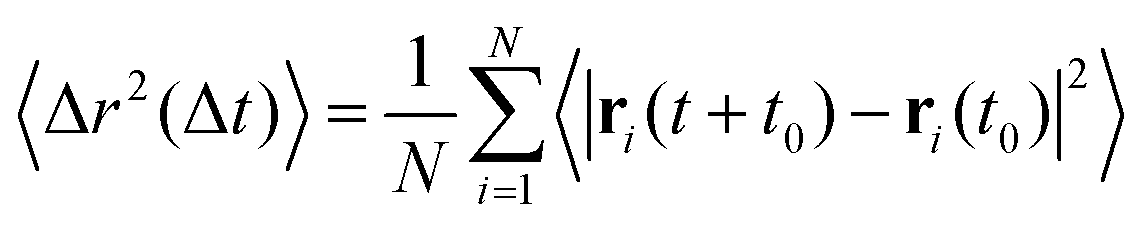 , where ri represents the position vector of the central crosslinker beads at a time t, and N is the total number of central crosslinker beads in each network. At short times (Fig. 2C), all the hybrid networks show similar mobility, which can be attributed to the thermal motion of the constituent network beads since the analysis is performed in the melt regime. Eventually, the thermoset (i.e., ξ = 0) reaches a rubbery plateau, represented by the leveling off of the MSD at longer times. The rigid network bonds restrict segmental motion and cannot undergo network topology alteration due to the absence of bond exchange. Although reactive beads are present in the networks with ξ = 0.5 and 0.1, their low concentration hinders any real segmental relaxation from occurring, and thus, their MSDs are marginally higher than that of the thermoset. The rapid increase in the frequency of bond exchange reactions due to ample dynamic links is described by the increase in the MSD for ξ ≥ 0.5, indicative of the liquid-like flow of the vitrimer networks. We infer that a hybrid network requires upwards of 50 mol% of reactive sites to retain its characteristic flow behavior required for reprocessing, positively correlating to existing studies on partial vitrimer networks.17
, where ri represents the position vector of the central crosslinker beads at a time t, and N is the total number of central crosslinker beads in each network. At short times (Fig. 2C), all the hybrid networks show similar mobility, which can be attributed to the thermal motion of the constituent network beads since the analysis is performed in the melt regime. Eventually, the thermoset (i.e., ξ = 0) reaches a rubbery plateau, represented by the leveling off of the MSD at longer times. The rigid network bonds restrict segmental motion and cannot undergo network topology alteration due to the absence of bond exchange. Although reactive beads are present in the networks with ξ = 0.5 and 0.1, their low concentration hinders any real segmental relaxation from occurring, and thus, their MSDs are marginally higher than that of the thermoset. The rapid increase in the frequency of bond exchange reactions due to ample dynamic links is described by the increase in the MSD for ξ ≥ 0.5, indicative of the liquid-like flow of the vitrimer networks. We infer that a hybrid network requires upwards of 50 mol% of reactive sites to retain its characteristic flow behavior required for reprocessing, positively correlating to existing studies on partial vitrimer networks.17
To quantify the heterogeneity in the dynamics of the hybrid networks and their deviation from the Gaussian behavior, we calculated the non-Gaussian parameter, α2, given by 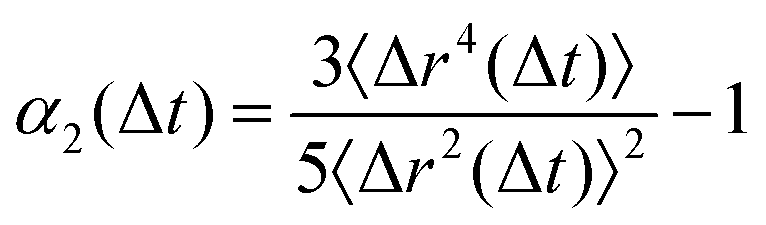 , where α2 = 0 highlights Fickian diffusion while higher values indicate a non-Gaussian behavior of the distribution of displacement.36 We observe that an increase in the concentration of dynamic bonds increases the α2 up to a limiting value of ξ = 0.3 which shows that hybrid networks with ξ ≤ 0.3 exhibit heterogeneous dynamics even within the melting regime. Similar to the MSD, an increase in the dynamic bonds concentration allows the network to show higher segmental relaxation resulting in a drastic drop in α2 and a transition towards Gaussian dynamics.37,38
, where α2 = 0 highlights Fickian diffusion while higher values indicate a non-Gaussian behavior of the distribution of displacement.36 We observe that an increase in the concentration of dynamic bonds increases the α2 up to a limiting value of ξ = 0.3 which shows that hybrid networks with ξ ≤ 0.3 exhibit heterogeneous dynamics even within the melting regime. Similar to the MSD, an increase in the dynamic bonds concentration allows the network to show higher segmental relaxation resulting in a drastic drop in α2 and a transition towards Gaussian dynamics.37,38
We simulate the tensile deformation by triaxial stretching the simulation box primarily in the x direction at a constant dimensionless strain rate of ![[small epsi, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a1.gif) = 10−3 while maintaining constant dimensions along the y and z directions. We investigate the effect of the applied stress on the mechanical and bond exchange characteristics of the hybrid networks by deforming them in the glassy regime. We report the tensile stress in the primary stretching direction σxx as a function of the strain ε, as seen in Fig. 3A. The representative stress–strain curves response for all ξ values demonstrate an initial linear elastic regime (Fig. 3A). The comparable magnitudes for the yield stress for all the hybrid networks indicates that the network strands were completely stretched and any further straining would induce plastic deformation in the network.39 Another notable inference from the comparable yield stress magnitudes was that the vitrimer networks have not yet experienced significant topological rearrangements which are corroborated by the cumulative bond exchanges (Fig. S1A of the ESI†).
= 10−3 while maintaining constant dimensions along the y and z directions. We investigate the effect of the applied stress on the mechanical and bond exchange characteristics of the hybrid networks by deforming them in the glassy regime. We report the tensile stress in the primary stretching direction σxx as a function of the strain ε, as seen in Fig. 3A. The representative stress–strain curves response for all ξ values demonstrate an initial linear elastic regime (Fig. 3A). The comparable magnitudes for the yield stress for all the hybrid networks indicates that the network strands were completely stretched and any further straining would induce plastic deformation in the network.39 Another notable inference from the comparable yield stress magnitudes was that the vitrimer networks have not yet experienced significant topological rearrangements which are corroborated by the cumulative bond exchanges (Fig. S1A of the ESI†).
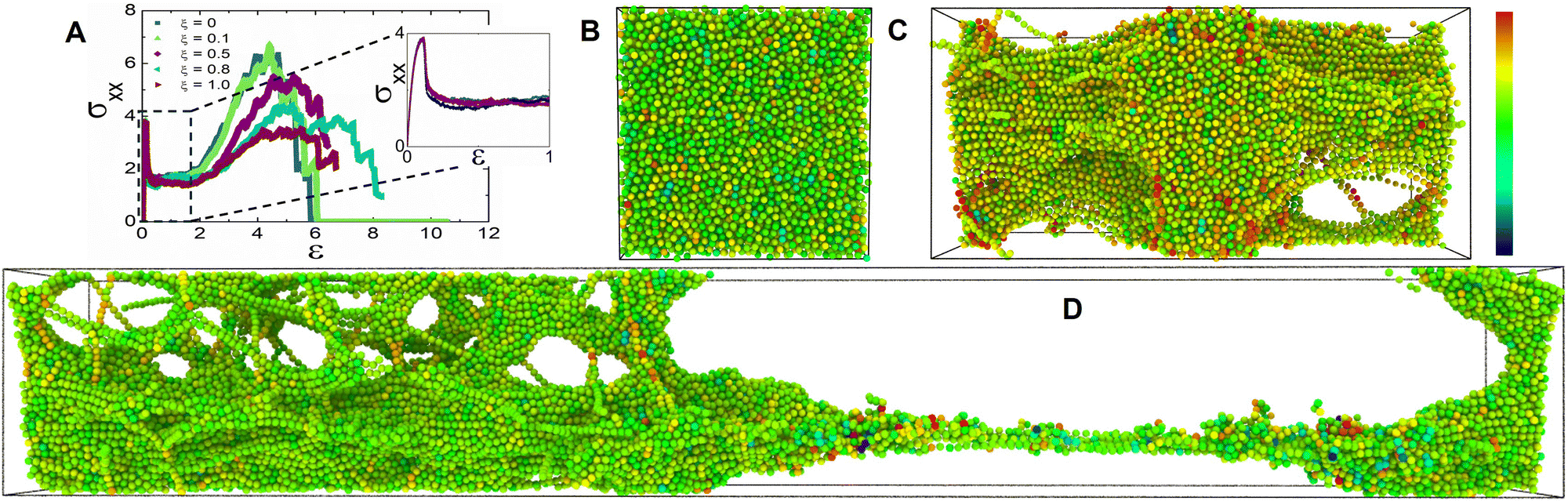 | ||
| Fig. 3 (A) σxx as a function of strain, ε. Inset shows σxx at lower strain. (B)–(D) Simulation box snapshots of the triaxial stretching experiment for a network with ξ = 0.5. The color of the beads is representative of the stress per particle, represented by a color scale bar on the right. Here, the blue color represents low stress per particle and the red represents higher stress per particle. (B) Represents the simulation box before the triaxial stretching experiment at ε ≅ 0. (C) Represents the nucleation of crazes in the network through cavitation ε ≅ 1. (D) Represents the eventual fracture in the network at ε ≅ 7. The chains under stress due to elongation are consecutively in red due to high per-particle stress. Due to the stress relaxed by bond exchange, blue beads representing low per-particle stress are observed in hybrid vitrimer networks. | ||
As the networks are strained further, the tensile stress decreases, marking the nucleation of crazes by virtue of cavitation.40–42 These crazes populate the bulk of the network and develop into craze fibrils on further elongation which is characterized by a plateau of σxx in the stress–strain curve (Fig. 3A). The termination of this plateau indicated that the crazes in the network were fully developed which is interestingly postponed for hybrid networks with ξ ≥ 0.5.21 The hybrid network's ability to relax the stress by topological rearrangements prevents crazes from breaking down to form cracks and greatly reduces the number of load-bearing strands, analogous to networks with loop defects.25,39,43,44
As we further stretch these networks, σxx increases indicating strain-hardening behavior. We infer from the nature of this stress–strain curve that glassy hybrid networks exhibit characteristics synonymous with glassy polymers above the critical entanglement density that dissipates energy through molecular disentanglement and molecular scission.43 Beyond this threshold, σxx drops to zero indicating the rupture of the inter and intra-chain network bonds (Fig. S1B, and C of the ESI†). The deformation energy added to the networks during the stretching experiment enables the bond exchange reactions. We observed that the networks with a higher ξ exhibit a diminished secondary peak σxx because these networks with 0.5 ≤ ξ ≤ 1 efficiently relax imposed stress, while also reaching higher strains before eventual fracture demonstrating increased stretchability typically observed in vitrimers whereas the fracture toughness in hybrid networks with 0 ≤ ξ ≤ 0.1 relies heavily on the imposed displacement rate.45,46 The dynamic nature of the crosslink sites provides a toughening mechanism to the vitrimer networks which can be compared to mechanochemically weak crosslinkers.47 We supported these interpretations by examining the cumulative bond exchanges and the bonds broken for 4 ≤ ε ≤ 6 (Fig. S1B, and C of the ESI†).
We perform the shear creep tests following the test procedures mentioned in our previous publication.31 The hybrid networks were subjected to low (σxy = 0.1), and high (σxy = 1.0) shear stresses to determine the shear creep compliance, J(t) = ε(t)/σxy, presented as a function of time (Fig. S2 of the ESI†). Under low stress, the deformation experienced by all hybrid networks is low as this stress is not significant enough to allow stress-induced bond exchanges (Fig. S3A of the ESI†). The phenomenon of stress-induced bond exchange and the additional model for network relaxation were seen clearly at higher stress. Thermosets are known to attain equilibrium creep compliance due to the constituent permanent linkages. Interestingly, increasing the ξ up to 0.5 induced a significant amount of bond exchanges (Fig. S3B of the ESI†) in the network, yet the creep compliance achieved a limiting value. Further, an increase in ξ increases the creep compliance indicative of steady deformation in the network. This inference, combined with the MSD and tensile deformation simulations, concludes that a ξ of 0.5 is ideal for enabling the hybrid network to retain its rubbery characteristics while exhibiting improved mechanical properties like enhanced stretchability, delayed fracture, and also suppressing shear-induced creep.15,17
We finish by noting that using hybrid MD-MC simulations,28–31,48 we studied the effect of varying concentrations of reactive sites on the dynamic and mechanical properties of hybrid vitrimer networks. The ability of vitrimer networks to effectively relax stress by bond exchange reactions has already been well understood and this property paves the way for interesting mechanical behavior. Understanding this relationship is crucial for designing hybrid vitrimer networks that can display desirable mechanical and creep-resistant properties, and still be reprocessable. Under triaxial stress, glassy hybrid vitrimer networks exhibit strain-hardening behavior similar to highly entangled polymer glasses. A high concentration of reactive sites effectively allows the hybrid networks to relax stress, delay craze development, and enhance elongation at fracture.
HP: conceptualization (supporting); formal analysis (lead); writing – original draft (lead); writing – review and editing (lead). FK: conceptualization (lead); investigation (lead); project administration (lead); writing – review and editing (lead).
This work was supported by the US National Science Foundation under Grant Number 2152210. The authors also acknowledge the Texas Advanced Computing Center (TACC) at the University of Texas at Austin for providing computational resources that contributed to the research results reported in this paper.
Data availability
Data are available upon reasonable request from the authors.Conflicts of interest
There are no conflicts to declare.References
- D. Montarnal, et al., Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Röttger, et al., Science, 2017, 356, 62–65 CrossRef PubMed.
- M. Capelot, et al., ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- M. K. McBride, et al., Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS PubMed.
- M. Chen, et al., ACS Macro Lett., 2019, 8, 255–260 CrossRef CAS PubMed.
- J. Dahlke, et al., Adv. Mater. Interfaces, 2018, 5, 1800051 CrossRef.
- Q. Chen, et al., Chem. Sci., 2017, 8, 724–733 RSC.
- J. Zheng, et al., Mater. Today, 2021, 51, 586–625 CrossRef CAS.
- J. Huang, et al., Macromolecules, 2023, 56, 1253–1262 CrossRef CAS.
- Z. Lyu and T. Wu, Macromol. Rapid Commun., 2020, 41, 2000265 CrossRef CAS PubMed.
- S. Wang, et al., J. Am. Chem. Soc., 2024, 146, 16112–16118 CrossRef CAS PubMed.
- S. Wang, et al., J. Am. Chem. Soc., 2024, 146, 9920–9927 CrossRef CAS PubMed.
- Y. Wang, et al., Macromolecules, 2022, 55, 7845–7855 CrossRef CAS.
- E. Du, et al., Macromolecules, 2024, 57, 672–681 CrossRef CAS.
- A. Breuillac, et al., Macromolecules, 2019, 52, 7102–7113 CrossRef CAS.
- C. Taplan, et al., Mater. Horiz., 2020, 7, 104–110 RSC.
- L. Li, et al., Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- F. Van Lijsebetten, et al., Angew. Chem., Int. Ed., 2022, 61, e202113872 CrossRef CAS PubMed.
- Z. Song, et al., Mech. Mater., 2021, 153, 103687 CrossRef.
- F. Meng, et al., Macromolecules, 2016, 49, 2843–2852 CrossRef CAS.
- F. Meng and E. M. Terentjev, Polymers, 2016, 8, 108 CrossRef PubMed.
- S. Ciarella and W. G. Ellenbroek, Coatings, 2019, 9, 114 CrossRef.
- G. Singh and V. Sundararaghavan, Chem. Phys. Lett., 2020, 760, 137966 CrossRef CAS.
- R. G. Ricarte and S. Shanbhag, Macromolecules, 2021, 54, 3304–3320 CrossRef CAS.
- S. Ciarella, et al., Phys. Rev. Lett., 2018, 121, 058003 CrossRef CAS PubMed.
- A. Jourdain, et al., Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- F. Van Lijsebetten, et al., Chem. Sci., 2022, 13, 12865–12875 RSC.
- A. Perego and F. Khabaz, Macromolecules, 2020, 53, 8406–8416 CrossRef CAS.
- A. Perego, et al., Macromolecules, 2022, 55, 7605–7613 CrossRef CAS.
- A. Perego and F. Khabaz, Macromol. Rapid Commun., 2023, 44, 2200313 CrossRef CAS PubMed.
- H. Pandya, et al., J. Appl. Polym. Sci., 2024, 141, e55039 CrossRef CAS.
- A. Stukowski, JOM, 2014, 66, 399–407 CrossRef CAS.
- Y. Yang, et al., Nat. Commun., 2019, 10, 3165 CrossRef PubMed.
- A. Arbe, et al., ACS Macro Lett., 2023, 12, 1595–1601 CrossRef CAS PubMed.
- N. Sadeghi, et al., Macromolecules, 2024, 57, 3937–3948 CrossRef CAS.
- J. P. Boon, et al. Molecular Hydrodynamics, Courier Corporation, 1991 Search PubMed.
- H. Zhao, et al., Macromolecules, 2023, 56, 9336–9349 CrossRef CAS.
- J. Xia, et al., Macromolecules, 2023, 56, 8080–8093 CrossRef CAS.
- A. Arora, et al., Macromolecules, 2020, 53, 7346–7355 CrossRef CAS.
- J. Wang, et al., Macromolecules, 2022, 55, 1267–1278 CrossRef CAS.
- J. Rottler and M. O. Robbins, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 68, 011801 CrossRef PubMed.
- T. Ge, et al., Macromolecules, 2017, 50, 459–471 CrossRef CAS.
- C. Bukowski, et al., Sci. Adv., 2021, 7, eabg9763 CrossRef CAS PubMed.
- M. Zhong, et al., Science, 2016, 353, 1264–1268 CrossRef CAS PubMed.
- C.-Y. Hui and R. Long, Soft Matter, 2012, 8, 8209–8216 RSC.
- H. Zhang, et al. , Adv. Mater., 2019, 31, 1904029 CrossRef CAS PubMed.
- S. Wang, et al., Science, 2023, 380, 1248–1252 CrossRef CAS PubMed.
- A. Perego and F. Khabaz, J. Polym. Sci., 2021, 59, 2590–2602 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Simulation details, bond characteristics during triaxial stretching experiment, shear creep experiment, bond characteristics during shear creep experiment. See DOI: https://doi.org/10.1039/d4cc03030c |
| This journal is © The Royal Society of Chemistry 2024 |